Two Decades of Negative Thermal Expansion Research: Where Do We Stand?


Department of Chemistry, the University of Toledo, Toledo, OH 43606, USA
Materials 2012, 5(6), 1125-1154; https://doi.org/10.3390/ma5061125
Submission received: 16 April 2012
/
Revised: 1 June 2012
/
Accepted: 15 June 2012
/
Published: 20 June 2012
(This article belongs to the Special Issue Negative Thermal Expansion Materials)
Abstract
:Negative thermal expansion (NTE) materials have become a rapidly growing area of research over the past two decades. The initial discovery of materials displaying NTE over a large temperature range, combined with elucidation of the mechanism behind this unusual property, was followed by predictions that these materials will find use in various applications through controlled thermal expansion composites. While some patents have been filed and devices built, a number of obstacles have prevented the widespread implementation of NTE materials to date. This paper reviews NTE materials that contract due to transverse atomic vibrations, their potential for use in controlled thermal expansion composites, and known problems that could interfere with such applications.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
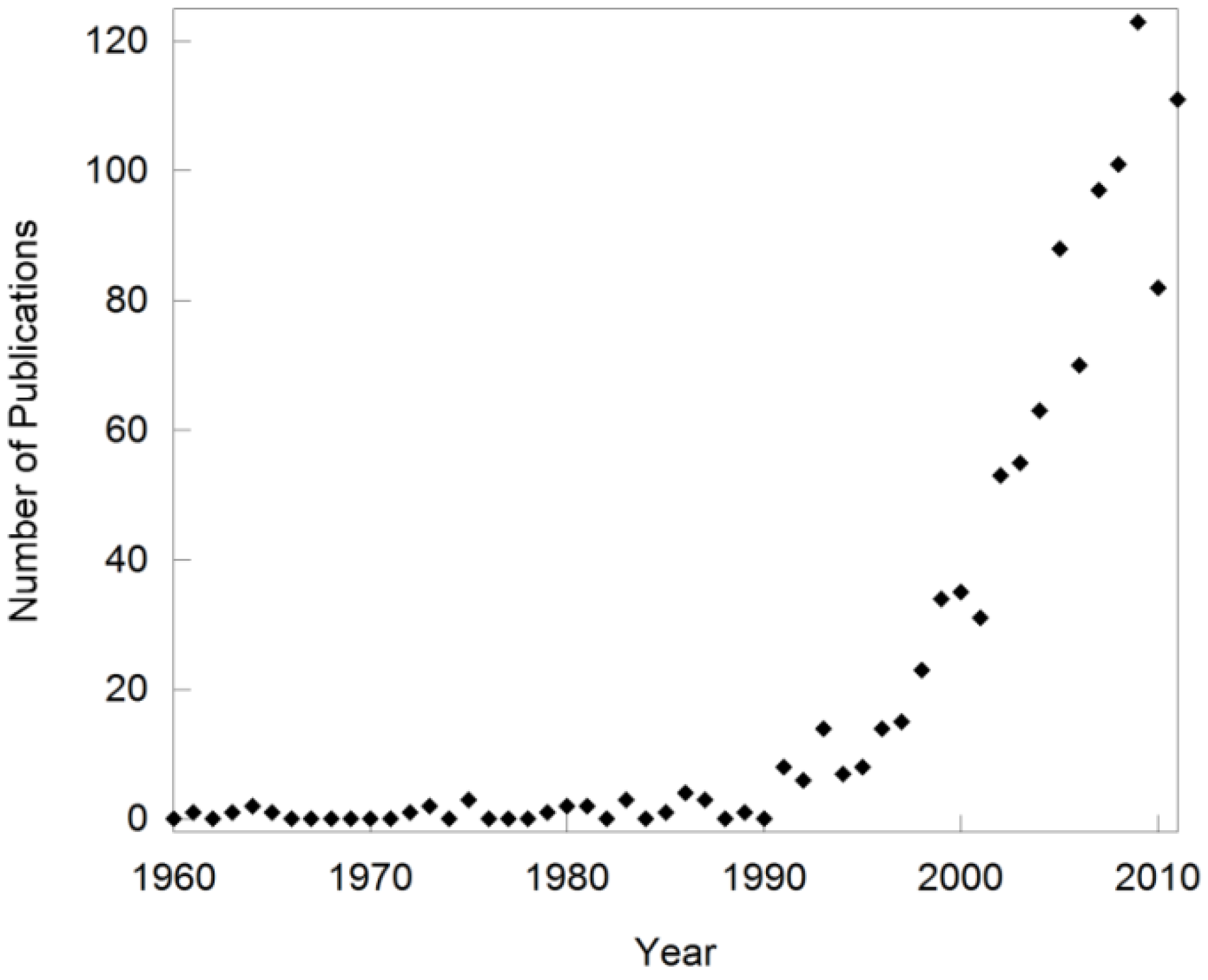
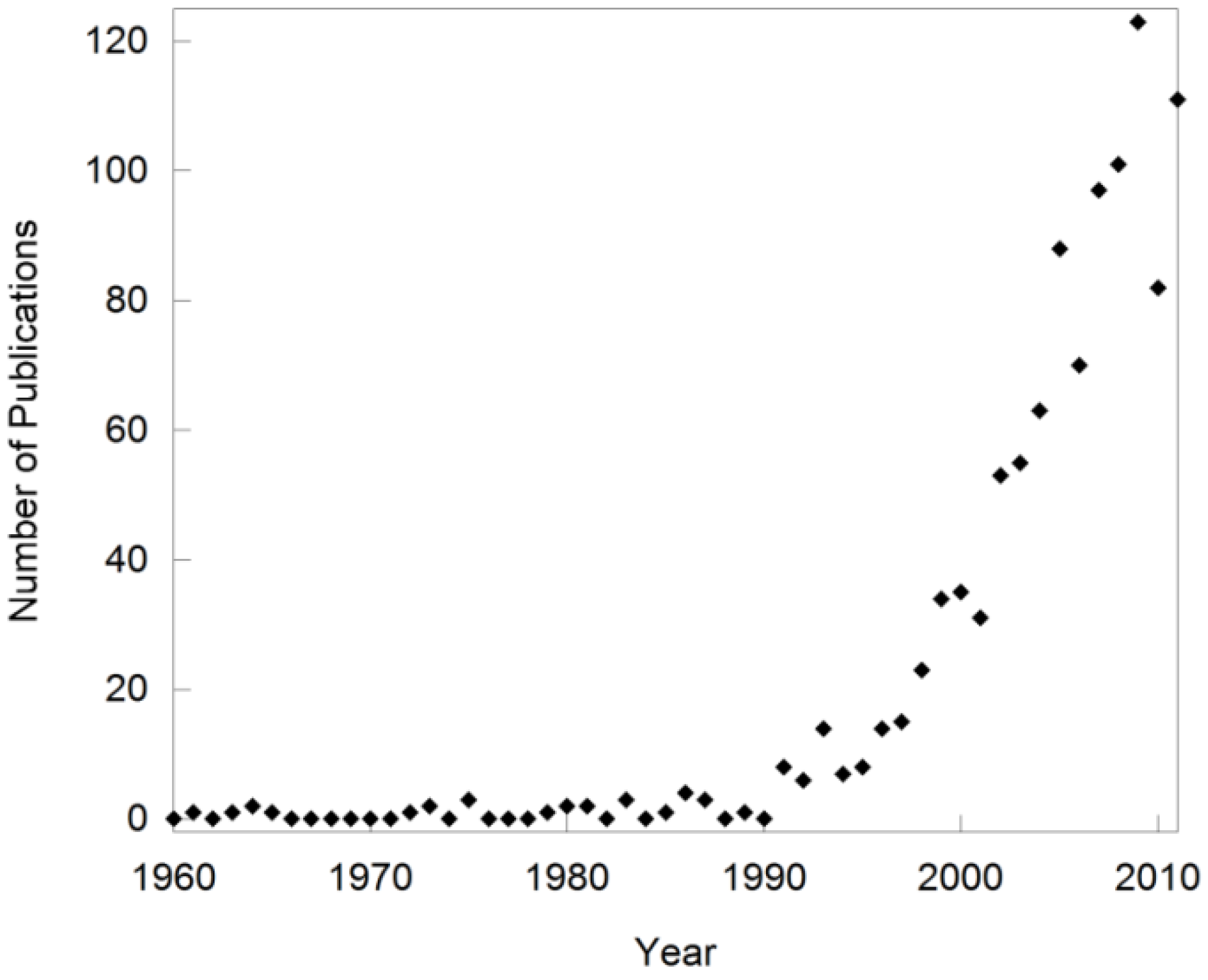
Over the past two decades, the field of negative thermal expansion (NTE) has rapidly expanded [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24]. This is evident when tracking the number of publications on “negative thermal expansion” over the past 50 years (Figure 1), and the fact that special issues of journals have been devoted to the topic [25]. Such sudden growth of new research fields is often related to the discovery of a new phenomenon or a new class of compounds. However, the first observation of compounds that contract upon heating dates back several hundred years to the discovery of the “density anomaly of water”. Shrinkage of a solid was first documented by Scheel in 1907 for quartz and vitreous silica at low temperatures [26,27], and additional reports of materials that contract over various temperature ranges appeared in the literature throughout the years. This included research on lithium aluminum silicates (LAS) in the 1950s [28,29], and the discovery of the sodium zirconate phosphate (NZP) family in the 1980s [30,31,32,33]. These materials can show either positive or negative volume expansion depending on composition, as contraction is observed along only one or two of the crystallographic axes. They were usually referred to as “low expansion ceramics” instead of “NTE materials”, and the term “NTE” was used only sporadically between the 1960s and the 1990s. Notably, the expansion behavior of ZrW2O8 [34], which has since become one of the most researched NTE compounds and is often used as the key representative of NTE, was measured over the temperature range 323 to 973 K in 1968 [35]. However, this behavior remained a peculiarity until the mid 1990’s, when Sleight’s group could show that the NTE behavior of several families of compounds was intimately related to their crystal structures [1,2,3,5]. This included the first observation of inherently isotropic NTE over a large temperature range in cubic ZrV2O7 [1] and ZrW2O8 [3]. Theoretical and experimental studies soon established sophisticated models that can be used to explain this unusual behavior for a number of framework compounds [6,8,11,12,15,16,21,36,37,38,39,40,41,42,43,44]. This opened up the targeted synthesis of new NTE compositions, and established NTE as a specialized field of research. Several new families of materials in which NTE is caused by different mechanisms have been discovered as well, but they are outside the scope of this review. Compounds belonging to the LAS and NZP families will not be discussed in detail either, as they were already well established as low expansion ceramics by the time NTE became a separate field of research.
Figure 1.
Number of publications per year based on a Web of Science search for “negative thermal expansion”. Note that early publications related to the field are missing as they did not use NTE as a keyword.
Figure 1.
Number of publications per year based on a Web of Science search for “negative thermal expansion”. Note that early publications related to the field are missing as they did not use NTE as a keyword.
The potential uses of NTE materials in controlled thermal expansion composites were readily recognized, and possible applications ranging from fiber optics coatings, electronics and mirror substrates to tooth fillings were proposed [9,45,46,47,48]. However, some limitations of different NTE materials became quickly apparent, one of which relates to the fact that many NTE compounds contain transition metals, which would increase product cost. In addition, problems with stability under processing and use conditions, and incompatibilities with other composite components were encountered [49,50,51,52]. These challenges have become active areas of research, and efforts are directed at the discovery of new NTE materials, improvements of properties of existing materials, modification of particles to achieve compatibility, and establishing processing conditions for formation of homogeneous composites.
2. Negative Thermal Expansion Due to Transverse Vibrations
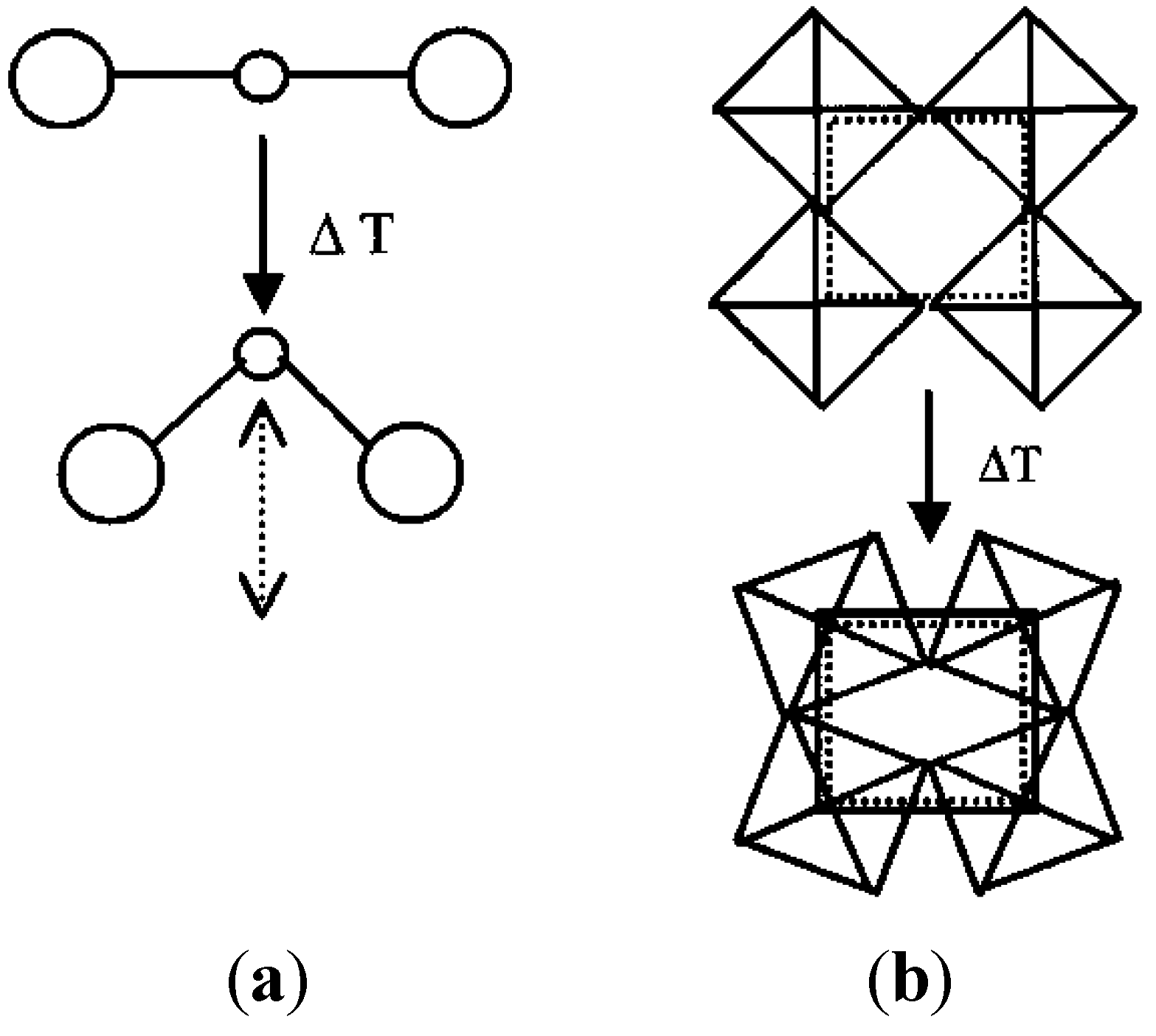
The expansion behavior of most NTE materials that were known or discovered in the 1990’s can be explained based on their crystal structure. These compounds are composed of rigid MO4 tetrahedra and/or MO6 octahedra, which are connected by corner-sharing oxygen atoms. Due to the corner-sharing nature of the frameworks, the polyhedra can undergo concerted tilting or rocking motions when transverse vibrations of the corner-sharing oxygen atoms are excited. For approximately linear M-O-M linkages, this process leads to a reduction of second-nearest-neighbor distances, and can result in linear or volume NTE (Figure 2). This mechanism operates in materials belonging to the zirconium tungstate family [3,53,54], scandium tungstate family [5,23], zirconium vanadate family [1], a number of zeolites and aluminum phosphates [24,55,56], Prussian blue analogs [57,58], and a few other materials [59]. Early theoretical models treated the polyhedra as rigid units, and referred to the concerted lattice vibrations as “rigid unit modes”, or RUMs [37,59,60,61,62].
The RUM model can adequately describe the NTE behavior observed in ZrW2O8 and some zeolites, however, the polyhedra in the ZrV2O7 and Sc2W3O12 families have been found to distort. These distortions have resulted in more varied values for expansion coefficients for the same structural family depending on the size and rigidity of the polyhedra. Similar behavior has also been observed in some cyanides, where the CN linkages undergo vibrations that lead to a shortening of metal-metal distances. Due to the greater flexibility of the two-atom linker, the expansion behavior can vary widely from strong NTE to positive expansion. In some cyanide frameworks, other mechanisms also contribute to NTE behavior.
