Ultrafast Transient Spectroscopy of Polymer/Fullerene Blends for Organic Photovoltaic Applications

Abstract
:1. Introduction

2. Experimental
2.1. Photoexcitations Dynamics in Pristine P3HT
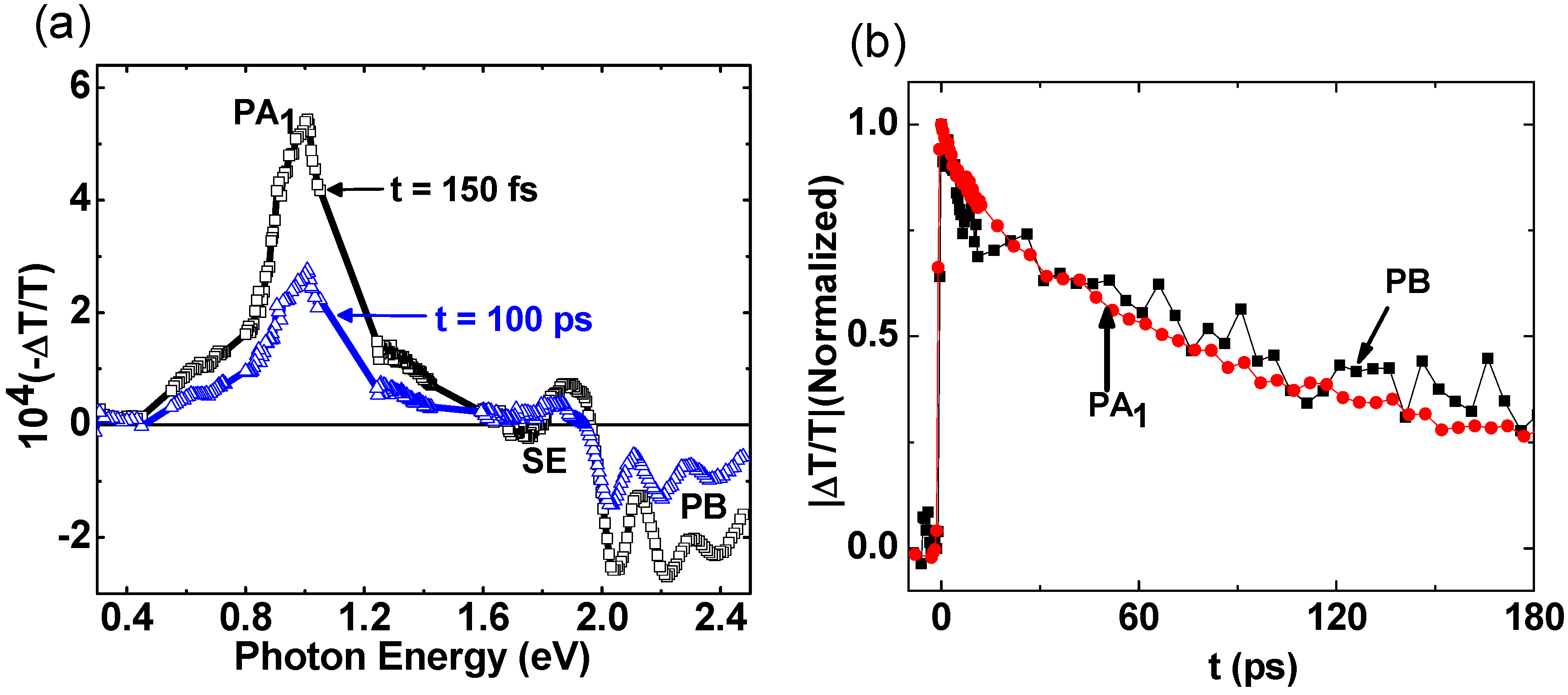
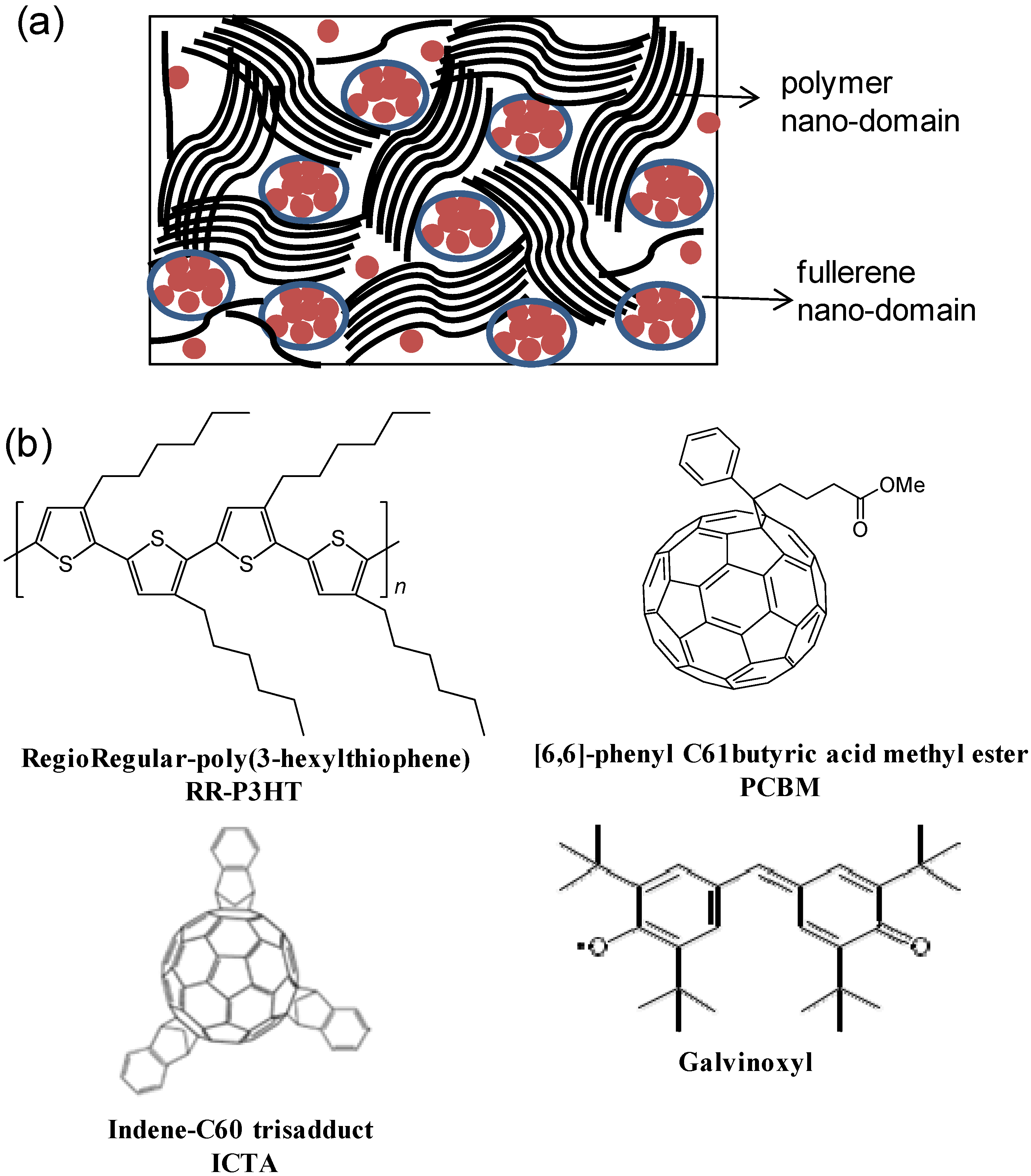
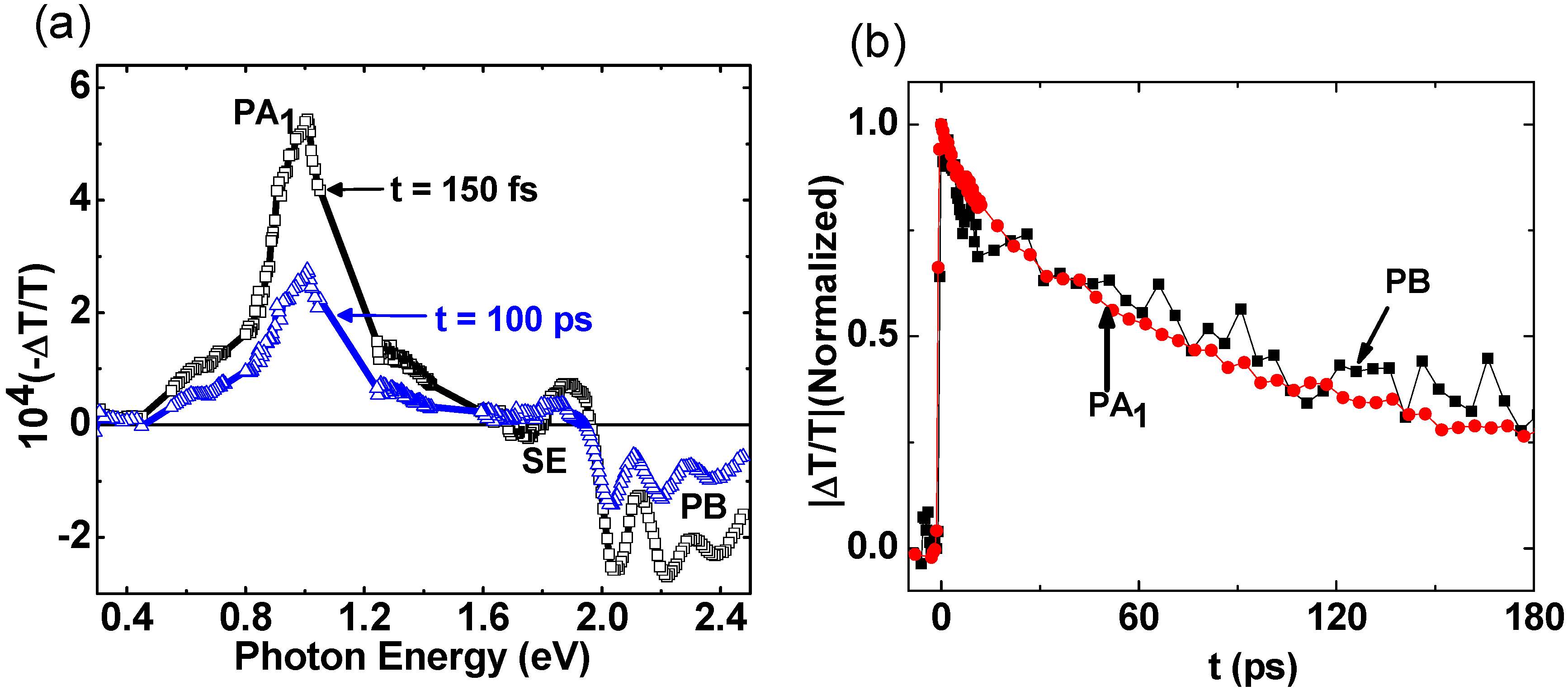
2.2. Photoexcitations Dynamics in P3HT/PCBM Blend

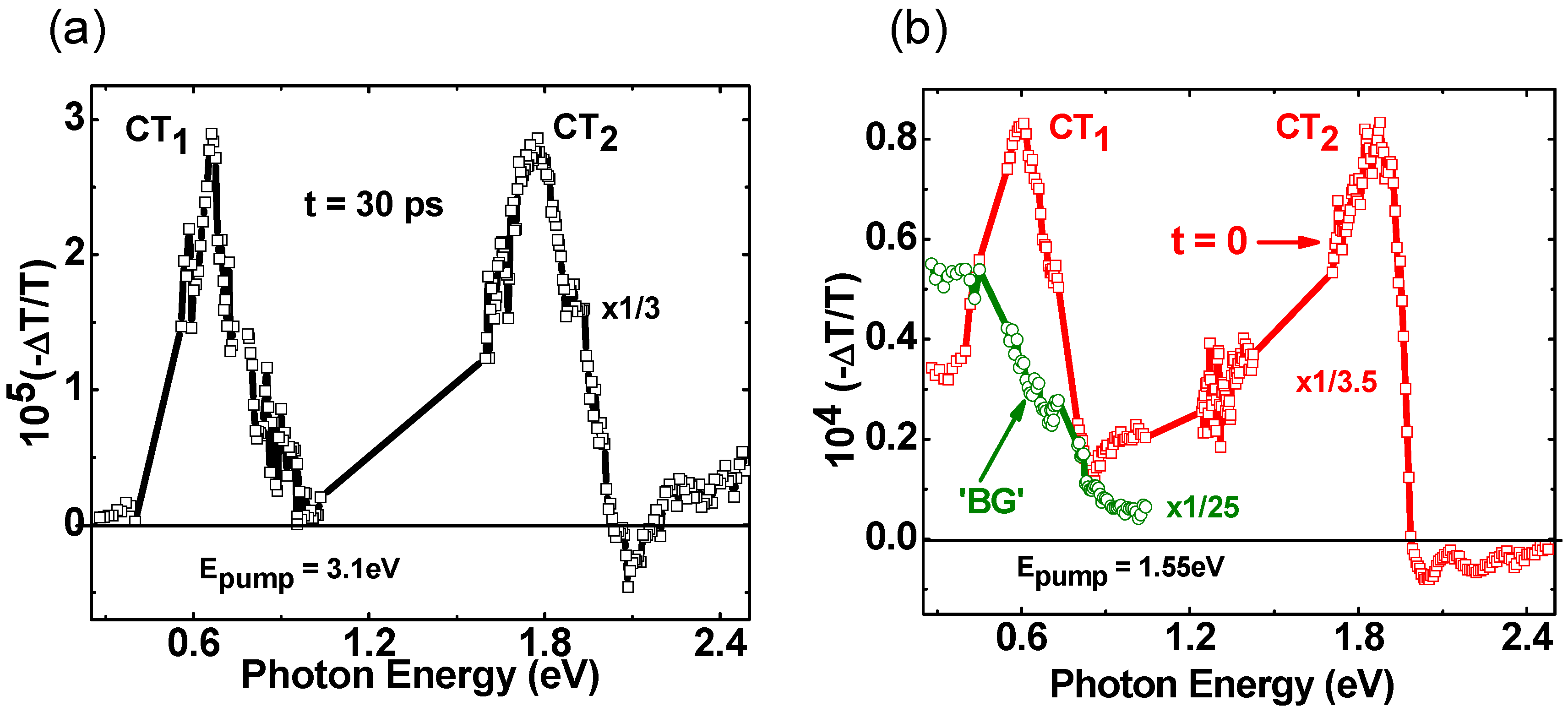
2.3. Photoexcitation Dynamics in P3HT/PCBM Blend Doped with Galvinoxyl Spin 1/2 Radicals


{kind=link}
{kind=link}
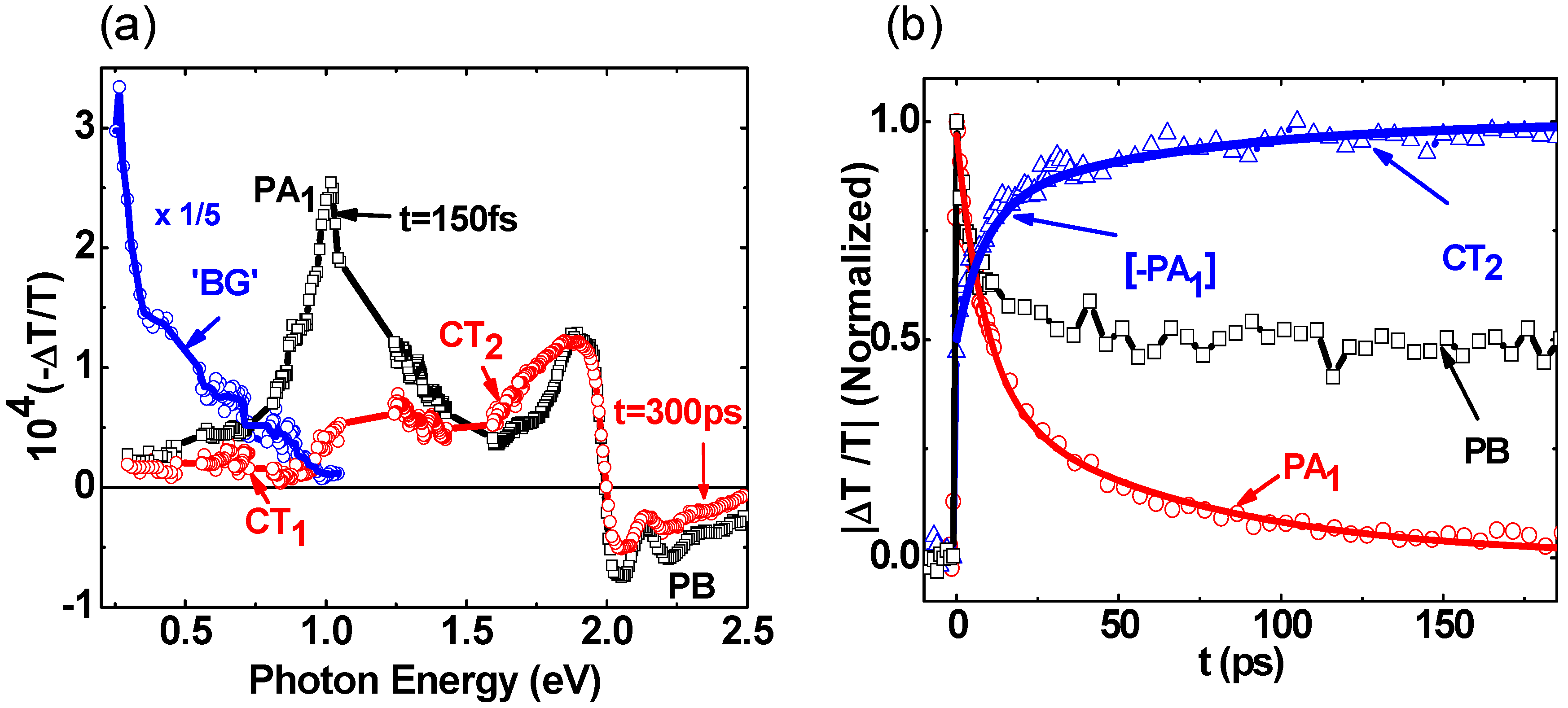{kind=link}
{kind=link}
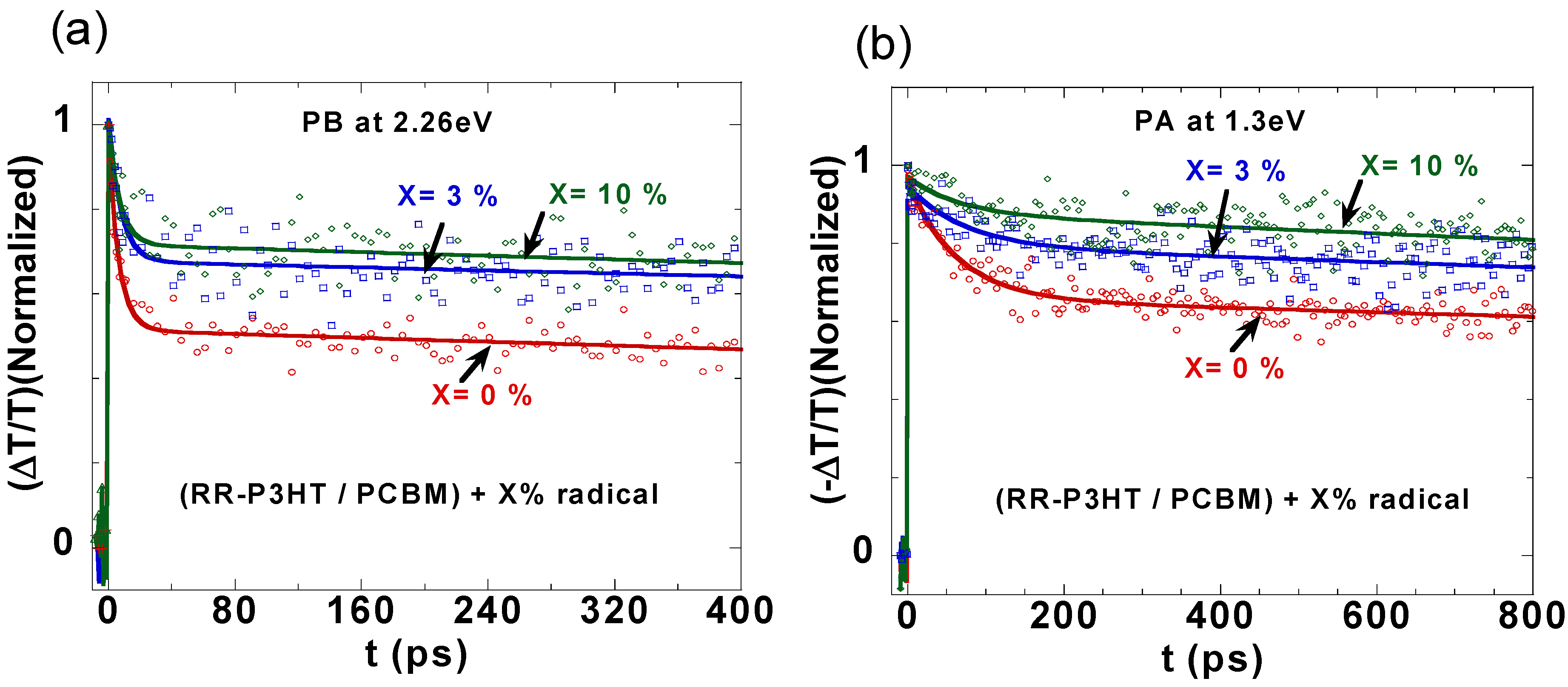{kind=link}
{kind=link}
| Galvinoxyl (%) | Transient Decay at 2.26 eV | Transient Decay at 1.3 eV | Jsc (mA/cm2) | Voc (V) | Fill-Factor (%) | Efficiency (%) |
|---|---|---|---|---|---|---|
| 0 | 7 ps (47%), >5 ns (53%) | 62 ps (32%), >5 ns (68%) | 11.3 | 0.57 | 55 | 3.46 |
| 3 | 8 ps (33%), >5 ns (67%) | 73 ps (16%), >5 ns (84%) | 12.7 | 0.58 | 60 | 4.20 |
| 10 | 7 ps (28%), >5 ns (72%) | 63 ps (10%), >5 ns (90%) | 11.2 | 0.61 | 53 | 3.55 |
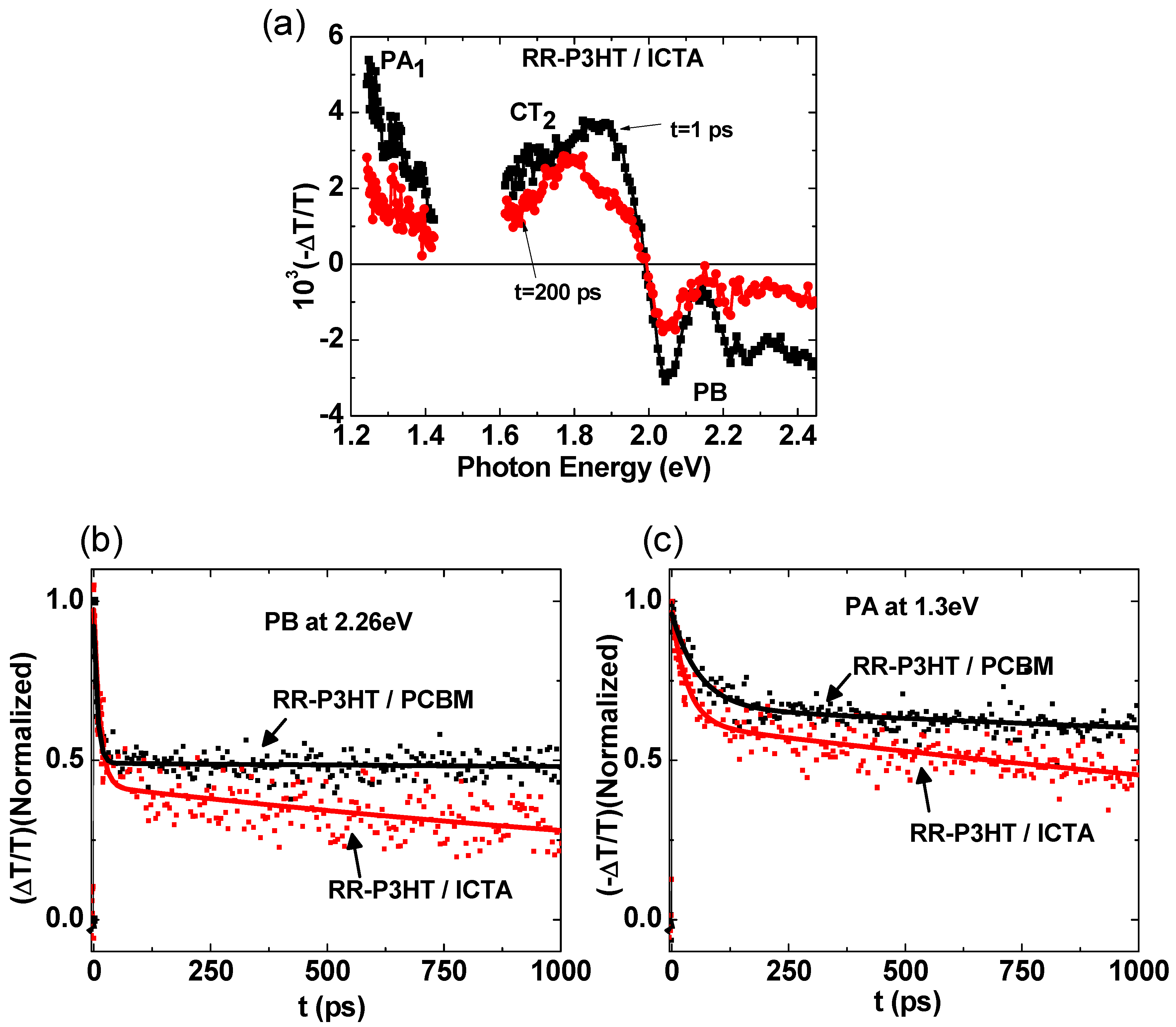
| Sample | Transient Decay at 2.26 eV | Transient Decay at 1.3 eV |
|---|---|---|
| RR-P3HT/PCBM | 7 ps (47%), >5 ns (53%) | 60 ps (32%), >5 ns (68%) |
| RR-P3HT/ICTA | 9 ps (57%), ~2.5 ns (43%) | 34 ps (36%), ~3.3 ns (64%) |
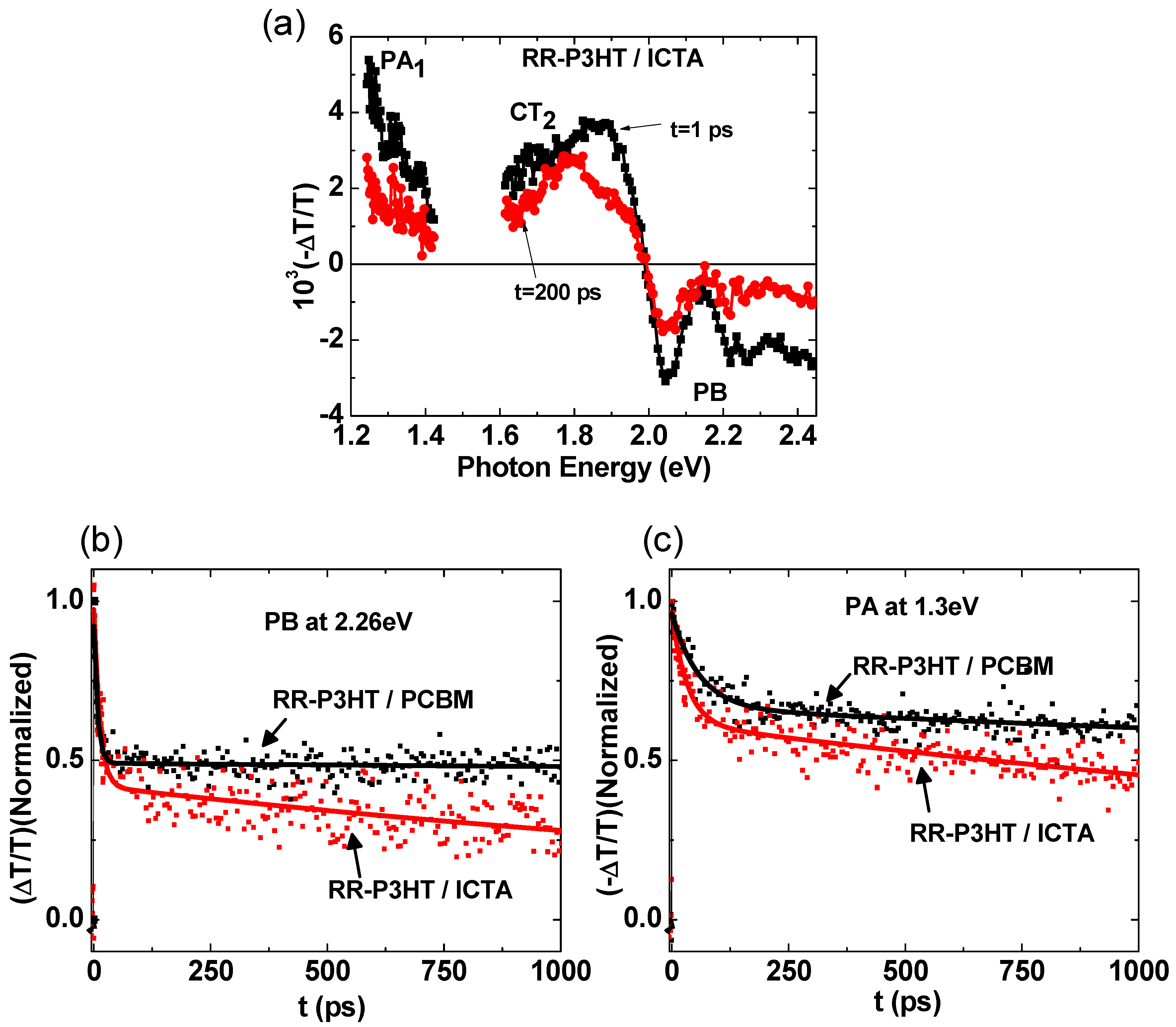
2.4. Photoexcitation Dynamics in P3HT/ICTA Blend
