1. Introduction
There have been many publications in recent years on the subject of hybrid polymer composites [
1,
2,
3,
4,
5,
6,
7,
8,
9]. The main reason for the interest in this area is the possibility of developing materials with higher rigidity and impact resistance. Well-designed hybrid composites utilize the advantages of the individual components so as to minimize the defects arising from their separate use.
Undoubtedly, the continued development of hybrid composite materials, mainly nanocomposites, is associated with a search for better modifiers with unique functional properties, whose presence in the composite in small quantities significantly improves properties, such as fire resistance and thermal stability. In addition to well-known flame retardants, polyhedral oligomeric silsesquioxanes (POSS) are particularly noteworthy. Important features that make this an interesting research material include the presence of an inorganic silicon-oxygen core and organic functional groups located at the corners, and its small size of about 0.5 nm, where the size of the whole molecule with substituents amounts to 1–3 nm. Because of the cost of synthesis of octasilsesquioxanes, research is being carried out into their use as modifiers of conventional fillers, such as aluminosilicates or silica, which can then be used as functional hybrid nanofillers of polymers.
The main advantage of layered aluminosilicate (LAS) modified with octasilsesquioxane salts is its high thermal stability, due to the relatively high decomposition temperature of the modifier, which typically exceeds 300 °C. In recent years, many papers have been published [
10,
11] in which oligomeric silsesquioxane was used as a modifier of montmorillonites. They describe a method of preparation of polylactide nanocomposites containing montmorillonite (MMT) modified by protonated aminopropylisobutylsilsesquioxane (POSS-NH
3+). Zhao
et al. [
12] used a quaternary ammonium salt of aminopropylisooctylsilsesquioxane to modify bentonite. The authors studied the effects of the type and amount of modifier on the degree of modification of bentonite, which was then used to prepare a composite based on polyamide 12. This case provided the greatest improvement in Young’s modulus (by about 60%), and a 10% increase in yield stress, compared with the unfilled polyamide 12. In turn, McLauchlin
et al. [
13] used a pair of surfactants in the form of aminopropylsilsesquioxane (AP-POSS) for modification of the bentonite Closite
® Na
+. The resulting filler was used to produce a nanomatrix of poly(butylene terephthalate). In another study, Liu
et al. [
14] described a method of modifying montmorillonite using octaaminopropylsilsesquioxane. The modified filler was used to obtain epoxy nanocomposites, which displayed improved thermal stability. Fu
et al. [
15] used a process of polymerization in emulsion to obtain polystyrene nanocomposites, using three types of modified LAS, the modifiers being
N,
N-dimethyl octadecylamine, 4-vinylbenzyl chloride and trisilanolisobutylsilsesquioxane. The use of these modifiers led to the largest separation of aluminosilicate plates, from 1.29 nm to about 3.95 nm, and consequently the obtaining of composite polystyrene with the best functional properties. In another paper [
16] the same authors performed a similar modification of bentonite. The resultant filler was used to obtain a polystyrene nanocomposite in which the glass transition temperature increased to 103–108 °C.
Silicas, like LAS, are very interesting fillers. They are used in many technological and industrial applications [
17,
18,
19]. Due to its unique mechanical, thermal and dielectric properties, amorphous silica has become a key material used, e.g., in microelectronics [
20], for the production of glass and ceramics, in nanotechnology and in the electro-chemical industry [
20,
21]. One of the most important properties of silicas is their significant sensitivity to chemical modifications. The modification of silica by organofunctional silanes has already been extensively tested and documented. A great deal of research has also been done on the chemistry of the formation of covalent bonds between the silanol groups of silica and a reactive group derived from organosilane or from a hydrolyzed group of a proadhesive compound [
22,
23,
24,
25]. Thus far, there are only limited reports in the literature on the use of POSS to functionalize the surface of silicas. For example, Carniato
et al. [
26] examined the immobilization of cellular titanosilsesquioxanes (Ti-POSS), both on the surface of ordered mesoporous silica (SBA-15) and on a silica with a disordered structure (SiO
2-Dav). The aim of the study was to obtain active heterogeneous catalysts. The silica surface was modified with 3-isocyanatopropyltriethoxysilane (TSIPI) and a POSS compound containing reactive aminopropyl groups and ethoxy groups (Ti-NH
2POSS). Bhagiyalakshmi
et al. [
27] presented an interesting example of bifunctionalization of the silica medium. On the surface of chlorofunctionalized mesoporous silica of type SBA-15 (Cl-SAB-15), octa(3-aminophenyl)octasilsesquioxanes (OAPS) were inoculated. As a result of their study, the authors concluded that this inoculation extends the range of possible applications of polyhedral oligomeric silsesquioxanes, and classified them as novel compounds used for the adsorption of carbon dioxide [
27]. SiO
2/POSS systems were also produced by Bianchini
et al. [
28]. Research was carried out to demonstrate the catalytic properties of metallocene silica modified by closed POSS structures in the process of ethylene polymerization. It was observed that the use of POSS-modified silica led to an increase in catalytic activity by about 50% compared with the unmodified medium. In addition, Szwarc-Rzepka
et al. [
29,
30] have proposed not only the permanent connection of silicas with POSS compounds using organosilanes, but also direct modification of the silica surface by silsesquioxanes [
31,
32]. The resulting hybrid systems were used, for instance, as fillers in gel polymer electrolytes [
33].
Our previous work on modified bentonite nanocomposites with a synthetic resin matrix [
34,
35,
36,
37] prompted us to undertake studies on the development of new epoxy composites with various fillers with different particle shapes. This provides a new way to combine POSS-modified bentonite (platelet shape) and POSS-modified silica (spherical shape), leading to functional hybrid composites. This was the aim of our recent patent notification [
38]. The objective of the present work is to investigate the impact of the new synthesized nanofillers—POSS-modified bentonite and POSS-modified silica—in improving the mechanical and flame retardant properties of epoxy-based composites.
2. Experimental Section
2.1. Materials
Bentonite from Russian deposits, provided by a representative of Bento Group Minerals Company Poland Sp. z o.o., Gdynia, denoted as B.
Epoxy resin Epidian 6—a product of the Organika-Sarzyna chemical plant in Nowa Sarzyna, Poland, denoted as EP.
Triethylenetetramine—a technical product of the Organika-Sarzyna chemical plant Nowa Sarzyna, Poland, denoted as Z-1.
SiO
2 filler precipitated in polar media, analogously as in the procedure previously described by Jesionowski
et al. [
39]. The silica filler surface (HS) was grafted with POSS modifier (POSS3), in a quantity of 20 parts by weight of SiO
2.
The bentonite and silica modifiers from the group of cellular silsesquioxanes are presented in
Table 1.
Table 1.
Polyhedral oligomeric silsesquioxanes (POSS) used for modification of bentonite and hydrated silica.
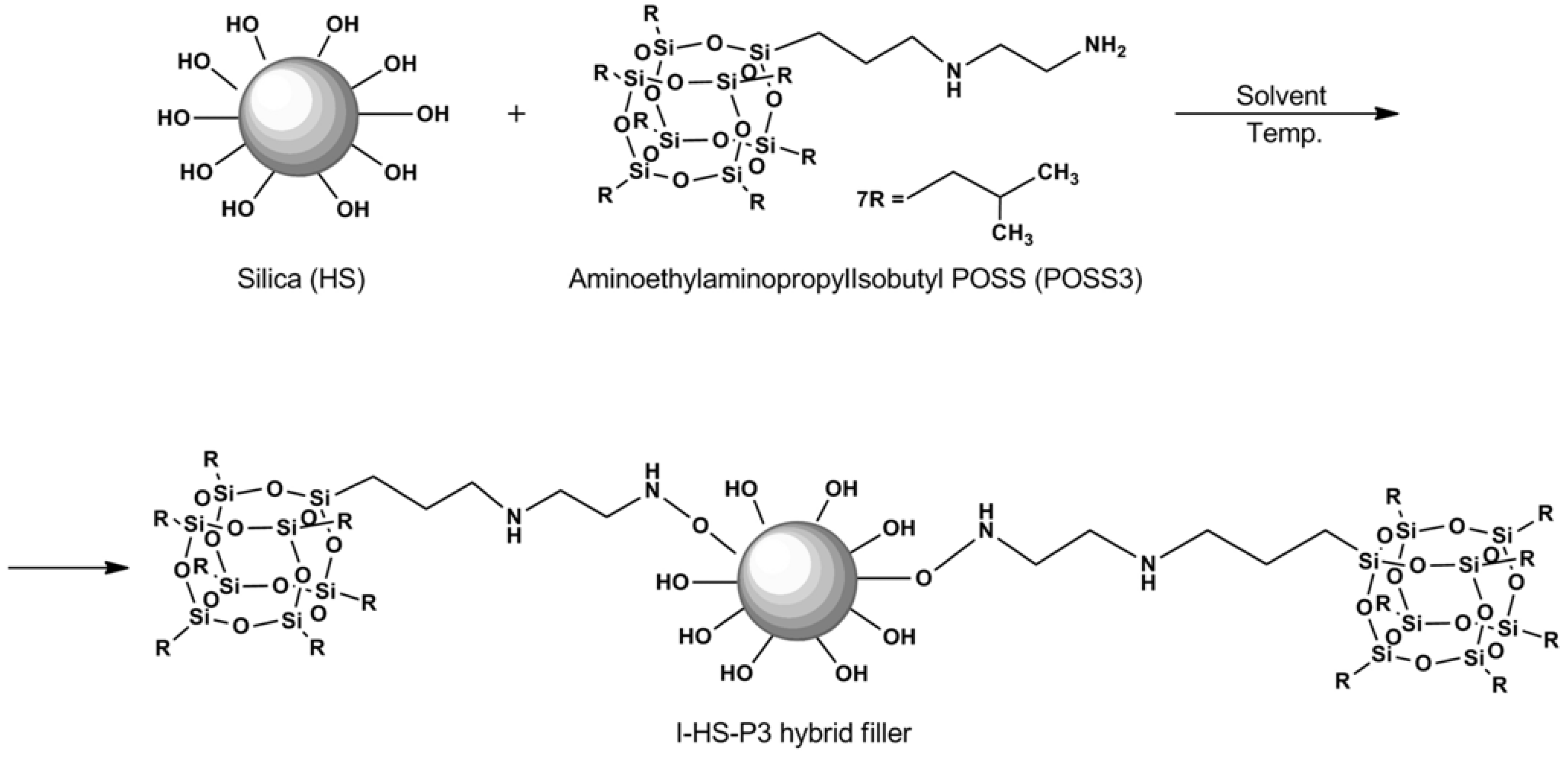
2.2. Preparation of Bentonite and SiO2 Fillers Modified with Silsesquioxane (POSS)
The study employed the procedure of modification of smectic clays using POSS, which has been previously patented [
37,
38] and described in the literature [
34,
35,
36].
Silica filler was subjected to functionalization by the method of immobilization in an organic solvent. In a reactor equipped with a high speed stirrer and heating mantle, an appropriate fraction of hydrated silica was introduced into toluene. Nitrogen was introduced to provide an inert atmosphere. When the temperature in the reactor reached 50 °C, the process of immobilization was begun. The modifying mixture was dosed with the use of a peristaltic pump. The mixture was then mixed in the reactor for 2 h (
ca. 800 rpm). After that time, the organic solvent was separated by distillation. The resulting powder material was dried in a convectional dryer at a temperature of 120 °C for 48 h. The procedure has been described in detail in a previous paper [
30].
2.3. Determination of Physicochemical Properties of the Fillers
Prior to the determination of particle size, all bentonites were sieved through a 0.06 mm mesh. The test was performed using a Mastersizer Hydro MU2000 apparatus (Malvern Instruments Ltd., Malvern, UK) at 20 °C. The particles of the filler were pre-dispersed ultrasonically in propanol-2-ol for more efficient measurement.
