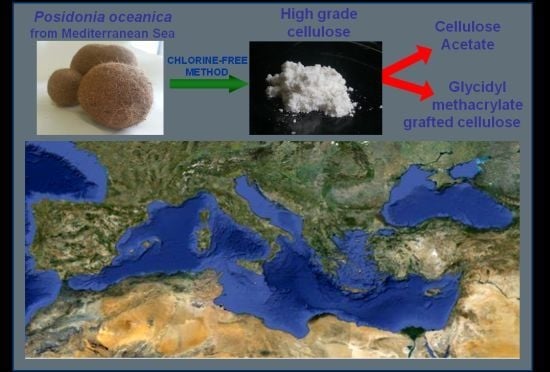
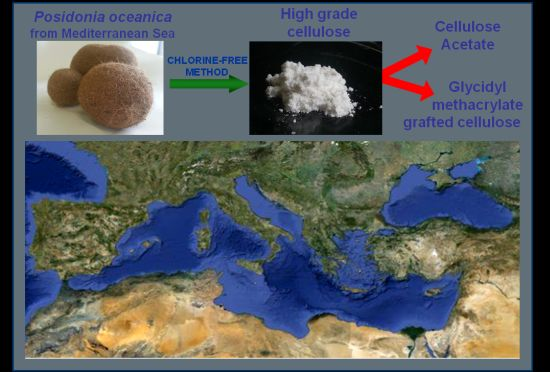
Posidonia oceanica as a Renewable Lignocellulosic Biomass for the Synthesis of Cellulose Acetate and Glycidyl Methacrylate Grafted Cellulose

Abstract
:
1. Introduction
- (i)
- Cellulose activation, which involves radical activation via physical (electron beam and plasma) or chemical (Fenton’s reaction) processes;
- (ii)
- Cellulose grafting, which involves radical quenching via GMA grafting to produce GMA-C.
2. Results and Discussion
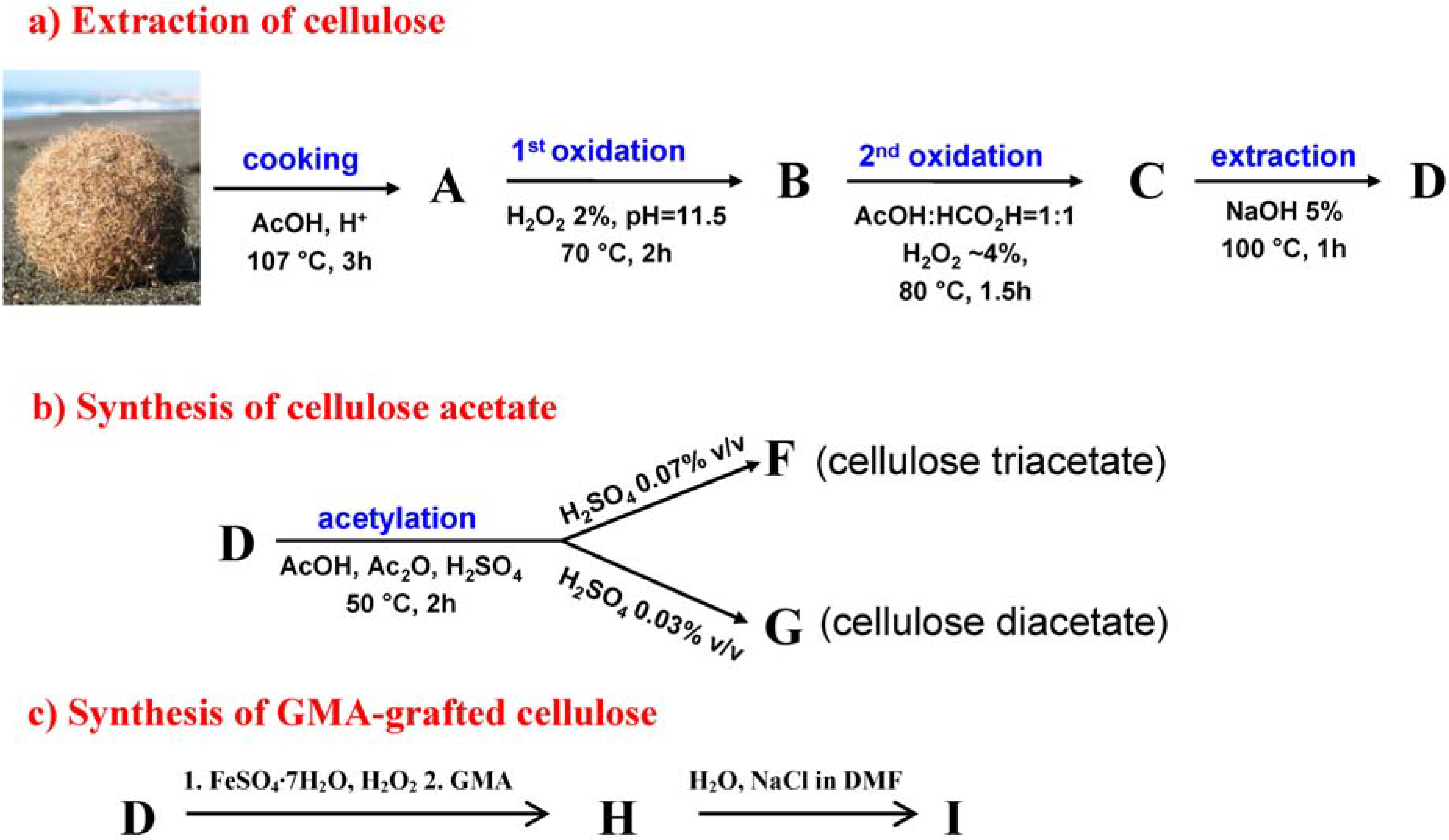
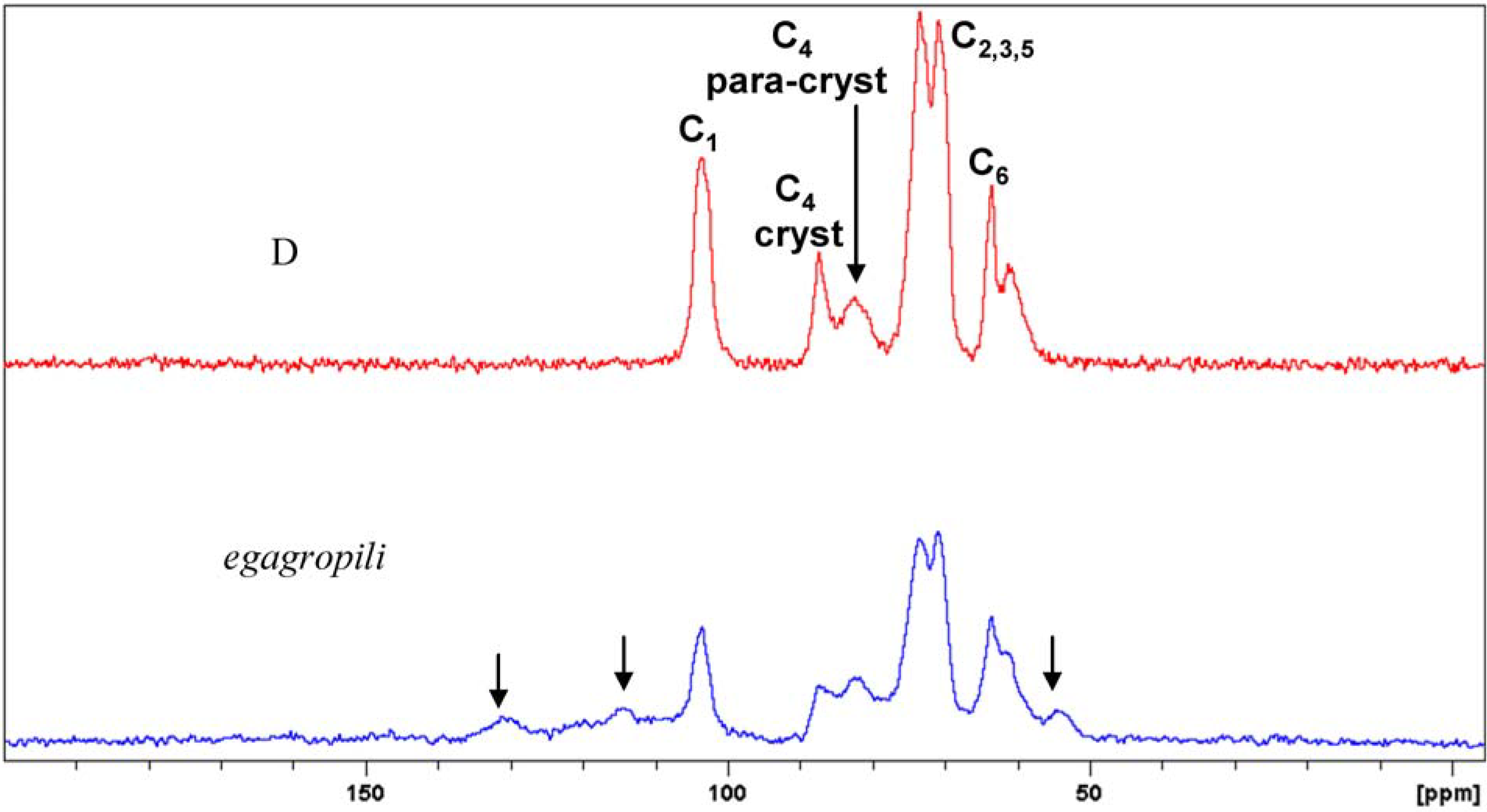
2.1. Extraction and Characterisation of α-Cellulose
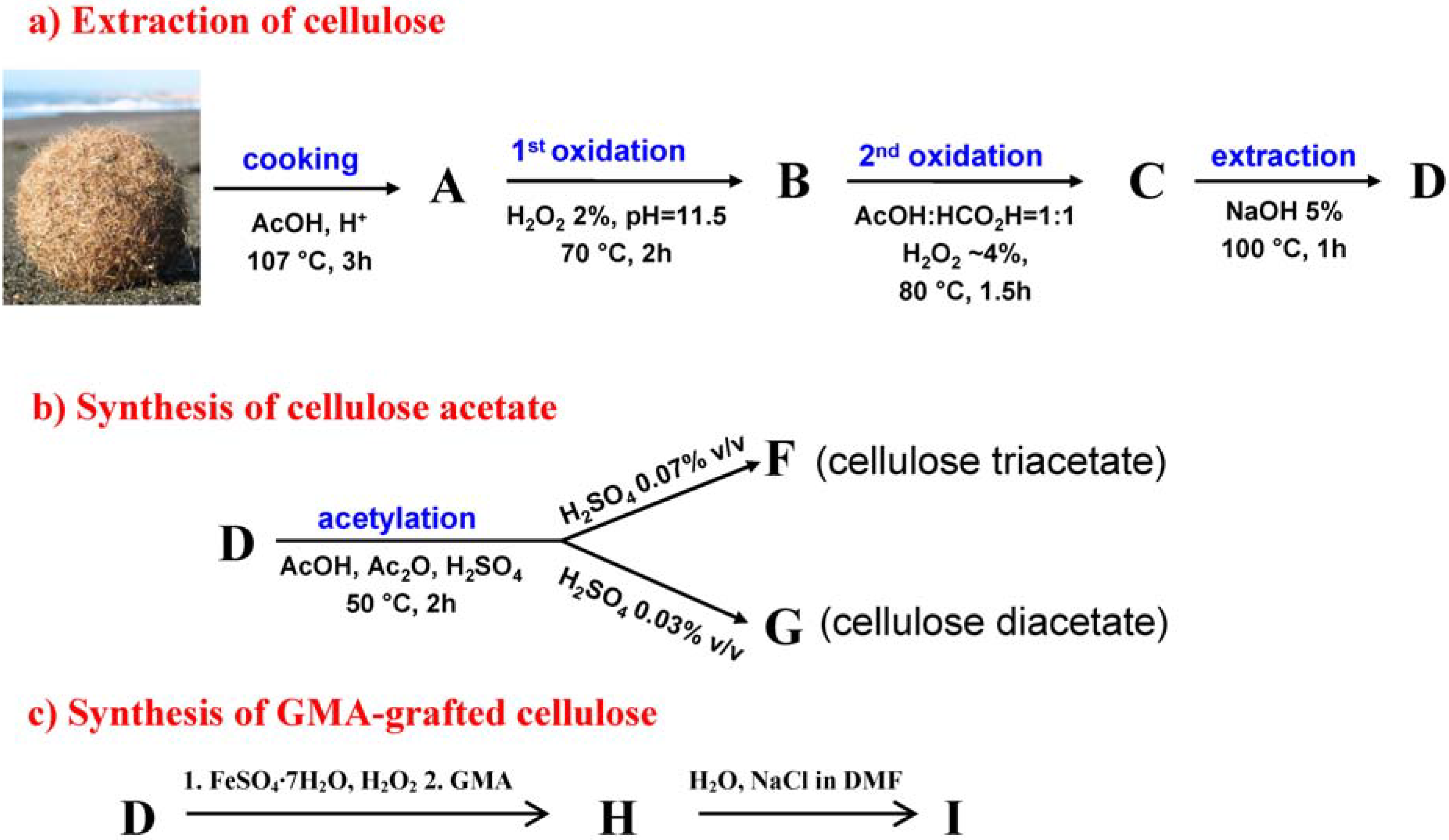
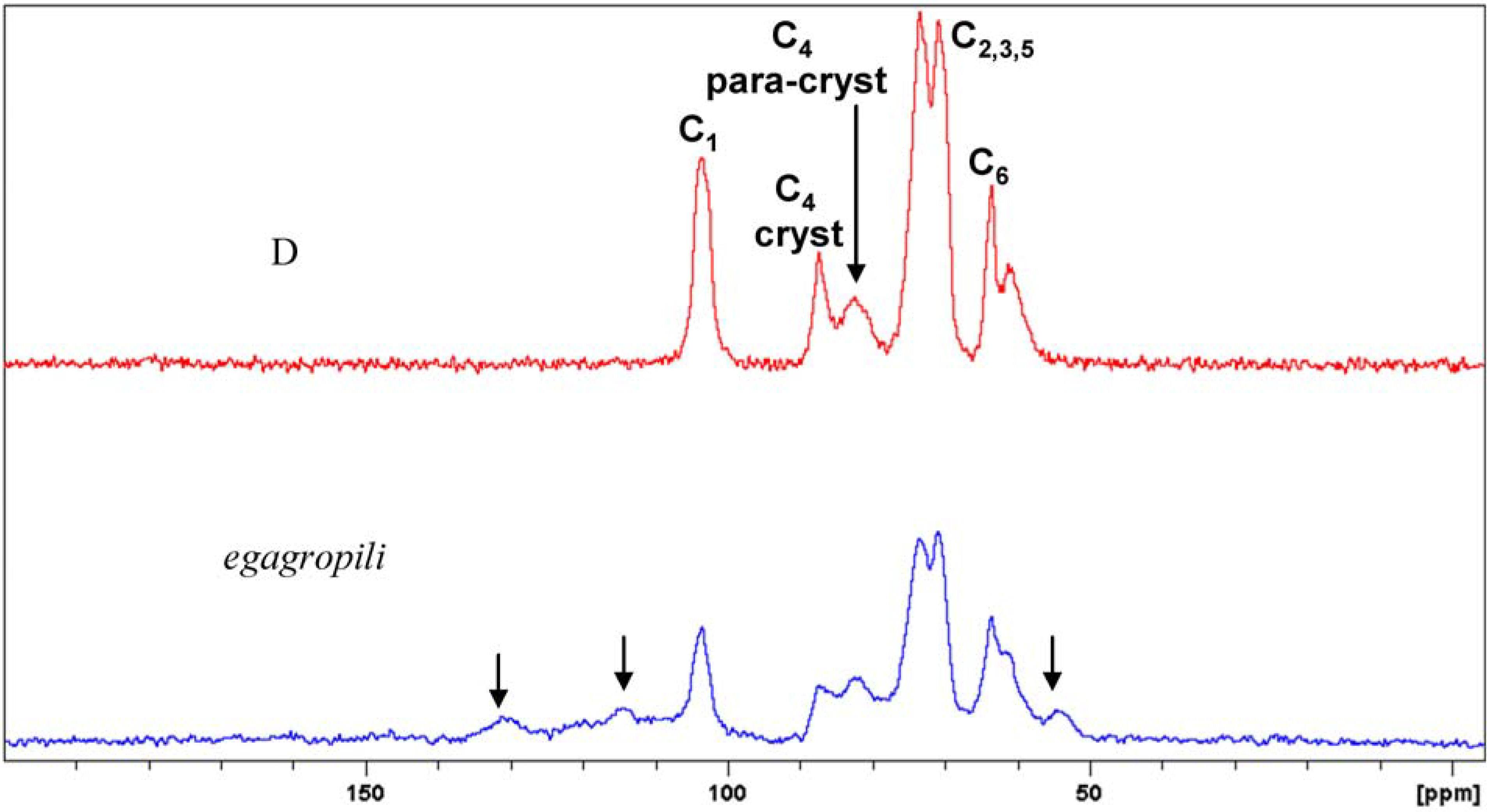
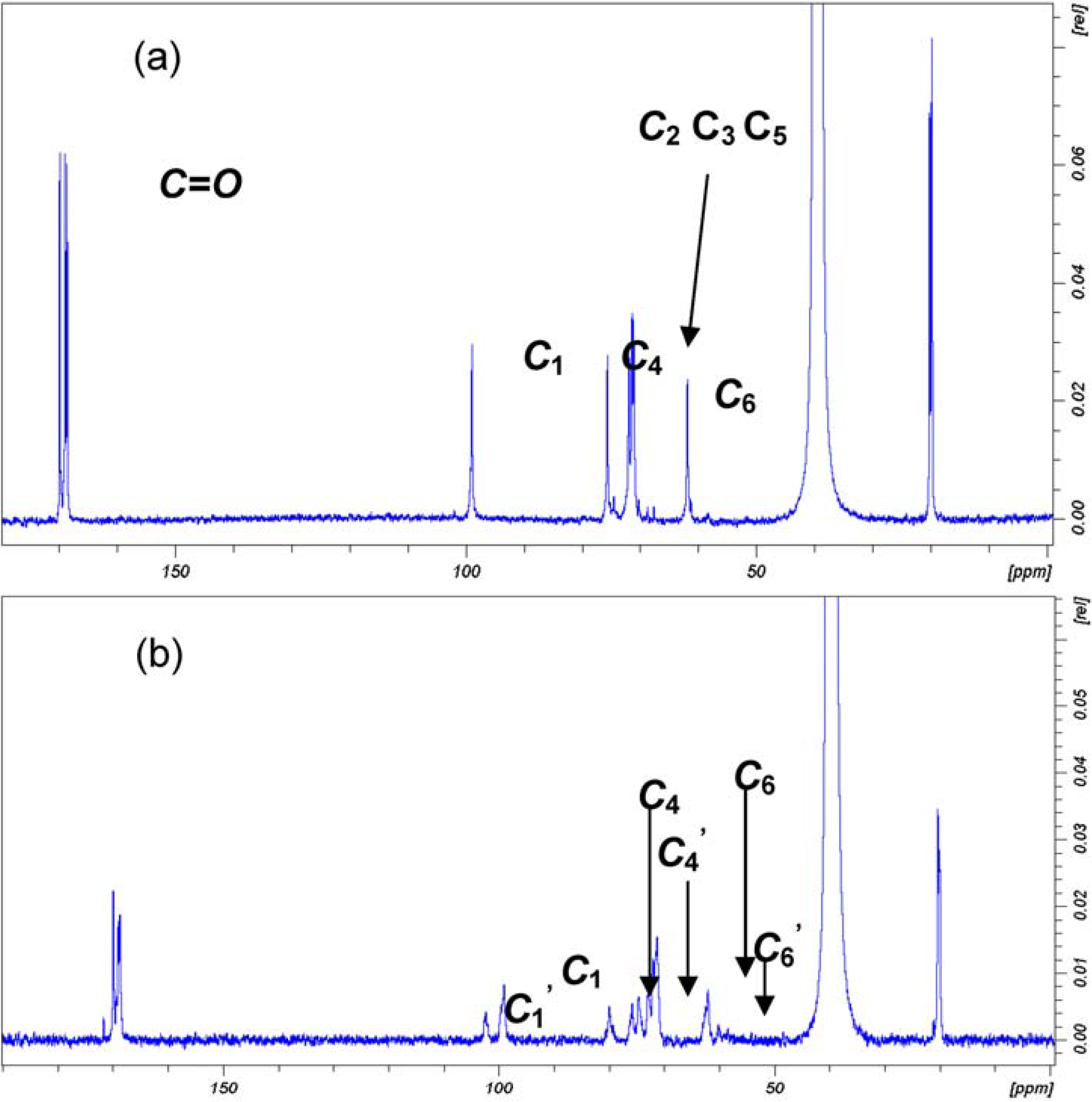
2.2. Synthesis and Characterisation of Cellulose Acetate (CA)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
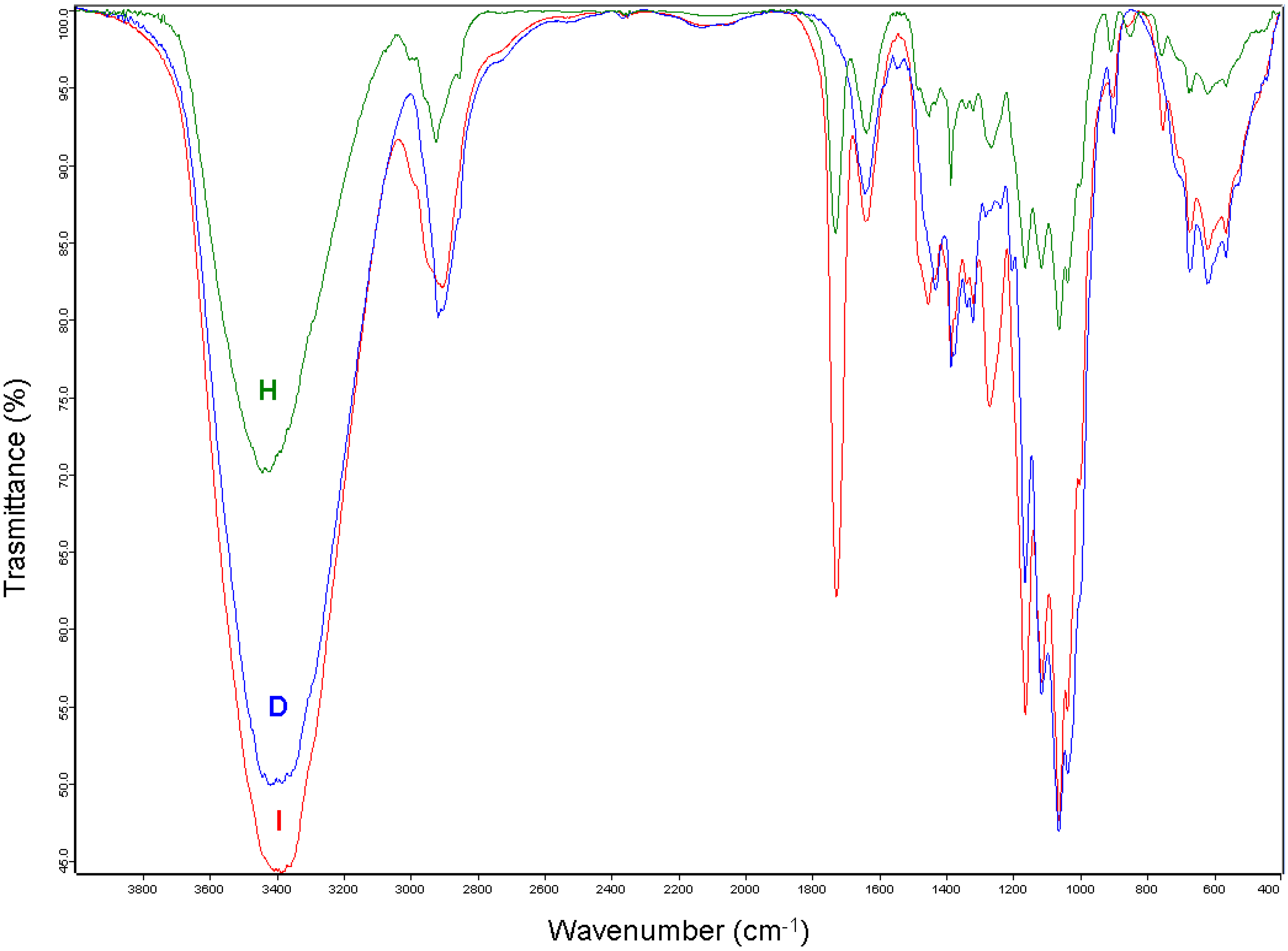
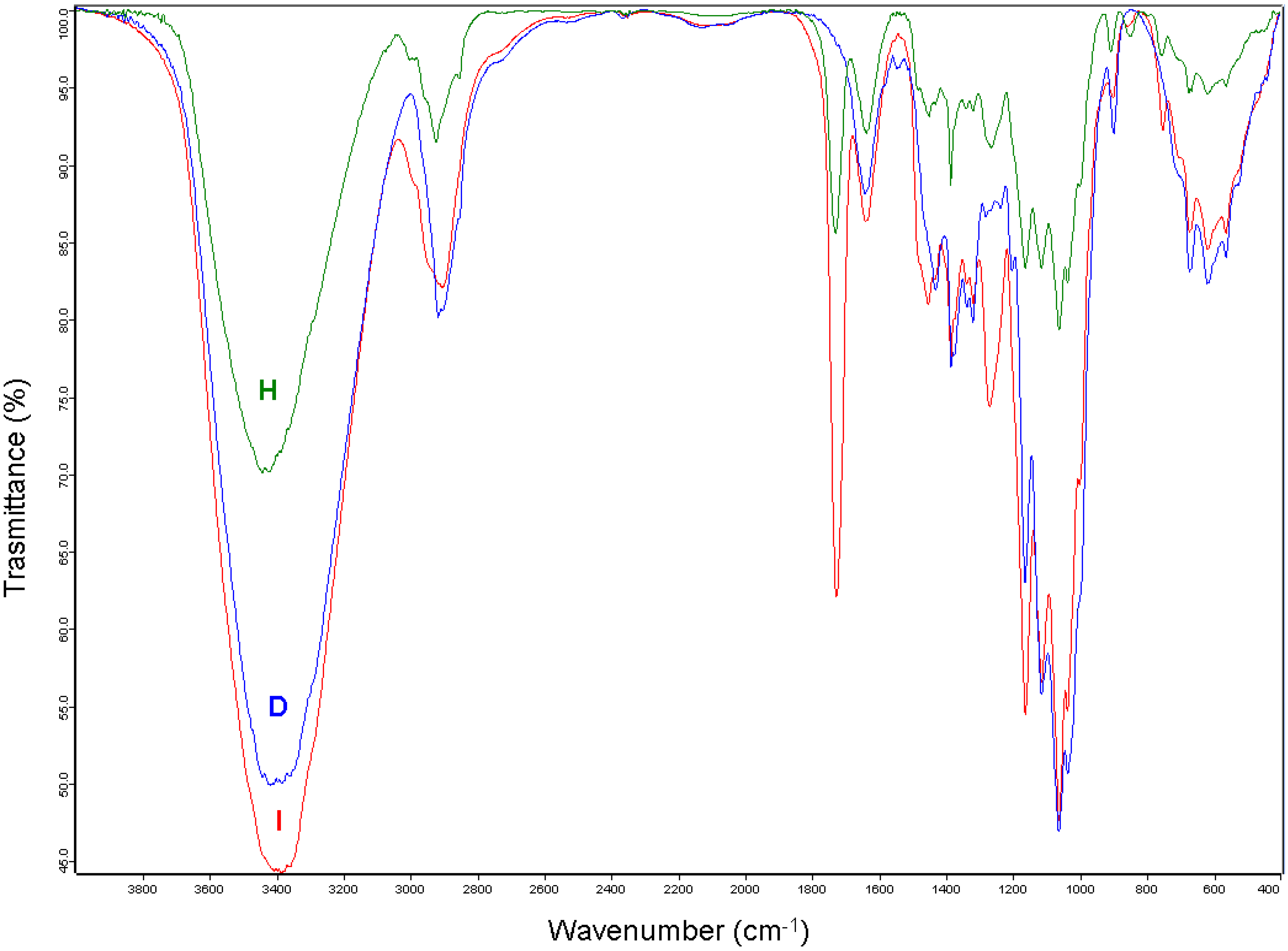{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | DStot | |||
|---|---|---|---|---|
| F | 3 | 3 | 3 | 3 |
