Native Cellulose: Structure, Characterization and Thermal Properties

Abstract
:1. Introduction
2. Experimental Section
2.1. Materials
2.2. Fourier Transform Infrared (FTIR) Spectroscopy
2.3. X-ray Diffraction



2.4. Thermogravimetric Analysis
3. Results and Discussion
3.1. Fibers Chemical Composition

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fibers | Cellulose (wt%) | Hemicellulose (wt%) | Lignin (wt%) | Pectin (wt%) | Waxes (wt%) | Extractives (wt%) |
|---|---|---|---|---|---|---|
| Holocellulose (wt%) | ||||||
| Eucalyptus grandis | 61.3–64 | 31–33 | – | – | 3.9–4.3 | |
| Pinus elliottii | 60–62.3 | 32.8–35 | – | – | 4.4–4.6 | |
| Dipteryx odorata | 56.5–57.7 | 30–31 | – | – | 11–11.2 | |
| Mezilaurus itauba | 56.8–58.8 | 27.7–28 | – | – | 13–14.3 | |
| Curaua | 71–74 | 9.9–21 | 7.5–11 | – | 0.79–0.9 | 2.5–2.8 |
| Jute | 45–71 | 13.6–21 | 12–26 | 0.2–10 | 0.5 | 2 |
| Kenaf | 31–72 | 20.3–23 | 9–19 | 3–5 | – | 2–5 |
| Ramie | 68.6–91 | 5–16.7 | 0.6–0.7 | 1.9–2 | 0.3 | 6 |
| Sisal | 65–67 | 12 | 9.9 | 2–10 | 0.3–2 | 0.8–2 |
| Buriti | 65–71 | 21–27 | – | – | 5.4–6.0 | |
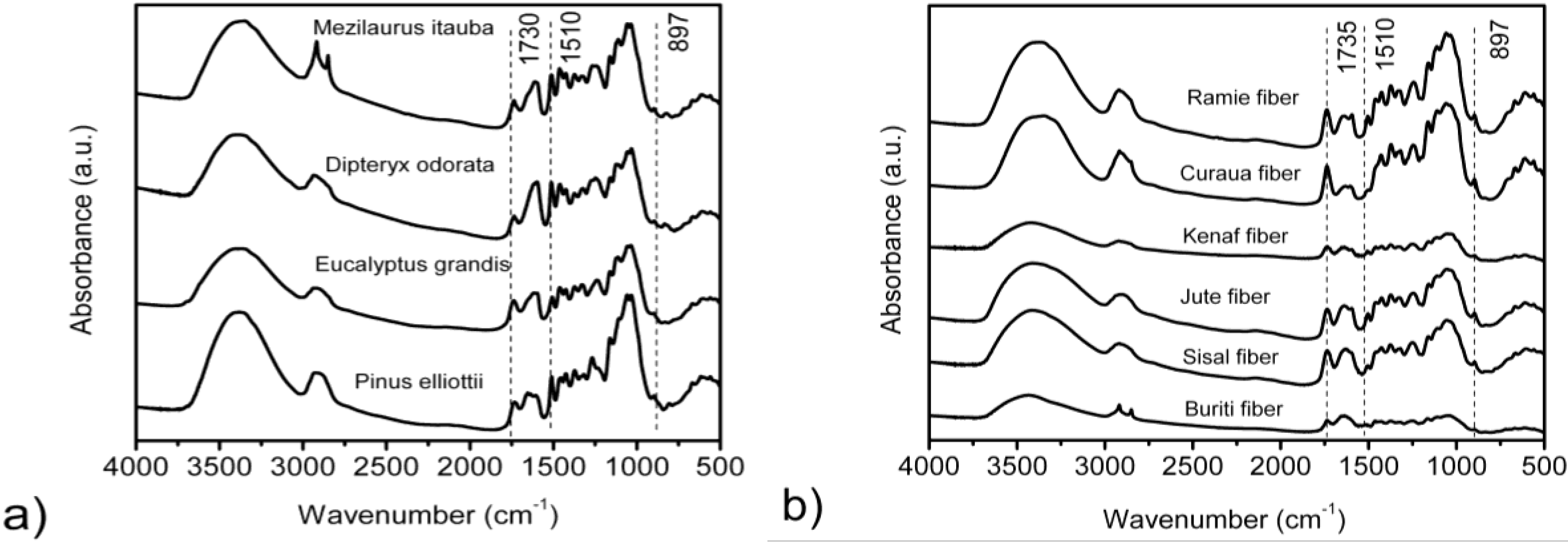
3.2. FTIR Spectroscopy
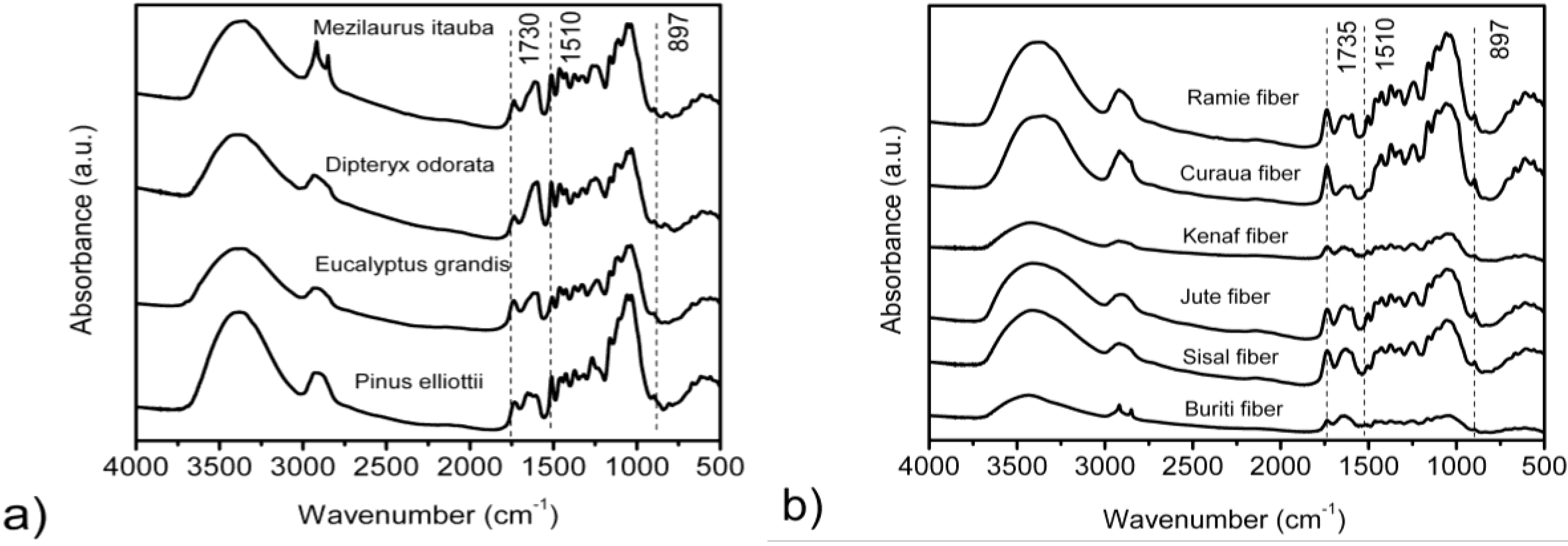

| Fibers | 3567 cm−1 | 3423 cm−1 | 3342 cm−1 | 3278 cm−1 | 3221 cm−1 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| EH (kJ) | R (Å) | EH (kJ) | R (Å) | EH (kJ) | R (Å) | EH (kJ) | R (Å) | EH (kJ) | R (Å) | |
| Eucalyptus grandis | 6.185 | 2.832 | 16.182 | 2.800 | 22.438 | 2.782 | 26.574 | 2.768 | 30.349 | 2.756 |
| Pinus elliottii | 6.329 | 2.831 | 16.757 | 2.799 | 22.007 | 2.782 | 26.394 | 2.768 | 30.314 | 2.757 |
| Dipteryx odorata | 6.401 | 2.831 | 16.613 | 2.799 | 22.438 | 2.781 | 26.610 | 2.768 | 30.874 | 2.754 |