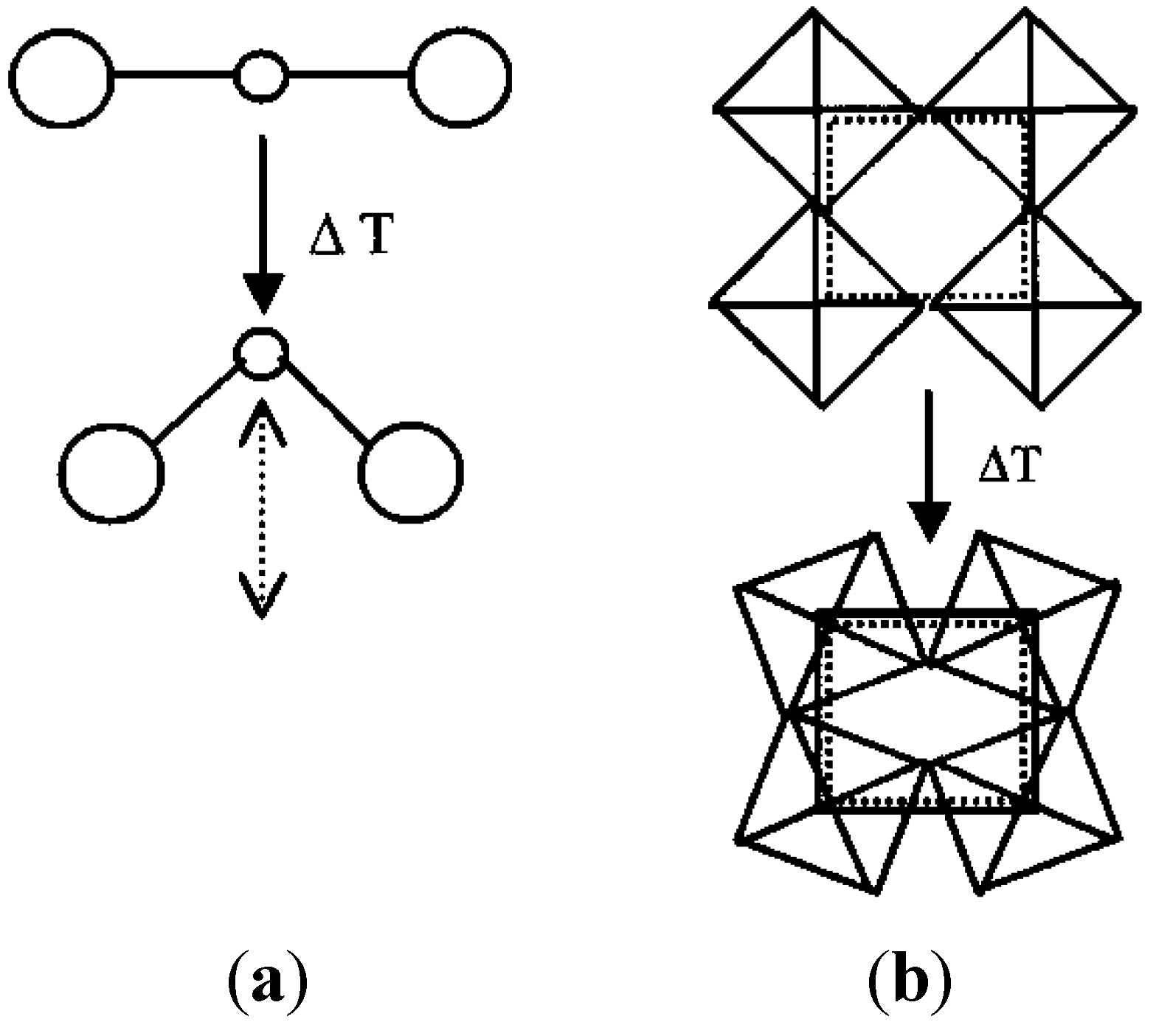
Figure 2.
Schematics of vibrational modes leading to NTE: (a) Transverse vibrational motion of an oxygen atom in a M-O-M linkage causing a decrease of the metal-metal distance; (b) cooperative rocking of polyhedra causing a decrease in average metal-metal distances.
Figure 2.
Schematics of vibrational modes leading to NTE: (a) Transverse vibrational motion of an oxygen atom in a M-O-M linkage causing a decrease of the metal-metal distance; (b) cooperative rocking of polyhedra causing a decrease in average metal-metal distances.
The transverse vibration mechanism can also be expressed in terms of low energy librational phonon modes with large, negative Grüneisen parameters (γ). The presence of such modes has been proven experimentally through specific heat [8,39,63], phonon density of states [6,10,39], and total neutron scattering studies [12,64,65] Because the overall expansion behavior of a compound depends on the relative contributions from all phonons, not all materials with low energy phonon modes with negative γi values will exhibit NTE behavior. A feature necessary for the occurrence of NTE is the presence of low energy phonons with negative γi values, and a phonon gap that separates these modes from the high energy phonons also present in the structure [21].
2.1. ZrW2O8 Family
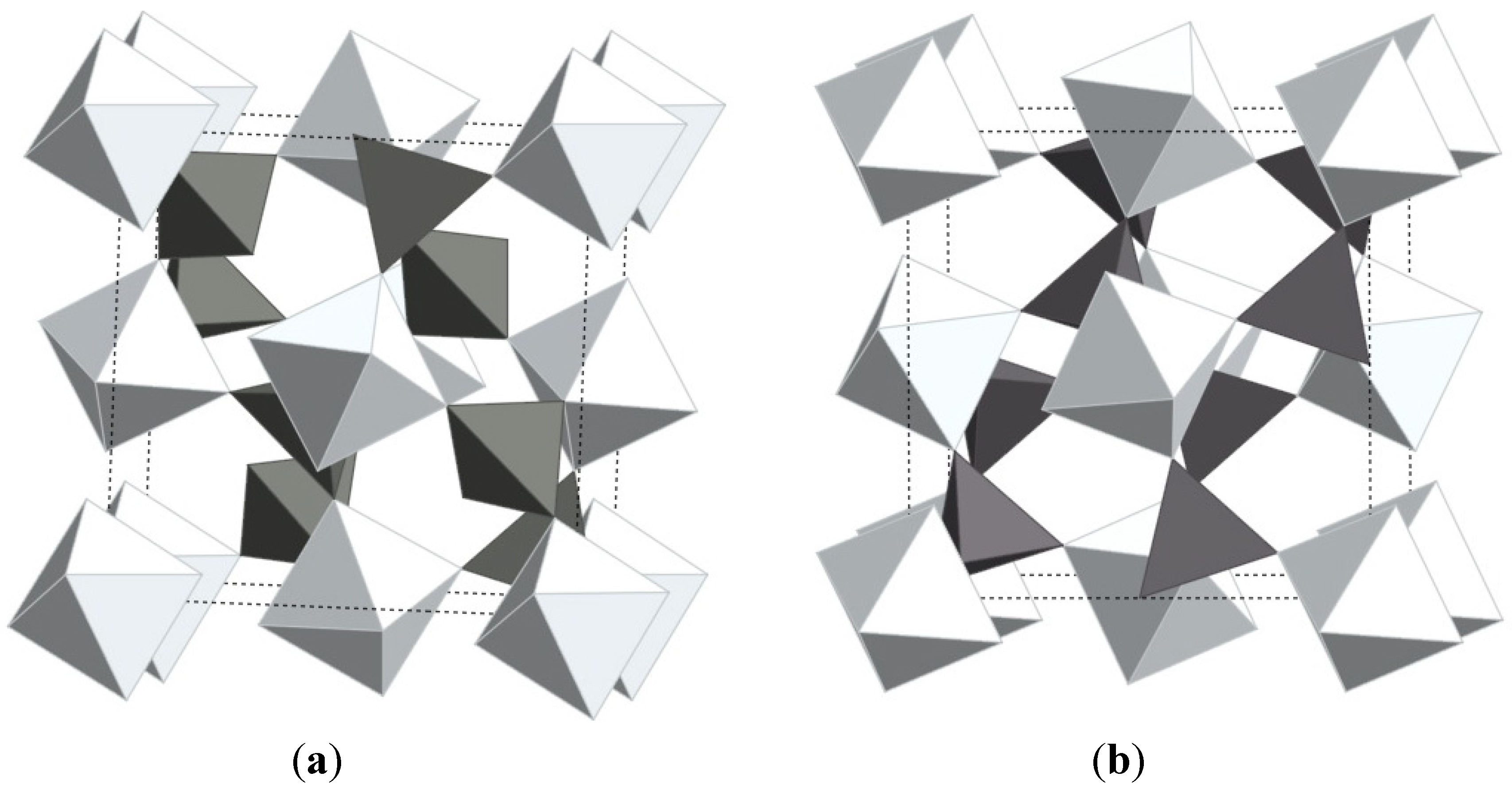
While a number of materials have been found to contract upon heating, the compound zirconium tungstate has become almost synonymous with the expression NTE. ZrW2O8 was first discovered in 1959 by Graham [34] and its unusual expansion behavior was documented by Martinek and Hummel in 1968 [35] At that time, the strong contraction was regarded as equally detrimental as strong positive expansion, and the search for zero expansion materials moved on to different materials. In 1995, Auray solved the crystal structure of ZrW2O8 [66], and in 1996, Sleight’s group showed that the structure is responsible for the strong NTE behavior observed from 0.3 to 1050 K [3]. The material is thermodynamically stable between 1378 and 1508 K, but can be quenched and remains metastable up to 1050 K. The structure is composed of corner-sharing ZrO6 octahedra and WO4 tetrahedra, with each ZrO6 connected to six WO4 units, while each tetrahedron is connected to only three octahedra, leaving one terminal oxygen. The WO4 units are oriented along the body diagonal of the cubic cell, and can be described as W2O8 units with one 4-coordinated tungsten, and a tungsten with 4+1 coordination due to a long range contact with an oxygen from the neighboring tungsten (Figure 3a).
Figure 3.
Crystal structures of (a) α-ZrW2O8; (b) ZrV2O7 (ideal high temperature structure); bright: ZrO6 octahedra; dark: WO4/VO4 tetrahedra. The structures only differ in the orientation and connectivity of the polyhedra.
Figure 3.
Crystal structures of (a) α-ZrW2O8; (b) ZrV2O7 (ideal high temperature structure); bright: ZrO6 octahedra; dark: WO4/VO4 tetrahedra. The structures only differ in the orientation and connectivity of the polyhedra.

The material’s contraction is inherently isotropic due to its cubic structure, with αl values of −9.1 × 10−6 K−1 below 350 K, and −5.0 × 10−6 K−1 above 450 K. The magnitude of expansion changes due to an order-disorder phase transition at 448 K (α-ZrW2O8 to β-ZrW2O8), but the cubic symmetry is preserved (space groups P213 and Pa3, respectively). The transition involves a reorientation of the WO4 tetrahedra, and causes a discontinuity in the otherwise linear expansion behavior. The ZrW2O8 structure supports rigid unit modes, which are responsible for the strong NTE over a large temperature range. Phonon density of states measurements revealed significant contributions from very low energy modes with negative Grüneisen parameters, and the presence of a gap between low and high energy phonons [6,8,39].
2.1.1. Substitution of ZrW2O8
Both metal sites in the ZrW2O8 family can be substituted. Shortly after the pivotal ZrW2O8 paper, NTE compounds prepared by substitution of Hf on the Zr site and Mo on the W site were reported [53,54,67]. The HfW2O8 analog shows essentially identical expansion behavior with respect to the magnitude of α, and only a small increase in the temperature of the order-disorder phase transition (Ttrs = 463 K) is observed. In contrast, ZrMo2O8 and HfMo2O8 do not undergo a transition to the ordered P213 structure, but adopt space group Pa3 at all temperatures. A transition from dynamic to static oxygen disorder occurs at low temperatures, which increases the magnitude of NTE (αl −8 × 10−6 K−1 below 200 K, and −5 × 10−6 K−1 from 200 to 600 K) [68]. Early research suggested that both molybdates were metastable at all temperatures and could only be obtained by dehydration and topotactic recrystallization from a hydrated precursor (AM2O7(OH)2·2(H2O); A = Zr, Hf; M = Mo, W) [54,69,70]. However, recent in situ diffraction experiments provide evidence that cubic ZrMo2O8 may be stable in sealed tubes above 1350 K. The disappearance of diffraction peaks at higher temperatures was interpreted as melting, as little to no formation of ZrO2 was observed at 1460 K, and the cubic phase recrystallized during quenching experiments [71]. The sealed environment is necessary to prevent evaporation of MoO3, which becomes volatile above 1000 K under atmospheric pressure.
The complete range of Zr1−xHfxW2−yMoyO8 solid solutions (0 ≤ x ≤ 1; 0 ≤ y ≤ 2) can be synthesized either from AM2O7(OH)2·2(H2O) precursors or by solid-state methods [72,73]. This is not surprising, as Zr4+ and Hf4+ (0.72 Å and 0.71 Å in octahedral coordination) and Mo6+ and W6+ (0.55 Å and 0.56 Å in tetrahedral coordination) have very similar ionic radii to each other. Like for the pure tungstates, hafnium substitution does not significantly change the expansion and phase transition behavior of the solid solutions. In contrast, the molybdenum content strongly influences formation of the ordered P213 phase. For compositions with more than 50% tungsten, the order-disorder (α to β) transition is observed, and the temperature varies linearly with composition. ZrMoWO8 remains in the Pa3 structure when rapidly cooled, but formation of the P213 polymorph was observed at about 270 K upon slow cooling [74]. No ordered phase has been reported for y > 1, although it is possible that ordering was not detected for some compositions due to slow kinetics. While ZrW2O8 adopts a fully ordered structure at low temperatures, some local disordered regions remain even to the lowest temperatures for ZrMoWO8.
Substitution of ZrW2O8 by elements other than Hf and Mo is also possible, although solubility is limited in all cases even when metals with identical charges are chosen. The highest substitution levels have been achieved with Sn4+ in Zr0.7Sn0.3W2O8 [75]. The solubility of Ti4+ is limited to about 5% due to its much smaller ionic radius (octahedrally coordinated Ti4+: 0.61 Å), which induces lattice strain [76] In both cases, a reduction in the α-β phase transition temperature was observed (400 K for Zr0.7Sn0.3W2O8 and 405 K for Zr0.95Ti0.05W2O8, respectively). The expansion coefficients show limited dependence on composition, and fall around −10 × 10−6 K−1 for materials in the α-phase, and −5 × 10−6 K−1 for materials in the β-phase.
Aliovalent ions can also be incorporated into the ZrW2O8 structure. Substitution of the Zr/Hf site by a number of trivalent ions (Sc, Y, In, Eu, Er, Yb, Lu) has been reported [77,78,79,80,81]. These systems show limited solubility, ranging from 1.6% for Eu3+ to 5% for Yb3+. However, even small amounts can lead to significant changes in Ttrs. For example, 4% substitution lowers Ttrs to 390 K for Y3+, 380 K for In3+, and 360 K for Sc3+ [80]. This clearly indicates that the trivalent substituents introduce disorder into the ZrW2O8 framework. Even at very low temperatures, only partial ordering is observed, similar to ZrMoWO8. The phase transition temperature can be correlated to the normalized saturated order parameter η. All trivalent cations investigated are larger than Zr4+ or Hf4+. In addition, an oxygen vacancy is created for every two A3+ cations, which is evident from the decrease of the lattice constant with increasing substitution by the larger cations. This results in a distortion of the AO6 octahedra, which in turn act as a local, spherical disturbance on the bonded MO4 tetrahedra. Yamamura et al. analyzed the anisotropic peak broadening observed for Sc, In and Y substituted ZrW2O8, and quantified the size of the distorted region as 1.3 to 1.7 nm, which is equivalent to 8 to 12 WO4 units [80]. The expansion behavior in the disordered high temperature phase was identical to β-ZrW2O8, regardless of identity and quantity of substituent, while slightly less negative expansion was observed with increasing A3+ content in the α-phase.
The only example of aliovalent substitution of the M site to date is the compound ZrV0.2W1.8O7.9 [82,83]. This material was reported to crystallize in space group Pa3, which is also adopted by both β-ZrW2O8 and ZrV2O7. The main difference between these structures lies in the fact that the V2O7 groups in ZrV2O7 are truly centrosymmetric, while the W2O8 groups are not, requiring equal amounts of opposite orientations to give an average centrosymmetric structure. A later publication on the same composition assigned space group P213 at room temperature, and reported a transition to the Pa3 polymorph between 358 and 400 K. Interestingly, the β-phase expansion is less negative (−1.6 × 10−6 K−1) than for most compositions (−5 × 10−6 K−1), while the α-phase expansion is identical to ZrW2O8. The solubility limit of vanadium in ZrW2O8 has not yet been determined.
2.1.2. High Pressure Behavior
Open framework compounds are prone to undergo pressure-induced phase transitions. As the preparation and use of composites is likely to expose NTE fillers to pressure, their application requires investigation of their high pressure behavior. In situ experiments and measurements on samples recovered from high pressure have been reported for ZrW2O8 [4,7,84,85,86,87], HfW2O8 [63,88,89], ZrMo2O8 [54,90,91,92], and HfMo2O8 [90], but no solid solutions have been studied under pressure to date.