3. Conclusions
Acknowledgement
References
- Dou, L.; You, J.; Yang, J.; Chen, C.; He, Y.; Murase, S.; Moriarty, T.; Emery, K.; Li, G.; Yang, Y. Tandem polymer solar cells featuring a spectrally matched low-bandgap polymer. Nat. Photon 2012, 6, 180–185. [Google Scholar] [CrossRef]
- Green, M.A.; Emery, K.; Hishikawa, Y.; Warta, W. Solar cell efficiency tables (version 37). Prog. Photovolt. 2011, 19, 84–92. [Google Scholar] [CrossRef]
- Green, M.A.; Emery, K.; Hishikawa, Y.; Warta, W.; Dunlap, E.W. Solar cell efficiency tables (version 39). Prog. Photovolt. 2012, 20, 12–20. [Google Scholar] [CrossRef]
- Yin, W.; Dadmun, M. A new model for the morphology of P3HT/PCBM organic photovoltaics from small-angle neutron scattering: Rivers and streams. ACS Nano 2011, 5, 4756–4768. [Google Scholar]
- Treat, N.D.; Brady, M.A.; Smith, G.; Toney, M.F.; Kramer, E.J.; Hawker, C.J.; Chabinyc, M.L. Interdiffusion of P3HT and PCBM revels miscibility in a photovoltaiclly active blend. Adv. Energy Mater. 2011, 1, 82–89. [Google Scholar] [CrossRef]
- Clarke, T.M.; Durrant, J.R. Charge photogeneration in organic solar cells. Chem. Rev. 2010, 110, 6736–6767. [Google Scholar] [CrossRef] [PubMed]