| Mezilaurus itauba | 6.473 | 2.831 | 16.325 | 2.800 | 22.295 | 2.781 | 26.753 | 2.767 | 30.493 | 2.756 |
| Curaua | 5.969 | 2.833 | 16.038 | 2.801 | 21.935 | 2.782 | 26.746 | 2.767 | 30.997 | 2.754 |
| Jute | 5.980 | 2.833 | 16.038 | 2.801 | 21.827 | 2.783 | 26.538 | 2.768 | 30.666 | 2.755 |
| Kenaf | 5.667 | 2.833 | 16.253 | 2.800 | 22.043 | 2.782 | 27.041 | 2.766 | 30.781 | 2.755 |
| Ramie | 6.048 | 2.832 | 16.325 | 2.800 | 22.100 | 2.782 | 26.782 | 2.767 | 31.140 | 2.754 |
| Sisal | 6.156 | 2.832 | 16.253 | 2.800 | 21.863 | 2.783 | 26.538 | 2.768 | 31.148 | 2.754 |
| Buriti | 6.185 | 2.832 | 16.253 | 2.800 | 22.366 | 2.781 | 27.041 | 2.766 | 31.184 | 2.753 |
| Fibers | IR crystallinity ratio | HBI | |
|---|---|---|---|
| H1372/H2900 (TCI) | H1429/H897 (LOI) | A3400/A1320 | |
| Eucalyptus grandis | 0.608 ± 0.01 | 3.172 ± 0.02 | 1.440 |
| Pinus elliottii | 0.474 ± 0.01 | 2.299 ± 0.04 | 1.598 |
| Dipteryx odorata | 0.389 ± 0.02 | 3.137 ± 0.03 | 1.508 |
| Mezilaurus itauba | 0.237 ± 0.03 | 2.060 ± 0.01 | 1.523 |
| Curaua | 1.300 ± 0.01 | 1.070 ± 0.01 | 1.132 |
| Jute | 1.150 ± 0.03 | 0.990 ± 0.01 | 1.207 |
| Kenaf | 1.190 ± 0.01 | 0.930 ± 0.02 | 1.119 |
| Ramie | 1.240 ± 0.01 | 1.050 ± 0.01 | 1.426 |
| Sisal | 1.150 ± 0.02 | 0.970 ± 0.03 | 1.625 |
| Buriti | 1.150 ± 0.01 | 0.780 ± 0.05 | 2.241 |
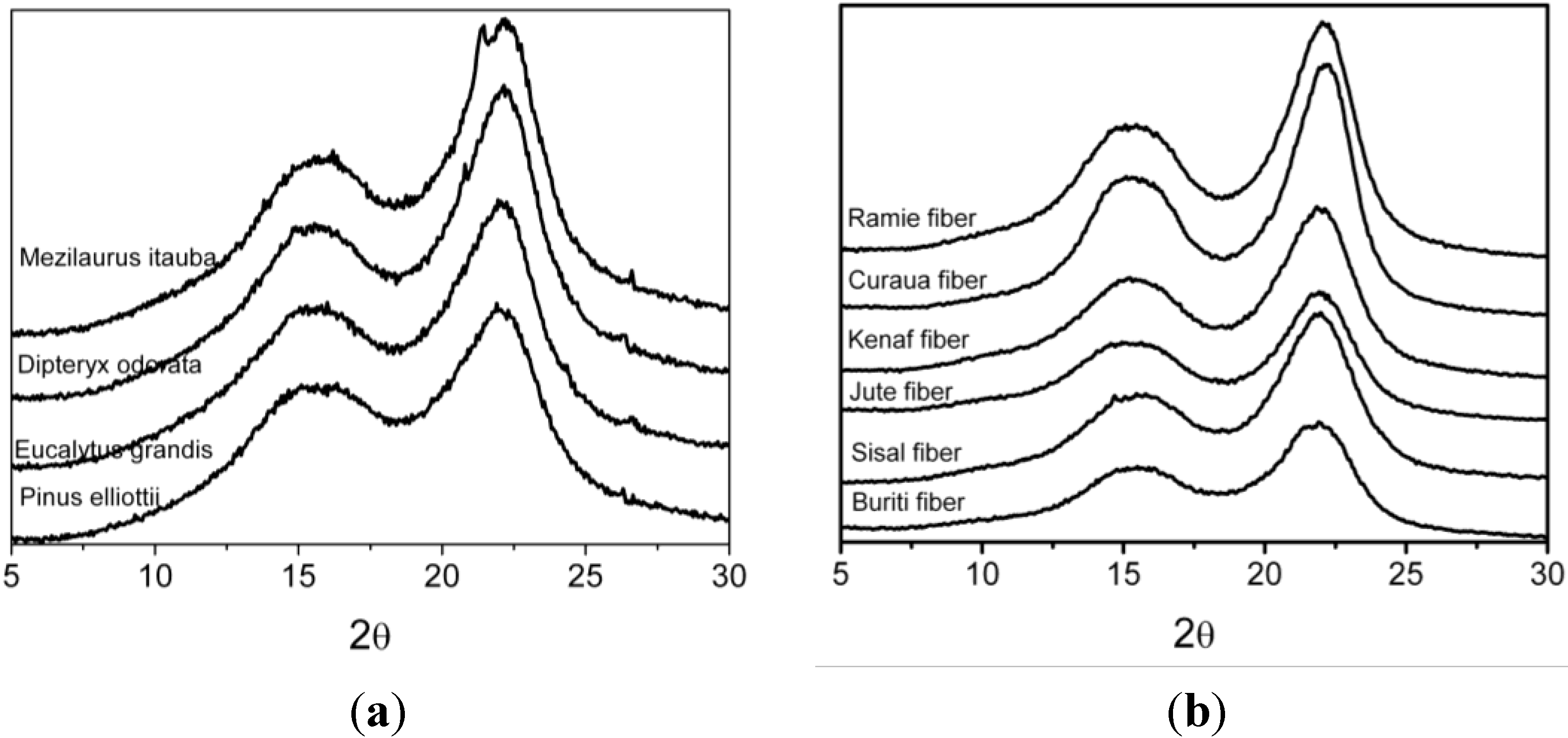
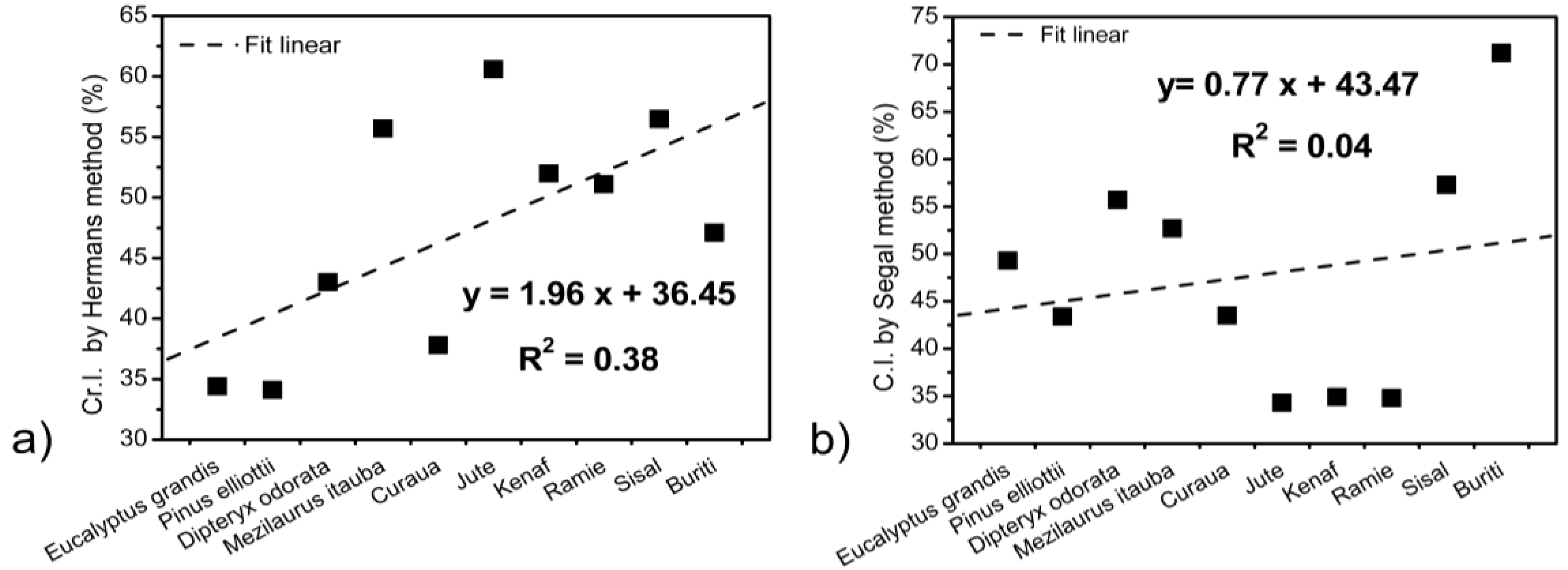
3.3. XRD Results

| Fibers | L (200) (nm) | Cr.I. a (%) | C.I. b (%) | Z-values |
|---|---|---|---|---|
| Eucalyptus grandis | 2.11 | 34.4 | 49.3 | −25.7 |
| Pinus elliottii | 1.92 | 34.1 | 43.4 | −32.9 |
| Dipteryx odorata | 2.18 | 43.0 | 55.7 | −31.6 |
| Mezilaurus itauba | 2.23 | 37.8 | 52.7 | −46.3 |
| Curaua | 3.43 | 60.6 | 43.5 | −40.8 |