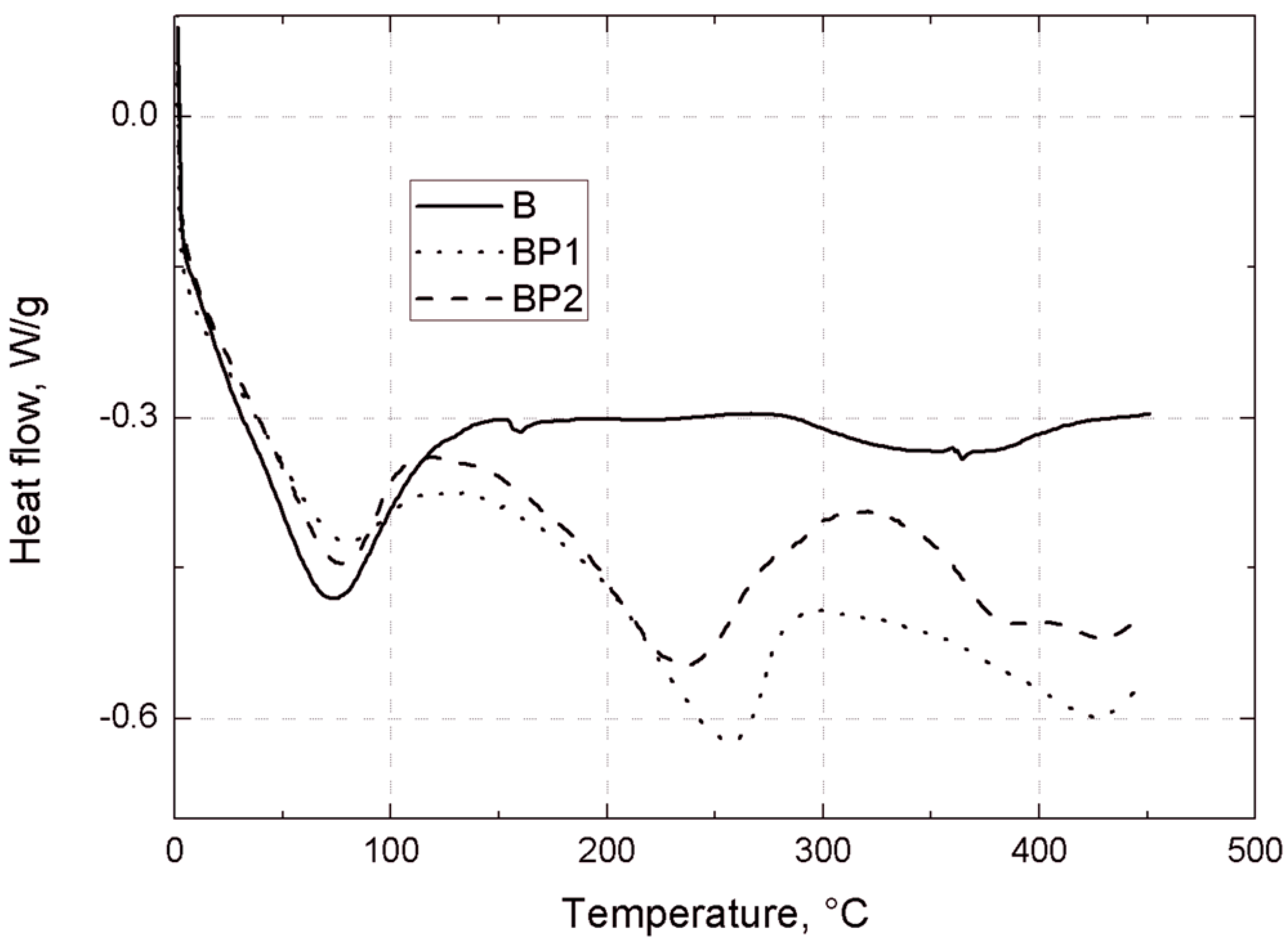
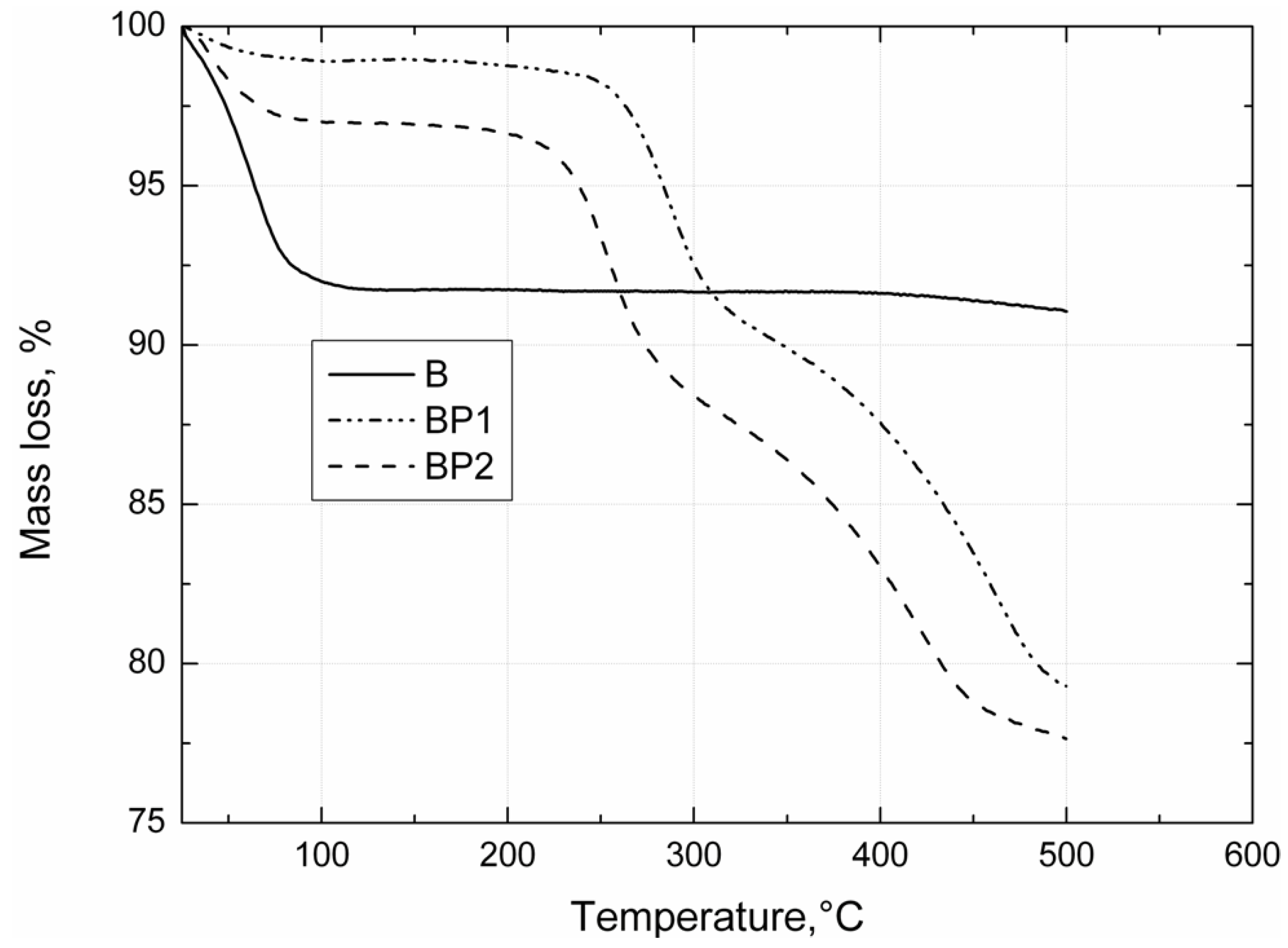
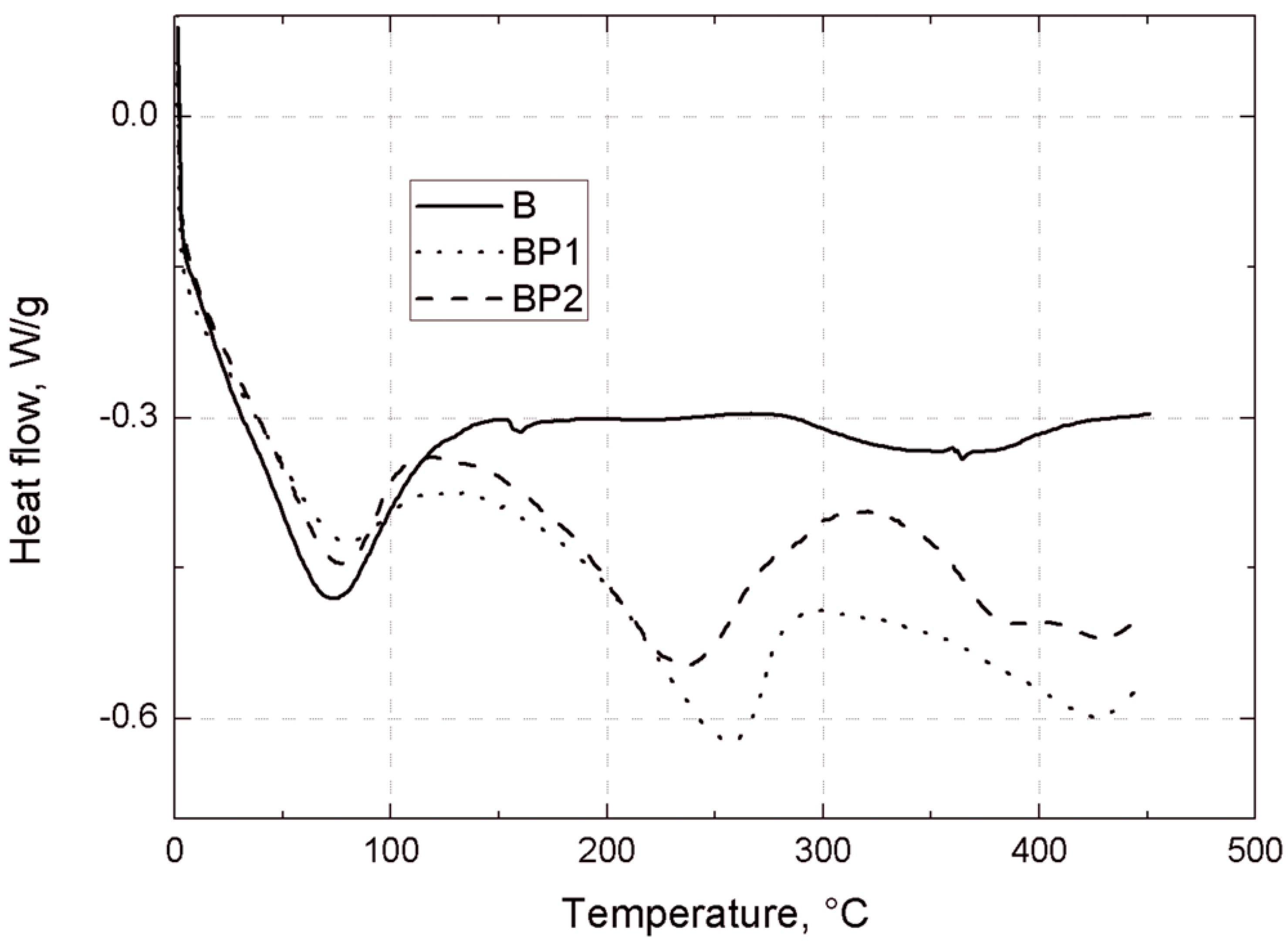
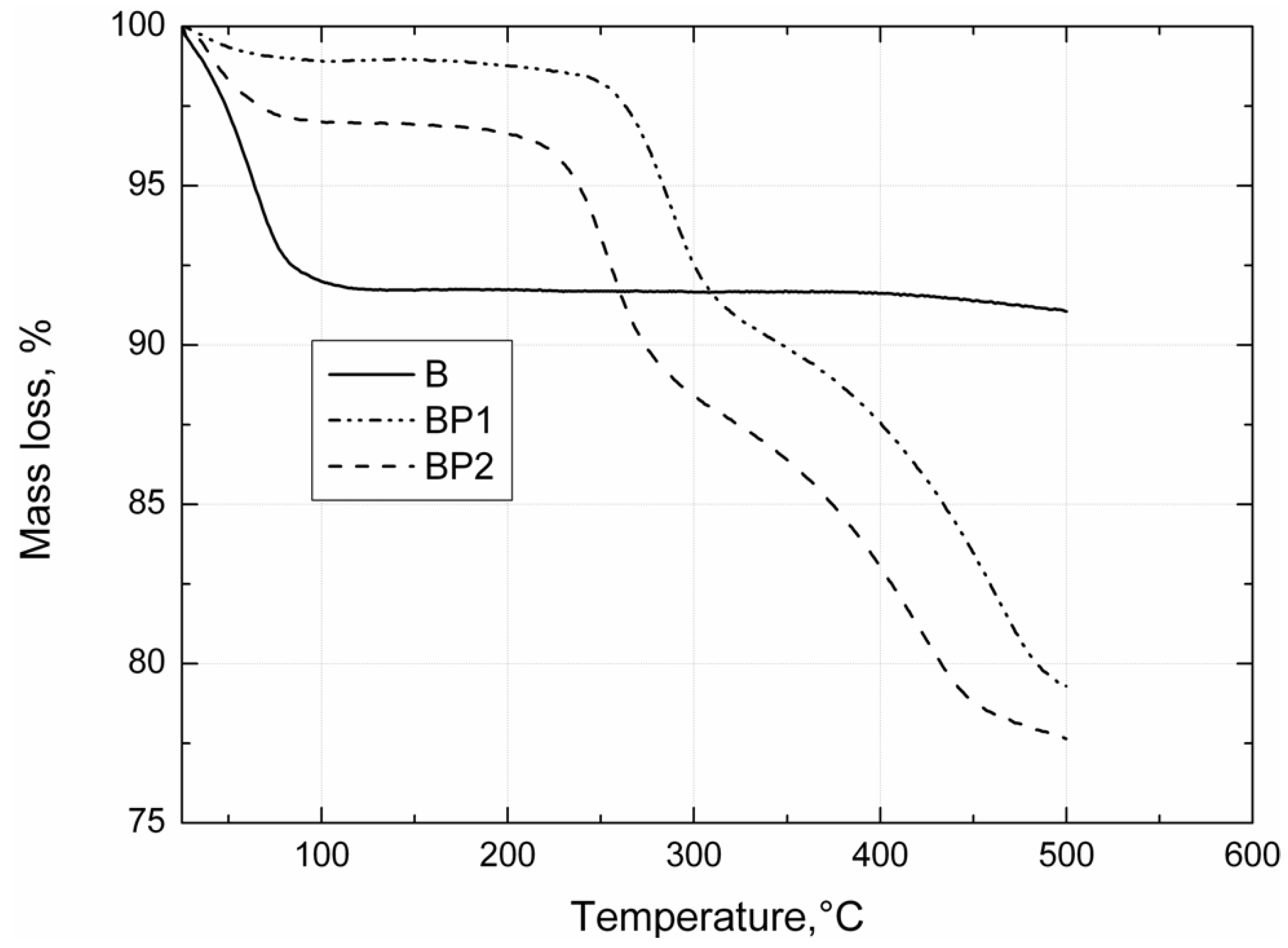
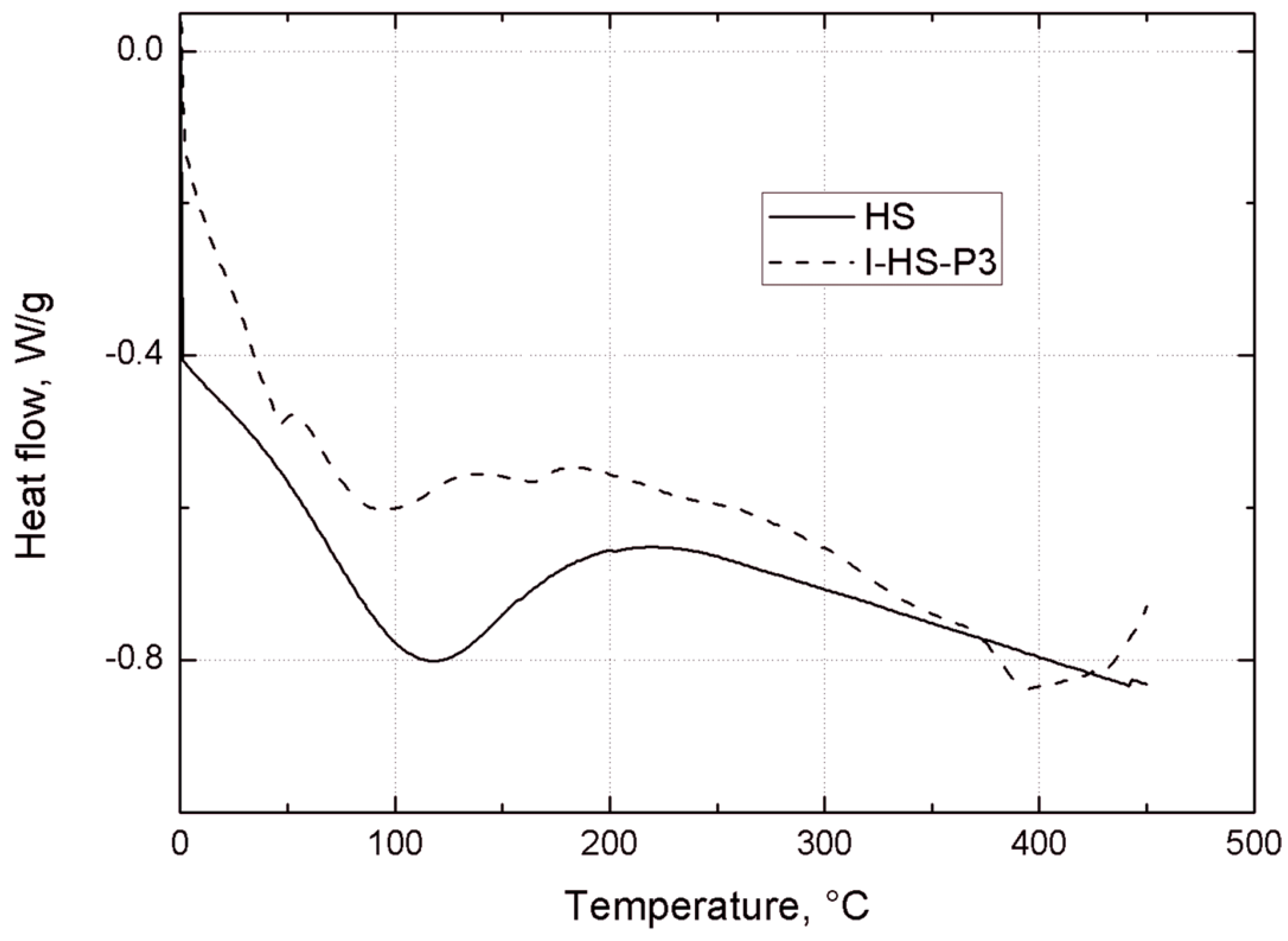
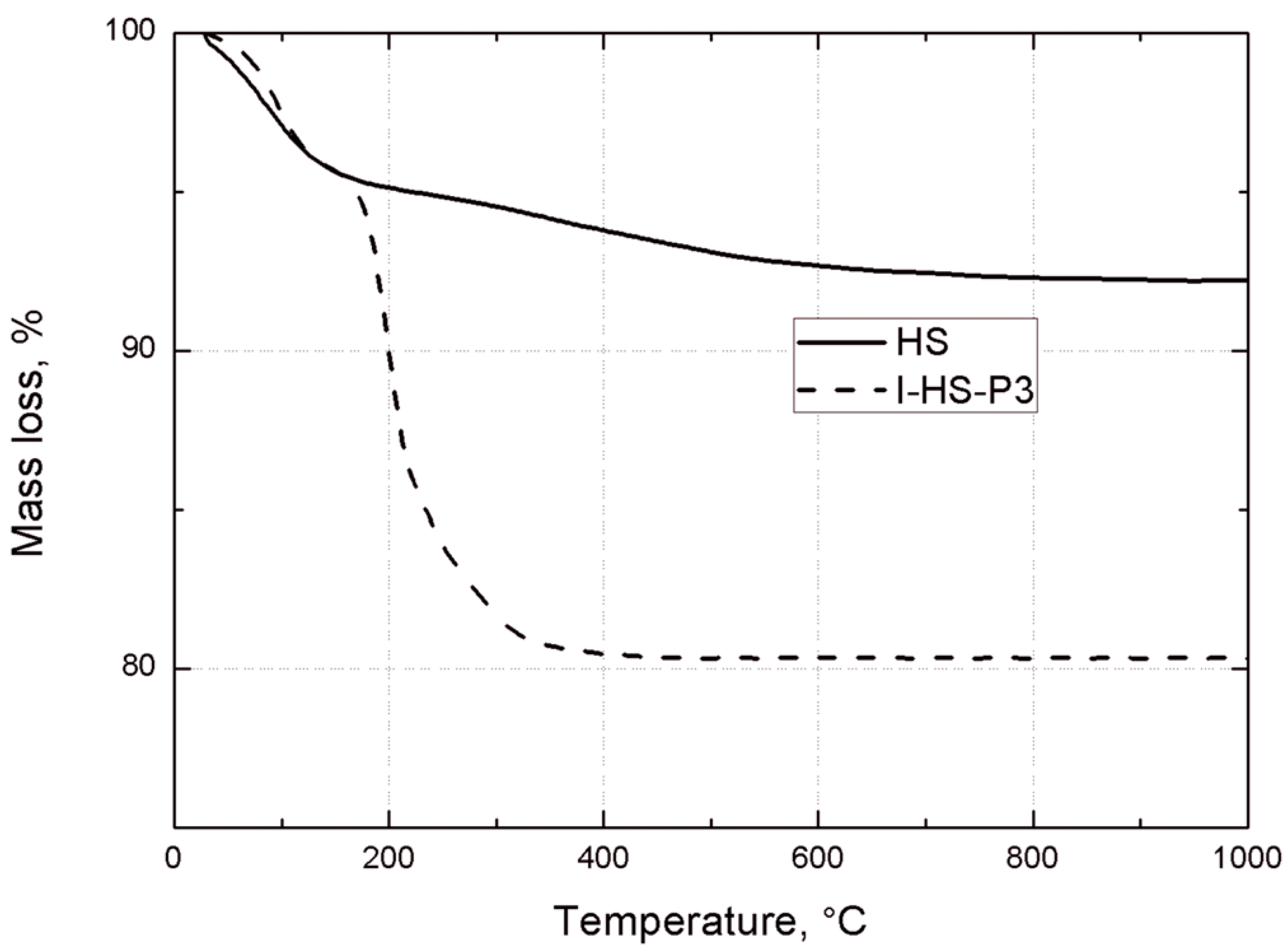
Thermogravimetric analysis of the bentonites was carried out under nitrogen, using a TGA/DSC1 thermobalance (Mettler Toledo, Greifensee, Switzerland). Measurements were made in the temperature range 25–450 °C, with a heating rate of 10 °C/min.