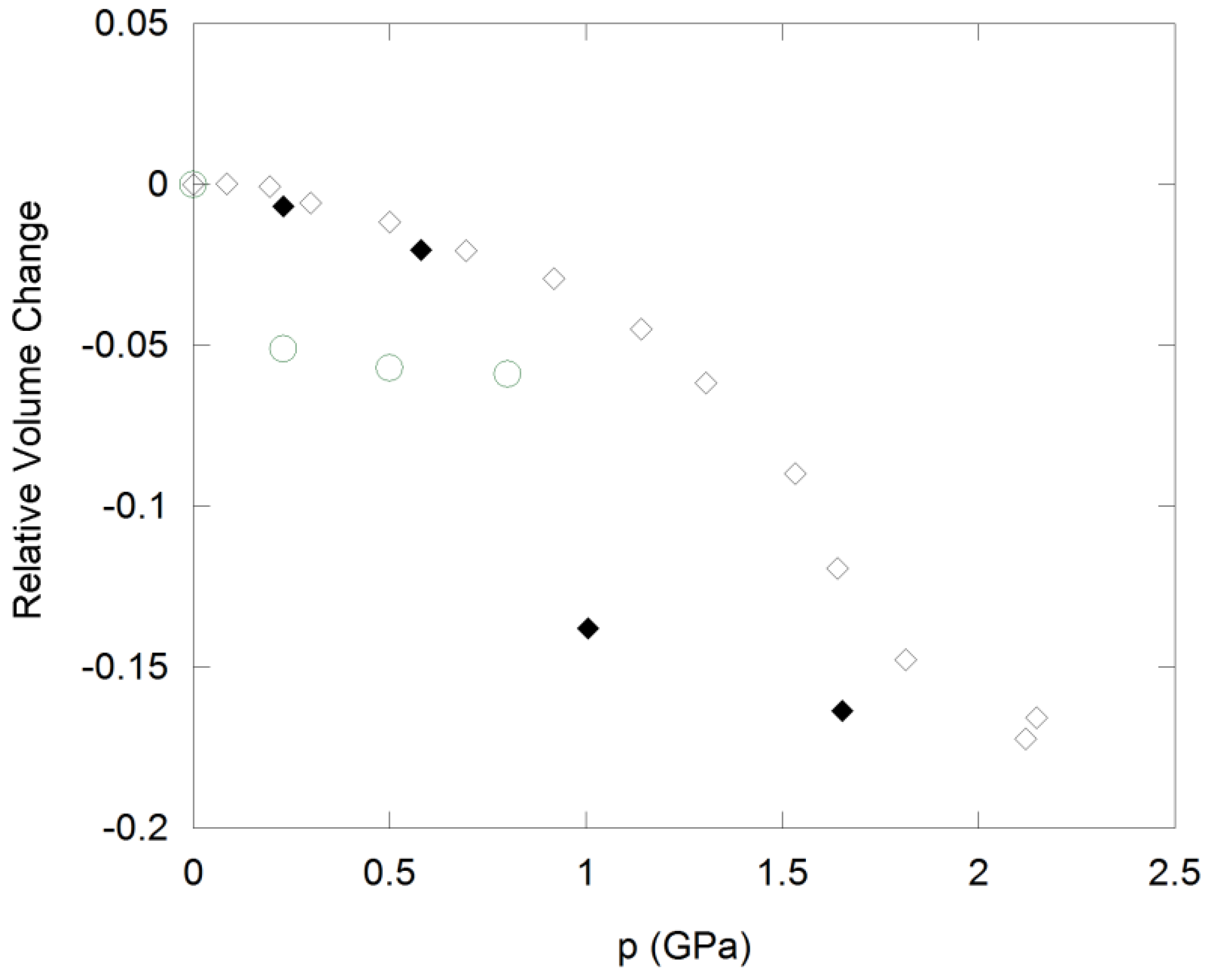
Cubic ZrW2O8 undergoes an irreversible phase transition to γ-ZrW2O8 at 0.2 to 0.3 GPa (Figure 4), which is accompanied by a 5% decrease in volume per formula unit. The structure of this phase is closely related to the α-polymorph, and involves a reorientation of one third of the W2O8 units, which results in tripling of one cell axis and lowering of the symmetry to the orthorhombic system [93]. γ-ZrW2O8 can be quenched to ambient conditions, and converts back to the cubic structure upon heating to 390 K. It shows weak NTE below 225 K, and positive expansion at higher temperatures. Further compression of ZrW2O8 results in pressure-induced amorphization between 1.9 and 2.4 GPa [7]. The amorphous phase is ~25% denser than the cubic starting material, and can be retained upon decompression. To recrystallize α-ZrW2O8 at ambient pressure, heating to 923 K is necessary. Both pressure-induced phase transitions are accompanied by an increase in tungsten coordination: The orthorhombic unit cell contains one 4-coordinated W, four W atoms in 4 + 1 coordination, and one W with a 5 + 1 environment. Further changes in W coordination towards more centrosymmetric environments have been observed in PDF and XANES/EXAFS experiments during amorphization [87,94]. Zr K-edge EXAFS data on samples recovered after compression also suggest that the Zr coordination number may increase to 7 at higher pressures. These observations are consistent with a mechanism of amorphization that involves disordering of existing structural polyhedra, which leads to formation of additional bonds that crosslink the polyhedra and increase the metal coordination.
HfW2O8 shows very similar behavior to ZrW2O8, except that the transition to the γ-polymorph occurs at higher pressure (0.63 GPa) and shows sluggish kinetics [89]. Full conversion is only achieved after 24 h at 0.63 GPa, while no transformation occurs even after 11 d at 0.52 GPa. Amorphization is observed at ~2 GPa. Both γ- and amorphous HfW2O8 are metastable under ambient conditions. The orthorhombic material can be converted back to the cubic phase by heating to 360 K. No reports are available on the recrystallization of amorphous HfW2O8.
Figure 4.
Compressibility of cubic ZrW2O8 (circles) and cubic ZrMo2O8 (diamonds) under hydrostatic conditions. Open symbols: Data collected upon compression; solid symbols: Data collected upon decompression.
Figure 4.
Compressibility of cubic ZrW2O8 (circles) and cubic ZrMo2O8 (diamonds) under hydrostatic conditions. Open symbols: Data collected upon compression; solid symbols: Data collected upon decompression.
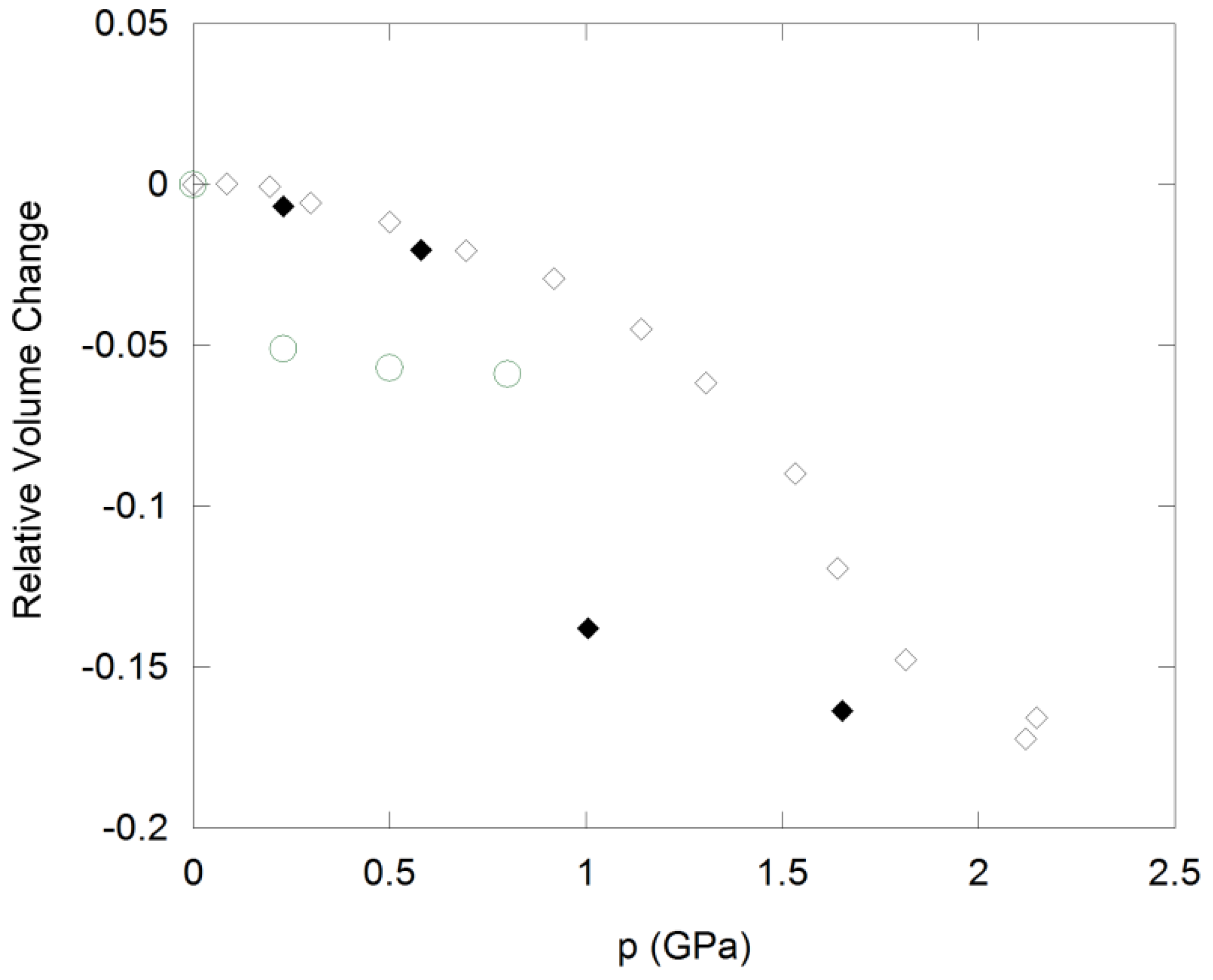
ZrMo2O8 and HfMo2O8 also undergo pressure-induced amorphization, and EXAFS/XANES studies on ZrMo2O8 suggest that the amorphous phase possesses similar local structures to ZrW2O8 [92]. The amorphous materials cannot be reconverted to the cubic structures. Instead, the more stable trigonal and monoclinic AM2O8 polymorphs are formed during heating at low and high pressure, respectively. Application of non-hydrostatic pressure results in amorphization at 0.5 to 1.5 GPa, while crystallinity is retained under hydrostatic conditions up to 3.0 GPa [90]. In hydrostatic environments, a crystalline-to-crystalline transition at 0.7–2.0 GPa precedes amorphization. This transformation is accompanied by a 10–11% decrease in cell volume (Figure 4), suggesting that the still unknown structure of the high pressure polymorph is different from γ-ZrW2O8. The data could be fitted in a pseudo-cubic cell, although subtle peak splitting indicative of symmetry lowering was evident. The transition is reversible upon decompression, although considerable hysteresis is observed, with the original cubic phases reforming below 1.0 GPa.
2.2. ZrV2O7 Family
Negative thermal expansion in the zirconium vanadate family was first reported in the mid 90’s [1]. ZrV2O7 is thermodynamically stable up to ~1075 K, where decomposition into binary oxides is observed. Like ZrW2O8, ZrV2O7 adopts a cubic structure, making its expansion behavior inherently isotropic. The ideal ZrV2O7 structure is closely related to the rocksalt structure, with Zr4+ as the cation, and (V2O7)4− as the anion. The V2O7 groups order along the threefold rotation axis, lowering the overall symmetry to Pa3, and constraining the V-O-V bond angles to be linear on average, although significant displacements of the oxygen atoms from the inversion centers have been reported. The cations are octahedrally coordinated by oxygen, and the octahedra and tetrahedra form a corner-sharing 3D network (Figure 3b). This structural arrangement can give rise to transverse vibrations of corner-sharing oxygen atoms, which may result in NTE. In contrast to ZrW2O8, such vibrational modes always involve distortions of the polyhedra. The vibrations can thus not be described by the RUM model, but may be regarded as “quasi-rigid unit modes”, or qRUMs [59]. ZrV2O7 shows strong NTE with αl values between −7 and −10 × 10−6 K−1 above 375 K, but undergoes phase transitions to an incommensurate phase, and to an ordered cubic 3 × 3 × 3 superstructure in space group Pa3 upon cooling to 375 and 350 K, respectively. In the room temperature superstructure, 2/3 of the V-O-V linkages are no longer constrained to be linear on average by symmetry, and the expansion coefficient becomes positive [95,96,97].
2.2.1. Substitution of ZrV2O7
The ZrV2O7 structure can accommodate a wide range of tetra- and pentavalent ions. For M = P, the A4+ cation can be Zr, Hf, Ti, U, Th, Pu, Np, Mo, W, Ce, Pb, Sn, Ge or Si [1], while for the vanadates only the Zr and Hf compounds [1], and for the arsenates only the Zr and Th compounds are known [98,99]. For a number of years, all compounds in the AM2O7 family were believed to adopt the ideal ZrV2O7 structure in space group Pa3 at high temperatures, and to transform to the cubic 3 × 3 × 3 superstructure at lower temperatures. This view has been challenged over the past decade. While SiP2O7, TiP2O7, and HfV2O7 indeed adopt a cubic 3 × 3 × 3 superstructure, NMR and high resolution synchrotron studies have shown that the symmetry is lower for many materials. For example, SnP2O7 and GeP2O7 were found to be monoclinic [100,101,102], CeP2O7 and AnP2O7 (An = U, Th, Pu, Np) were reported as triclinic [103,104], and ZrP2O7 and HfP2O7 exhibit an orthorhombic distortion [105,106]. The structure of the latter compounds was solved independently from synchrotron single crystal and powder diffraction data by the Birkedal and Evans groups, and consists of a 136 atom unit cell in space group Pbca. All P-O-P bond angles are significantly smaller than 180°, eliminating the possibility of transverse oxygen vibrations that could lead to a unit cell contraction [105,106]. However, high resolution studies of AnP2O7 have shown that NTE can be observed at high temperature in materials with bent P-O-P units [104]. Interestingly, SnP2O7 does not adopt the ideal ZrV2O7 structure at all, but undergoes a series of different distortions up to its decomposition temperature. Insufficient data are available to unambiguously address whether other AM2O7 compounds undergo symmetry-lowering distortions.
The magnitude of expansion of AM2O7 compounds depends on the identity of the A and M cations, which determine the size of the polyhedra. Larger polyhedra can accommodate the distortions required for transverse oxygen vibrations more easily, and as a result, most phosphates show positive expansion at all temperatures, while the vanadates show strong NTE in the high temperature phase. Phosphates with large A4+ cations (CeP2O7, AnP2O7) show a change from positive to negative expansion with increasing temperature.
The phase transition temperature for the ideal ZrV2O7 can be suppressed by formation of solid solutions Zr1−xHfxV2−yPyO7 [1] While substitution of Hf for Zr has a limited effect on the phase transition temperature, the incorporation of P on the M site strongly influences the phase transition behavior. For small amounts of mixing, the transition temperatures for both phase changes are lowered, and for values of 0.4 < y < 1.6, the materials adopt the high temperature ZrV2O7 structure at room temperature. Expansion coefficients range from small positive to strongly negative depending on composition.
Reports of substitution of the ZrV2O7 structure by aliovalent cations are rare, and are limited to formation of A0.53+A’0.55+P2O7 (A = Bi, Nb, Nd, Eu, Al, Fe, Ga, In, Y; A’ = Nb, Ta) [107,108], Nb0.05Y0.05Zr0.9P2−xVxO7 [109], and ZrV2-xMoxO7+x/2 (0 ≤ x ≤ 0.8) [110]. Positive expansion was observed at all temperatures for the phosphates. The incorporation of Y3+ and Nb5+ into ZrP2-xVxO7 reduced the positive expansion in the low temperature superstructure, but did not significantly alter the phase transition temperature and magnitude of NTE in the high temperature phase. Similarly to V-substituted ZrW2O8, the Mo-substituted ZrV2O7 crystallizes in space group Pa3, and the structure is closely related to the ZrMo2O8 and ZrV2O7 parent structures. The expansion behavior determined for a single crystal with 25% Mo substitution is similar to that of ZrV2O7.
2.2.2. High Pressure Behavior
High pressure studies in the ZrV2O7 family have been limited to TiP2O7, ZrP2O7, CeP2O7, ZrV2O7 and HfV2O7 [103,111,112,113,114]. Interestingly, in situ diffraction experiments show no evidence for pressure-induced phase transitions or amorphization for TiP2O7 and ZrP2O7, both of which are positive thermal expansion compounds. Raman studies suggest that subtle structural changes could be occurring upon compression, but cell volumes extracted from X-ray data showed smooth compression up to 40 and 20 GPa, respectively [111].
In contrast, CeP2O7, ZrV2O7 and HfV2O7 undergo phase transitions to crystalline high pressure phases at 0.65 GPa, 1.6 GPa and 3.7 GPa. Peak splitting indicates lowering of the cubic symmetry in all cases. For CeP2O7, a second crystalline high pressure phase is observed above 5 GPa, while pressure-induced amorphization occurs above 4.0 GPa for ZrV2O7. HfV2O7 also progressively amorphizes, but traces of crystallinity are still observed at 42 GPa. Both transitions in CeP2O7 are reversible upon decompression. Raman studies on the vanadates suggest that amorphization is irreversible, whereas the crystalline high pressure phase reverts back to the ambient pressure polymorph upon decompression.