| Jute | 2.94 | 52.0 | 34.3 | −18.0 |
| Kenaf | 2.71 | 51.1 | 34.9 | −21.4 |
| Ramie | 3.31 | 56.5 | 34.8 | −9.4 |
| Sisal | 3.37 | 47.1 | 57.3 | −34.7 |
| Buriti | 3.70 | 45.1 | 71.2 | −34.4 |

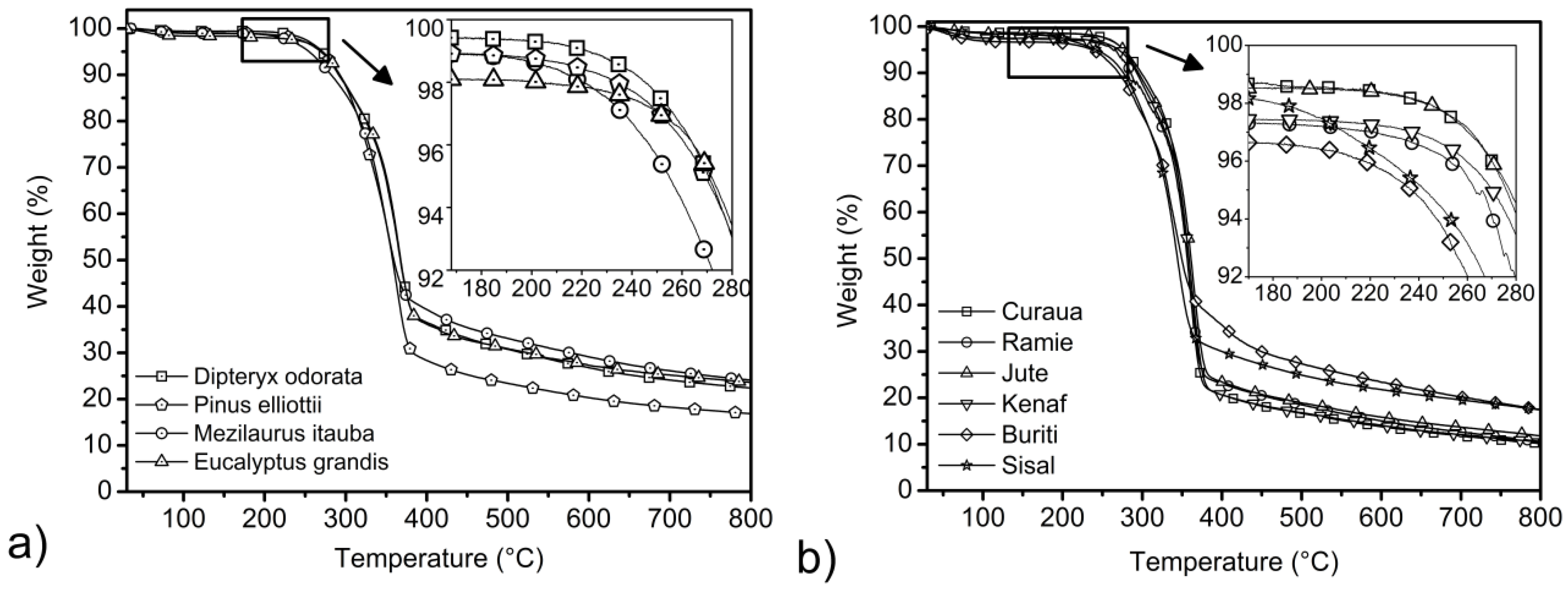
3.4. Thermogravimetric Results
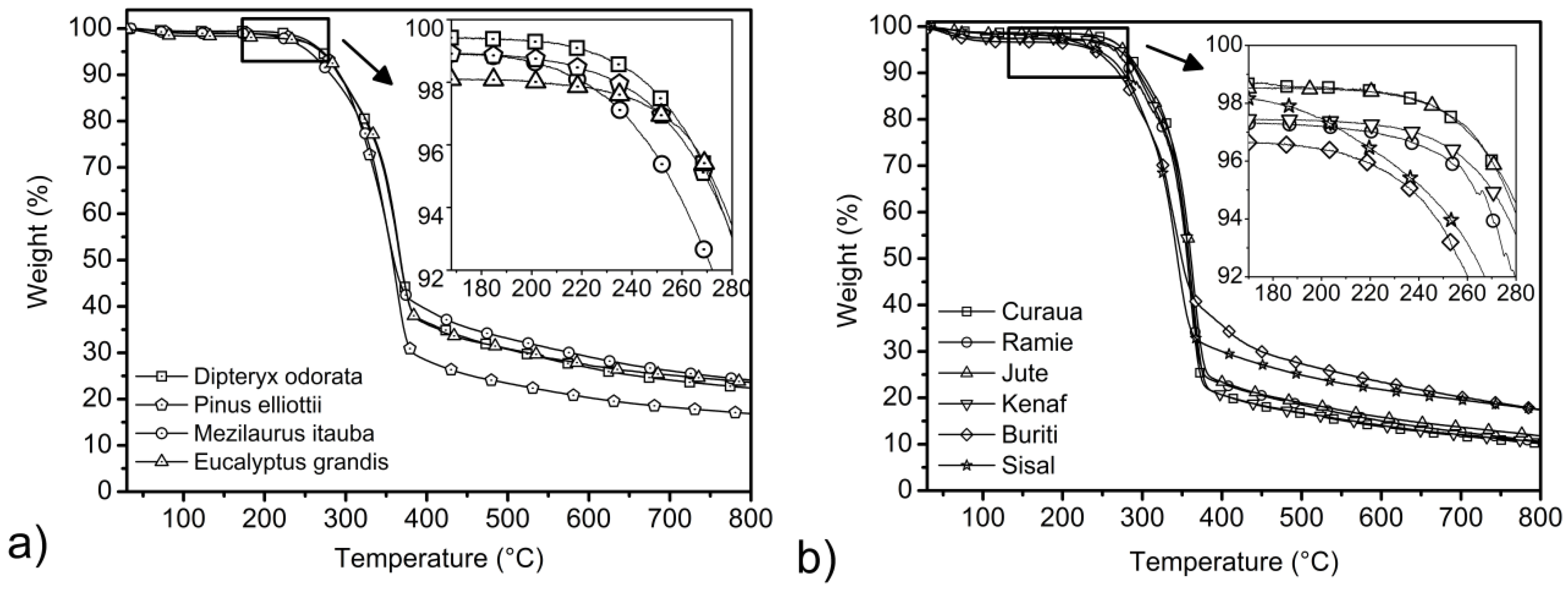
| Fibers | Ti (°C) 3 wt% loss | T shoulder (°C) | DTG peak (°C) | Residue at 800 °C(%) |
|---|---|---|---|---|
| Eucalyptus grandis | 250 | 291 | 364 | 23.6 |
| Pinus elliottii | 251 | 322 | 367 | 16.8 |
| Dipteryx odorata | 257 | 289 | 368 | 22.4 |
| Mezilaurus itauba | 237 | 275 | 350 | 24.1 |
| Curaua | 260 | 283 | 344 | 10.1 |
| Jute | 262 | 297 | 365 | 11.8 |
| Kenaf | 238 | 298 | 364 | 10.5 |
| Ramie | 221 | 289 | 357 | 10.6 |
| Sisal | 209 | 295 | 347 | 17.4 |
| Buriti | 92 | 291 | 334 | 17.5 |
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Poletto, M.; Zeni, M.; Zattera, A.J. Effects of wood flour addition and coupling agent content on mechanical properties of recycled polystyrene/wood flour composites. J. Thermoplast. Compos. Mater. 2012, 25, 821–833. [Google Scholar] [CrossRef]
- Ornaghi, H.L., Jr.; Bolner, A.S.; Fiorio, R.; Zattera, A.J.; Amico, S.C. Mechanical and dynamic mechanical analysis of hybrid composites molded by resin transfer molding. J. Appl. Polym. Sci. 2010, 118, 887–896. [Google Scholar]
- Romanzini, D.; Ornaghi, H.L., Jr.; Amico, S.C.; Zattera, A.J. Influence of fiber hybridization on the dynamic mechanical properties of glass/ramie fiber-reinforced polyester composites. J. Reinf. Plast. Compos. 2012, 31, 1652–1661. [Google Scholar] [CrossRef]
- Han, G.; Huan, S.; Han, J.; Zhang, Z.; Wu, Q. Effect of acid hydrolysis conditions on the properties of cellulose nanoparticle-reinforced polymethylmethacrylate compostes. Materials 2014, 7, 16–29. [Google Scholar]