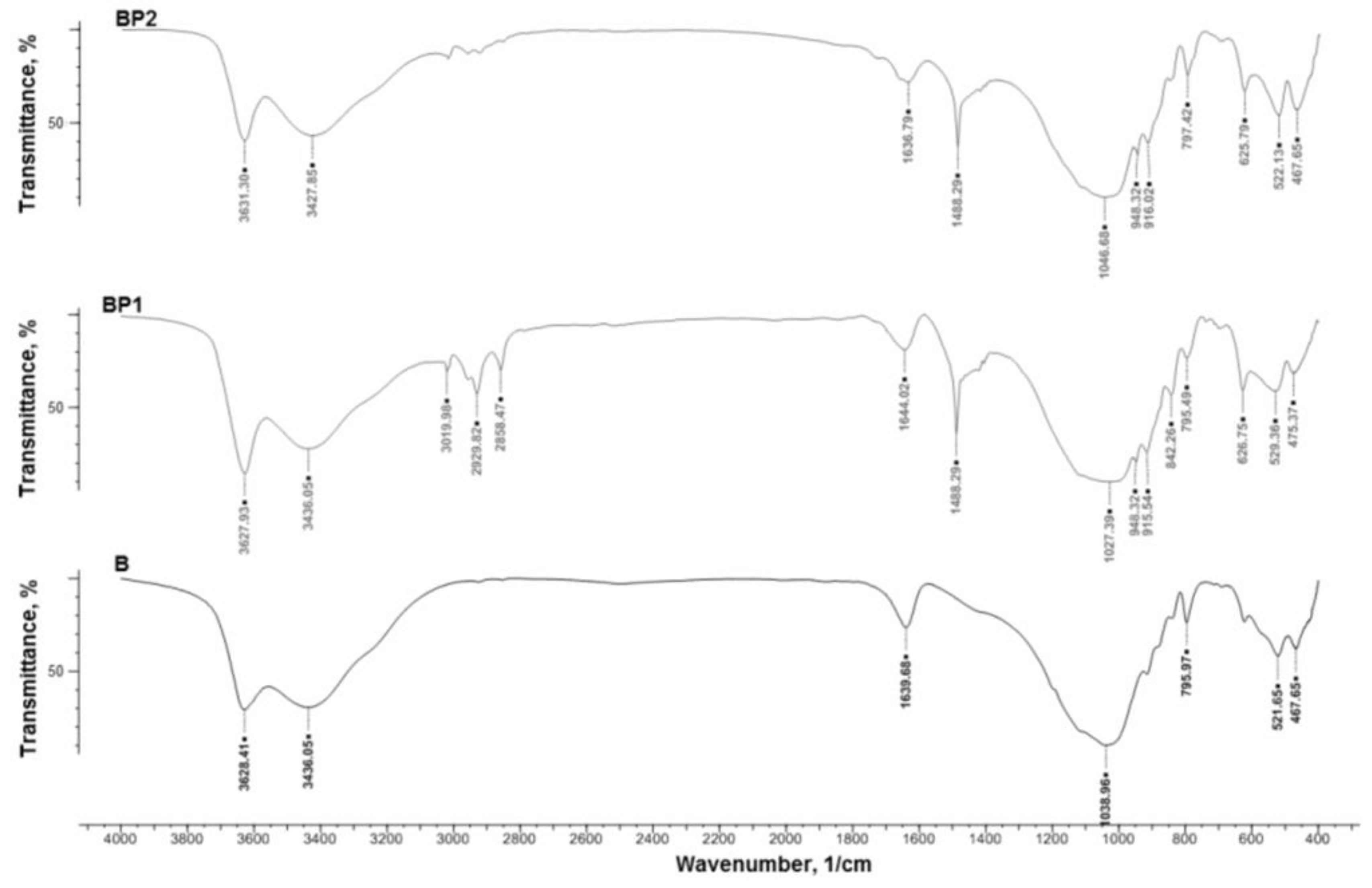
The IR spectra of the bentonites were recorded on a Nicolet 8700 spectrophotometer (Thermo Electron Scientific Instruments LLC, Madison, WI, USA), in the range 4000–4550 cm−1, using the KBr pellet technique (1 mg sample/200 mg KBr).
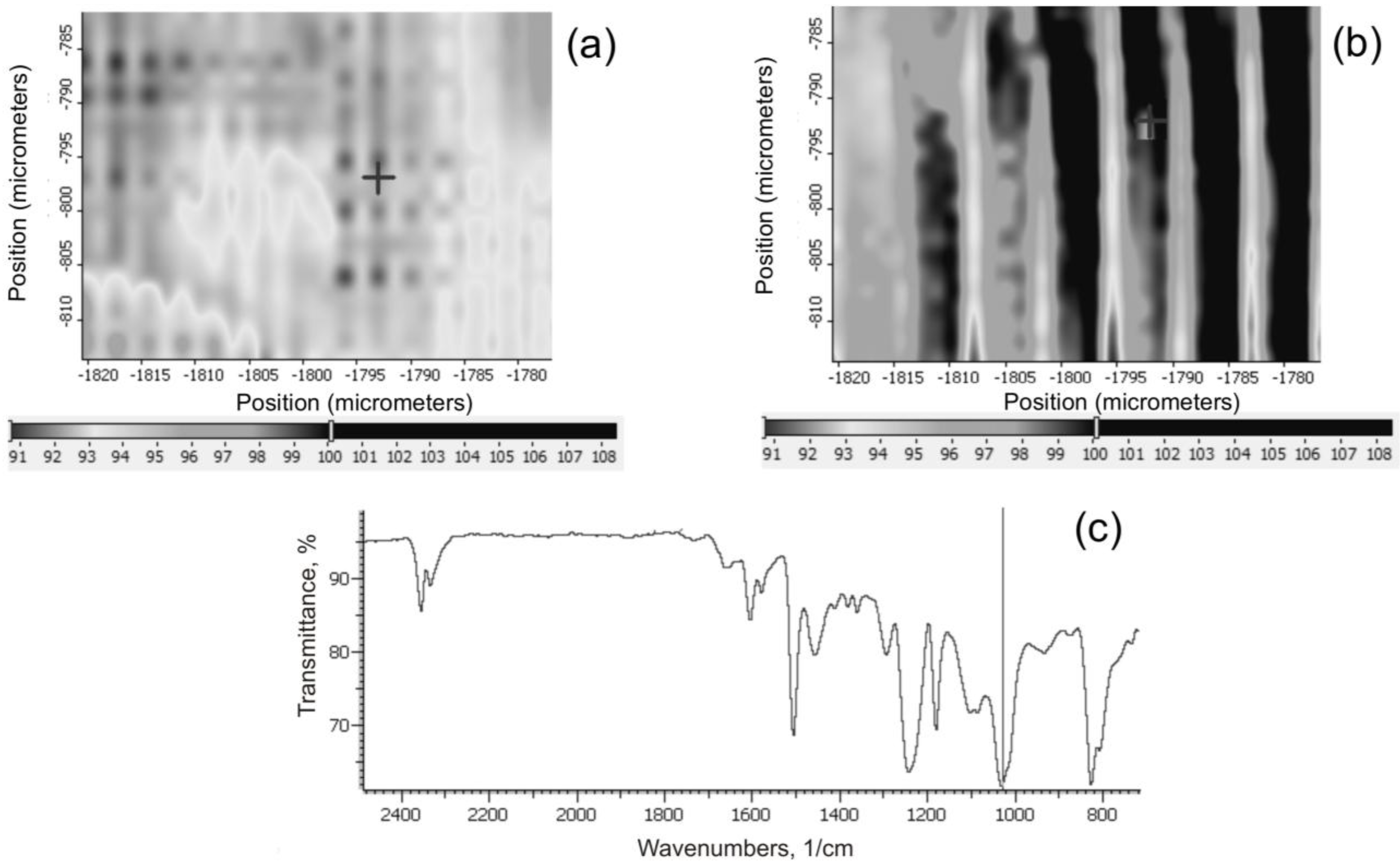
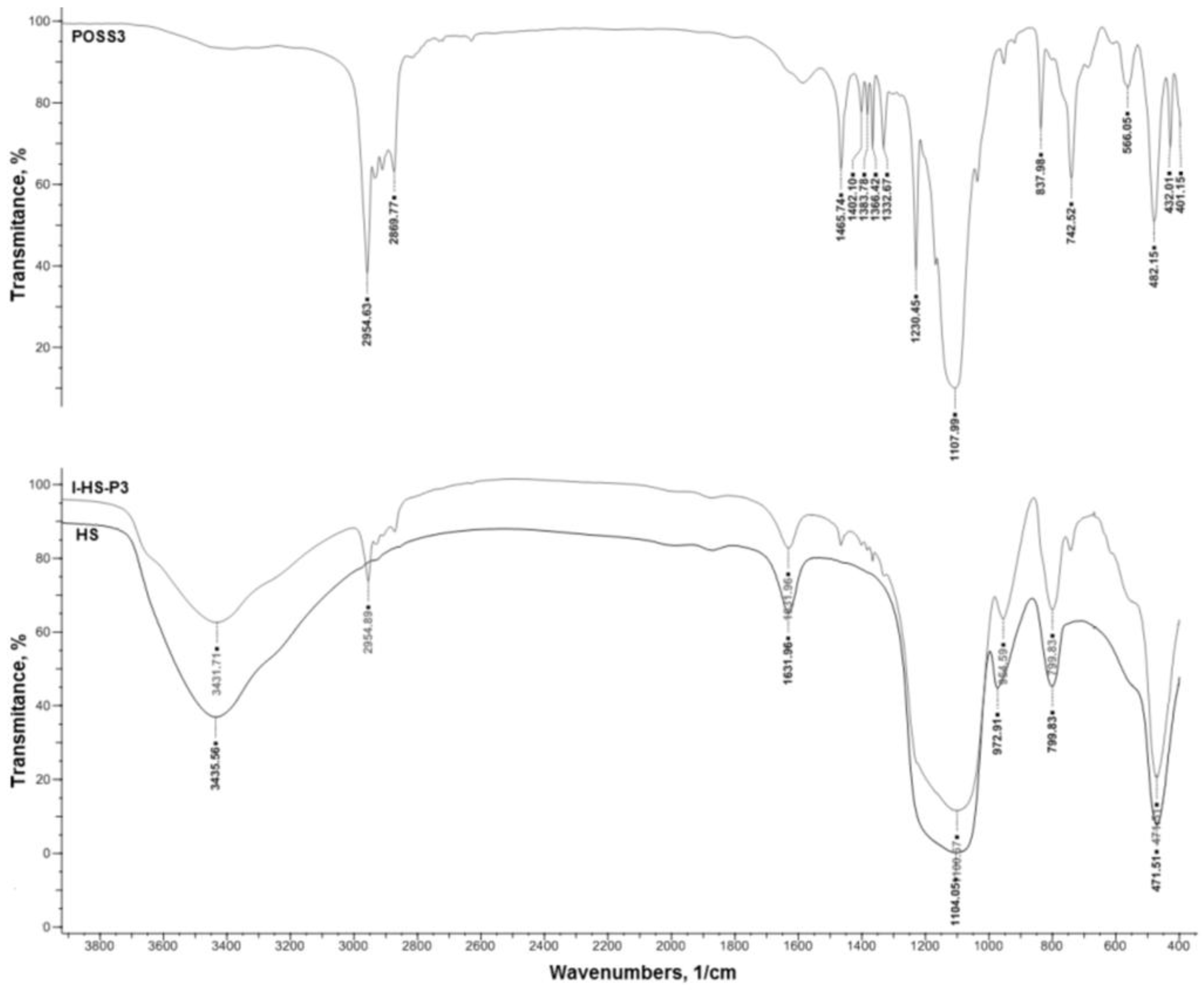
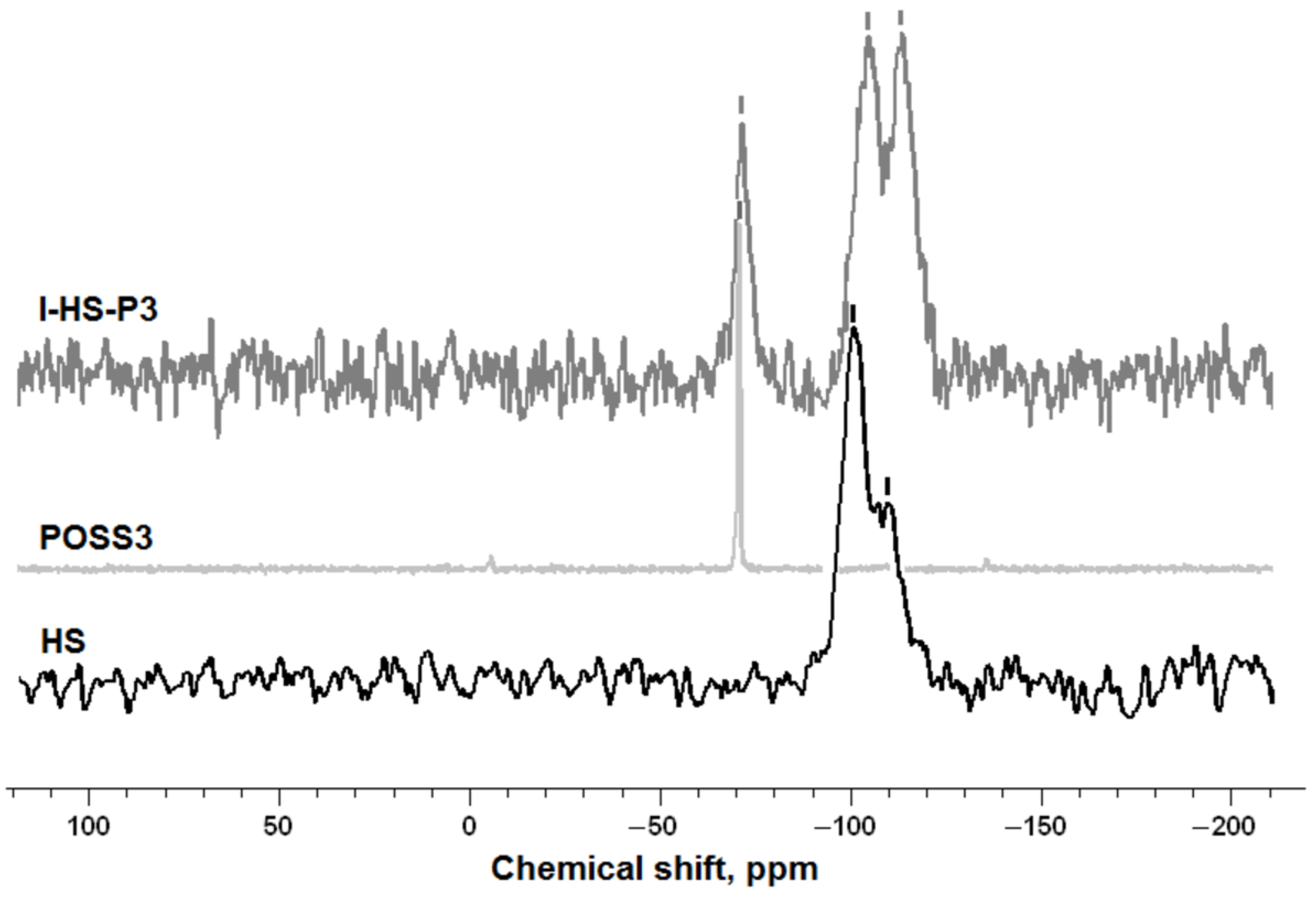
The final silica and SiO2/POSS3 hybrid filler were analyzed by a number of methods. The effectiveness and degree of functionalization of SiO2 with the POSS compound were estimated using an FTIR IFS 66 v/S spectrophotometer (Bruker Optik GmbH, Ettlingen, Germany). The samples were prepared by mixing with KBr and then pressing into small tablets. FTIR spectra were obtained in the transmission mode between 4000 and 400 cm−1.