- Deibel, C.; Strobel, T.; Dyakonov, V. Role of the charge transfer state in organic donor-acceptor solar cells. Adv. Mater. 2010, 22, 4097–4111. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Pandit, B.; Basel, T.P.; Li, S.; Laird, D.; Vardeny, Z.V. Two-step charge photogeneration dynamics in polymer/fullerene blends for photovoltaic applications. Phys. Rev. B 2012, 85, 205206:1–205206:7. [Google Scholar]
- Singh, S.; Pandit, B.; Hukic-Markosian, G.; Tek, P.B.; Vardeny, Z.V.; Li, S.; Laird, D. Ultrafast transient spectroscopy of nano-domains of polymer/fullerene blend for organic photovoltaic applications. J. Appl. Phys. 2012, 112, 123505:1–123505:6. [Google Scholar]
- Banerji, N.; Cowan, S.; Vauthey, E.; Heeger, A.J. Ultrafast relaxation of the Poly(3-hexylthiophene) emission spectrum. J. Phys. Chem. C 2011, 115, 9726–9739. [Google Scholar] [CrossRef]
- Hwang, I.-W.; Moses, D.; Heeger, A.J. Photoinduced carrier generation in P3HT/PCBM bulk heterojunction materials. J. Phys. Chem. C 2008, 112, 4350–4354. [Google Scholar] [CrossRef]
- Howard, I.A.; Mauer, R.; Meister, M.; Laquai, F. Effect of morphology on ultrafast free carrier generation in polythiophene:fullerene organic solar cells. J. Am. Chem. Soc. 2010, 132, 14866–14876. [Google Scholar] [CrossRef] [PubMed]
- Ayzner, A.L.; Tassone, C.J.; Tolbert, S.H.; Schwartz, B.J. Reappraising the need for bulk heterojunctions in polymer-fullerene photovoltaics: The role of carrier transport in all-solution-processed P3HT/PCBM bilayer solar cells. J. Phys. Chem. C 2009, 113, 20050–20060. [Google Scholar] [CrossRef]