2.3. Sc2W3O12 Family
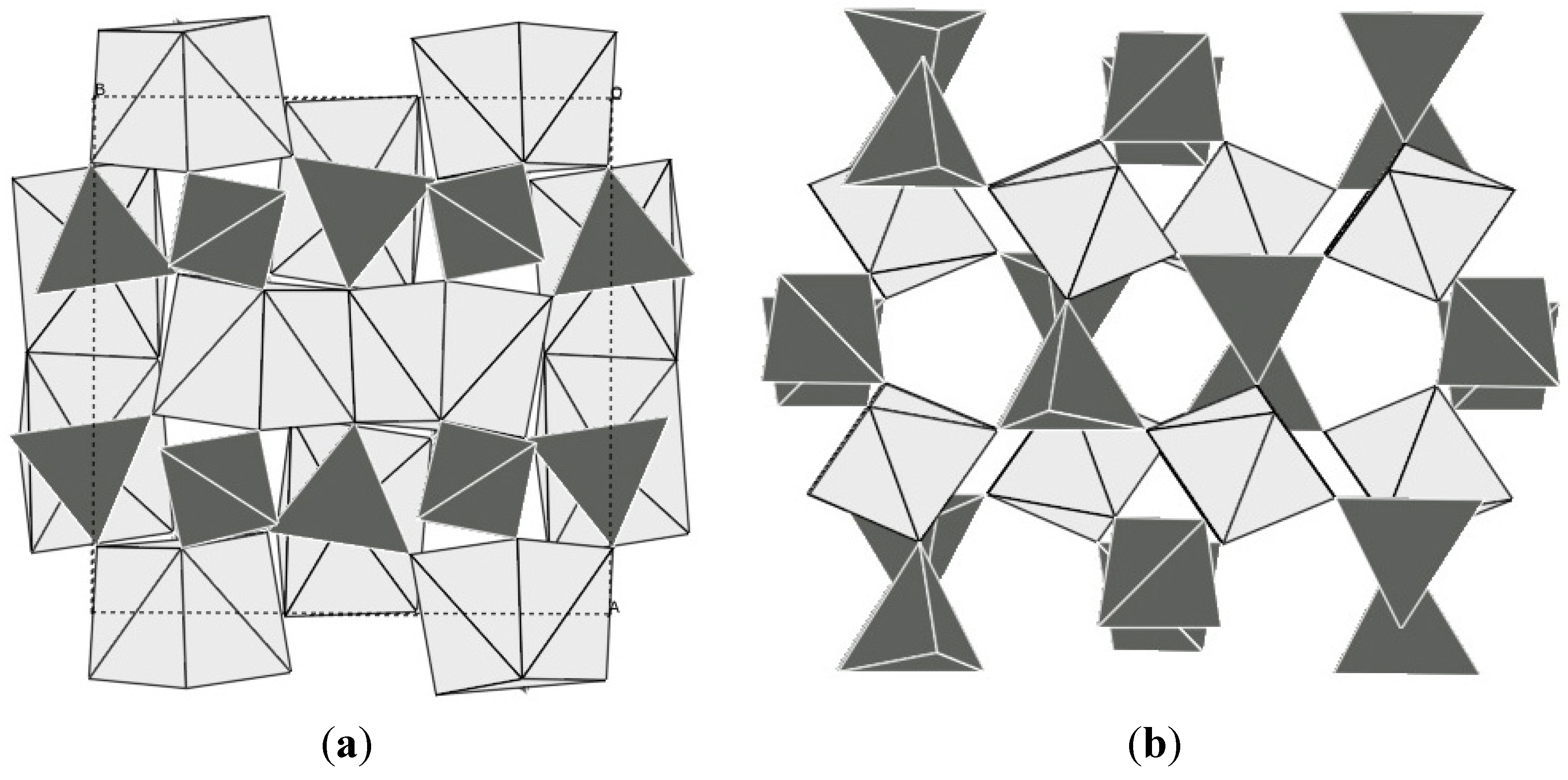
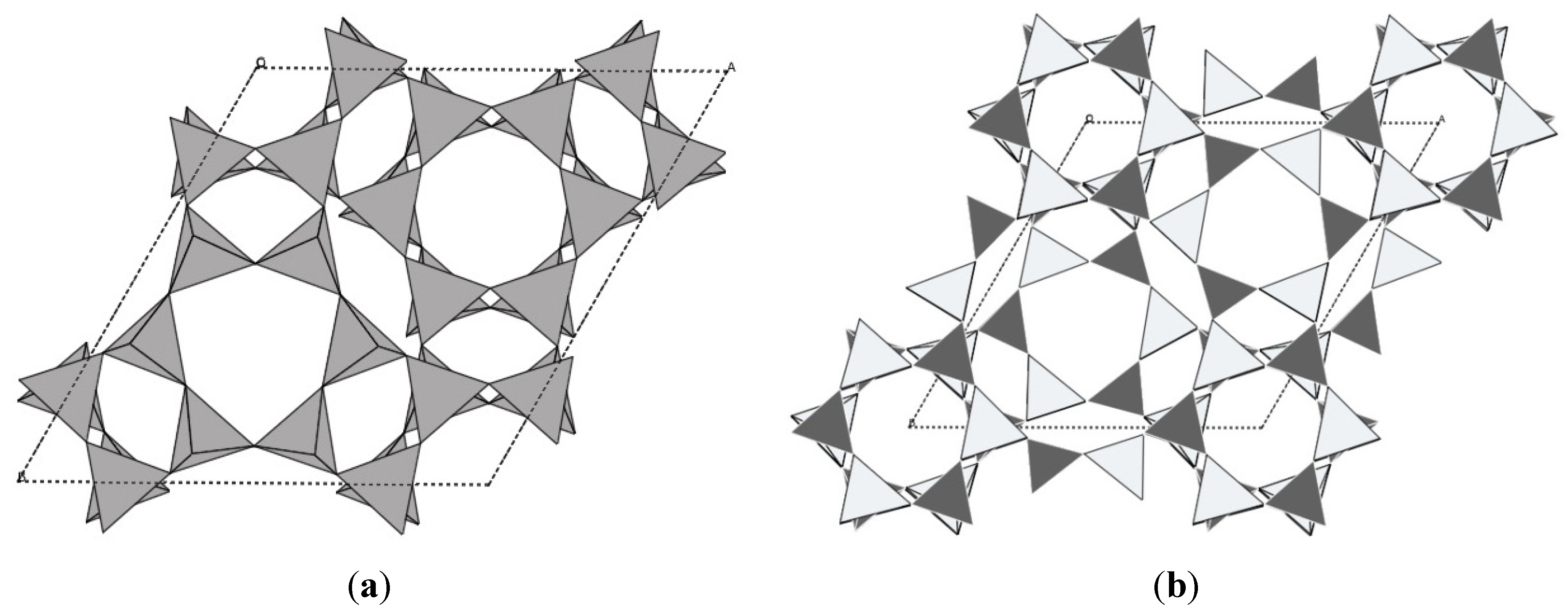
The scandium tungstate family has also attracted a lot of attention, as it offers a wide range of possible compositions, and the potential to tune the expansion behavior of the resulting compounds [5,23]. Sc2W3O12 is thermodynamically stable over a wide temperature range, allowing straightforward preparation by traditional ceramic methods. Unlike ZrW2O8 and ZrV2O7, it does not adopt a cubic structure, but crystallizes in the orthorhombic space group Pnca [115]. This gives rise to anisotropic expansion. The crystal structure is composed of a corner-sharing network of ScO6 octahedra and WO4 tetrahedra (Figure 5a). Negative volume expansion is observed from 10 to 1300 K [115,116], and an average αl value of −2.2 × 10−6 K−1 was determined from variable diffraction data for the range 50–450 K. Dilatometry on ceramic bars gave significantly more negative values of −6 to −11 × 10−6 K−1 which was attributed to the presence of microcracks in the bars combined with the anisotropic expansion of the three unit cell axes, where the a and c axis contract, while the b axis expands. Detailed structural analysis showed that there were only small changes in bond distances and angles within the polyhedra as a function of temperature, but large amplitudes of vibration were observed for the corner-sharing oxygen atoms. While later theoretical studies showed that no true RUMs are present in the structure [59], the atomic displacement parameters extracted from diffraction data clearly demonstrate that transverse oxygen vibrations give rise to the observed NTE. While Sc2W3O12 remains orthorhombic at all temperatures, other compounds in this family undergo a symmetry-lowering displacive phase transition to a denser monoclinic polymorph at low temperatures (Figure 5b). In general, the orthorhombic Sc2W3O12 structure is classified as an NTE polymorph, while positive expansion has been reported for monoclinic phases.
Figure 5.
Crystal structures of (a) orthorhombic; (b) monoclinic Al2Mo3O12; bright: AlO6 octahedra; dark: MoO4 tetrahedra.
Figure 5.
Crystal structures of (a) orthorhombic; (b) monoclinic Al2Mo3O12; bright: AlO6 octahedra; dark: MoO4 tetrahedra.

The phonon density of state (DOS) for Sc2W3O12 has been determined from specific heat measurements. Like for ZrW2O8, very low energy phonons (~5 meV) with negative γi values, and a gap between the low and high energy phonons were observed [21]. Interestingly, a similar DOS distribution was found for Sc2Mo3O12, which adopts a monoclinic structure at low temperature. This suggests that the monoclinic structure has the potential to exhibit NTE as well. While the higher energy phonon contributions with positive γi values outweigh the effect of the low energy phonons with negative Grüneisen parameters at most temperatures, negative expansion of the b axis (4–60 K) and very small negative volume expansion (4–30 K, αv = −3.7 × 10−7 K−1) were observed at very low temperatures [117].
2.3.1. Substitution of Sc2W3O12
The scandium tungstate structure shows excellent tolerance towards ionic substitution of both metal sites. In most A2M3O12 compounds, the M site is occupied by Mo or W. In these cases, the A site can be substituted by any trivalent cation ranging in size from Al3+ (roct = 0.54 Å) to Ho3+ (roct = 0.90 Å). Ln2M3O12 compositions with larger lanthanides crystallize in structures that adopt higher coordination numbers (7 or 8) for the A-site. However, for solid solutions of two trivalent cations, significant amounts of larger Ln3+ can be incorporated into the scandium tungstate structure, as evidenced by reports of Er2−xCexW3O12 (x ≤ 0.4) [118], Er2−xSmxW3O12 (x ≤ 0.5) [119], Er2−xNdxW3O12 (x ≤ 0.5) [120], Er2−xDyxW3O12 (x ≤ 0.7) [121], Y2−xDyxW3O12 (x ≤ 1.0) [121], and Y2−xNdxW3O12 (x ≤ 0.4) [122]. In addition, substitution with aliovalent cations has been achieved in AP2MO12 [123,124] and MgAM3O12 (A = Zr, Hf; M = Mo, W) [125,126,127]. AP2MO12 adopts the same structure as Sc2W3O12 with an ordered arrangement of P and M [124], while MgAM3O12 crystallizes in a different orthorhombic structure in space group Pnma or Pna21 [125,126].
Many A2M3O12 compositions adopt a monoclinic structure at low temperatures, and transform to the orthorhombic Pnca phase upon heating [117]. The temperature for this phase transition is generally higher for molybdates than for tungstates, and increases with increasing electronegativity of the A-site cation [128]. This observation can be explained by the fact that oxygen-oxygen repulsive interactions make the denser monoclinic phase less favorable. More electronegative A-site elements reduce the partial charge on the oxygen atoms and thus the repulsive forces. However, shifts in the temperature or the complete absence of the phase transition for some mixed A-site compounds suggest that entropic factors also play a role. As the monoclinic phase generally displays positive expansion, this phase transition is undesirable for any potential uses of these NTE compounds in composites.
The expansion coefficient of the orthorhombic phase depends strongly on the identity of the A-site cation. As stated earlier, the A2M3O12 structure does not support RUMs, and distortion of the polyhedra occurs during the transverse vibrations of oxygen atoms. Larger A-site cations give rise to softer octahedra that distort more easily. As a result, expansion tends to become more negative with increasing size of A3+, reaching values of −7.0 × 10−6 K−1 in Y2W3O12 [129] and −9.3 × 10−6 K−1 in Y2Mo3O12. Additional slight decreases have been achieved in solid solutions of these compounds with larger lanthanides [118,119,120,121,122]. In contrast, Al2W3O12 has been reported to show low positive expansion with an αl value of +2.2 × 10−6 K−1 [5]. Reports on expansion coefficients vary not only between αl values extracted from variable temperature diffraction data and dilatometry, but also between different diffraction measurements. These discrepancies may in some cases arise from averaging over different temperature ranges, as the volume expansion of many A2M3O12 compounds is not completely linear. Nevertheless, the accumulated knowledge on the expansion behavior of different compositions can be used to design materials with desired α values, including zero expansion compounds. The first report of a solid solution with close to zero expansion around room temperature (InAlW3O12) was published in 1999 by Mary and Sleight [23]. More sophisticated solid solutions like Al2x(MgHf)1−xW3O12 have since been reported [130].
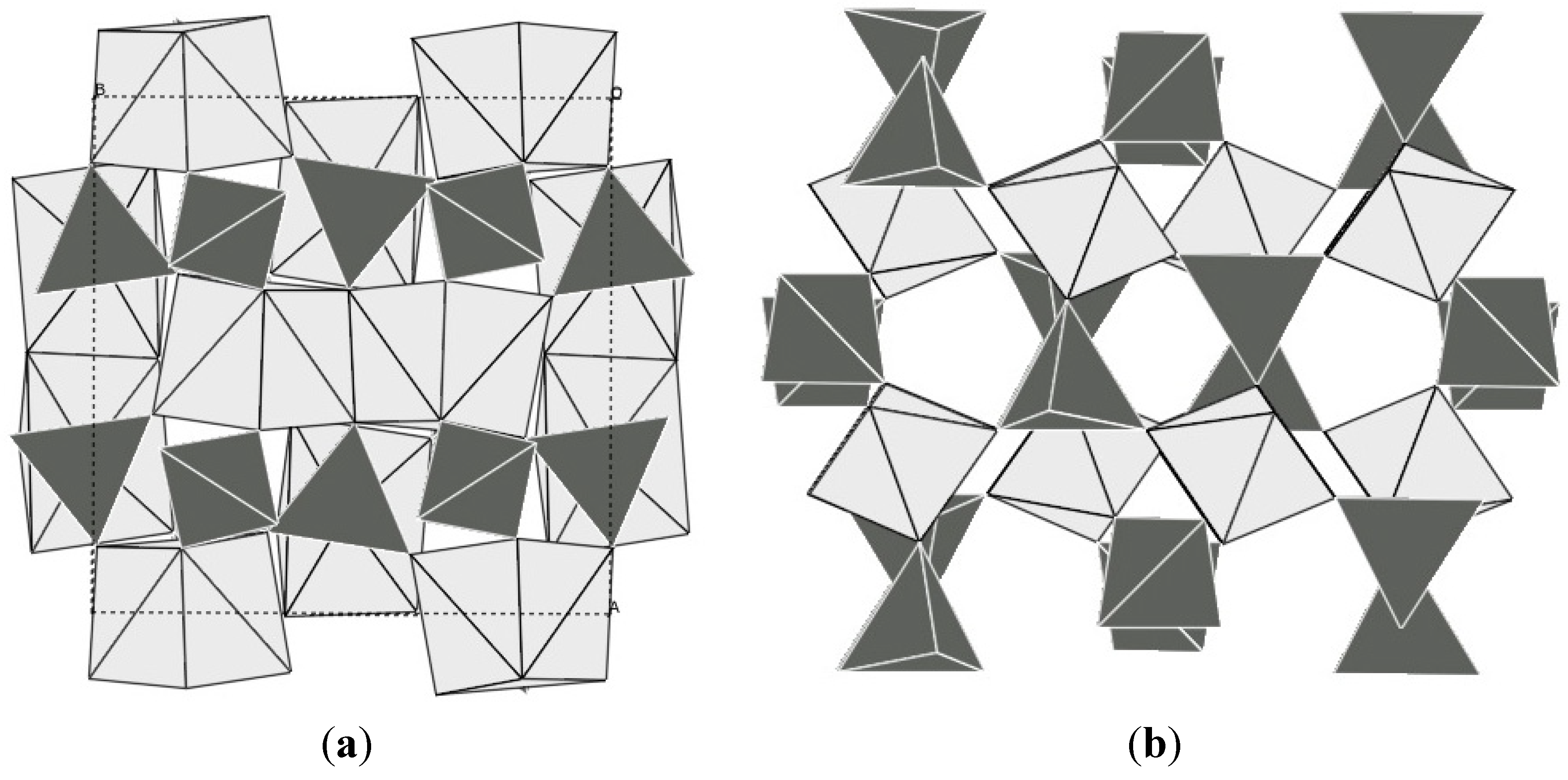
While the record negative expansion coefficients in the A2M3O12 family have been achieved with rare earth elements or the “pseudo-lanthanide” Y, it is necessary to point out that all of these compositions suffer from hygroscopicity. Under ambient conditions, a trihydrate is formed by absorption of moisture from the atmosphere [118,119,120,121,122,131,132]. This limits the usefulness of these compositions to temperatures above the dehydration point or sealed systems. In addition, several of these compounds could be on the borderline of thermodynamic stability of the Pnca-NTE polymorph, as it was shown that this structure is only thermodynamically stable above 823 K for Y2Mo3O12, and metastable with respect to a denser Pba2-polymorph at lower temperatures [133] (Figure 6).
Figure 6.
Crystal structures of (a) Pba2-Y2Mo3O12; (b) Pbcn-Y2Mo3O12; bright: YOn polyhedral; dark: MoO4 tetrahedra. In the denser Pba2 polymorph, the coordination number of yttrium is increased, and the polyhedra share edges.
Figure 6.
Crystal structures of (a) Pba2-Y2Mo3O12; (b) Pbcn-Y2Mo3O12; bright: YOn polyhedral; dark: MoO4 tetrahedra. In the denser Pba2 polymorph, the coordination number of yttrium is increased, and the polyhedra share edges.
2.3.2. High Pressure Behavior
A number of A2M3O12 materials have been studied under pressure by diffraction and spectroscopic methods. Orthorhombic compounds tend to undergo a transition to the denser monoclinic polymorph at very low pressures (<0.6 GPa) [134,135,136,137,138]. Most compounds investigated in situ undergo at least one additional crystalline-to-crystalline phase transition [135,137,138,139,140,141,142,143,144,145,146,147]. Pressure-induced amorphization is observed at pressures ranging from 5 GPa to 20 GPa. Similar high pressure diffraction patterns were observed for Sc2W3O12 [136], Sc2Mo3O12 [135], Al2W3O12 [143], and Ga2Mo3O12 [146], but due to limited data quality, the structures have not been characterized.