| G | 0.71 | 0.64 | 0.85 | 2.2 |

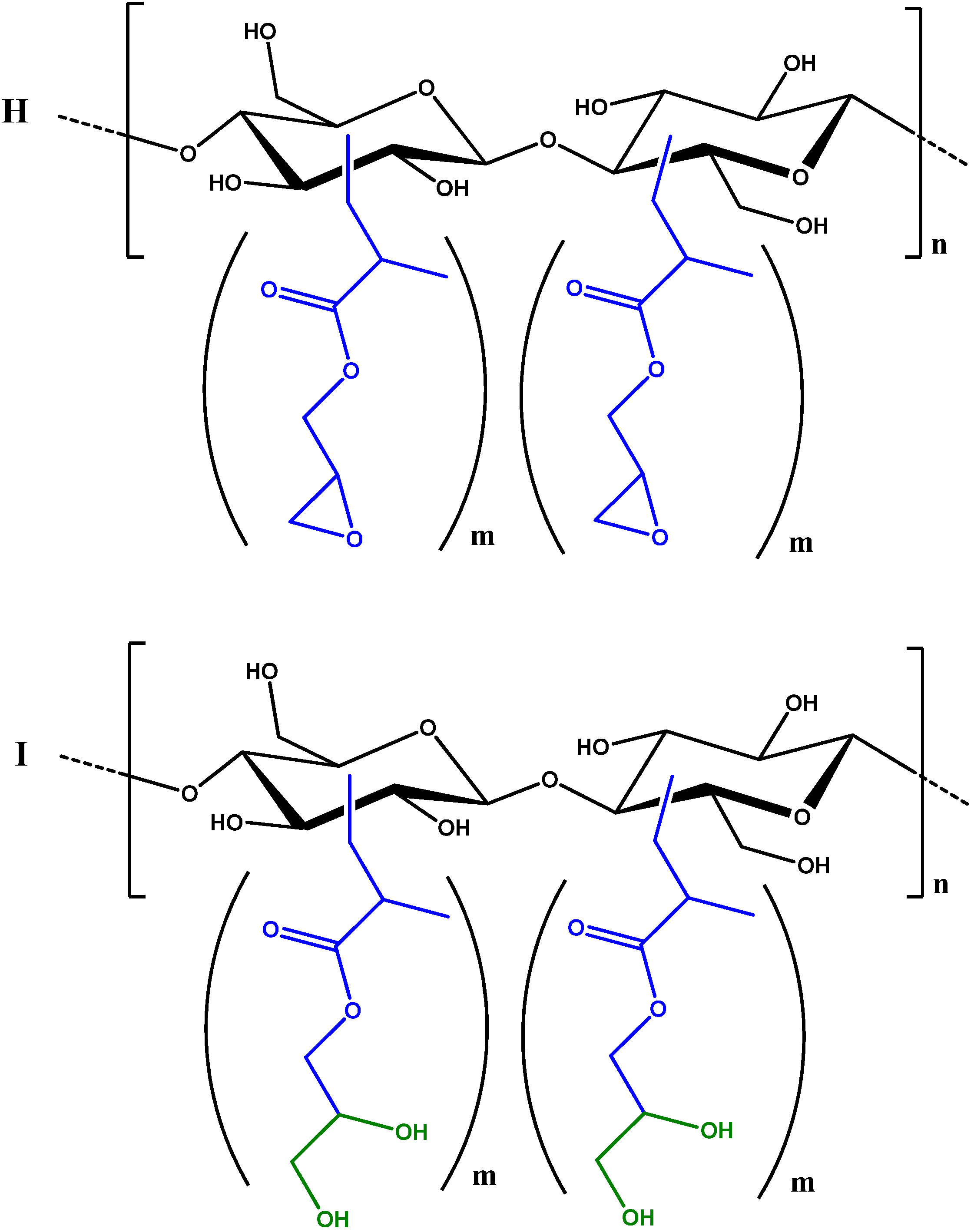
2.3. Synthesis and Characterisation of GMA Grafted Cellulose (GMA-C)
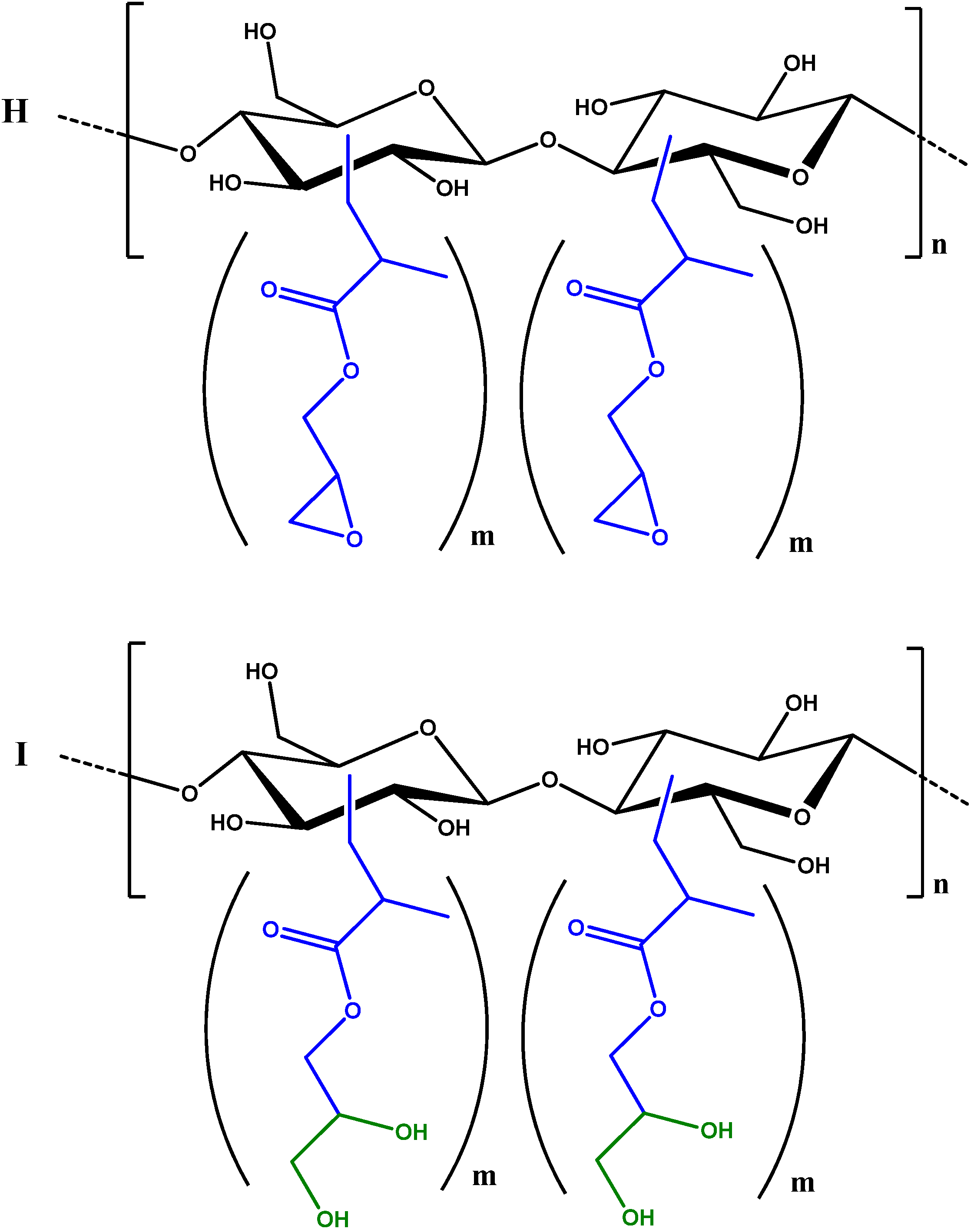
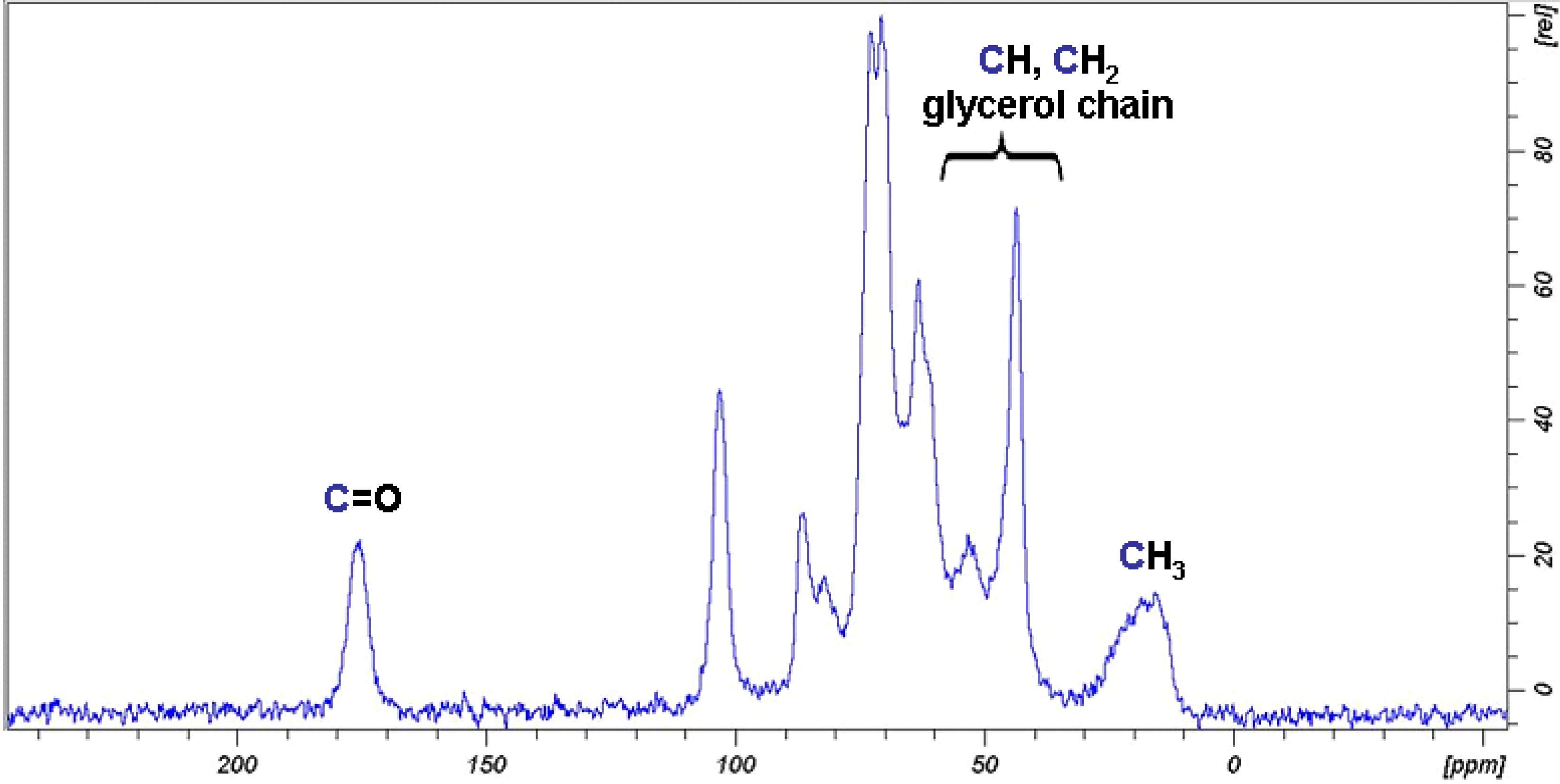
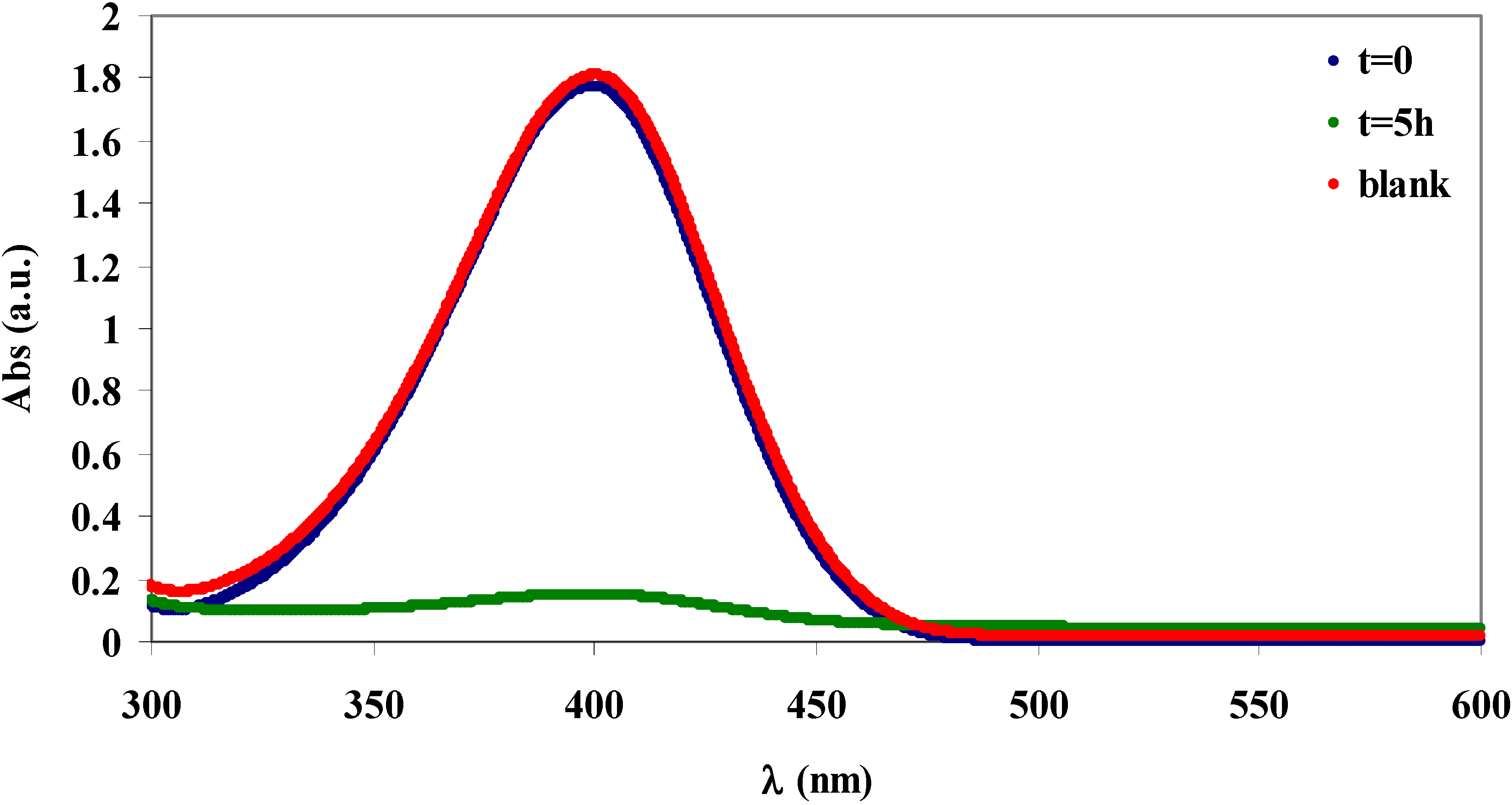
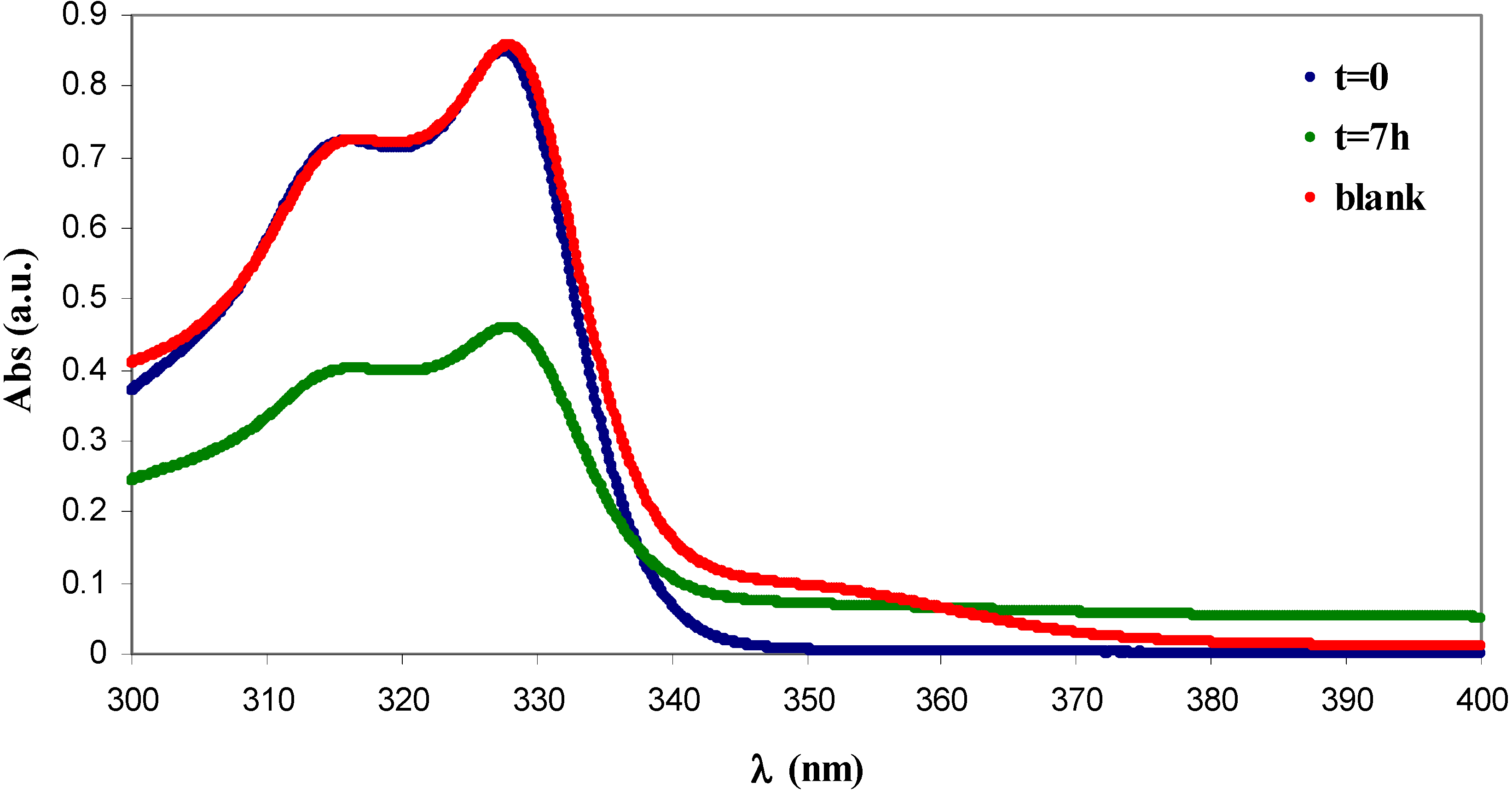
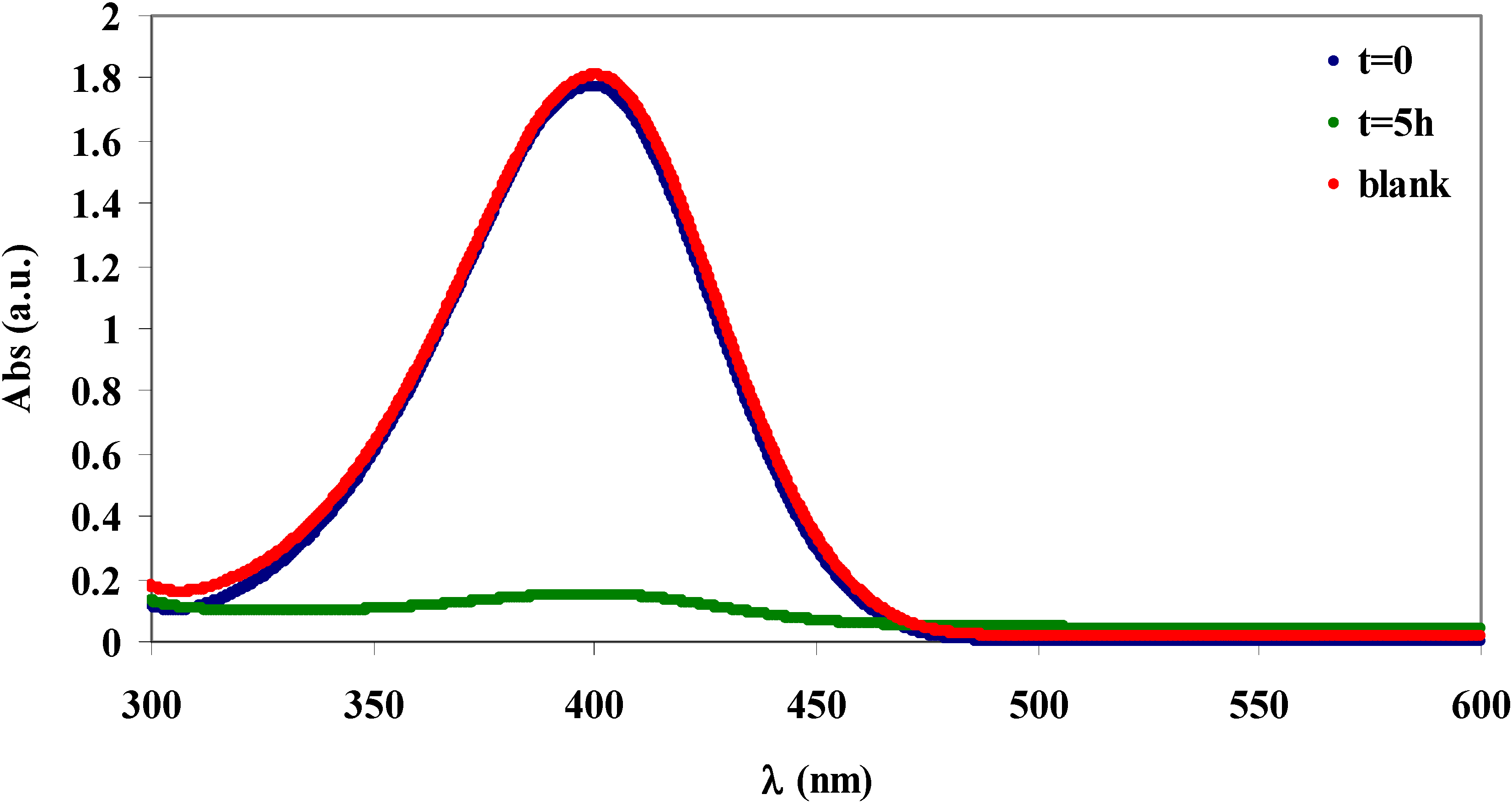
2.4. Absorption of β-naphthol and of p-nitrophenol
3. Experimental Section
3.1. Materials and Methods
3.2. Extraction of Cellulose
3.3. Determination of α-Cellulose Content
3.4. Solid State 13C CP MAS NMR
3.5. Synthesis of Cellulose Acetate
3.6. Solution 13C NMR
3.7. Functionalisation of Cellulose for the Adsorption of Organic Aromatic Pollutants
3.8. Quantification of GMA Grafting by FT-IR Spectroscopy
3.9. Absorption Experiments
4. Conclusions
Acknowledgements
References
- Fornes, A.; Basterretxea, G.; Orfila, A.; Jordi, A.; Alvarez, A.; Tintore, J. Mapping Posidonia oceanica from IKONOS. ISPRS J. Photogramm. 2006, 60, 315–322. [Google Scholar] [CrossRef]