- Klemm, D.; Heublein, B.; Fink, H.-P.; Bohn, A. Cellulose: Fascinating biopolymer and sustainable raw material. Angew. Chem. Int. Ed. 2005, 44, 3358–3393. [Google Scholar] [CrossRef]
- kerholm, M.; Hinterstoisser, B.; Salmén, L. Characterization of the crystalline structure of cellulose using static and dynamic FT-IR spectroscopy. Carbohydr. Res. 2004, 339, 569–578. [Google Scholar] [CrossRef]
- Oh, S.Y.; Yoo, D.I.; Shin, Y.; Kim, H.C.; Kim, H.Y.; Chung, Y.S.; Park, W.H.; Youk, J.H. Crystalline structure analysis of cellulose treated with sodium hydroxide and carbon dioxide by means of X-ray diffraction and FTIR spectroscopy. Carbohydr. Res. 2005, 340, 2376–2391. [Google Scholar] [CrossRef]
- Wada, M.; Okano, T. Localization of Iα and Iβ phases in algal cellulose revealed by acid treatments. Cellulose 2001, 8, 183–188. [Google Scholar] [CrossRef]
- Popescu, M.-C.; Popescu, C.-M.; Lisa, G.; Sakata, Y. Evaluation of morphological and chemical aspects of different wood species by spectroscopy and thermal methods. J. Mol. Struct. 2011, 988, 65–72. [Google Scholar] [CrossRef]
- Gümüskaya, E.; Usta, M.; Kirei, H. The effects of various pulping conditions on crystalline structure of cellulose in cotton linters. Polym. Degrad. Stab. 2003, 81, 559–564. [Google Scholar] [CrossRef]
- Faruk, O.; Bledzki, A.K.; Fink, H.; Sain, M. Biocomposites reinforced with natural fibers: 2000–2010. Prog. Polym. Sci. 2012, 37, 1552–1596. [Google Scholar] [CrossRef]
- Bledzki, A.K.; Gassan, J. Composites reinforced with cellulose based fibres. Prog. Polym. Sci. 1999, 24, 221–274. [Google Scholar] [CrossRef]