2.4. Other NTE Oxides
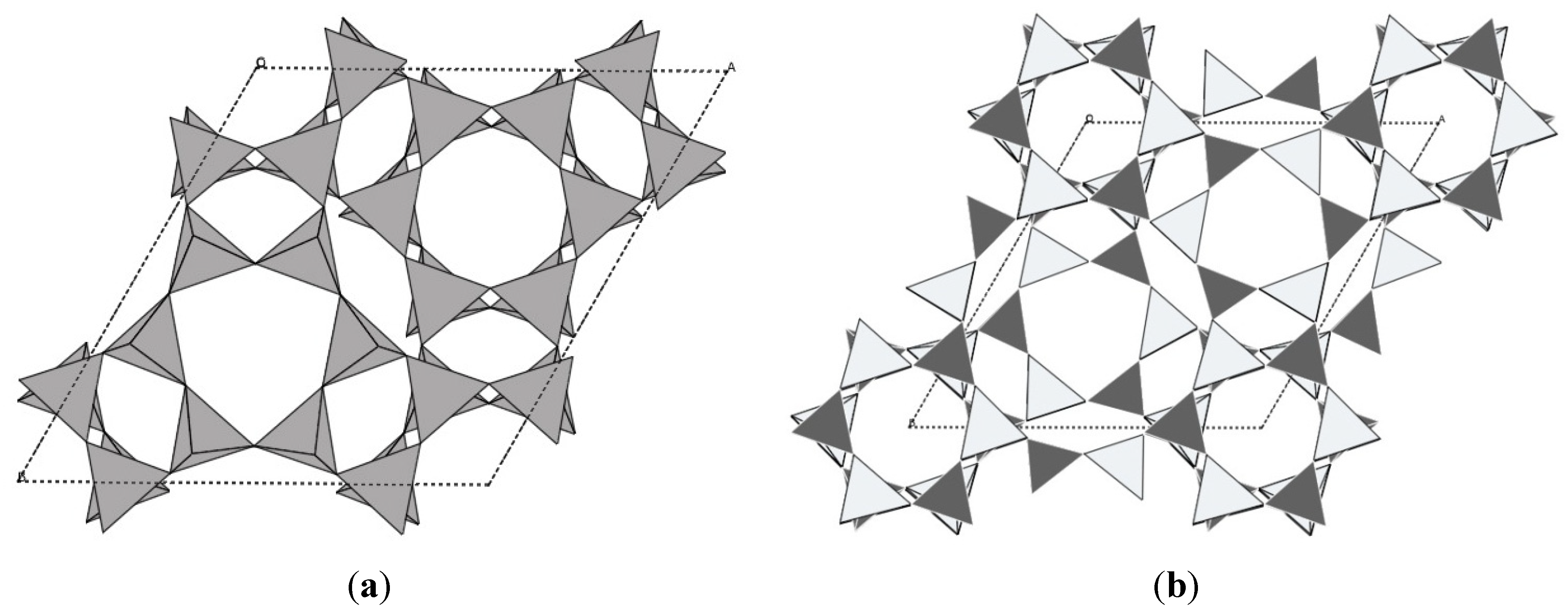
In addition to the ZrW2O8, ZrV2O7 and Sc2W3O12 families discussed so far, NTE due to transverse atomic vibrations has been observed in several other oxide structures. Many zeolites and zeolitic frameworks (AlPOs, GaPOs) exhibit this unusual property, which is not surprising considering that their structures are composed of corner-sharing tetrahedral networks [24,148]. In “normal” zeolites that contain a mixture of Si and Al, the presence of counter ions and water interferes with NTE, as the amount of empty space is reduced. This makes pure siliceous zeolites and group 13 phosphates better targets for studying NTE in zeolites. The magnitudes of NTE differ widely from close to zero in chabazite to the record of αl = −11.7 × 10−6 K−1 in AlPO-17 [24,55,149] (Figure 7).
Figure 7.
Crystal structures of (a) chabazite; (b) AlPO-17.
Another interesting example is found in Ag2O and Cu2O, in which the cations are linearly coordinated by two oxygens, and occupy the corners of the tetrahedral network of OM4 units. The overall structure consists of two independent, interpenetrating OM4 networks. NTE in Cu2O at low temperatures was first reported in 1985 [150], while this behavior was only documented in Ag2O in 2002 [13] EXAFS studies indicate significant distortion of the OM4 units, showing that a RUM model is not adequate to describe their expansion behavior [13,151]. A recent PDF study suggests that the distortion can lead to a shortening of the tetrahedral edge length and thus contribute to the NTE arising from transverse vibrations [43].
NTE has also been reported in materials with the delafossite structure [152,153,154], AIV2O(PO4)2 (A = Zr, Hf, U, Th) [155,156,157,158,159], ReO3 [160], NbOPO4 [161,162,163], NbVO5 [164], and TaVO5 [165]. While not an oxide, strong NTE in ScF3, which adopts the simple ReO3 structure, was reported by Greve et al. in 2010 [166]. This compound constitutes the first example of NTE over a wide temperature range in a purely octahedral framework.
2.5. Metal Cyanide Networks
Negative thermal expansion in metal cyanide frameworks was first reported for Zn(CN)2 by Williams et al. in 1997 [167]. This compound adopts a purely tetrahedral framework similar to silicates, but instead of a corner-sharing oxygen, the tetrahedra are connected by a diatomic cyanide linker. Fe[Co(CN)6] was the first purely octahedral metal cyanide network reported to display NTE in 2004 [58]. Its structure is related to the ReO3 structure by replacing the corner-sharing oxygen atoms of the octahedra by a diatomic cyanide bridge. Such frameworks can accommodate the same polyhedral rocking motions that cause NTE in the oxide families discussed so far, but the diatomic bridges give the structures more flexibility, which favors low frequency vibrational modes and increases the number of possible RUMs [168] The transverse vibrations of the CN linkers could be directly observed by pair distribution function analysis [15].
Metal cyanide frameworks quickly became the record holders for strongest NTE behavior with reports of αl values as low as −33 × 10−6 K−1 for Cd(CN)2 [20], −40 × 10−6 K−1 for Zn3[Fe(CN)6]2, Fe3[Co(CN)6]2 and Co3[Co(CN)6]2 [169], and −48 × 10−6 K−1 for Mn3[Co(CN)6]2 [169]. It was also recognized that like in zeolites, the presence of guest molecules in the open frameworks interferes with the transverse vibrations of the linker groups, thus reducing the NTE coefficients [57]. Additional materials were synthesized, and composition dependent NTE was reported for a number of metal ions [18,170].
In 2008, metal cyanide networks set another record when colossal positive and negative (colossal: |α| ≥ 100 × 10−6 K−1) expansion behavior was observed in Ag3[Co(CN)6] along different unit cell axes [19]. The a axis of the trigonal unit cell expands with +130 × 10−6 K−1 ≤ αa ≤ +150 × 10−6 K−1, while the c axis shows a similar magnitude of contraction with −130 × 10−6 K−1 ≤ αc ≤ −120 × 10−6 K−1. Transverse vibrations of cyanide groups, which are observed in the structure, cannot account for the observed expansion behavior. The simultaneous observation of colossal positive and negative expansion points to the crystal structure as the source of this unusual behavior [171]. This material is composed of sheets of Co(CN)6 octahedra that are separated by layers of Ag+ ions. Each Ag+ is linearly coordinated by two cyanide groups, one from each adjacent layer. Ag···Ag contacts close to the sum of van der Waals radii are observed, which suggest argentophilic interactions. These Ag···Ag and the corresponding [Co(CN)6]···[Co(CN)6] contacts increase rapidly with temperature. In order to preserve the Co-CN-Ag-NC-Co bridges in the material, the strong expansion along the a axis must be coupled to equally strong contraction along the c axis, which has been described as similar to “garden lattice fencing”. It was later established that the argentophilic interactions between silver atoms are the major driving force for the observed colossal expansion behavior, as isostructural D3[Co(CN)6] did not display colossal behavior [172]. However, strong metallophilic interactions can reduce expansion coefficients [44].
3. Composites
One of the reasons why NTE materials have attracted significant interest from the science and engineering communities lies in their potential applications as fillers in controlled thermal expansion composites. Mismatches in thermal expansion of device components have long been recognized as a serious problem, whether in the expansion of road surfaces, railroad tracks and bridges, or in thin films where lattice matching at deposition and use temperatures is important. Precision positioning is critical in electronic devices, and nanotechnology only amplifies the level of control necessary for proper function.
In most modern applications, very specific materials’ properties are required, which may include conductivity, magnetic and optical properties, hardness, ductility or more. This can make it difficult to achieve zero expansion or match the expansion coefficient of another device component. In such cases, use of composites is an attractive alternative, which can allow preservation of desirable material properties while modifying unfavorable ones or adding new, advantageous ones. NTE materials can theoretically be used to reduce or offset the expansion of any other material through preparation of controlled thermal expansion composites. In practice, the preparation of high quality composites poses a number of interesting engineering challenges.
3.1. Desirable Properties of Filler Materials
The best filler material for any controlled expansion composite is the least expensive compound that will achieve the desired reduction in expansion and can easily be prepared in the necessary quantities. In addition, stability under processing and use conditions, and compatibility with other system components, is necessary. In situ phase formation may aid in achieving homogeneous composites, alternatively, small particle sizes will improve mixing. Scaling up processes to industrial scale will likely bring additional challenges. Materials with isotropic NTE offer the advantage that filler particle orientation does not affect expansion, making isotropic behavior desirable in many cases. However, if the expansion of a highly oriented matrix or device needs to be compensated, anisotropic NTE may be preferable.
The exact optimum properties of a filler material can change depending on the exact nature of the composite. For example, compatibility will depend on the identity and properties of other device components. Similarly, the targeted application for a device will often dictate processing and conditions (temperature, pressure, atmosphere/environment etc.). Nevertheless, there are certain known properties of NTE materials that could impede their usefulness in controlled thermal expansion composites, which will be discussed in the following section.
3.2. Potential Problems with Known NTE Filler Materials
With the exception of zeolitic materials, all NTE materials discussed in this review contain transition metals, which generally increases the price of the starting materials. For high end applications where other device components or manufacturing processes already have a significant cost, the added expense of small amounts of NTE materials becomes negligible. However, for mass production of cheaper devices, the NTE filler cost may prove prohibitive. Zr, V, Mo and W based starting materials are significantly more expensive than zeolites, and many Sc2W3O12-type compounds that show NTE around room temperature contain expensive lanthanides or pseudo-lanthanides. Commercial availability is another important consideration. Zeolites are readily available, but ZrW2O8 is the only NTE material discussed in Section 2.1 through Section 2.3 that can be purchased. The commercially available ZrW2O8 powder has a median particle size of 15-20 μm, and may contain small ZrO2 and WO3 impurities.
The ease of preparation of NTE fillers depends on their thermodynamic stability and whether particle size control is necessary. Most compounds in the ZrV2O7 and Sc2W3O12 families are thermodynamically stable, and can be obtained by traditional ceramic methods [1,5]. In contrast, the ZrW2O8-type materials are metastable at room temperature, requiring rapid quenching and thus limiting batch sizes for traditional solid state synthesis [34,71]. Preparation of significant quantities of ZrMo2O8 or HfMo2O8 by high temperature approaches may not be feasible. In addition, P2O5, V2O5, MoO3 and WO3 show considerable volatility at high temperatures, making extended heating of binary oxide starting mixtures unfavorable. Solution based routes to all families of NTE compounds have been reported, which offer the added advantage of particle size control [173,174]. However, synthesis conditions must be carefully optimized to ensure preparation of stoichiometric, homogeneous NTE compounds, especially for complex solid solutions. For the ZrW2O8 family, low temperature routes require topotactic recrystallization at a temperature where the compounds are metastable, which further complicates the preparation of phase pure samples, especially for the molybdates [69]. AM2O8 compounds and other NTE materials with an upper limit of thermal stability are not suitable for ceramic composites that require sintering at high temperature.
The temperature range over which a material displays NTE is important. For most applications, this range should include room temperature, and a wider range of NTE can be considered beneficial. This makes several zeolites, the ZrV2O7 family and Sc2W3O12-type materials with smaller A3+ cation less attractive.
A major drawback of many NTE materials is their instability under moderate pressure. Composite preparation has to avoid pressures at which the filler irreversibly transforms to a different phase. In addition, pressure-induced phase transitions can cause problems during thermal cycling of composites if localized pressures on individual filler particles exceed the transition pressure. ZrW2O8 and HfW2O8 show the most detrimental behavior in this respect, with irreversible phase transitions at 0.2 and 0.6 GPa, respectively [84,89]. While many orthorhombic A2M3O12 compounds transform to a denser monoclinic phase at 0.3 to 0.7 GPa as well [135,136,143], this transition is reversible upon decompression. Zr2WP2O12 transforms to the denser monoclinic polymorph as well, but only above 1.4 GPa [145].
Another problem arises from the instability of some NTE compounds under ambient conditions unless they are used in a sealed system. A2M3O12 compositions are very hydroscopic when A is a lanthanide or yttrium, forming a trihydrate A2M3O12·3H2O within minutes of exposure to atmospheric moisture. While the water can be removed by heating, repeated hydration and dehydration is likely to accelerate composite deterioration. Solid solutions Zr1−xHfxMo2−yWyO8 with 30 to 90% tungsten content also incorporate water into their crystal structures [175,176], although the absorption is slower and does not lead to a significant change in crystal structure. Recently, this autohydration behavior was also observed in nanosized ZrW2O8 [177], while micron sized particles required hydrothermal treatment at 180 °C to force water into the structure [175].
Lastly, compatibility and mixing of filler and matrix are important for the preparation of composites. While other components in ceramic composites are usually compatible with NTE fillers, reactivity towards metals has been reported [49], and poor interaction between as-prepared filler particles and matrix has been observed in polymer composites [178,179,180].
3.3. Literature Reports on Composites with NTE Fillers
To date, literature reports on composites using the NTE fillers discussed in this review article are sparse. Zeolitic materials have not been utilized in attempts to reduce other materials’ expansion, despite their commercial availability and lower cost. This may be related to the fact that their expansion behavior is strongly influenced by guest molecules, making their behavior in composites less predictable. Interestingly, the NTE observed in many zeolites has been reported as a problem, as their shrinkage leads to crack formation and delamination between zeolite membranes and alumina supports [181,182]. Recently, use of a zeolite-based support instead of alumina was proposed to overcome this expansion mismatch [183].
With two exceptions, NTE composite research has focused on ZrW2O8. A ceramic FexSc2−xMo3O12/MoO3 composite with close to zero thermal expansion was prepared by melt reaction [184]. The final composite contained significant void space and was brittle, which was attributed to evaporation of MoO3. In addition, a magnesium composite containing 20 vol% Zr2WP2O12 was reported [185]. The overall expansion coefficient was only slightly reduced.
ZrW2O8 has been incorporated into metal, ceramic and polymer composites. Most of the metal composite work focused on Cu composites [49,50,186,187,188,189,190]. Cu is used as a heat sink in microelectronics, and copper composites that match the expansion coefficient of Si (4 × 10−6 K−1) or Al2O3 (7 × 10−6 K−1) could find widespread applications [49]. However, in all cases, formation of significant amounts of orthorhombic γ-ZrW2O8 was observed during thermal cycling. It was found that the copper matrix exerts a pressure of ~0.45 GPa on the filler particles [187,189], which is high enough to induce the cubic to orthorhombic phase transition. Attempts to reduce the local stress through precoating of particles with Cu did not succeed in suppressing the transformation [190]. It should be possible to overcome this problem by using HfW2O8 as a filler instead of ZrW2O8, however, no such attempts have been reported to date. An exploratory investigation on a ZrW2O8/low expansion steel composite (αsteel = 1.5 × 10−6 K−1) still observed formation of γ-ZrW2O8, clearly indicating that the application of ZrW2O8 will be limited to environments that will not exert significant pressure on the filler particles [191].
The exploratory synthesis of ZrW2O8/SiO2 [192] and ZrW2O8/cement composites [193] have been reported. However, most ZrW2O8/ceramic composites have used ZrO2 [51,52,194,195,196,197,198,199,200] or Zr2WP2O12 [201,202,203] as the second component. Due to the similarity of the two components, good compatibility was observed. Formation of dense ceramic bodies (up to 95% dense) was achieved by addition of small amounts of Al2O3 [194,195], and control of thermal expansion ranging from negative to positive values was achieved for ZrO2, while all ZrW2O8/Zr2WP2O12 mixtures exhibited negative expansion. The α- to β-transition of ZrW2O8 was suppressed for composites with 75% Zr2WP2O12.
ZrW2O8 has been incorporated into several polymer systems, including phenolic resins [204], epoxy resins [205] and polyimides [178,179,180]. In contrast to oxide ceramics, ZrW2O8 is not readily compatible with most polymer systems, and surface modification of filler particles is necessary to achieve good interaction with the matrix [178,179,205]. Significant reductions in expansion have been observed in all cases, ranging from 30% reduction for phenolic resins with 52 vol% and polyimides with 22 vol% filler to 60% reduction in epoxy composites for 40 vol% loading. Significant particle agglomeration was observed when large filler particles were used. In addition, large particles are prone to settling at the bottom of any polymer composite films during film formation. Nanoparticles can be used to overcome this problem [178,179], however, a compromise between particle size and kinetics of the recently reported autohydration [177] will have to be found. Optimization of processing conditions like the use of reprecipitation blending reported for polyimide composites may allow formation of homogeneous ZrW2O8/polymer composites with intermediate particle size [179].
4. Conclusions
Negative thermal expansion has been established as a specialized field of research since the mid 1990s. New materials belonging to previously identified families of compounds, new families of NTE materials, and new insights into mechanisms are added to the literature every year. The initially predicted widespread use of NTE materials as fillers in a variety of controlled thermal expansion composites has not yet been achieved, but some promising preliminary results on ZrW2O8/ZrO2 and ZrW2O8/polymer systems have been reported. Exploration of other filler materials that are less pressure sensitive or do not autohydrate under ambient conditions will be necessary to further the applications of NTE fillers, and will likely lead to interesting application as the field matures.
Acknowledgments
Some of the work presented in this review was supported by the National Science Foundation under grant DMR-0545517.