- Dalla Via, J.; Sturmbauer, C.; Schönweger, G.; Sötz, E.; Mathekowitsch, S.; Stifter, M.; Rieger, R. Light gradients and meadow structure in Posidonia oceanica: Ecomorphological and functional correlates. Mar. Ecol. Progr. Ser. 1998, 163, 267–278. [Google Scholar]
- De Falco, G.; Simeone, S.; Baroli, M. Management of beach-cast posidonia oceanica seagrass on the island of sardinia (Italy, Western Mediterranean). J. Coast. Res. 2008, 24, 69–75. [Google Scholar]
- Khiari, R.; Mhenni, M.F.; Belgacem, M.N.; Mauret, E. Chemical composition and pulping of date palm rachis and Posidonia oceanic—A comparison with other wood and non-wood fibre sources. Bioresour. Technol. 2010, 101, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Khiari, R.; Mhenni, M.F.; Belgacem, M.N.; Mauret, E. Valorisation of vegetal wastes as a source of cellulose and cellulose derivatives. J. Polym. Environ. 2011, 19, 80–89. [Google Scholar] [CrossRef]
- Aguir, C.; Mhenni, M.F. Experimental study on carboxymethylation of cellulose extracted from Posidonia oceanica. J. Appl. Polym. Sci. 2006, 99, 1808–1816. [Google Scholar] [CrossRef]