- Aarnio, H.; Sehati, P.; Braun, S.; Nyman, M.; de Jong, M.P.; Fahlman, M.; Osterbacka, R. Spontaneous charge transfer and dipole formation at the interface between P3HT and PCBM. Adv. Energy Mater. 2011, 1, 792–797. [Google Scholar] [CrossRef]
- Lee, J.; Vandewal, K.; Yost, S.R.; Bhalke, M.E.; Goris, L.; Baldo, M.A.; Manca, J.V.; Voorhis, T.V. Charge transfer state versus hot exciton dissociation in polymer-fullerene blended solar cells. J. Am. Chem. Soc. 2010, 132, 11878–11880. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.; Katoh, R.; Furube, A. Ultrafast studies of charge generation in PCBM:P3HT blend films following excitation of the fullerene PCBM. J. Phys. Chem. C 2009, 113, 2547–2552. [Google Scholar] [CrossRef]
- Grancini, G.; Polli, D.; Fazzi, D.; Cabanilas-Gonzalez, J.; Cerullo, G.; Lanzani, G. Transient absorption imaging of P3HT:PCBM photovoltaic blend: Evidence for interfacial charge transfer state. J. Phys. Chem. Lett. 2011, 2, 1099–1105. [Google Scholar] [CrossRef]
- Howard, I.A.; Laquai, F. Optical probes of charge generation and recombination in bulk heterojunction organic solar cells. Macromol. Chem. Phys. 2010, 211, 2063–2070. [Google Scholar] [CrossRef]
- Veldman, D.; Meskers, S.C.J.; Janssen, R.A.J. The energy of charge-transfer states in electron donor-acceptor blends: Insight into the energy losses in organic solar cells. Adv. Funct. Mater. 2009, 19, 1939–1948. [Google Scholar] [CrossRef]