References and Notes
- Korthuis, V.; Khosrovani, N.; Sleight, A.W.; Roberts, N.; Dupree, R.; Warren, W.W. Negative thermal expansion and phase transitions in the ZrV2−xPxO7 series. Chem. Mater. 1995, 7, 412–417. [Google Scholar] [CrossRef]
- Sleight, A.W. Thermal Contraction. Endeavour 1995, 19, 64–68. [Google Scholar] [CrossRef]
- Mary, T.A.; Evans, J.S.O.; Vogt, T.; Sleight, A.W. Negative thermal expansion from 0.3 to 1050 Kelvin in ZrW2O8. Science 1996, 272, 90–92. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Hu, Z.; Jorgensen, J.D.; Argyriou, D.N.; Short, S.; Sleight, A.W. Compressibility, phase transitions, and oxygen migration in zirconium tungstate, ZrW2O8. Science 1997, 275, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.S.O.; Mary, T.A.; Sleight, A.W. Negative thermal expansion in a large molybdate and tungstate family. J.Solid State Chem. 1997, 133, 580–583. [Google Scholar] [CrossRef]
- Ernst, G.; Broholm, C.; Kowach, G.R.; Ramirez, A.P. Phonon density of states and negative thermal expansion in ZrW2O8. Nature 1998, 396, 147–149. [Google Scholar] [CrossRef]
- Perottoni, C.A.; da Jornada, J.A.H. Pressure-induced amorphization and negative thermal expansion in ZrW2O8. Science 1998, 280, 886–889. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.P.; Kowach, G.R. Large low temperature specific heat in the negative thermal expansion compound ZrW2O8. Phys. Rev. Lett. 1998, 80, 4903–4906. [Google Scholar] [CrossRef]
- Sleight, A.W. Negative thermal expansion materials. Curr. Opin. Solid State Mater. Sci. 1998, 3, 128–131. [Google Scholar] [CrossRef]
- Ravindran, T.R.; Arora, A.K.; Mary, T.A. High pressure behavior of ZrW2O8: Gruneisen parameter and thermal properties. Phys. Rev. Lett. 2000, 84, 3879–3882. [Google Scholar] [CrossRef] [PubMed]
- Reisner, B.A.; Lee, Y.; Hanson, J.C.; Jones, G.A.; Parise, J.B.; Corbin, D.R.; Toby, B.H.; Freitag, A.; Larese, J.Z.; Kahlenberg, V. Understanding negative thermal expansion and ‘trap door’ cation relocations in zeolite rho. Chem. Commun. 2000, 22, 2221–2222. [Google Scholar] [CrossRef]
- Mittal, R.; Chaplot, S.L.; Schober, H.; Mary, T.A. Origin of negative thermal expansion in cubic ZrW2O8 revealed by high pressure inelastic neutron scattering. Phys. Rev. Lett. 2001, 86, 4692–4695. [Google Scholar] [CrossRef] [PubMed]
- Beccara, S.A.; Dalba, G.; Fornasini, P.; Grisenti, R.; Sanson, A. Local thermal expansion in a cuprite structure: The case of Ag2O. Phys. Rev. Lett. 2002, 89, 025503:1–025503:4. [Google Scholar] [CrossRef]
- Birkedal, H.; Schwarzenbach, D.; Pattison, P. Observation of uniaxial negative thermal expansion in an organic crystal. Angew. Chem. 2002, 114, 780–782. [Google Scholar] [CrossRef]
- Chapman, K.W.; Chupas, P.J.; Kepert, C.J. Direct observation of a transverse vibrational mechanism for negative thermal expansion in Zn(CN)2: An atomic pair distribution function analysis. J. Am. Chem. Soc. 2005, 127, 15630–15636. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.P.; Kieffer, J. Structural origin of negative thermal expansion in high-temperature silica polymorphs. Phys. Rev. Lett. 2005, 95, 215901:1–215901:4. [Google Scholar]
- Tucker, M.G.; Goodwin, A.L.; Dove, M.T.; Keen, D.A.; Wells, S.A.; Evans, J.S.O. Negative thermal expansion in ZrW2O8: Mechanisms, rigid unit modes, and neutron total scattering. Phys. Rev. Lett. 2005, 95, 255501:1–255501:4. [Google Scholar] [CrossRef]
- Chapman, K.W.; Chupas, P.J.; Kepert, C.J. Compositional dependence of negative thermal expansion in the Prussian blue analogues (MPtIV)-PtII(CN)6 (M = Mn, Fe, Co, Ni, Cu, Zn, Cd). J. Am. Chem. Soc. 2006, 128, 7009–7014. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, A.L.; Calleja, M.; Conterio, M.J.; Dove, M.T.; Evans, J.S.O.; Keen, D.A.; Peters, L.; Tucker, M.G. Colossal positive and negative thermal expansion in the framework material Ag3[Co(CN)6]. Science 2008, 319, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.E.; Goodwin, A.L.; Halder, G.J.; Southon, P.D.; Kepert, C.J. Nanoporosity and exceptional negative thermal expansion in single-network cadmium cyanide. Angew. Chem. 2008, 120, 1418–1421. [Google Scholar] [CrossRef]
- Yamamura, Y.; Ikeuchi, S.; Saito, K. Characteristic Phonon spectrum of negative thermal expansion materials with framework structure through calorimetric study of Sc2M3O12 (M = W and Mo). Chem. Mater. 2009, 21, 3008–3016. [Google Scholar] [CrossRef]
- Azuma, M.; Chen, W.T.; Seki, H.; Czapski, M.; Olga, S.; Oka, K.; Mizumaki, M.; Watanuki, T.; Ishimatsu, N.; Kawamura, N.; Ishiwata, S.; Tucker, M.G.; Shimakawa, Y.; Attfield, J.P. Colossal negative thermal expansion in BiNiO3 induced by intermetallic charge transfer. Nature Commun. 2011, 2. [Google Scholar] [CrossRef]
- Mary, T.A.; Sleight, A.W. Bulk thermal expansion for tungstate and molybdates of the type A2M3O12. J. Mater. Res. 1999, 14, 912–915. [Google Scholar] [CrossRef]
- Lightfoot, P.; Woodcock, D.A.; Maple, M.J.; Villaescusa, L.A.; Wright, P.A. The widespread occurrence of negative thermal expansion in zeolites. J. Mater. Chem. 2001, 11, 212–216. [Google Scholar] [CrossRef]
- Negative or Zero Thermal Expansion Materials ; (Special Issue Name); Journal of the Chinese Ceramic Society: Beijing, China, 2009; Volume 37, pp. 651–759.
- Scheel, K. Versuche ueber die ausdehnung fester koerper, insbesondere von quarz in richtung der hauptachse, platin, palladium und quarzglas bei der temperatur der fluessigen Luft. Verh. Deutsch. Phys. Ges. 1907, 9, 3–23. [Google Scholar]
- Scheel, K. Ueber die ausdehnung des quarzglases. Verh. Deutsch. Phys. Ges. 1907, 9, 719–721. [Google Scholar]
- Hummel, F.A. Thermal expansion properties of natural Lithia minerals. Foote Prints 1948, 20, 3–11. [Google Scholar]
- Hummel, F.A. Termal expansion properties of some synthetic lithia minerals. J. Am. Ceram. Soc. 1951, 34, 235–239. [Google Scholar] [CrossRef]
- Roy, R.; Agrawal, D.K.; McKinstry, H.A. Very low thermal expansion coefficient materials. Annu. Rev. Mater. Sci. 1989, 19, 59–81. [Google Scholar] [CrossRef]
- Boilot, J.P.; Salanie, J.P. Phase transformation in Na1+xSixZr2P3−xO12 compounds. Mater. Res. Bull. 1979, 14, 1469–1477. [Google Scholar] [CrossRef]
- Limaye, S.Y.; Agrawal, D.K.; Roy, R.; Mehrotra, Y. Synthesis, sintering and thermal-expansion of Ca1−xSrxZr4P6O24—An ultra-low thermal-expansion ceramic system. J. Mater. Sci. 1991, 26, 93–98. [Google Scholar] [CrossRef]
- Lenain, G.E.; McKinstry, H.A.; Alamo, J.; Agrawal, D.K. Structural model for thermal expansion in MZr2P3O12 (M = Li, Na, K, Rb, Cs). J. Mater. Sci. 1987, 22, 17–22. [Google Scholar] [CrossRef]
- Graham, J.; Wadsley, A.D.; Weymouth, J.H.; Williams, L.S. A new ternary oxide, ZrW2O8. J. Am. Ceram. Soc. 1959, 42, 570. [Google Scholar] [CrossRef]
- Martinek, C.; Hummel, F.A. Linear thermal expansion of three tungstates. J. Am. Ceram. Soc. 1968, 51, 227–228. [Google Scholar] [CrossRef]
- Pryde, A.K.A.; Hammonds, K.D.; Dove, M.T.; Heine, V.; Gale, J.D.; Warren, M.C. Origin of the negative thermal expansion in ZrW2O8 and ZrV2O7. J. Phys. Condens. Matter 1996, 8, 10973–10982. [Google Scholar] [CrossRef]
- Pryde, A.K.A.; Hammonds, K.D.; Dove, M.T.; Heine, V.; Gale, J.D.; Warren, M.C. Rigid unit modes and the negative thermal expansion in ZrW2O8. Phase Transit. 1997, 61, 141–153. [Google Scholar] [CrossRef]
- Stillinger, F.H.; Stillinger, D.K. Negative thermal expansion in the Gaussian core model. Phys. A 1997, 244, 358–369. [Google Scholar] [CrossRef]
- David, W.I.F.; Evans, J.S.O.; Sleight, A.W. Direct evidence for a low-frequency phonon mode mechanism in the negative thermal expansion compound ZrW2O8. Europhys. Lett. 1999, 46, 661–666. [Google Scholar] [CrossRef]
- Heine, V.; Welche, P.R.L.; Dove, M.T. Geometrical origin and theory of negative thermal expansion in framework structures. J. Am. Ceram. Soc. 1999, 82, 1793–1802. [Google Scholar] [CrossRef]
- Ramirez, A.P.; Broholm, C.L.; Cava, R.J.; Kowach, G.R. Geometrical frustration, spin ice and negative thermal expansion—The physics of underconstraint. Phys. B Condens. Matter 2000, 280, 290–295. [Google Scholar] [CrossRef]
- Boerio-Goates, J.; Stevens, R.; Lang, B.; Woodfield, B.F. Heat capacity calorimetry—Detection of low frequency modes in solids and an application to negative thermal expansion materials. J. Therm. Anal. Calorim. 2002, 69, 773–783. [Google Scholar] [CrossRef]
- Chapman, K.W.; Chupas, P.J. Anomalous thermal expansion of cuprites: A combined high resolution pair distribution function and geometric analysis. Chem. Mater. 2009, 21, 425–431. [Google Scholar] [CrossRef]
- Korcok, J.L.; Katz, M.J.; Leznoff, D.B. Impact of metallophilicity on “colossal” positive and negative thermal expansion in a series of isostructural dicyanometallate coordination polymers. J. Am. Chem. Soc. 2009, 131, 4866–4871. [Google Scholar] [CrossRef] [PubMed]
- Sleight, A.W.; Thundathil, M.A.; Evans, J.S.O. Negative Thermal Expansion Materials. U.S. Patent 5,514,360, 7 May 1996. [Google Scholar]
- Sleight, A.W. Compounds that contract on heating. Inorg. Chem. 1998, 37, 2854–2860. [Google Scholar] [CrossRef]
- Sleight, A.W. Isotropic negative thermal expansion. Ann. Rev. Mater. Sci. 1998, 28, 29–43. [Google Scholar] [CrossRef]
- Fleming, D.A.; Johnson, D.W.; Lemaire, P.J. Article Comprising a Temperature Compensated Optical Fiber Refractive Index Grating. U.S. Patent 5,694,503, 2 December 1997. [Google Scholar]
- Verdon, C.; Dunand, D.C. High-temperature reactivity in the ZrW2O8-Cu system. Scr. Mater. 1997, 36, 1075–1080. [Google Scholar] [CrossRef]
- Balch, D.K.; Dunand, D.C. Copper-zirconium tungstate composites exhibiting low and negative thermal expansion influenced by reinforcement phase transformations. Metall. Mater. Trans. A 2004, 35, 1159–1165. [Google Scholar] [CrossRef]
- De Buysser, K.; Lommens, P.; de Meyer, C.; Bruneel, E.; Hoste, S.; van Driessche, I. ZrO2-ZrW2O8 composites with tailor-made thermal expansion. Ceram. Silik. 2004, 48, 139–144. [Google Scholar]
- Lommens, P.; de Meyer, C.; Bruneel, E.; de Buysser, K.; van Driessche, I.; Hoste, S. Synthesis and thermal expansion of ZrO2/ZrW2O8 composites. J. Eur. Ceram. Soc. 2005, 25, 3605–3610. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Mary, T.A.; Vogt, T.; Subramanian, M.A.; Sleight, A.W. Negative thermal expansion in ZrW2O8 and HfW2O8. Chem. Mater. 1996, 8, 2809–2823. [Google Scholar] [CrossRef]
- Lind, C.; Wilkinson, A.P.; Hu, Z.B.; Short, S.; Jorgensen, J.D. Synthesis and properties of the negative thermal expansion material cubic ZrMo2O8. Chem. Mater. 1998, 10, 2335–2337. [Google Scholar] [CrossRef]
- Attfield, M.P.; Sleight, A.W. Exceptional negative thermal expansion in AlPO4–17. Chem. Mater. 1998, 10, 2013–2019. [Google Scholar] [CrossRef]
- Attfield, M.P.; Sleight, A.W. Strong negative thermal expansion in siliceous faujasite. Chem. Commun. 1998, 601–602. [Google Scholar]
- Goodwin, A.L.; Chapman, K.W.; Kepert, C.J. Guest-dependent negative thermal expansion in nanoporous Prussian Blue analogues MIIPtIV(CN)6 {H2O} (0 ≤ x ≤ 2; M = Zn, Cd). J. Am. Chem. Soc. 2005, 127, 17980–17981. [Google Scholar] [CrossRef] [PubMed]
- Margadonna, S.; Prassides, K.; Fitch, A.N. Zero thermal expansion in a Prussian blue analogue. J. Am. Chem. Soc. 2004, 126, 15390–15391. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.Z.; Sleight, A.W. The role of rigid unit modes in negative thermal expansion. J. Solid State Chem. 2003, 173, 442–448. [Google Scholar] [CrossRef]
- Bieniok, A.; Hammonds, K.D. Rigid unit modes and the phase transition and structural distortions of zeolite rho. Microporous Mesoporous Mat. 1998, 25, 193–200. [Google Scholar] [CrossRef]
- Dove, M.T.; Hammonds, K.D.; Harris, M.J.; Heine, V.; Keen, D.A.; Pryde, A.K.A.; Trachenko, K.; Warren, M.C. Amorphous silica from the Rigid unit mode approach. Miner. Mag. 2000, 64, 377–388. [Google Scholar] [CrossRef]
- Dove, M.T.; Trachenko, K.O.; Tucker, M.G.; Keen, D.A. Rigid unit modes in framework structures: Theory, experiment and applications. Rev. Mineral. Geochem. 2000, 39, 1–33. [Google Scholar] [CrossRef]
- Yamamura, Y.; Nakajima, N.; Tsuji, T.; Iwasa, Y.; Saito, K.; Sorai, M. Heat capacity and Gruneisen functions of negative thermal expansion compound HfW2O8. Solid State Commun. 2002, 121, 213–217. [Google Scholar] [CrossRef]
- Mittal, R.; Chaplot, S.L. Phonon density of states and thermodynamic properties in cubic and orthorhombic phases of ZrW2O8. Solid State Commun. 2000, 115, 319–322. [Google Scholar] [CrossRef]
- Mittal, R.; Chaplot, S.L.; Schober, H.; Kolesnikov, A.I.; Loong, C.K.; Lind, C.; Wilkinson, A.P. Negative thermal expansion in cubic ZrMo2O8: Inelastic neutron scattering and lattice dynamical studies. Phys. Rev. B 2004, 70, 214303:1–214303:6. [Google Scholar] [CrossRef]
- Auray, M.; Quarton, M. Zirconium tungstate. Acta Crystallogr. C 1995, 51, 2210–2213. [Google Scholar] [CrossRef]
- Closmann, C.; Sleight, A.W.; Haygarth, J.C. Low-temperature synthesis of ZrW2O8 and Mo-substituted ZrW2O8. J. Solid State Chem. 1998, 139, 424–426. [Google Scholar] [CrossRef]
- Allen, S.; Evans, J.S.O. Negative thermal expansion and oxygen disorder in cubic ZrMo2O8. Phys. Rev. B 2003, 68, 134101:1–134101:3. [Google Scholar] [CrossRef]
- Lind, C.; Wilkinson, A.P.; Rawn, C.J.; Payzant, E.A. Preparation of the negative thermal expansion material cubic ZrMo2O8. J. Mater. Chem. 2001, 11, 3354–3359. [Google Scholar] [CrossRef]
- Lind, C.; Wilkinson, A.P.; Rawn, C.J.; Payzant, E.A. Kinetics of the cubic to trigonal transformation in ZrMo2O8 and their dependence on precursor chemistry. J. Mater. Chem. 2002, 12, 990–994. [Google Scholar] [CrossRef]
- Readman, J.E.; Lister, S.E.; Peters, L.; Wright, J.; Evans, J.S.O. Direct synthesis of cubic ZrMo2O8 followed by ultrafast in situ powder diffraction. J. Amer. Chem. Soc. 2009, 131, 17560–17562. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Hanson, P.A.; Ibberson, R.M.; Duan, N.; Kameswari, U.; Sleight, A.W. Low-temperature oxygen migration and negative thermal expansion in ZrW2−xMoxO8. J. Am. Chem. Soc. 2000, 122, 8694–8699. [Google Scholar] [CrossRef]
- Kameswari, U.; Sleight, A.W.; Evans, J.S.O. Rapid synthesis of ZrW2O8 and related phases, and structure refinement of ZrWMoO8. Int. J. Inorg. Mater. 2000, 2, 333–337. [Google Scholar] [CrossRef]