- Cordeiro, N.C.; Gouveia, A.G.O.; Moraes, A.M.; Amico, S.C. Natural fibers characterization by inverse gas chromatography. Carbohyd. Polym. 2011, 84, 110–117. [Google Scholar] [CrossRef]
- Satyanarayana, K.G.; Guimarães, J.L.; Wypych, F. Studies on lignocellulosic fibers of Brazil. Part I: Source, production, morphology, properties and applications. Compos. Part A 2007, 38, 1694–1709. [Google Scholar] [CrossRef]
- Ornaghi, H.L., Jr.; Poletto, M.P.; Zattera, A.J.; Amico, S.C. Correlation of the thermal stability and the decomposition kinetics of six different vegetal fibers. Cellulose 2014, 21, 177–188. [Google Scholar] [CrossRef]
- Poletto, M.P.; Zattera, A.J.; Santana, R.M.C. Structural differences between wood species: Evidence from chemical composition, FTIR spectroscopy, and thermogravimetric analysis. J. Appl. Polym. Sci. 2012, 126, E336–E343. [Google Scholar]
- Popescu, C.-M.; Singurel, G.; Popescu, M.-C.; Vasile, C.; Argyropoulos, D.S.; Willför, S. Vibrational spectroscopoy and X-ray diffraction methods to establish the differences between hardwood and softwood. Carbohydr. Polym. 2009, 77, 851–857. [Google Scholar] [CrossRef]
- Yokoi, H.; Nakase, T.; Goto, K.; Ishida, Y.; Ohtani, H.; Tsuge, S.; Sonoda, T.; Ona, T. Rapid characterization of wood extractives in wood by thermal desorption-gas chromatography in the presence of tetramethylammonium acetate. J. Anal. Appl. Pyrolysis 2003, 67, 191–200. [Google Scholar] [CrossRef]
- Ishida, Y.; Goto, K.; Yokoi, H.; Tsuge, S.; Ohtani, H.; Sonoda, T.; Ona, T. Direct analysis of phenolic extractives in wood by thermochemolysis-gas chromatography in the presence of tetrabutylammonium hydroxide. J. Anal. Appl. Pyrolysis 2007, 78, 200–206. [Google Scholar] [CrossRef]