- Vandewal, K.; Tvingstedt, K.; Gadisa, A.; Inganas, O.; Manca, J.V. On the origin of the open-circuit voltage of polymer-fullerene solar cells. Nat. Mater. 2009, 8, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Keivanidis, P.E.; Clarke, T.M.; Lilliu, S.; Agostinelli, T.; Macdonald, J.E.; Durrant, J.R.; Bradley, D.D.C.; Nelson, J. Dependence of charge separation efficiency on film microstructure in poly(3-hexylthiophene-2,5-diyl):[6,6]-Phenyl-C-61 butyric acid methyl ester blend films. J. Phys. Chem. Lett. 2010, 1, 734–738. [Google Scholar] [CrossRef]
- Snedden, E.W.; Monkman, A.P.; Dias, F.B. Photophysics of charge generation in organic photovoltaic materials: Kinetic studies of geminate and free polarons in a model donor/acceptor system. J. Phys. Chem. C 2012, 116, 86–97. [Google Scholar] [CrossRef]
- Parkinson, P.; Lloyd-Hughes, J.; Johnston, M.B.; Herz, L.M. Efficient generation of charges via below-gap photoexcitation of polymer-fullerene blend films investigated by terahertz spectroscopy. Phys. Rev. B 2008, 78, 115321:1–115321:11. [Google Scholar] [CrossRef]
- Bredas, J.-L.; Norton, J.E.; Cornil, J.; Coropceanu, V. Molecular understanding of organic solar cells: The challenges. Acc. Chem. Res. 2009, 42, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Sariciftci, N.S.; Smilowitz, L.; Heeger, A.J.; Wudl, F. Photoinduced electron transfer from a conducting polymer to buckminsterfullerene. Science 1992, 258, 1474–1476. [Google Scholar] [CrossRef] [PubMed]