- Allen, S.; Evans, J.S.O. The kinetics of low-temperature oxygen migration in ZrWMoO8. J. Mater. Chem. 2004, 14, 151–156. [Google Scholar] [CrossRef]
- De Meyer, C.; Bouree, F.; Evans, J.S.O.; de Buysser, K.; Bruneel, E.; van Driessche, I.; Hoste, S. Structure and phase transition of Sn-substituted Zr1−xSnxW2O8. J. Mater. Chem. 2004, 14, 2988–2994. [Google Scholar]
- De Buysser, K.; van Driessche, I.; Putte, B.V.; Vanhee, P.; Schaubroeck, J.; Hoste, S. Study of negative thermal expansion and shift in phase transition temperature in Ti4+- and Sn4+-substituted ZrW2O8 materials. Inorg. Chem. 2008, 47, 736–741. [Google Scholar]
- Nakajima, N.; Yamamura, Y.; Tsuji, T. Synthesis and physical properties of negative thermal expansion materials Zr1−xMxW2O8-y (M = Sc, In, Y) substituted for Zr(IV) sites by M(III) ions. Solid State Commun. 2003, 128, 193–196. [Google Scholar] [CrossRef]
- Hashimoto, T.; Kuwahara, J.; Yoshida, T.; Nashimoto, M.; Takahashi, Y.; Takahashi, K.; Morito, Y. Thermal conductivity of negative-thermal-expansion oxide, Zr1−xYxW2O8 (x = 0.00, 0.01): Temperature dependence and effect of structural phase transition. Solid State Commun. 2004, 131, 217–221. [Google Scholar] [CrossRef]
- Tsuji, T.; Yamamura, Y.; Nakajima, N. Thermodynamic properties of negative thermal expansion materials ZrW2O8 substituted for Zr site. Thermochim. Acta 2004, 416, 93–98. [Google Scholar] [CrossRef]
- Yamamura, Y.; Nakajima, N.; Tsuji, T.; Kojima, A.; Kuroiwa, Y.; Sawada, A.; Aoyagi, S.; Kasatani, H. Drastic lowering of the order-disorder phase transition temperatures in Zr1−xMxW2O8−y (M = Sc,Y,In) solidsolutions. Phys. Rev. B 2004, 70, 104107:1–104107:6. [Google Scholar] [CrossRef]
- Yamamura, Y.; Masago, K.; Kato, M.; Tsuji, T. Comprehensive interpretation of a substitution effect on an order-disorder phase transition in A1−xMxW2O8−y (A = Zr, Hf; M = trivalent cations) and other ZrW2O8-based solid solutions. J. Phys. Chem. B 2007, 111, 10118–10122. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tao, J.; Ma, H.; Zhao, X. Disordered structure of ZrW1.8V0.2O7.9 from a combined X-ray and neutron powder diffraction study at 530 K. Acta Crystallogr. C 2009, 65, 74–76. [Google Scholar] [CrossRef]
- Chen, X.; Deng, X.; Ma, H.; Tao, J.; Zhao, X. Hydrothermal synthesis and thermal properties of a novel cubic ZrW1.80V0.20O7.90 solid solution. J. Solid State Chem. 2011, 184, 1090–1095. [Google Scholar] [CrossRef]
- Hu, Z.; Jorgensen, J.D.; Teslic, S.; Short, S.; Argyriou, D.N.; Evans, J.S.O.; Sleight, A.W. Pressure-induced phase transformation in ZrW2O8: Compressibility and thermal expansion of the orthorhombic phase. Physica B 1997, 241, 370–372. [Google Scholar] [CrossRef]
- Jorgensen, J.D.; Hu, Z.; Teslic, S.; Argyriou, D.N.; Short, S.; Evans, J.S.O.; Sleight, A.W. Pressure-induced cubic-to-orthorhombic phase transition in ZrW2O8. Phys. Rev. B 1999, 59, 215–225. [Google Scholar] [CrossRef]
- Grzechnik, A.; Crichton, W.A.; Syassen, K.; Adler, P.; Mezouar, M. A new polymorph of ZrW2O8 synthesized at high pressures and high temperatures. Chem. Mater. 2001, 13, 4255–4259. [Google Scholar] [CrossRef]
- Keen, D.A.; Goodwin, A.L.; Tucker, M.G.; Dove, M.T.; Evans, J.S.O.; Crichton, W.A.; Brunelli, M. Structural description of pressure-induced amorphization in ZrW2O8. Phys. Rev. Lett. 2007, 98, 225501:1–225501:4. [Google Scholar] [CrossRef]
- Chen, B.; Muthu, D.V.S.; Liu, Z.X.; Sleight, A.W.; Kruger, M.B. High-pressure optical study of HfW2O8. J. Phys. Condens. Matter 2002, 14, 13911–13916. [Google Scholar] [CrossRef]
- Jorgensen, J.D.; Hu, Z.; Short, S.; Sleight, A.W.; Evans, J.S.O. Pressure-induced cubic-to-orthorhombic phase transformation in the negative thermal expansion material HfW2O8. J. Appl. Phys. 2001, 89, 3184–3188. [Google Scholar] [CrossRef]
- Lind, C.; VanDerveer, D.G.; Wilkinson, A.P.; Chen, J.H.; Vaughan, M.T.; Weidner, D.J. New high-pressure form of the negative thermal expansion materials zirconium molybdate and hafnium molybdate. Chem. Mater. 2001, 13, 487–490. [Google Scholar] [CrossRef]
- Grzechnik, A.; Crichton, W.A. Structural transformations in cubic ZrMo2O8 at high pressures and high temperatures. Solid State Sci. 2002, 4, 1137–1141. [Google Scholar] [CrossRef]
- Varga, T.; Wilkinson, A.P.; Lind, C.; Bassett, W.A.; Zha, C.S. Pressure-induced amorphization of cubic ZrMo2O8 studied in situ by X-ray absorption spectroscopy and diffraction. Solid State Commun. 2005, 135, 739–744. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Jorgensen, J.D.; Short, S.; David, W.I.F.; Ibberson, R.M.; Sleight, A.W. Thermal expansion in the orthorhombic gamma phase of ZrW2O8. Phys. Rev. B 1999, 60, 14643–14648. [Google Scholar] [CrossRef]
- Varga, T.; Wilkinson, A.P.; Jupe, A.C.; Lind, C.; Bassett, W.A.; Zha, C.S. Pressure-induced amorphization of cubic ZrW2O8 studied in situ and ex situ by synchrotron X-ray diffraction and absorption. Phys. Rev. B 2005, 72, 024117:1–024117:10. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Hanson, J.C.; Sleight, A.W. Room-temperature superstructure of ZrV2O7. Acta Crystallogr. B 1998, 54, 705–713. [Google Scholar] [CrossRef]
- Khosrovani, K.; Sleight, A.W.; Vogt, T. Structure of ZrV2O7 from −263 to 470 degrees C. J. Solid State Chem. 1997, 132, 355–360. [Google Scholar] [CrossRef]
- Withers, R.L.; Evans, J.S.O.; Hanson, J.C.; Sleight, A.W. An in-situ tempertaure dependent electron and X-ray diffraction study of the structural phase transitions in ZrV2O7. J. Solid State Chem. 1998, 137, 161–167. [Google Scholar] [CrossRef]
- Onken, H. Ueber zirkonpyroarsenat. Naturwissenschaften 1965, 52, 344. [Google Scholar] [CrossRef]
- Le Flem, G.; Lamic, J.; Hagenmuller, P. As2O5-ThO2 system. Bull. Soc. Chim. Fr. 1966, 6, 1880–1883. [Google Scholar]
- Losilla, E.R.; Cabeza, A.; Bruque, S.; Aranda, M.A.G.; Sanz, J.; Iglesias, J.E.; Alonso, J.A. Syntheses, structures, and thermal expansion of germanium pyrophosphates. J. Solid State Chem. 2001, 156, 213–219. [Google Scholar] [CrossRef]
- Fayon, F.; King, I.J.; Harris, R.K.; Gover, R.K.B.; Evans, J.S.O.; Massiot, D. Characterization of the room-temperature structure of SnP2O7 by P31 through-space and through-bond NMR correlation spectroscopy. Chem. Mater. 2003, 15, 2234–2239. [Google Scholar] [CrossRef]
- Gover, R.K.B.; Withers, N.D.; Allen, S.; Withers, R.L.; Evans, J.S.O. Structure and phase transitions of SnP2O7. J. Solid State Chem. 2002, 166, 42–48. [Google Scholar] [CrossRef]
- White, K.M.; Lee, P.L.; Chupas, P.J.; Chapman, K.W.; Payzant, E.A.; Jupe, A.C.; Bassett, W.A.; Zha, C.S.; Wilkinson, A.P. Synthesis, symmetry, and physical properties of cerium pyrophosphate. Chem. Mater. 2008, 20, 3728–3734. [Google Scholar] [CrossRef]
- Wallez, G.; Raison, P.E.; Dacheux, N.; Clavier, N.; Bykov, D.; Delevoye, L.; Popa, K.; Bregiroux, D.; Fitch, A.N.; Konings, R.J.M. Triclinic-cubic phase transition and negative expansion in the actinide IV (Th, U, Np, Pu) diphosphates. Inorg. Chem. 2012, 51, 4314–4322. [Google Scholar] [CrossRef] [PubMed]
- Stinton, G.W.; Hampson, M.R.; Evans, J.S.O. The 136-atom structure of ZrP2O7 and HfP2O7 from powder diffraction data. Inorg. Chem. 2006, 45, 4352–4358. [Google Scholar] [CrossRef] [PubMed]
- Birkedal, H.; Andersen, A.M.K.; Arakcheeva, A.; Chapuis, G.; Norby, P.; Pattison, P. The room-temperature superstructure of ZrP2O7 is orthorhombic: There are no unusual 180° P-O-P bond angles. Inorg. Chem. 2006, 45, 4346–4351. [Google Scholar] [CrossRef] [PubMed]
- Oyetola, S.; Verbaere, A.; Guyomard, D.; Crosnier, M.P.; Piffard, Y.; Tournoux, M. New ZrP2O7-like diphosphates of either mixed (M1/2(III)M’1/2(V)) cations (M = Sb, Bi, Nd, Eu-M’ = Sb, Nb, Ta) or M’V cations (M’ = Ta, Nb)—Synthesis and structure. Eur. J. Solid State Inorg. Chem. 1991, 28, 23–36. [Google Scholar]
- Varga, T.; Wilkinson, A.P.; Haluska, M.S.; Payzant, E.A. Preparation and thermal expansion of (MIII0.5M’V0.5)P2O7 with the cubic ZrP2O7 structure. J. Solid State Chem. 2005, 178, 3541–3546. [Google Scholar] [CrossRef]
- Yanase, I.; Kojima, T.; Kobayashi, H. Effects of Nb and Y substitution on negative thermal expansion of ZrV2−xPxO7 (0 ≤ X ≤ 0.8). Solid State Commun. 2011, 151, 595–598. [Google Scholar] [CrossRef]
- Sahoo, P.P.; Sumithra, S.; Madras, G.; Row, T.N.G. Synthesis, structure, negative thermal expansion, and photocatalytic property of Mo doped ZrV2O7. Inorg. Chem. 2011, 50, 8774–8781. [Google Scholar] [CrossRef] [PubMed]
- Carlson, S.; Andersen, A.M.K. High-pressure properties of TiP2O7, ZrP2O7 and ZrV2O7. J. Appl. Crystallogr. 2001, 34, 7–12. [Google Scholar] [CrossRef]
- Hemamala, U.L.C.; El-Ghussein, F.; Muthu, D.V.S.; Andersen, A.M.K.; Carlson, S.; Ouyang, L.; Kruger, M.B. High-pressure Raman and infrared study of ZrV2O7. Solid State Commun. 2007, 141, 680–684. [Google Scholar] [CrossRef]
- Lipinska-Kalita, K.E.; Kruger, M.B.; Carlson, S.; Andersen, A.M.K. High-pressure studies of titanium pyrophosphate by Raman scattering and infrared spectroscopy. Physica B 2003, 337, 221–229. [Google Scholar] [CrossRef]
- Petruska, E.A.; Muthu, D.V.S.; Carlson, S.; Andersen, A.M.K.; Ouyang, L.; Kruger, M.B. High-pressure Raman and infrared spectroscopic studies of ZrP2O7. Solid State Commun. 2010, 150, 235–239. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Mary, T.A.; Sleight, A.W. Negative thermal expansion in Sc2(WO4)3. J. Solid State Chem. 1998, 137, 148–160. [Google Scholar] [CrossRef]
- Zhou, Y.; Adams, S.; Rao, R.P.; Edwards, D.D.; Neiman, A.; Pestereva, N. Charge transport by polyatomic anion diffusion in Sc2(WO4)3. Chem. Mater. 2008, 20, 6335–6345. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Mary, T.A. Structural phase transitions and negative thermal expansion in Sc2(MoO4)3. Int. J. Inorg. Mater. 2000, 2, 143–151. [Google Scholar] [CrossRef]
- Wu, M.M.; Cheng, Y.Z.; Peng, J.; Xiao, X.L.; Chen, D.F.; Kiyanagi, R.; Fieramosca, J.S.; Short, S.; Jorgensen, J.; Hu, Z.B. Synthesis of solid solution Er2−xCexW3O12 and studies of their thermal expansion behavior. Mater. Res. Bull. 2007, 42, 2090–2098. [Google Scholar] [CrossRef]
- Wu, M.M.; Peng, J.; Cheng, Y.Z.; Xiao, X.L.; Hao, Y.M.; Hu, Z.B. Thermal expansion in solid solution Er2−xSmxW3O12. Mater. Sci. Eng. B 2007, 137, 144–148. [Google Scholar] [CrossRef]
- Wu, M.M.; Peng, J.; Cheng, Y.Z.; Wang, H.; Yu, Z.X.; Chen, D.F.; Hu, Z.B. Structure and thermal expansion properties of solid solution Nd2−xErxW3O12 (0.0 ≤ x ≤ 0.6 and 1.5 ≤ x ≤ 2.0). Solid State Sci. 2006, 8, 665–670. [Google Scholar] [CrossRef]
- Xiao, X.L.; Peng, J.; Wu, M.M.; Cheng, Y.Z.; Chen, D.F.; Hu, Z.B. The crystal structure and thermal expansion properties of solid solutions Ln2−xDyxW3O12 (Ln = Er and Y). J. Alloy. Compd. 2008, 465, 556–561. [Google Scholar] [CrossRef]
- Peng, J.; Wu, M.M.; Wang, H.; Hao, Y.M.; Hu, Z.; Yu, Z.X.; Chen, D.F.; Kiyanagic, R.; Fieramosca, J.S.; Short, S.; Jorgensen, J. Structures and negative thermal expansion properties of solid solutions YxNd2−xW3O12 (x = 0.0–1.0, 1.6–2.0). J. Alloy. Compd. 2008, 453, 49–54. [Google Scholar] [CrossRef]
- Evans, J.S.O.; Mary, T.A.; Sleight, A.W. Structure of Zr2(WO4)(PO4)2 from powder X-ray data: Cation ordering with no superstructure. J. Solid State Chem. 1995, 120, 101–104. [Google Scholar] [CrossRef]
- Cetinkol, M.; Wilkinson, A.P.; Lee, P.L. Structural changes accompanying negative thermal expansion in Zr2(MoO4)(PO4)2. J. Solid State Chem. 2009, 182, 1304–1311. [Google Scholar] [CrossRef]
- Suzuki, T.; Omote, A. Negative thermal expansion in (HfMg)(WO4)3. J. Am. Ceram. Soc. 2004, 87, 1365–1367. [Google Scholar] [CrossRef]
- Gindhart, A.M.; Lind, C.; Green, M. Polymorphism in the negative thermal expansion material magnesium hafnium tungstate. J. Mater. Res. 2008, 23, 210–213. [Google Scholar] [CrossRef]
- Marinkovic, B.A.; Jardim, P.M.; Ari, M.; de Avillez, R.R.; Rizzo, F.; Ferreira, F.F. Low positive thermal expansion in HfMgMo3O12. Phys. Status Solidi B 2008, 245, 2514–2519. [Google Scholar] [CrossRef]
- Sleight, A.W.; Brixner, L.H. A New Ferroelastic transition in some A2(MO4)3 molybdates and tungstates. J. Solid State Chem. 1973, 7, 172–174. [Google Scholar] [CrossRef]
- Forster, P.M.; Sleight, A.W. Negative thermal expansion in Y2W3O12. Int. J. Inorg. Mater. 1999, 1, 123–127. [Google Scholar] [CrossRef]
- Suzuki, T.; Omote, A. Zero thermal expansion in (Al2x(HfMg)1−x)(WO4)3. J. Am. Ceram. Soc. 2006, 89, 691–693. [Google Scholar] [CrossRef]
- Marinkovic, B.A.; Jardim, P.M.; de Avillez, R.R.; Rizzo, F. Negative thermal expansion in Y2Mo3O12. Solid State Sci. 2005, 7, 1377–1383. [Google Scholar] [CrossRef]
- Sumithra, S.; Tyagi, A.K.; Umarji, A.M. Negative thermal expansion in Er2W3O12 and Yb2W3O12 by high temperature X-ray diffraction. Mater. Sci. Eng. B 2005, 116, 14–18. [Google Scholar] [CrossRef]
- Gates, S.D.; Lind, C. Polymorphism in yttrium molybdate Y2Mo3O12. J. Solid State Chem. 2007, 180, 3510–3514. [Google Scholar] [CrossRef]
- Varga, T.; Wilkinson, A.P.; Jorgensen, J.D.; Short, S. Neutron powder diffraction study of the orthorhombic to monoclinic transition in Sc2W3O12 on compression. Solid State Sci. 2006, 8, 289–295. [Google Scholar] [CrossRef]
- Varga, T.; Wilkinson, A.P.; Lind, C.; Bassett, W.A.; Zha, C.S. High pressure synchrotron X-ray powder diffraction study of Sc2Mo3O12 and Al2W3O12. J. Phys. Condens. Matter 2005, 17, 4271–4283. [Google Scholar] [CrossRef]
- Varga, T.; Wilkinson, A.P.; Lind, C.; Bassett, W.A.; Zha, C.S. In situ high-pressure synchrotron X-ray diffraction study of Sc2W3O12 at up to 10 GPa. Phys. Rev. B 2005, 71, 214106:1–214106:8. [Google Scholar]
- Garg, N.; Murli, C.; Tyagi, A.K.; Sharma, S.M. Phase transitions in Sc2(WO4)3 under high pressure. Phys. Rev. B 2005, 72, 064106:1–064106:7. [Google Scholar]
- Garg, N.; Panchal, V.; Tyagi, A.K.; Sharma, S.M. Pressure-induced phase transitions in Al2(WO4)3. J. Solid State Chem. 2005, 178, 998–1002. [Google Scholar] [CrossRef]
- Achary, S.N.; Mukherjee, G.D.; Tyagi, A.K.; Vaidya, S.N. Preparation, thermal expansion, high pressure and high temperature behavior of Al2(WO4)3. J. Mater. Sci. 2002, 37, 2501–2509. [Google Scholar] [CrossRef]
