Chemical Bonding of AlH3 Hydride by Al-L2,3 Electron Energy-Loss Spectra and First-Principles Calculations

Abstract
:1. Introduction
2. Results and Discussion
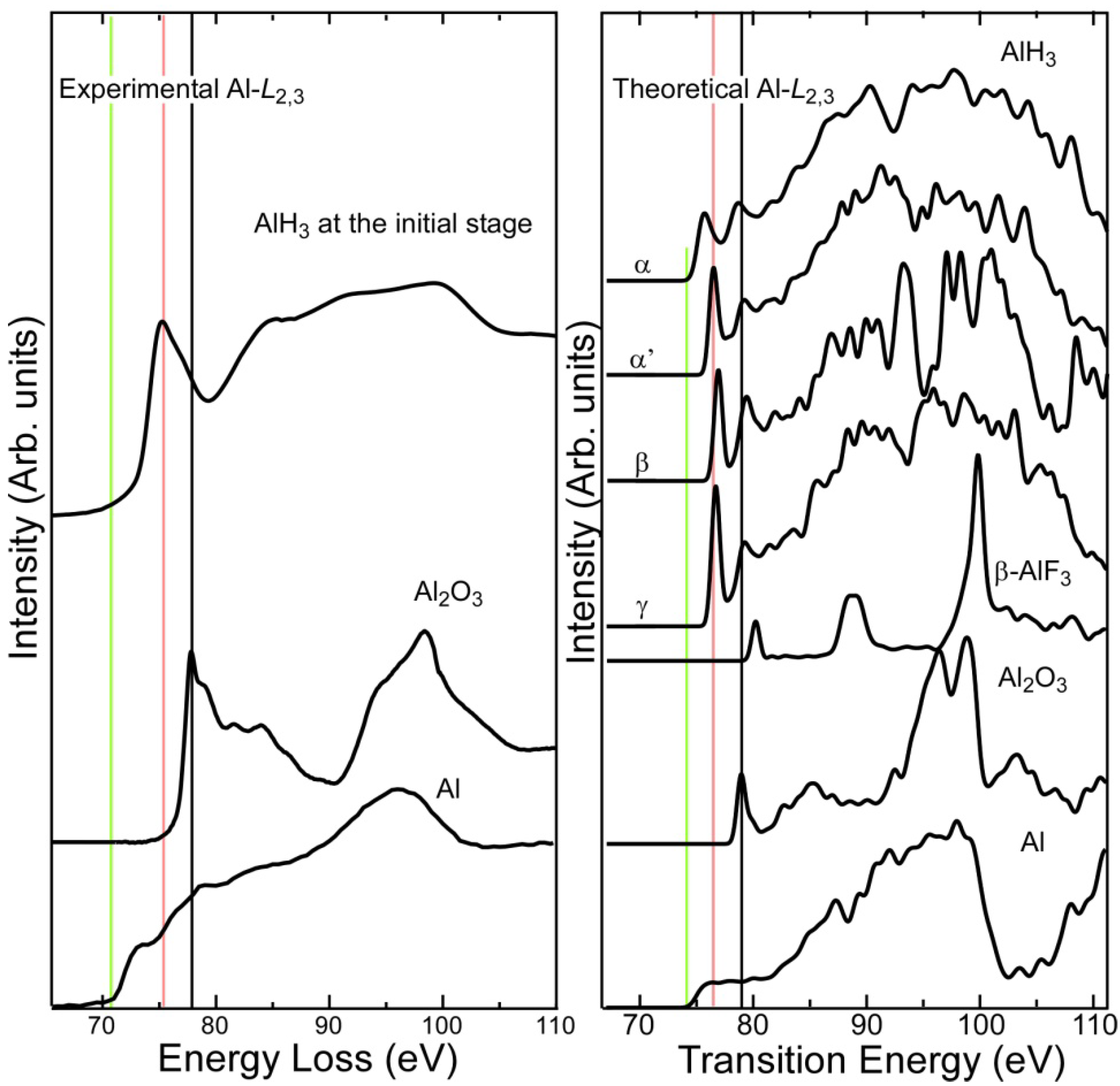
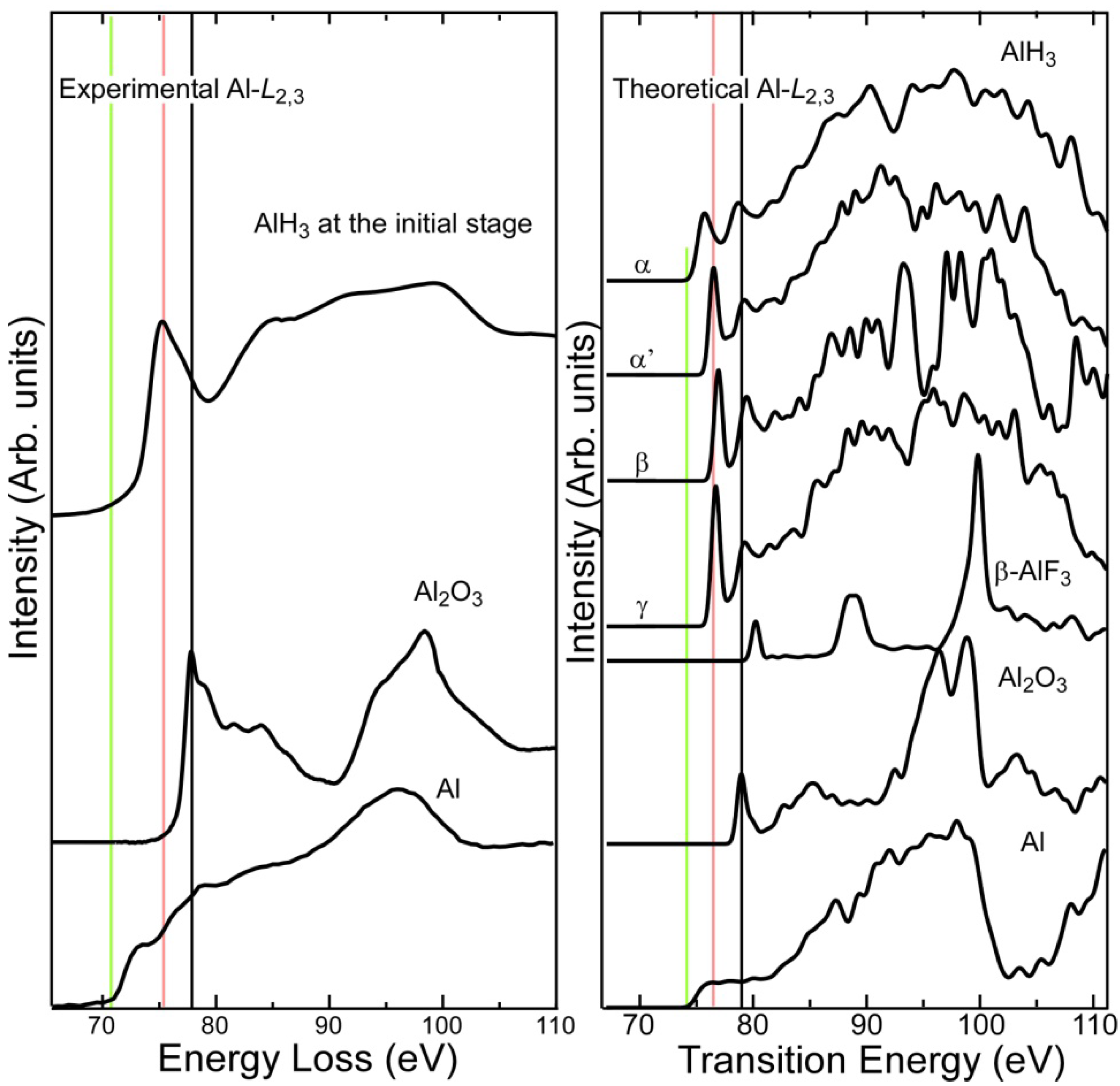
2.1. Al-L2,3 EELS
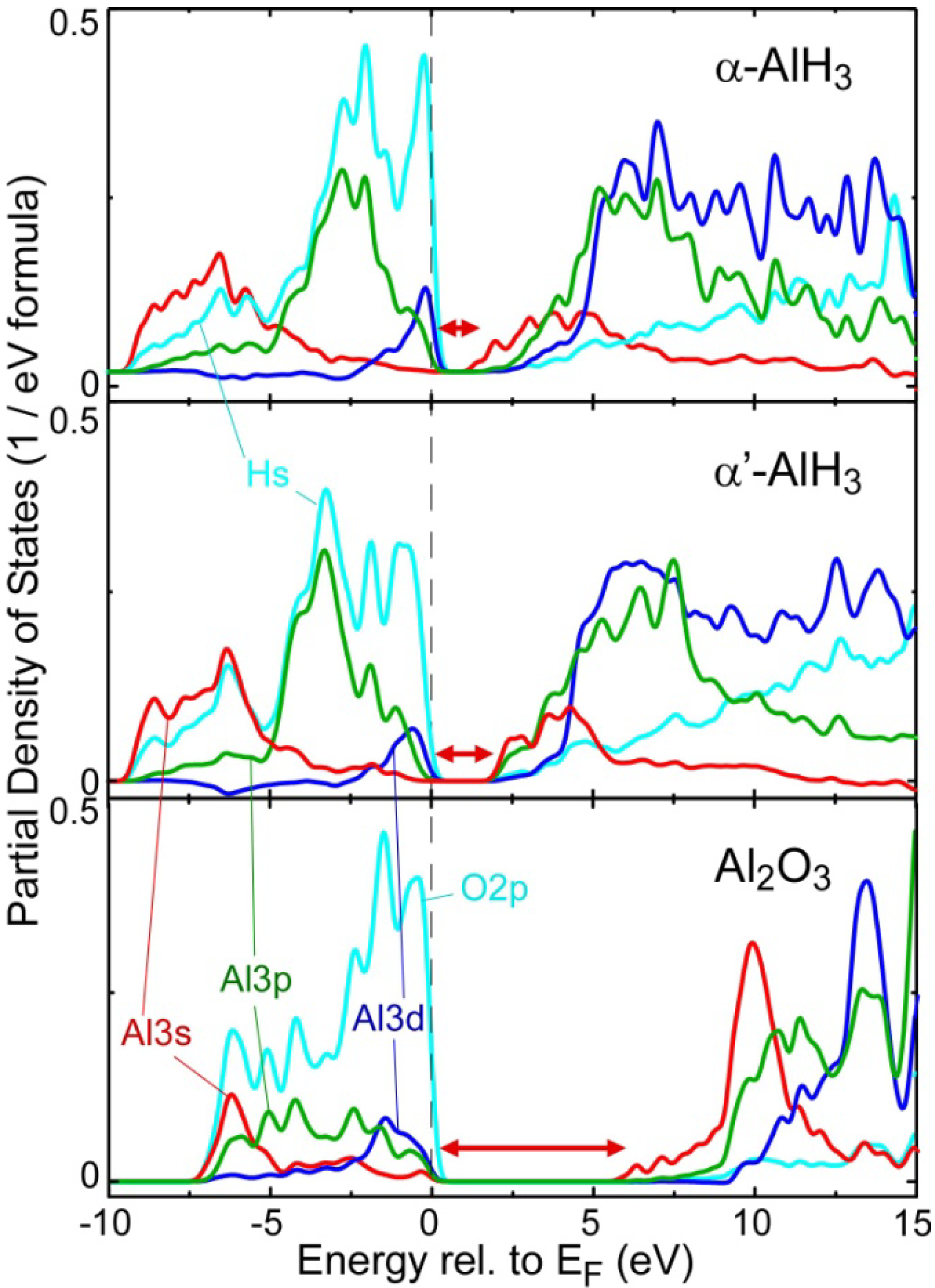
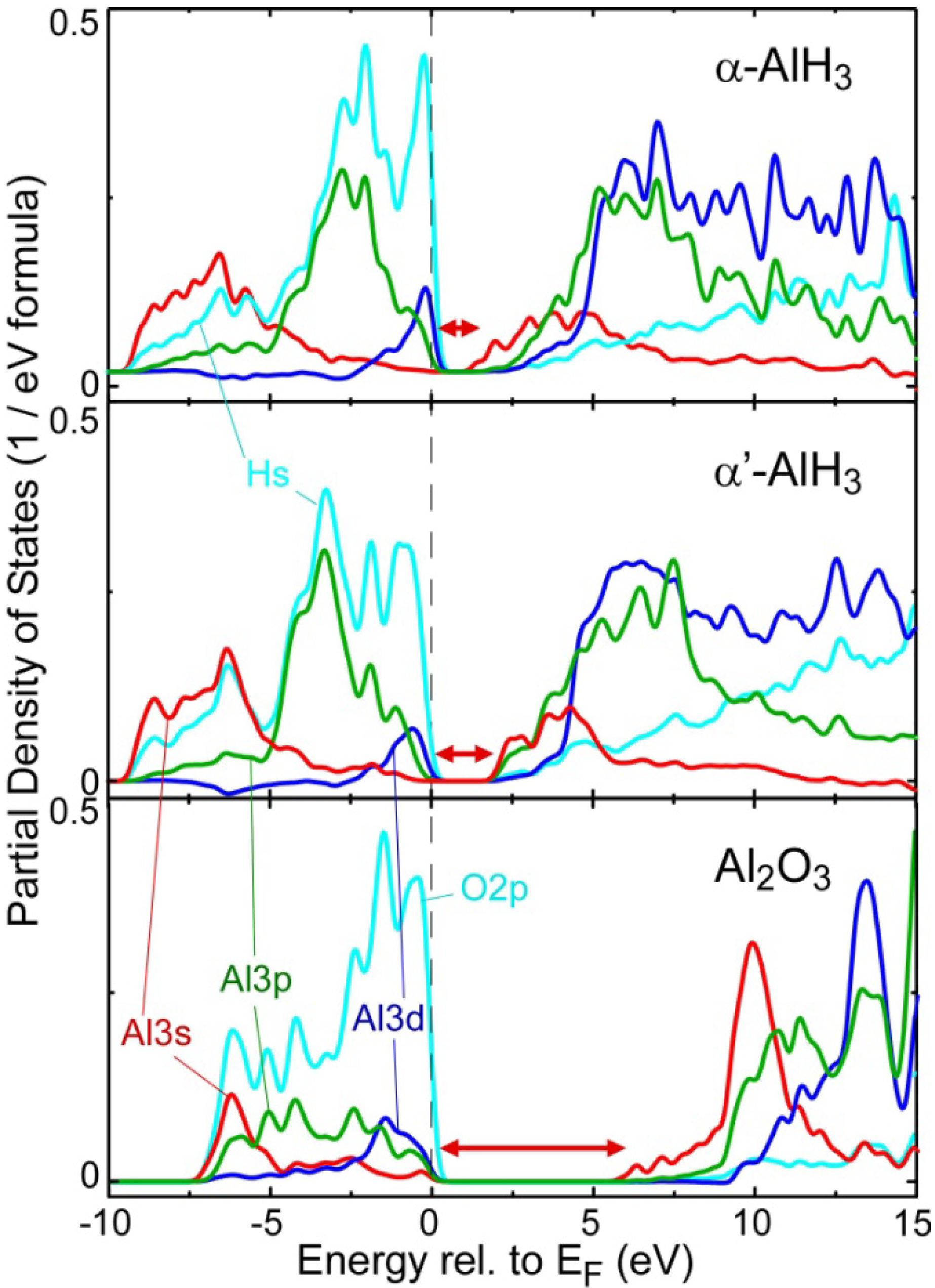