- Goris, L.; Poruba, A.; Hod’akova, L.; Vanecek, M.; Haenen, K.; Nesladek, M.; Wagner, P.; Vanderzande, D.; Schepper, L.D.; Manca, J.V. Observation of the subgap optical absorption in polymer-fullerene blend solar cells. Appl. Phys. Lett. 2006, 88, 052113:1–052113:3. [Google Scholar] [CrossRef]
- Drori, T.; Sheng, C.X.; Ndobe, A.; Singh, S.; Holt, J.; Vardeny, Z.V. Below-gap excitation of π-conjugated polymer-fullerene blends: Implications for bulk organic heterojunction solar cells. Phys. Rev. Lett. 2008, 101, 037401:1–037401:4. [Google Scholar] [CrossRef]
- Benson-Smith, J.J.; Goris, L.; Vandewal, K.; Haenen, K.; Manca, J.V.; Vanderzande, D.; Bradley, D.D.C.; Nelson, J. Formation of a ground-state charge-transfer complex in polyfluorene/[6,6]-phenyl-C61 butyric acid methyl ester (PCBM) blend films and its role in the function of polymer/PCBM solar cells. Adv. Funct. Mater. 2007, 17, 451–457. [Google Scholar] [CrossRef]
- Bakulin, A.A.; Rao, A.; Pavelyev, V.G.; van Loosdrecht, P.H.M.; Pshenichnikov, M.S.; Niedzialek, D.; Cornil, J.; Beljonne, D.; Friend, R.H. The role of driving energy and delocalized states for charge separation in organic semiconductors. Science 2012, 335, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Holt, J.; Singh, S.; Drori, T.; Zheng, Y.; Vardeny, Z.V. Optical probes of π-conjugated polymer blends with strong acceptor molecules. Phys. Rev. B 2009, 79, 195210:1–195210:8. [Google Scholar] [CrossRef]