- Arora, A.K.; Nithya, R.; Yagi, T.; Miyajima, N.; Mary, T.A. Two-stage amorphization of scandium molybdate at high pressure. Solid State Commun. 2004, 129, 9–13. [Google Scholar] [CrossRef]
- Baiz, T.I.; Heinrich, C.P.; Banek, N.A.; Vivekens, B.L.; Lind, C. In-situ non-ambient X-ray diffraction studies of indium tungstate. J. Solid State Chem. 2012, 187, 195–199. [Google Scholar] [CrossRef]
- Liu, H.; Secco, R.A.; Imanaka, N.; Rutter, M.D.; Adachi, G.; Uchida, T. Ionic to electronic dominant conductivity in Al2(WO4)3 at high pressure and high temperature. J. Phys. Chem. Solids 2003, 64, 287–294. [Google Scholar] [CrossRef]
- Mukherjee, G.D.; Vijaykumar, V.; Achary, S.N.; Tyagi, A.K.; Godwal, B.K. Phase transitions in Al2(WO4)3: High pressure investigations of low frequency dielectric constant and crystal structure. J. Phys. Condens. Matter 2004, 16, 7321–7330. [Google Scholar] [CrossRef]
- Secco, R.A.; Liu, H.; Imanaka, N.; Adachi, G.; Rutter, M.D. Electrical conductivity and amorphization Of Sc2(WO4)3 at high pressures and temperatures. J. Phys. Chem. Solids 2002, 63, 425–431. [Google Scholar] [CrossRef]
- Cetinkol, M.; Wilkinson, A.P.; Lind, C. In situ high-pressure synchrotron X-ray diffraction study of Zr2(WO4)(PO4)2 up to 16 GPa. Phys. Rev. B 2009, 79, 224118:1–224118:10. [Google Scholar] [CrossRef]
- Gates, S.D.; Colin, J.A.; Lind, C. Non-hydrolytic sol-gel synthesis, properties, and high-pressure behavior of gallium molybdate. J. Mater. Chem. 2006, 16, 4214–4219. [Google Scholar] [CrossRef]
- Karmakar, S.; Deb, S.K.; Tyagi, A.K.; Sharma, S.M. Pressure-induced amorphization in Y2(WO4)3: In situ X-ray diffraction and Raman studies. J. Solid State Chem. 2004, 177, 4087–4092. [Google Scholar] [CrossRef]
- Miller, W.; Smith, C.W.; Mackenzie, D.S.; Evans, K.E. Negative thermal expansion: A review. J. Mater. Sci. 2009, 44, 5441–5451. [Google Scholar] [CrossRef]
- Woodcock, D.A.; Lightfoot, P.; Villaescusa, L.A.; Diaz-Cabanas, M.J.; Camblor, M.A.; Engberg, D. Negative thermal expansion in the siliceous zeolites chabazite and ITQ-4: A neutron powder diffraction study. Chem. Mater. 1999, 11, 2508–2514. [Google Scholar] [CrossRef]
- Taylor, D. Thermal expansion data V. Miscellaneous binary oxides. Trans. J. Br. Ceram. Soc. 1985, 84, 9–14. [Google Scholar]
- Sanson, A.; Rocca, F.; Dalba, G.; Fornasini, P.; Grisenti, R.; Dapiaggi, M.; Artioli, G. Negative thermal expansion and local dynamics in Cu2O and Ag2O. Phys. Rev. B 2006, 73, 214305:1–214305:13. [Google Scholar] [CrossRef]
- Li, J.; Yokochi, A.; Amos, T.G.; Sleight, A.W. Strong negative thermal expansion along the O-Cu-O linkage in CuScO2. Chem. Mater. 2002, 14, 2602–2606. [Google Scholar] [CrossRef]
- Li, J.; Sleight, A.W.; Jones, C.Y.; Toby, B.H. Trends in negative thermal expansion behavior for AMO2 (A = Cu or Ag; M = Al, Sc, In, or La) compounds with the delafossite structure. J. Solid State Chem. 2005, 178, 285–294. [Google Scholar] [CrossRef]
- Ahmed, S.I.; Dalba, G.; Fornasini, P.; Vaccari, M.; Rocca, F.; Sanson, A.; Li, J.; Sleight, A.W. Negative thermal expansion in crystals with the delafossite structure: An extended X-ray absorption fine structure study of CuScO2 and CuLaO2. Phys. Rev. B 2009, 79, 104302:1–104302:8. [Google Scholar]
- Barreteau, C.; Bregiroux, D.; Laurent, G.; Wallez, G. Reducing ultra-low thermal expansion of beta-Zr2O(PO4)2 by substitutions? Mater. Res. Bull. 2010, 45, 1996–2000. [Google Scholar] [CrossRef]
- Clavier, N.; Wallez, G.; Dacheux, N.; Bregiroux, D.; Quarton, M.; Beaunier, P. Synthesis, Raman and Rietveld analysis of thorium diphosphate. J. Solid State Chem. 2008, 181, 3352–3356. [Google Scholar] [CrossRef]
- Wallez, G.; Bregiroux, D.; Quarton, M. Mechanism of the low thermal expansion in α-Hf2O(PO4)2 and its zirconium analog. J. Solid State Chem. 2008, 181, 1413–1418. [Google Scholar] [CrossRef]
- Wallez, G.; Clavier, N.; Dacheux, N.; Bregiroux, D. Negative thermal expansion in Th2O(PO4)2. Mater. Res. Bull. 2011, 46, 1777–1780. [Google Scholar] [CrossRef]
- Wallez, G.; Launay, S.; Quarton, M.; Dacheux, N.; Soubeyroux, J.L. Why does uranium oxide phosphate contract on heating? J. Solid State Chem. 2004, 177, 3575–3580. [Google Scholar] [CrossRef]
- Dapiaggi, M.; Fitch, A.N. Negative (and very low) thermal expansion in ReO3 from 5 to 300 K. J. Appl. Crystallogr. 2009, 42, 253–258. [Google Scholar] [CrossRef]
- Amos, T.G.; Sleight, A.W. Negative thermal expansion in orthorhombic NbOPO4. J. Solid State Chem. 2001, 160, 230–238. [Google Scholar] [CrossRef]
- Amos, T.G.; Yokochi, A.; Sleight, A.W. Phase transition and negative thermal expansion in tetragonal NbOPO4. J. Solid State Chem. 1998, 141, 303–307. [Google Scholar] [CrossRef]
- Mukherjee, G.D.; Vijaykumar, V.; Karandikar, A.S.; Godwal, B.K.; Achary, S.N.; Tyagi, A.K.; Lausi, A.; Busetto, E. Compressibility anomaly and amorphization in the anisotropic negative thermal expansion material NbOPO4 under pressure. J. Solid State Chem. 2005, 178, 8–14. [Google Scholar] [CrossRef]
- Wang, J.; Deng, J.; Yu, R.; Chen, J.; Xing, X. Coprecipitation synthesis and negative thermal expansion of NbVO5. Dalton Trans. 2011, 40, 3394–3397. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, Q.; Deng, J.; Yu, R.; Chen, J.; Xing, X. Phase transformation and negative thermal expansion in TaVO5. Inorg. Chem. 2011, 50, 2685–2690. [Google Scholar] [CrossRef] [PubMed]
- Greve, B.K.; Martin, K.L.; Lee, P.L.; Chupas, P.J.; Chapman, K.W.; Wilkinson, A.P. Pronounced negative thermal expansion from a simple structure: Cubic ScF3. J. Am. Chem. Soc. 2010, 132, 15496–15498. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.J.; Partin, D.E.; Lincoln, F.J.; Kouvetakis, J.; O’Keeffe, M. The disordered crystal structures of Zn(CN)2 and Ga(CN)3. J. Solid State Chem. 1997, 134, 164–169. [Google Scholar] [CrossRef]
- Goodwin, A.L.; Kepert, C.J. Negative thermal expansion and low-frequency modes in cyanide-bridged framework materials. Phys. Rev. B 2005, 71, 140301:1–140301:4. [Google Scholar] [CrossRef]
- Adak, S.; Daemen, L.L.; Hartl, M.; Williams, D.; Summerhill, J.; Nakotte, H. Thermal expansion in 3d-metal Prussian blue analogs—A survey study. J. Solid State Chem. 2011, 184, 2854–2861. [Google Scholar] [CrossRef]
- Goodwin, A.L.; Kennedy, B.J.; Kepert, C.J. Thermal expansion matching via framework flexibility in Zinc dicyanometallates. J. Am. Chem. Soc. 2009, 131, 6334–6335. [Google Scholar] [CrossRef] [PubMed]
- Conterio, M.J.; Goodwin, A.L.; Tucker, M.G.; Keen, D.A.; Dove, M.T.; Peters, L.; Evans, J.S.O. Local structure in Ag3[Co(CN)6]: Colossal thermal expansion, rigid unit modes and argentophilic interactions. J. Phys. Condens. Matter 2008, 20. [Google Scholar] [CrossRef]
- Goodwin, A.L.; Keen, D.A.; Tucker, M.G.; Dove, M.T.; Peters, L.; Evans, J.S.O. Argentophilicity-dependent colossal thermal expansion in extended Prussian blue analogues. J. Am. Chem. Soc. 2008, 130, 9660–9661. [Google Scholar] [CrossRef] [PubMed]
- Kozy, L.C.; Tahir, M.N.; Lind, C.; Tremel, W. Particle size and morphology control of the negative thermal expansion material cubic zirconium tungstate. J. Mater. Chem. 2009, 19, 2760–2765. [Google Scholar] [CrossRef]
- Nikolov, V.; Koseva, I.; Stoyanova, R.; Zhecheva, E. Conditions for preparation of nanosized Al2(WO4)3. J. Alloy. Compd. 2010, 505, 443–449. [Google Scholar] [CrossRef]
- Duan, N.; Kameswari, U.; Sleight, A.W. Further contraction of ZrW2O8. J. Am. Chem. Soc. 1999, 121, 10432–10433. [Google Scholar] [CrossRef]
- Lind, C. Negative Thermal Expansion Materials Related to Cubic Zirconium Tungstate. Ph.D. Dissertation, Georgia Institute of Technology, Atlanta, GA, USA, 2001. [Google Scholar]
- Banek, N.A.; Baiz, H.I.; Latigo, A.; Lind, C. Autohydration of nanosized cubic zirconium tungstate. J. Amer. Chem. Soc. 2010, 132, 8278–8279. [Google Scholar] [CrossRef]
- Sullivan, L.M.; Lukehart, C.M. Zirconium tungstate (ZrW2O8)/polyimide nanocomposites exhibiting reduced coefficient of thermal expansion. Chem. Mater. 2005, 17, 2136–2141. [Google Scholar] [CrossRef]
- Sharma, G.R.; Coleman, M.R.; Lind, C. Polyimide nanocomposites for tunable coefficient of thermal expansion. In Proceedings of the 40th International SAMPE Technical Conference, Memphis, TN, USA, 8–11 September 2008.
- Lind, C.; Coleman, M.R.; Kozy, L.C.; Sharma, G.R. Zirconium tungstate/polymer nanocomposites: Challenges and opportunities. Phys. Status Solidi B 2011, 248, 123–129. [Google Scholar] [CrossRef]
- Cho, C.H.; Oh, K.Y.; Kim, S.K.; Yeo, J.G.; Lee, Y.M. Improvement in thermal stability of NaA zeolite composite membrane by control of intermediate layer structure. J. Membr. Sci. 2011, 366, 229–236. [Google Scholar] [CrossRef]
- Cho, C.H.; Oh, K.Y.; Yeo, J.G.; Kim, S.K.; Lee, Y.M. Synthesis, ethanol dehydration and thermal stability of NaA zeolite/alumina composite membranes with narrow non-zeolitic pores and thin intermediate layer. J. Membr. Sci. 2010, 364, 138–148. [Google Scholar] [CrossRef]
- Akhtar, F.; Ojuva, A.; Wirawan, S.K.; Hedlund, J.; Bergstrom, L. Hierarchically porous binder-free silicalite-1 discs: A novel support for all-zeolite membranes. J. Mater. Chem. 2011, 21, 8822–8828. [Google Scholar] [CrossRef]
- Tran, K.D.; Groshens, T.J.; Nelson, J.G. Fabrication of near-zero thermal expansion (FexSc1−x)2Mo3O12-MoO3 ceramic composite using the reaction sintering process. Mater. Sci. Eng. A 2001, 303, 234–240. [Google Scholar] [CrossRef]
- Watanabe, H.; Tani, J.; Kido, H.; Mizuuchi, K. Thermal expansion and mechanical properties of pure magnesium containing zirconium tungsten phosphate particles with negative thermal expansion. Mater. Sci. Eng. A 2008, 494, 291–298. [Google Scholar] [CrossRef]
- Holzer, H.; Dunand, D.C. Phase transformation and thermal expansion of Cu/ZrW2O8 metal matrix composites. J. Mater. Res. 1999, 14, 780–789. [Google Scholar] [CrossRef]
- Yilmaz, S.; Dunand, D.C. Finite-element analysis of thermal expansion and thermal mismatch stresses in a Cu-60vol%ZrW2O8 composite. Compos. Sci. Technol. 2004, 64, 1895–1898. [Google Scholar] [CrossRef]
- Yilmaz, S. Phase transformations in thermally cycled Cu/ZrW2O8 composites investigated by synchrotron X-ray diffraction. J. Phys. Condens. Matter 2002, 14, 365–375. [Google Scholar] [CrossRef]
- Yilmaz, S. Thermal mismatch stress development in Cu-ZrW2O8 composite investigated by synchrotron X-ray diffraction. Compos. Sci. Technol. 2002, 62, 1835–1839. [Google Scholar] [CrossRef]
- Yan, X.; Cheng, X.; Xu, G.; Wang, C.; Sun, S.; Riedel, R. Preparation and thermal properties of zirconium tungstate/copper composites. Mater. Werkst. 2008, 39, 649–653. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, J.F.; Zhang, Y.H.; Zhang, J.L.; Lu, F.S.; Wang, X.L. Synthesis and thermal expansion of 4J36/ZrW2O8 composites. Rare Met. 2010, 29, 371–375. [Google Scholar] [CrossRef]
- Kanamori, K.; Kineri, T.; Fukuda, R.; Kawano, T.; Nishio, K. Low-temperature sintering of ZrW2O8-SiO2 by spark plasma sintering. J. Mater. Sci. 2009, 44, 855–860. [Google Scholar] [CrossRef]
- Kofteros, M.; Rodriguez, S.; Tandon, V.; Murr, L.E. A preliminary study of thermal expansion compensation in cement by ZrW2O8 additions. Scr. Mater. 2001, 45, 369–374. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, X.; Yan, X.; Yang, J.; Fu, T.; Qiu, J. Synthesis of ZrO2/ZrW2O8 composites with low thermal expansion. Compos. Sci. Technol. 2007, 67, 1167–1171. [Google Scholar] [CrossRef]
- Yang, X.; Xu, J.; Li, H.; Cheng, X.; Yan, X. In situ synthesis of ZrO2/ZrW2O8 composites with near-zero thermal expansion. J. Am. Ceram. Soc. 2007, 90, 1953–1955. [Google Scholar] [CrossRef]
- Zhang, Z.P.; Liu, H.F.; Cheng, X.N. Study on the technology of ZrO2-ZrW2O8 composites prepared by co-precipitation method. J. Inorg. Mater. 2008, 23, 991–995. [Google Scholar] [CrossRef]
- Yang, X.; Cheng, X.; Li, H.; Xu, J.; Sun, X. Thermal and electric conductivity of near-zero thermal expansion ZrW2O8/ZrO2 composites. J. Ceram. Soc. Jpn. 2008, 116, 471–474. [Google Scholar] [CrossRef]
- Sun, L.; Kwon, P. ZrW2O8/ZrO2 composites by in situ synthesis of ZrO2 + WO3: Processing, coefficient of thermal expansion, and theoretical model prediction. Mater. Sci. Eng. A 2009, 527, 93–97. [Google Scholar] [CrossRef]
- Sun, L.; Kwon, P. ZrW2O8-ZrO2 continuous functionally graded materials fabricated by in situ reaction of ZrO2 and WO3. J. Am. Ceram. Soc. 2010, 93, 703–708. [Google Scholar] [CrossRef]
- Sun, L.; Sneller, A.; Kwon, P. ZrW2O8-containing composites with near-zero coefficient of thermal expansion fabricated by various methods: Comparison and optimization. Compos. Sci. Technol. 2008, 68, 3425–3430. [Google Scholar] [CrossRef]
- Tani, J.-I.; Takahashi, M.; Kido, H. Fabrication and thermal expansion properties of ZrW2O8/Zr2WP2O12 composites. J. Eur. Ceram. Soc. 2010, 30, 1483–1488. [Google Scholar] [CrossRef]
- Isobe, T.; Kato, Y.; Mizutani, M.; Ota, T.; Daimon, K. Pressureless sintering of negative thermal expansion ZrW2O8/Zr2WP2O12 composites. Mater. Lett. 2008, 62, 3913–3915. [Google Scholar] [CrossRef]
- Isobe, T.; Umezome, T.; Kameshima, Y.; Nakajima, A.; Okada, K. Preparation and properties of negative thermal expansion Zr2WP2O12 ceramics. Mater. Res. Bull. 2009, 44, 2045–2049. [Google Scholar] [CrossRef]
- Tani, J.-I.; Kimura, H.; Hirota, K.; Kido, H. Thermal expansion and mechanical properties of phenolic resin/ZrW2O8 composites. J. Appl. Polym. Sci. 2007, 106, 3343–3347. [Google Scholar] [CrossRef]
- Chu, X.; Huang, R.; Yang, H.; Wu, Z.; Lu, J.; Zhou, Y.; Li, L. The cryogenic thermal expansion and mechanical properties of plasma modified ZrW(2)O(8) reinforced epoxy. Mater. Sci. Eng. A 2011, 528, 3367–3374. [Google Scholar] [CrossRef]
© 2012 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Lind, C. Two Decades of Negative Thermal Expansion Research: Where Do We Stand? Materials 2012, 5, 1125-1154. https://doi.org/10.3390/ma5061125
AMA Style
Lind C. Two Decades of Negative Thermal Expansion Research: Where Do We Stand? Materials. 2012; 5(6):1125-1154. https://doi.org/10.3390/ma5061125
Chicago/Turabian StyleLind, Cora. 2012. "Two Decades of Negative Thermal Expansion Research: Where Do We Stand?" Materials 5, no. 6: 1125-1154. https://doi.org/10.3390/ma5061125