- Mészáros, E.; Jakab, E.; Várhegyi, G. TG/MS, Py-GC/MS and THM-GC/MS study of the composition and thermal behavior of extractive components of Robinia pseudoacacia. J. Anal. Appl. Pyrolysis 2007, 79, 61–70. [Google Scholar] [CrossRef]
- Popescu, C.-M.; Popescu, M.-C.; Vasile, C. Structural changes in biodegraded lime wood. Carbohydr. Polym. 2010, 79, 362–372. [Google Scholar] [CrossRef]
- Kondo, T. The assignment of IR absorption bands due to free hydroxyl groups in cellulose. Cellulose 1997, 4, 281–292. [Google Scholar] [CrossRef]
- Struszczyk, H. Modification of lignins III. Reaction of lignosulfonates with chlorophosphazenes. J. Macromol. Sci. 1986, 23, 973–992. [Google Scholar] [CrossRef]
- Pimentel, G.C.; Sederholm, C.H. Correlation of infrared stretching frequencies and hydrogen bon distances in crystals. J. Chem. Phys. 1956, 24, 639. [Google Scholar] [CrossRef]
- Popescu, C.-M.; Popescu, M.-C.; Singurel, G.; Vasile, C.; Argyropoulos, D.S.; Willför, S. Spectral characterization of eucalyptus wood. Appl. Spectrosc. 2007, 61, 1168–1177. [Google Scholar] [CrossRef]
- Nelson, M.L.; O’Connor, R.T. Relation of certain infrared bands to cellulose crystallinity and crystal lattice type. Part I. Spectra of types I, II, III and of amorphous cellulose. J. Appl. Polym. Sci. 1964, 8, 1311–1324. [Google Scholar] [CrossRef]
- Oh, S.Y.; Yoo, D.I.; Shin, Y.; Seo, G. FTIR analysis of cellulose treated with sodium hydroxide and carbon dioxide. Carbohydr. Res. 2005, 340, 417–428. [Google Scholar]
- Carrilo, F.; Colom, X.; Suñol, J.J.; Saurina, J. Strucutral FTIR analysis and the thermal characterization of lyocell and viscose-type fibers. Eur. Polym. J. 2004, 40, 2229–2234. [Google Scholar] [CrossRef]