- Drori, T.; Holt, J.; Vardeny, Z.V. Optical studies of the charge transfer complex in polythiophene/fullerene blends for organic photovoltaic applications. Phys. Rev. B 2010, 82, 075207:1–075207:8. [Google Scholar] [CrossRef]
- Singh, S.; Zhang, Y.; Vardeny, Z.V. Effect of spin 1/2 radicals on the ultrafast photoexcitation dynamics in RR-P3HT/PCBM blends for photovoltaic applications. Synth. Meter. 2010, 160, 311–313. [Google Scholar] [CrossRef]
- Zhang, Y.; Basel, T.P.; Gautam, B.R.; Yang, X.; Mascaro, D.J.; Liu, F.; Vardeny, Z.V. Spin-enhanced organic bulk heterojunction photovoltaic solar cells. Nat. Commun. 2012, 3, 1043:1–1043:7. [Google Scholar]
- Li, G.; Zhu, R.; Yang, Y. Polymer solar cells. Nat. Photon. 2012, 6, 153–161. [Google Scholar] [CrossRef]
- Sheng, C.X.; Tong, M.; Singh, S.; Vardeny, Z.V. Experimental determination of the charge/neutral branching ratio η in the photoexcitation of π-conjugated polymers by broadband ultrafast spectroscopy. Phys. Rev. B 2007, 75, 085206:1–085206:7. [Google Scholar]
- Holt, J. Ultrafast optical measurements of charge generation and transfer mechnisms of π-conjugated polymers for solar cell applications. Ph.D. Thesis, University of Utah, UT, USA, 2009. [Google Scholar]