- Khiari, R.; Ferchichi, I.; Mhenni, M.F. Study of liquids absorption and retention capacities of new cellulosic materials and sodium cellulose carboxylmethylate prepared from posidonia. Fibre Polym. 2010, 11, 593–597. [Google Scholar] [CrossRef]
- Keijsers, E.R.P.; Yılmaz, G.; van Dam, J.E.G. The cellulose resource matrix. Carbohydr. Polym. 2013, 93, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Liebert, T.F.; Heinze, T.J.; Edgar, K.J. Cellulose Solvents: For Analysis, Shaping and Chemical Modification; ACS Symposium Series 1033; American Chemical Society: Washington, DC, USA, 2010. [Google Scholar]
- Saka, S.; Matsumura, H. Wood pulp manifacturing and quality characteristics. Macromol. Symp. 2004, 208, 37–48. [Google Scholar] [CrossRef]
- Vismara, E.; Gastaldi, G.; Valerio, A.; Bertini, S.; Cosentino, C.; Eisele, G. Alpha cellulose from industrial and agricultural renewable sources like short flax fibres, ears of corn and wheat-straw and its transformation into cellulose acetates. J. Mater. Chem. 2009, 19, 8678–8686. [Google Scholar] [CrossRef]
- Rustemeyer, P. History of CA and evaluation of the markets. Macromol. Symp. 2004, 208, 1–6. [Google Scholar] [CrossRef]
- Mohanty, A.K.; Misra, M.; Drzal, L.T. Sustainable bio-composites from renewable resources: Opportunities and challenges in the green materials world. J. Polym. Environ. 2002, 10, 19–25. [Google Scholar] [CrossRef]
- Saka, S. How Raw Materials Influence the Quality of Cellulose Diacetate. Abstracts of Papers. In Proceedings of the 229th ACS National Meeting, San Diego, CA, USA, 13–17 March 2005.
- Mondal, M.I.H.; Alam, A.B.M.F. Utilization of cellulosic wastes in textile and garment industries: 2. Synthesis and characterisation of cellulose acetate from knitted rag. J. Polym. Environ. 2013, 21, 280–285. [Google Scholar] [CrossRef]
- He, J.; Zhang, M.; Cui, S.; Wang, S.-Y. High-quality cellulose triacetate prepared from bamboo dissolving pulp. J. Appl. Polym. Sci. 2009, 113, 456–465. [Google Scholar] [CrossRef]
- Torri, G.; Vismara, E.; Alberti, A.; Bertini, S.; Ciardelli, G.; Gastaldi, G.; Nesti, S. Free-Radical Functionalised Polysaccharides. US Patent 7,754,878 B2, 13 July 2010. [Google Scholar]