- Corgié, S.C.; Smith, H.M.; Walker, L.P. Enzymatic transformations of cellulose assessed by quantitative high-throughput fourier transform infrared spectroscopy (QHT-FTIR). Biotechnol. Bioeng. 2011, 108, 1509–1520. [Google Scholar] [CrossRef]
- Kim, U.-J.; Eom, S.H.; Wada, M. Thermal decomposition of native cellulose: Influence on crystallite size. Polym. Degrad. Stab. 2010, 95, 778–781. [Google Scholar] [CrossRef]
- Xu, F.; Shi, Y.-C.; Wang, D. X-ray scattering studies of lignocellulosic biomass: A review. Carbohydr. Polym. 2013, 94, 904–917. [Google Scholar] [CrossRef]
- Poletto, M.; Pistor, V.; Zeni, M.; Zattera, A.J. Crystalline properties and decomposition kinetics of cellulose fibers in wood pulp obtained by two pulping process. Polym. Degrad. Stab. 2011, 96, 679–685. [Google Scholar] [CrossRef]
- Poletto, M.; Zattera, A.J.; Forte, M.M.C.; Santana, R.M.C. Thermal decomposition of wood: Influence of wood components and cellulose crystallite size. Bioresour. Technol. 2012, 109, 148–153. [Google Scholar] [CrossRef]
- Poletto, M.; Zattera, A.J.; Santana, R.M.C. Thermal decomposition of wood: Kinetics and degradation mechanisms. Bioresour. Technol. 2012, 126, 7–12. [Google Scholar] [CrossRef]
- Tserki, V.; Matzinos, P.; Kokkou, S.; Panayiotou, C. Novel biodegradable composites based on treated lignocellulosic waste flour as filler. Part I. Surface chemical modification and characterization of waste flour. Compos. Part A 2005, 36, 965–974. [Google Scholar] [CrossRef]
- Kim, H.-S.; Kim, S.; Kim, H.-J.; Yang, H.-S. Thermal properties of bio-flour-filled polyolefin composites with different compatibilizing agent type and content. Thermochim. Acta 2006, 451, 181–188. [Google Scholar] [CrossRef]
- John, M.J.; Thomas, S. Biofibres and biocomposites. Carbohydr. Polym. 2008, 71, 343–364. [Google Scholar] [CrossRef]