- Hukic-Markosian, G.; Basel, T.; Singh, S.; Vardeny, Z.V.; Li, S.; Laird, D. Study of photoexcitations in poly(3-hexylthiophene) for photovoltaic applications. Appl. Phys. Lett. 2012, 100, 213903:1–213903:5. [Google Scholar] [CrossRef]
- Osterbacka, R.; An, C.P.; Jiang, X.M.; Vardeny, Z.V. Two-dimensional electronic excitations in self-assembled conjugated polymer nanocrystals. Science 2000, 287, 839–842. [Google Scholar] [CrossRef] [PubMed]
- Forster, T. Transfer mechanisms of electronic excitation. Discuss. Faraday. Soc. 1959, 27, 7–17. [Google Scholar] [CrossRef]
- Coffey, D.C.; Ferguson, A.J.; Kopidakis, N.; Rumbles, G. Photovoltaic charge generation in organic semiconductors based on long-range energy transfer. ACS Nano 2010, 4, 5437–5445. [Google Scholar] [CrossRef] [PubMed]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Singh, S.; Vardeny, Z.V. Ultrafast Transient Spectroscopy of Polymer/Fullerene Blends for Organic Photovoltaic Applications. Materials 2013, 6, 897-910. https://doi.org/10.3390/ma6030897
Singh S, Vardeny ZV. Ultrafast Transient Spectroscopy of Polymer/Fullerene Blends for Organic Photovoltaic Applications. Materials. 2013; 6(3):897-910. https://doi.org/10.3390/ma6030897
Chicago/Turabian StyleSingh, Sanjeev, and Zeev Valy Vardeny. 2013. "Ultrafast Transient Spectroscopy of Polymer/Fullerene Blends for Organic Photovoltaic Applications" Materials 6, no. 3: 897-910. https://doi.org/10.3390/ma6030897