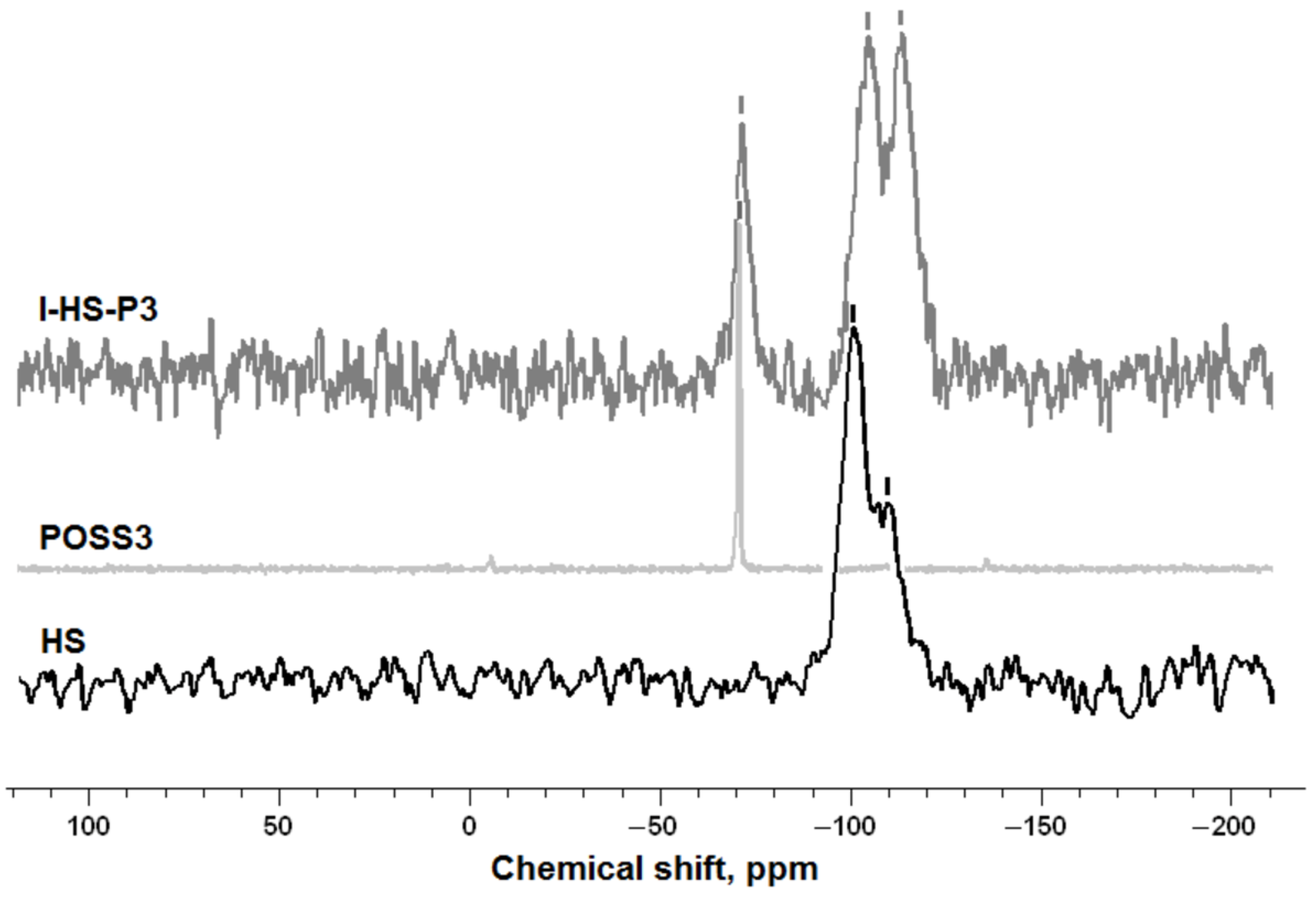
The structure of the silica surface, before and after modification with POSS, was examined using cross-polarization nuclear magnetic resonance tests. 29Si and 13C CP MAS NMR measurements were carried out using a DSX spectrometer (Bruker BioSpin GmbH, Rheinstetten, Germany). For the determination of NMR spectra, a sample of about 100 mg was placed in a ZrO2 rotator with diameter 4 mm, which enabled spinning of the sample. Centrifugation at the magic angle was performed at a spinning frequency of 8 kHz. 29Si CP MAS NMR spectra were recorded at pulse duration 4.5 µs, contact time 1.5 ms, and pulse spacing 6 s. The 13C CP MAS NMR spectra were recorded at 100.63 MHz in a standard 4 mm MAS probe using single pulse excitation with high power proton.
The particle size distributions of the silica samples were measured using a Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, UK), enabling measurements in the range 0.6–6000 nm, by the NIBS method. The microstructures of the samples were analyzed using transmission electron microscopy images (Joel 1200 EX II, JEOL Ltd., Tokyo, Japan).
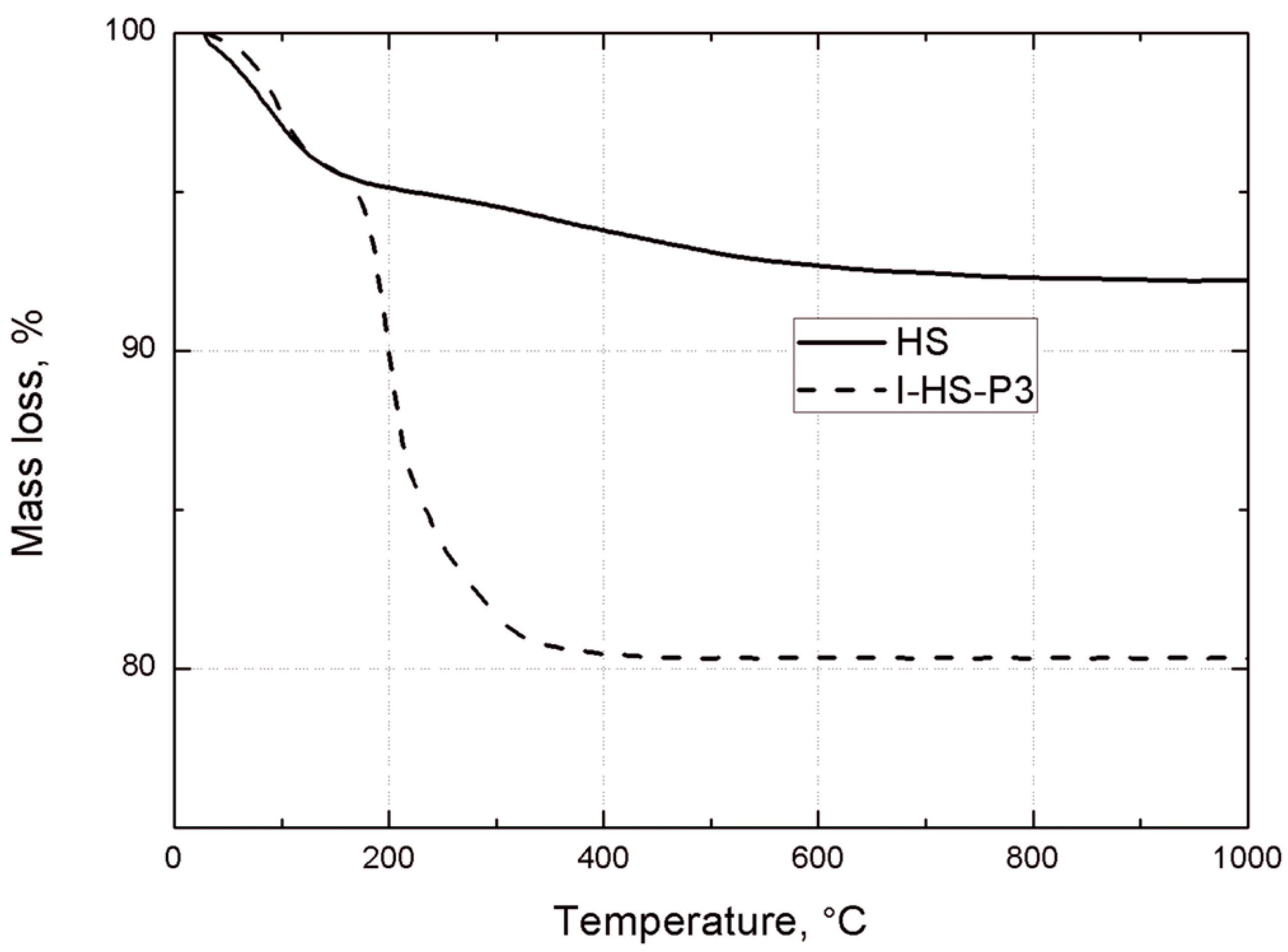
Thermogravimetric analysis was performed using a Jupiter STA 449 F3 (Netzsch GmbH, Selb, Germany). Samples weighing approximately 10.0 mg were placed in an Al2O3 crucible, and heated at a rate of 10 °C/min from 30 °C to 1000 °C in a nitrogen atmosphere.
2.4. Preparation of Composite of Epoxy Resin Filled with POSS-Modified Silica and Bentonite
The modified bentonite and modified silica prepared in this way, in proportions of 1:1 by weight, were introduced successively in quantities of 1.5–4.5 wt.% to the synthetic resin liquid mixture, and then homogenized in a multistage process. First a modified bentonite was introduced to the epoxy resin, and dispersed until a homogeneous suspension was formed, then modified silica was introduced and dispersed. In both cases, three-stage homogenization was used, based on: (I) pre-mixing by means of a mechanical stirrer at slow rotation at room temperature for 20 min; (II) stirring for 15 min using an ultrasonic homogenizer preheated to a temperature of 50 °C; (III) mixing in a high shear mixer with a turbine stirrer in the vessel, also at 50 °C, with a stirrer speed of 10,000 min−1, the time of homogenization in the mixer being 30 min; and (IV) final homogenization in a cylindrical vessel with a small gap of 0.75 mm and with the rotary speed of the cylinder equal to 6,000 min−1 to provide high shear. The duration of this operation was 15 min.