- Yang, H.; Yan, R.; Chen, H.; Zheng, C.; Lee, D.H.; Liang, D.T. In-Depth investigation of biomass pyrolysis based on three major components: Hemicellulose, cellulose and lignin. Energy Fuels 2006, 20, 388–393. [Google Scholar] [CrossRef]
- Hidaka, H.; Kim, U.-J.; Wada, M. Synchrotron X-ray fiber diffraction study on the termal expansion behavior of cellulose crystals in tension wood of Japanese poplar in the low-temperature region. Holzforsghung 2010, 64, 167–171. [Google Scholar]
- Poletto, M.; Dettenborn, J.; Pistor, V.; Zeni, M.; Zattera, A.J. Materials produced from plant biomass. Part I: Evaluation of thermal stability and pyrolysis of wood. Mater. Res. 2010, 13, 375–379. [Google Scholar] [CrossRef]
- Yao, F.; Wu, Q.; Lei, Y.; Guo, W.; Xu, Y. Thermal decomposition kinetics of natural fibers: Activation energy with dynamic thermogravimetric analysis. Polym. Degrad. Stab. 2008, 93, 90–98. [Google Scholar] [CrossRef]
- Di Blasi, C. Modeling chemical and physical process of wood and biomass pyrolysis. Prog. Energy Combust. Sci. 2008, 34, 47–90. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Poletto, M.; Ornaghi, H.L., Júnior; Zattera, A.J. Native Cellulose: Structure, Characterization and Thermal Properties. Materials 2014, 7, 6105-6119. https://doi.org/10.3390/ma7096105
Poletto M, Ornaghi HL Júnior, Zattera AJ. Native Cellulose: Structure, Characterization and Thermal Properties. Materials. 2014; 7(9):6105-6119. https://doi.org/10.3390/ma7096105
Chicago/Turabian StylePoletto, Matheus, Heitor L. Ornaghi, Júnior, and Ademir J. Zattera. 2014. "Native Cellulose: Structure, Characterization and Thermal Properties" Materials 7, no. 9: 6105-6119. https://doi.org/10.3390/ma7096105