- Vismara, E.; Melone, L.; Gastaldi, G.; Cosentino, C.; Torri, G. Surface functionalisation of cotton cellulose with glycidyl methacrylate and its application for the absorption of aromatic pollutants from wastewaters. J. Hazard. Mater. 2009, 170, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Anastas, P.; Eghbali, N. Green chemistry: Principles and practice. Chem. Soc. Rev. 2010, 39, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Zeronian, S.H.; Inglesby, M.K. Bleaching of cellulose by hydrogen peroxide. Cellulose 1995, 2, 265–272. [Google Scholar] [CrossRef]
- Yannoni, C.S. High-resolution NMR in solids: The CPMAS experiment. Acc. Chem. Res. 1982, 15, 201–208. [Google Scholar] [CrossRef]
- Atalla, R.H.; VanderHart, D.L. The role of solid state 13C NMR spectroscopy in studies of the nature of native celluloses. Solid State Nucl. Magn. Reson. 1999, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.X.; Sun, X.F.; Zhao, H.; Suna, R.C. Isolation and characterisation of cellulose from sugarcane bagasse. Polym. Degrad. Stable 2004, 84, 331–339. [Google Scholar] [CrossRef]
- Focher, B.; Palma, M.T.; Canetti, M.; Torri, G.; Cosentino, C.; Gastaldi, G. Structural differences between non-wood plant cellulose: Evidence from solid state NMR, vibrational spectroscopy and X-ray diffractometry. Ind. Crops Prod. 2001, 13, 193–208. [Google Scholar] [CrossRef]
- Steinmeier, H. Chemistry of cellulose acetylation. Macromol. Symp. 2004, 208, 49–60. [Google Scholar] [CrossRef]
- Edgar, K.J.; Buchanan, C.M.; Debenham, J.S.; Rundquist, P.A.; Seiler, B.D.; Shelton, M.C.; Tindall, D. Advances in cellulose ester performance and application. Prog. Polym. Sci. 2001, 26, 1605–1688. [Google Scholar] [CrossRef]
- Rodrigues Filho, G.; Monteiro, D.S.; Meireles, C.S.; de Assunção, R.M.N.; Cerqueira, D.A.; Silva Barud, H.; Ribeiro, S.J.L.; Massadeq, Y. Synthesis and characterisation of cellulose acetate produced from recycled newspaper. Carbohydr. Polym. 2008, 73, 74–82. [Google Scholar] [CrossRef]
- Viera, R.G.P.; Filho, G.R.; de Assunção, R.M.N.; Meireles, C.S.; Vieira, J.; de Oliveira, G.S. Synthesis and characterisation of methylcellulose from sugar cane bagasse cellulose. Carbohydr. Polym. 2007, 67, 182–189. [Google Scholar] [CrossRef]
- Miyamoto, T.; Sato, Y.; Shibata, T.; Inagaki, H.; Tanahashi, M. 13C nuclear magnetic resonance studies of cellulose acetate. J. Polym. Sci. Polym. Chem. Ed. 1984, 22, 2363–2370. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Coletti, A.; Valerio, A.; Vismara, E. Posidonia oceanica as a Renewable Lignocellulosic Biomass for the Synthesis of Cellulose Acetate and Glycidyl Methacrylate Grafted Cellulose. Materials 2013, 6, 2043-2058. https://doi.org/10.3390/ma6052043
Coletti A, Valerio A, Vismara E. Posidonia oceanica as a Renewable Lignocellulosic Biomass for the Synthesis of Cellulose Acetate and Glycidyl Methacrylate Grafted Cellulose. Materials. 2013; 6(5):2043-2058. https://doi.org/10.3390/ma6052043
Chicago/Turabian StyleColetti, Alessia, Antonio Valerio, and Elena Vismara. 2013. "Posidonia oceanica as a Renewable Lignocellulosic Biomass for the Synthesis of Cellulose Acetate and Glycidyl Methacrylate Grafted Cellulose" Materials 6, no. 5: 2043-2058. https://doi.org/10.3390/ma6052043