The composites prepared in this way were stored at about 4 °C to prevent possible sedimentation of the fillers. The specific compositions of the test samples are given in
Table 2.
Table 2.
Composition of test samples based on epoxy resin.
Table 2.
Composition of test samples based on epoxy resin.
| Composite symbol | Content BP1 1, wt.% | Content BP2 2, wt.% | Hybrid content I-HS-P3 3, wt.% |
|---|
| EP | 0.0 | 0.0 | 0.0 |
| EPBP1-1.5 | 1.5 | | - |
| EPBP1-3.0 | 3.0 | | - |
| EPBP1-4.5 | 4.5 | | - |
| EPIHSP3-1.5 | - | | 1.5 |
| EPIHSP3-3.0 | - | | 3.0 |
| EPIHSP3-4.5 | - | | 4.5 |
| EPBP1IHSP-1.5 | 0.75 | | 0.75 |
| EPBP1IHSP-3.0 | 1.5 | | 1.5 |
| EPBP1IHSP-4.5 | 2.25 | | 2.25 |
| EPBP2-1.5 | | 1.5 | - |
| EPBP2-3.0 | | 3.0 | - |
| EPBP2-4.5 | | 4.5 | - |
| EPBP2IHSP-1.5 | | 0.75 | 0.75 |
| EPBP2IHSP-3.0 | | 1.5 | 1.5 |
| EPBP2IHSP-4.5 | | 2.25 | 2.25 |
2.5. Obtaining Molded Pieces from the Composites for Structural, Strength and Flammability Tests
Composites based on the resin matrix Epidian 6 were cured using Z-1 (13 wt.%), according to the resin manufacturer’s instructions. The composites were then vented in a Vakuum UHG 400 laboratory vacuum chamber (Schuechl, Bawaria, Germany) and cast at 40 °C in silicone molds prepared in accordance with ISO 527-1:1998. The molded pieces were cured at room temperature for 24 h and then post-cured at 100 °C for 6 h. After two days, the molded pieces were tested in accordance with the relevant standards.
2.6. Study of Mechanical Properties of Composites
Tensile strength and Young’s modulus were determined according to ISO 527-1:1998 using an INSTRON 5967 testing machine equipped with an advanced videoextensometer. “Dog-bone” specimens (type 2 according to the standard) were used. Young’s modulus was obtained as the tangent to the linear segment of the stress-elongation curve. A rate of elongation of 2 mm/min was used for the linear stress-elongation curve segment, and after that 20 mm/min was used. The measurement temperature was 23 °C.
Charpy impact strength was determined according to DIN EN ISO 179-1 with a PSW4J camera (Gerhard Zorn, Berlin, Germany), using a hammer impact energy of 1 J with a digital result readout. Unnotched bars: 100 mm long, 10 mm width and 4 mm height were used.
Rockwell hardness was measured using a Zwick 3106 durometer (Zwick/Roell, Ulm, Germany), in accordance with EN 10109-1. The applied indenter load was equal to 358 N. Similar specimens as for Charpy impact testing were used.
The measurement temperature used for mechanical tests was 23 °C. All final results given for the mechanical properties of the studied composites are mean values from 10 specimen tests. Standard deviations were also calculated.
2.7. Study of the Morphology and Structure of Composites
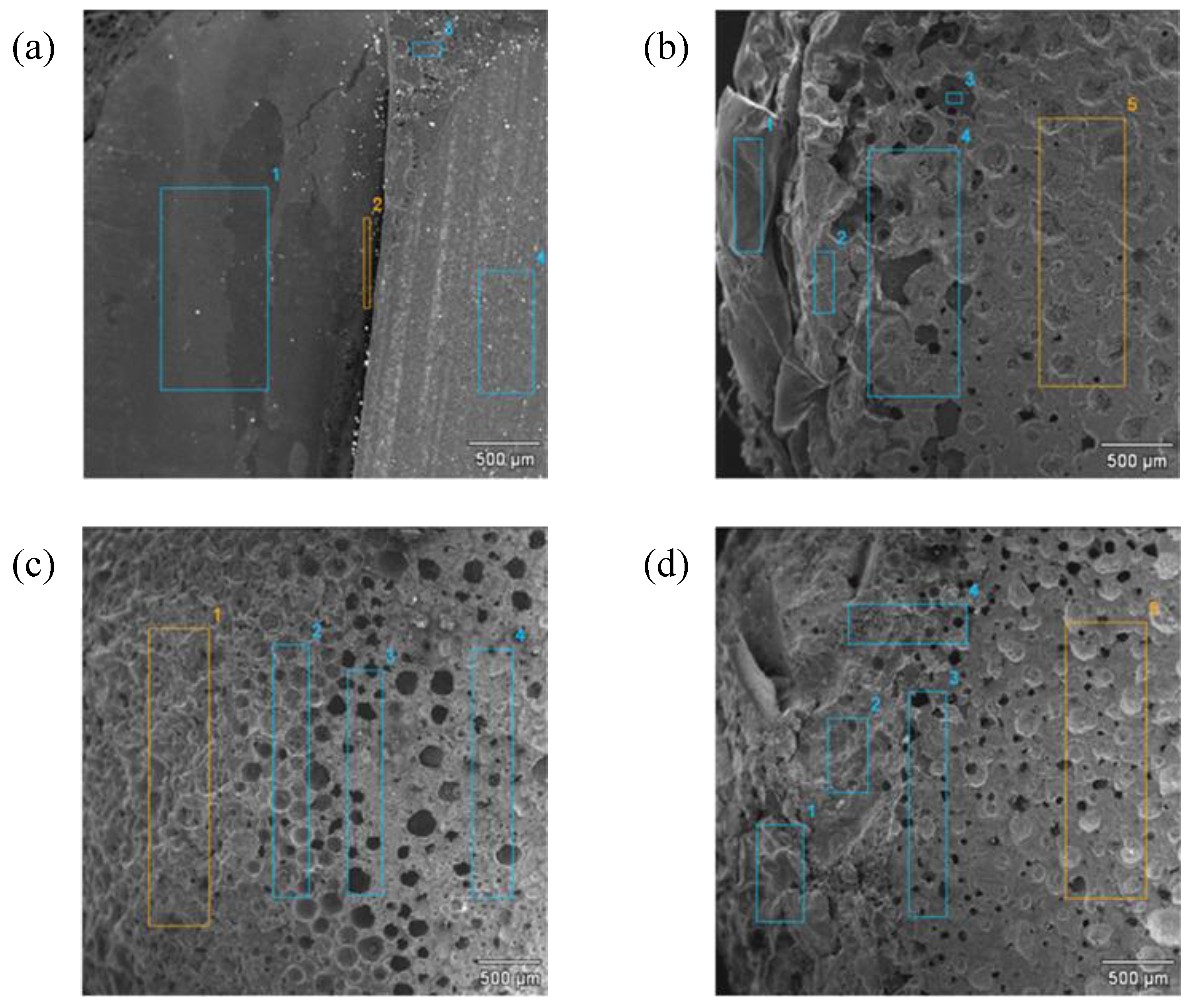
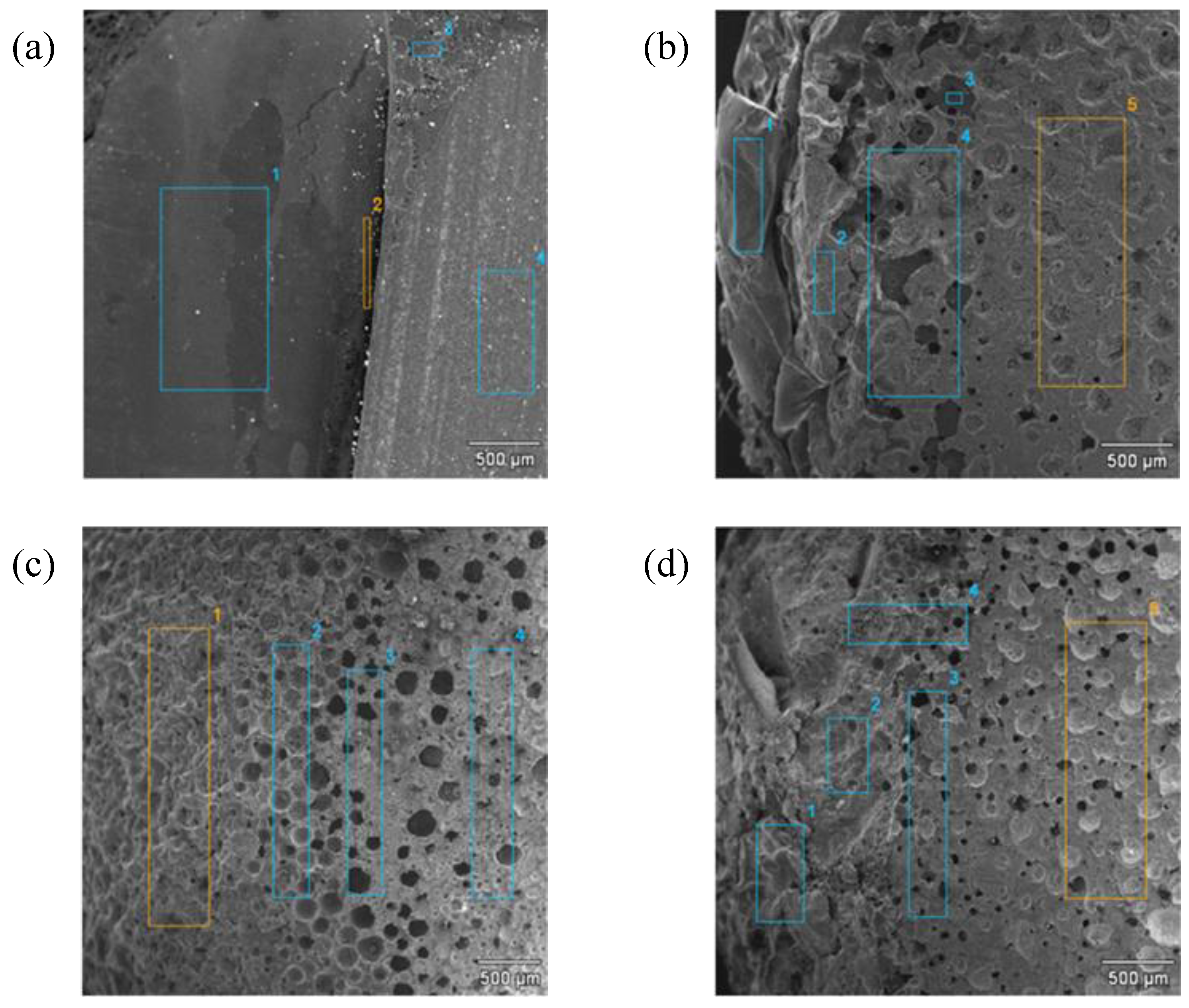
The brittle fracture morphology of the composites was analyzed using scanning electron microscopy (SEM 234a, JEOL Ltd., Tokyo, Japan). Fractured profiles were obtained after cooling in dry ice and impact-break. The microstructure of the composites was also observed using a Tecnai G2 SpiritTwin type 12 transmission electron microscope (TEM) (FEI Company, Hilsboro, NE, USA) at an accelerating voltage of 120 kV. Ultra-thin cuttings were performed at room temperature using a Tesla ultramicrotome (Tescan Orsay Holding, a.s, Brno, Czech Republic) with glass knives. They were collected on the surface of 10% aqueous acetone and placed on standard microscope copper grids.
Imaging of the surface of the samples was performed using an atomic force microscope (AFM) (Bruker Nano Surfaces Division, Santa Barbara, CA, USA) by the QNM technique. On the basis of local changes in the Young’s modulus of the sample surface, rigid areas that correspond to the presence of fillers, and areas with less rigidity associated with the polymer matrix, were observed. This enabled estimation of the degree of homogenization of the filler on the surface of the polymer matrix, and of the size of its particles. The tests were performed using a Nanoscope V microscope (Bruker Nano Surfaces Division, Santa Barbara, CA, USA) with an RTESPA scanning needle, with a resonance frequency of 270 kHz. The scanning speed was 1 kHz, and the resolution 256 lines.
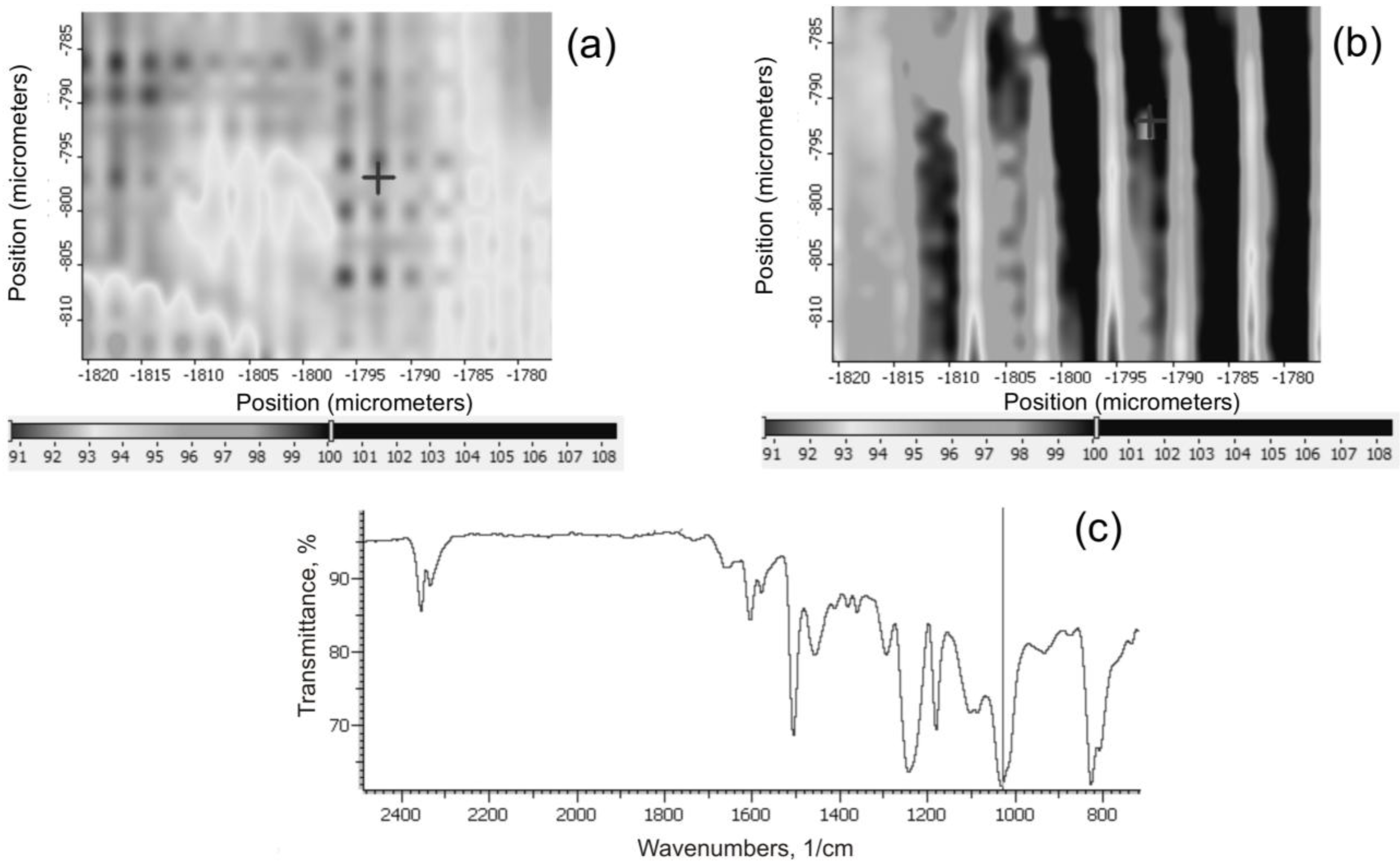
IR maps of the surface of the composites were made using a Nicolet FTIR MX IN10 microscope (Thermo Electron Scientific Instruments LLC, Madison, WI, USA), to determine the intensity of distribution of the characteristic Si–O–Si functional groups. This served to assess the uniformity of dispersion of grains of the filler in the composite.
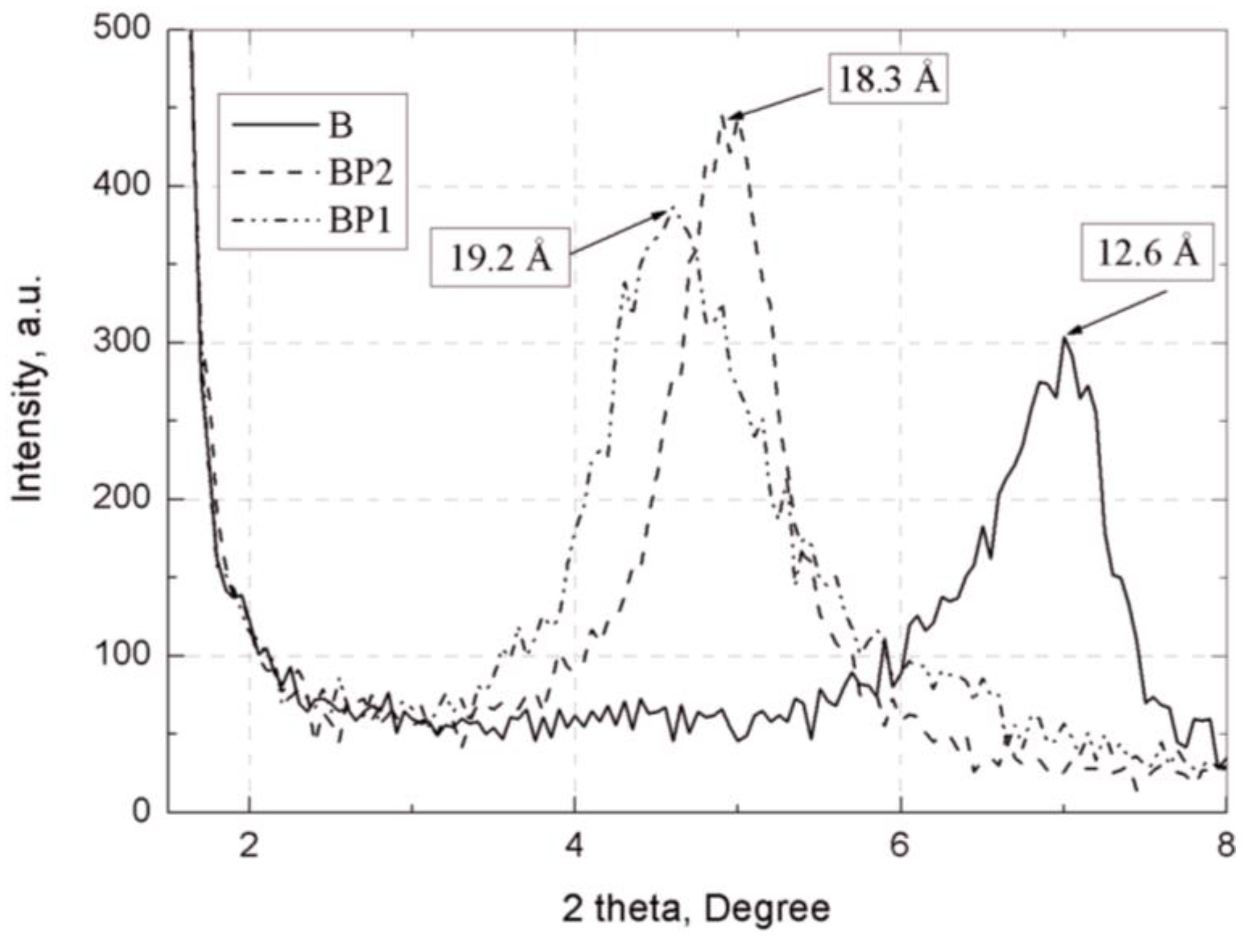
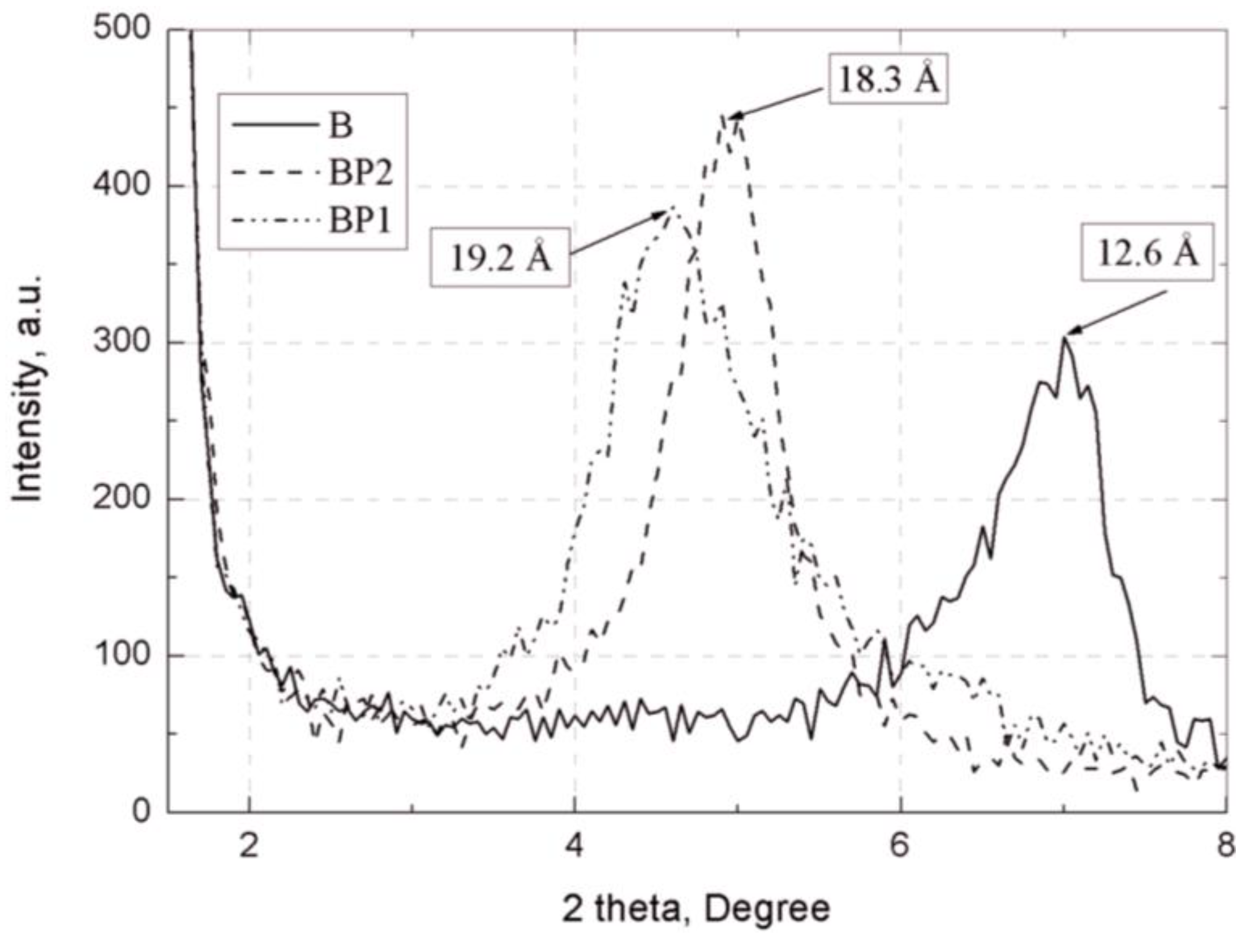
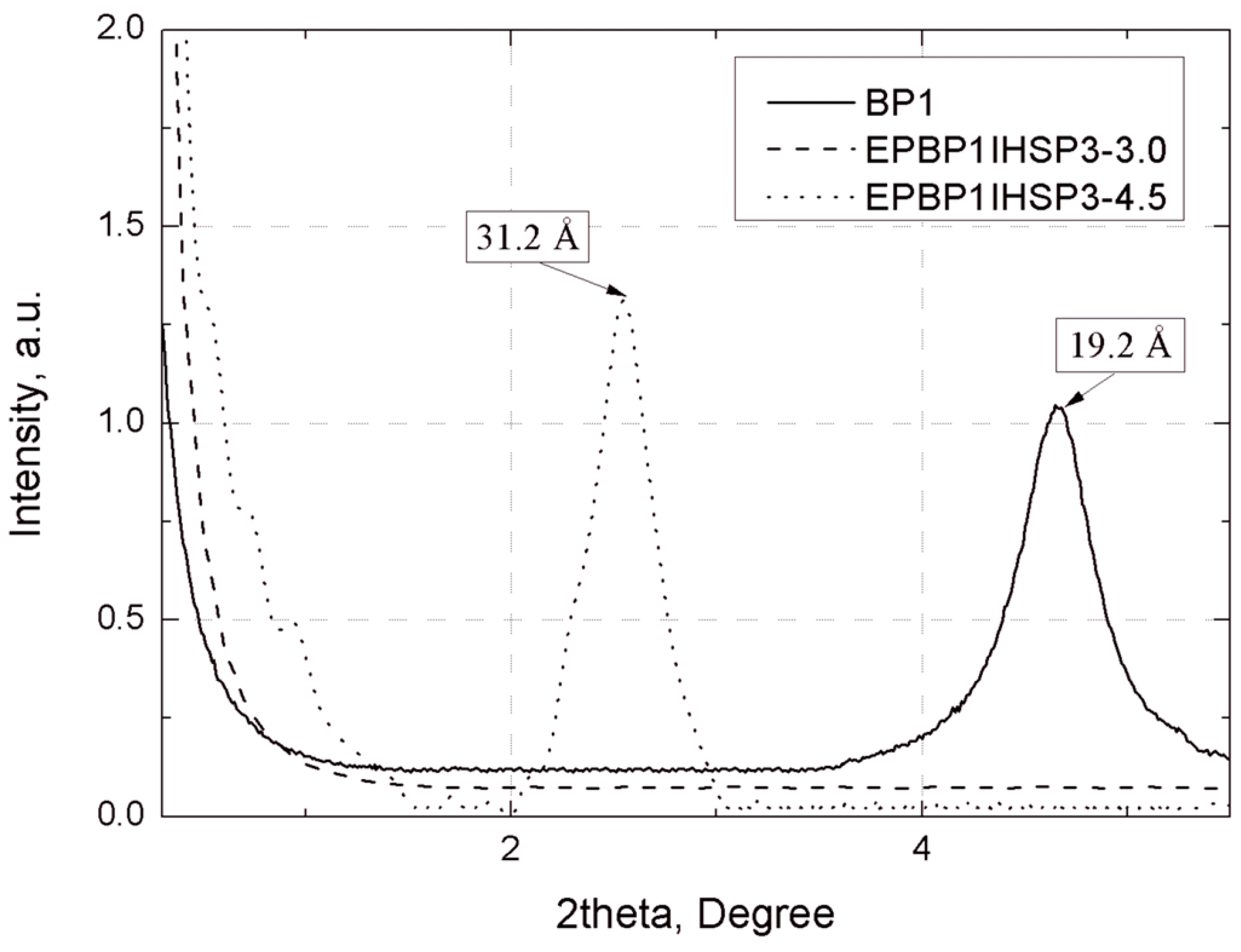
The separation of plates in bentonites and their EP composites was assessed by wide-angle X-ray scattering (WAXS), using Braggs’ law [
40]. The measurements were performed using a Bruker Nanostar diffractometer (Bruker AXS, Inc. Madison, WI, USA) with Cu lamp, for the bandwidth Kα. The samples were in the form of disks 25 mm in diameter and 2 mm in thickness, cast from the tested composites. The bentonite samples were tested in powder form.
2.8. Study of Flammability of Composites
Determination of the oxygen index (LOI) was performed at 25 °C according to EN ISO 4589-3, using apparatus made by Fire Testing Technology Ltd. (East Grinstead, UK).
UL 94 flammability testing was performed in a chamber designed for such tests (Fire Testing Technology Ltd., East Grinstead, UK). The measurements were made in accordance with PN-EN 60695-11-10.
Morphological and elemental analysis of burnt composite samples was performed using a Hitachi S-3400N scanning electron microscope (SEM) (Tokyo, Japan) equipped with an adapter for microanalysis (EDS) of chemical composition. Measurements were performed using a detector of secondary electrons (SE) (Thermo Scientific, Karlsruhe, Germany) and backscattered electrons (BSE) (Thermo Scientific, Karlsruhe, Germany), with an accelerating voltage of 15 kV and spot size <10 nm.
2.9. Determination of Gelation Time of the Hybrid Composites
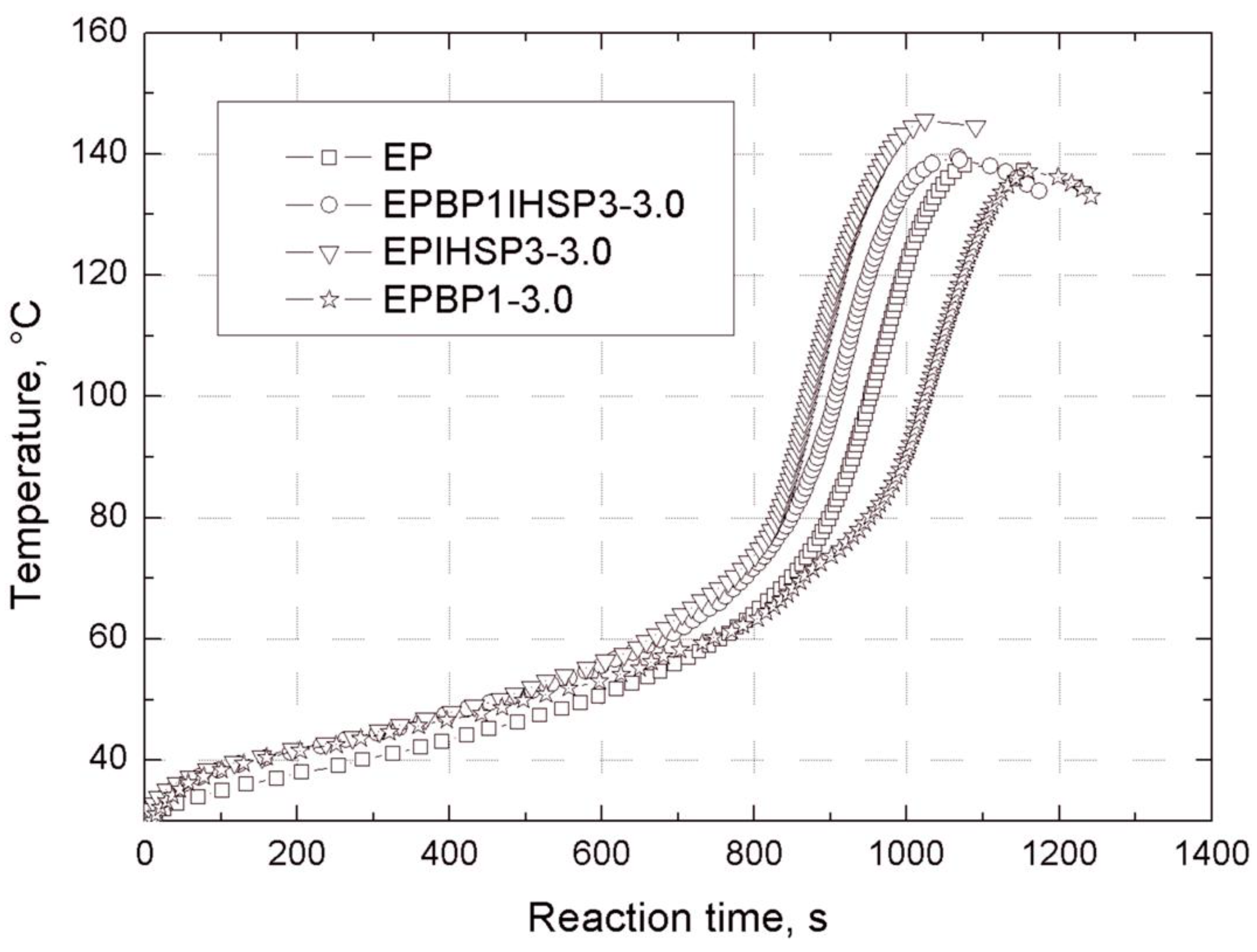
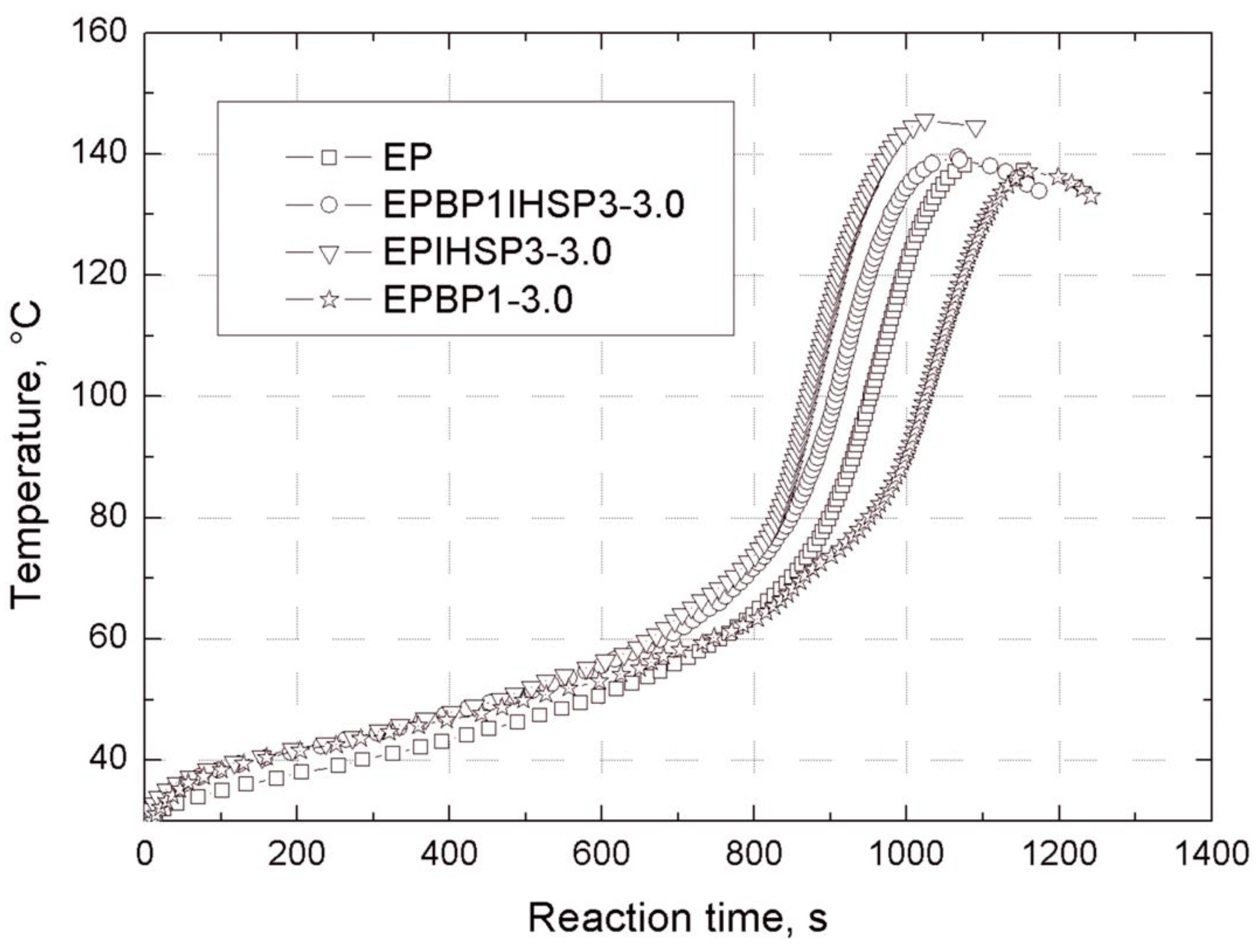
Gelation time after the addition of Z-1 hardener was measured at 25 °C according to PN-EN ISO 2535, using a WB-2 gel penetrator device (Rzeszow University of Technology, Rzeszow, Poland) of our own design and construction. The apparatus enables the tracking of temperature changes during the curing of a reaction mixture.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}