2.2. Electronic Structure near the Band Gap
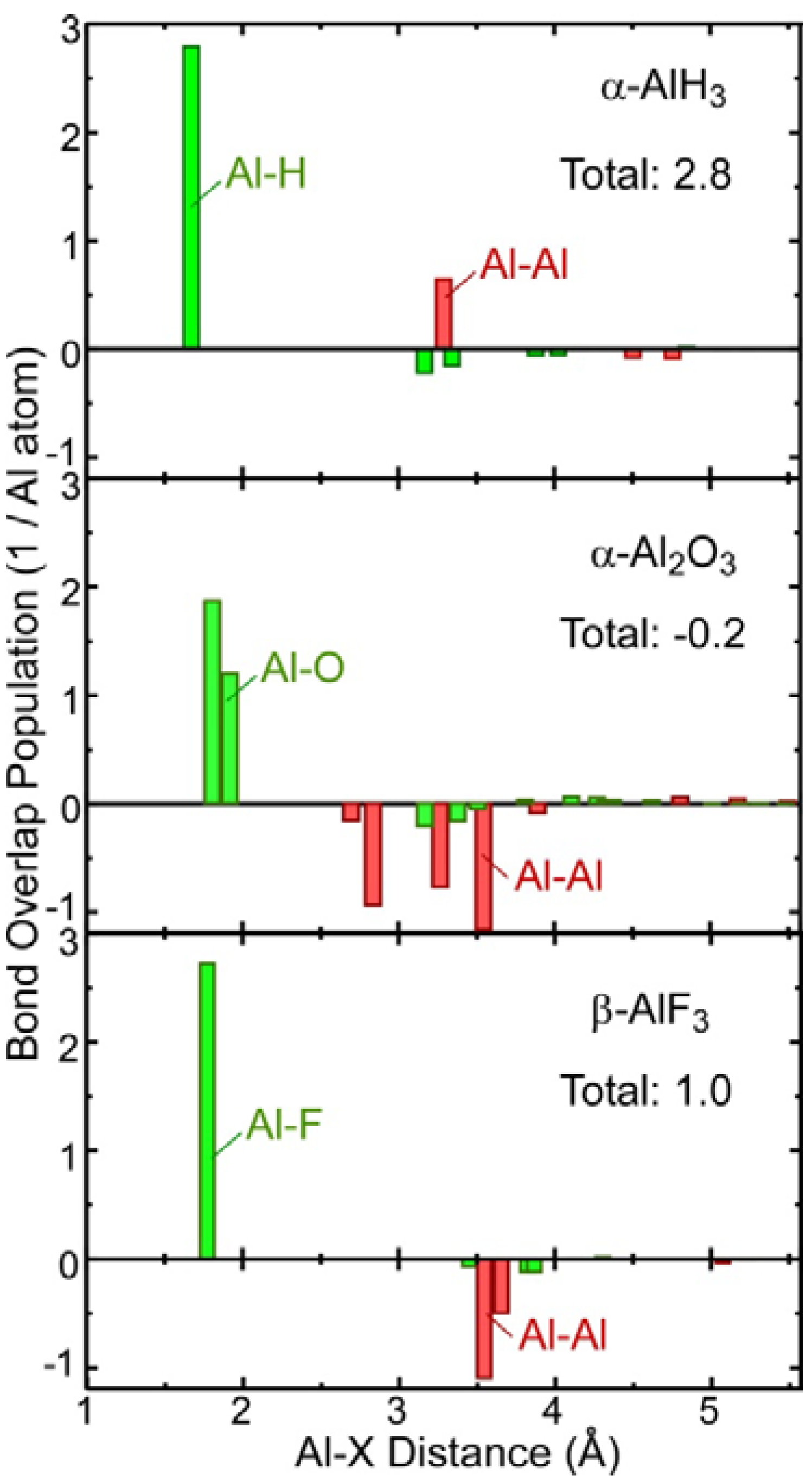
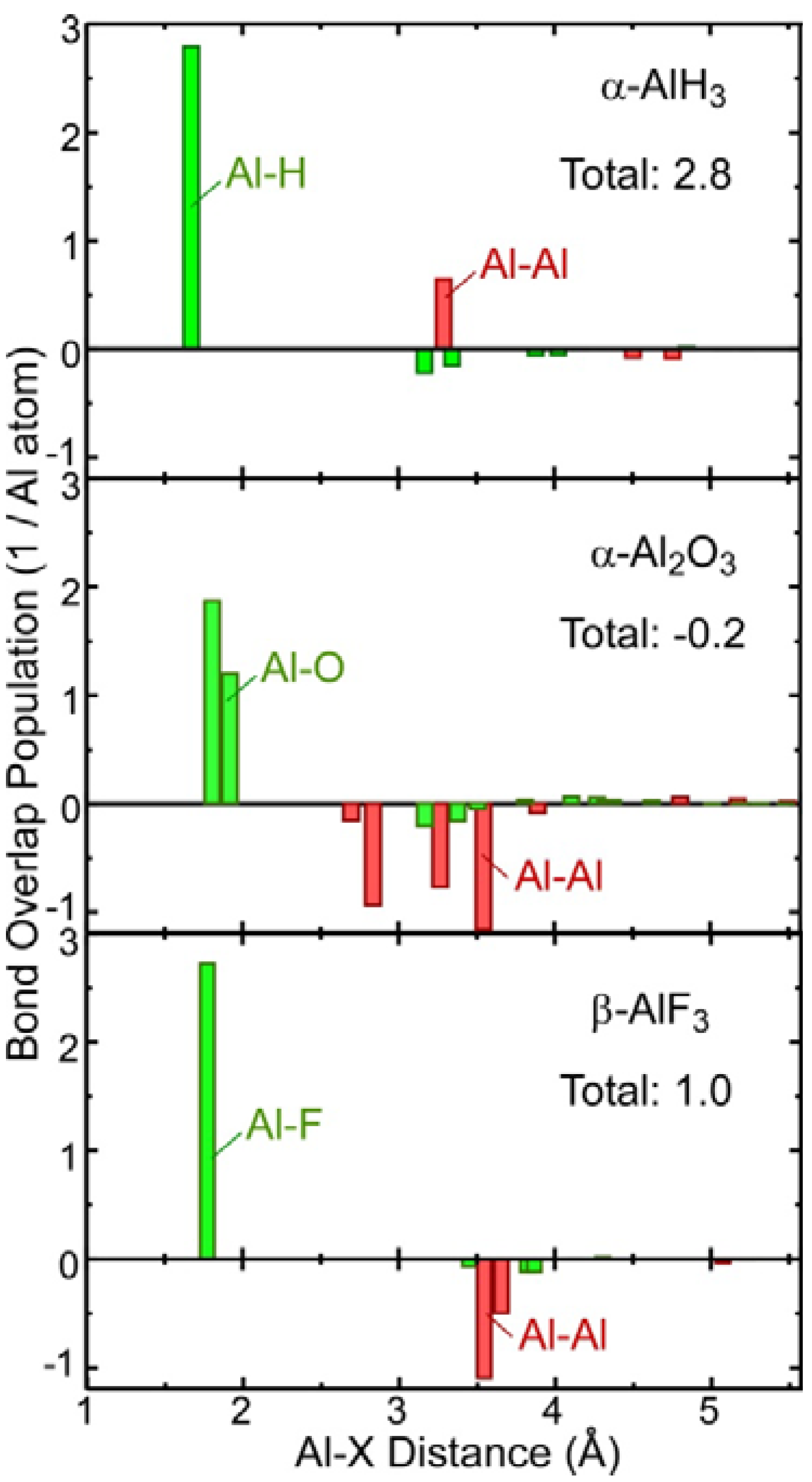
2.3. Covalent Bond Strength
2.4. Comparison of Chemical Bonding between the Al-Containing Compounds

{kind=link}
{kind=link}
{kind=link}
| Al | AlH3 (α, α') | Al2O3 | AlF3 | |
|---|---|---|---|---|
| LDA band gap (eV) | 0.0 | 1.4, 2.1 | 5.9 | 7.2 |
| Al effective charge | 3.0 | 2.8, 2.8 | 2.1 | 1.8 |
| Sum of BOP Al-X (1/Al atom) | 3.6 | 2.9, 2.7 | −0.2 | 1.0 |
3. Experimental and Theoretical Section
3.1. EELS Measurements
3.2. Theoretical Calculations
4. Conclusions
- (1)
- Experimental Al-L2,3 ELNES exhibited clear chemical shifts between metallic Al, AlH3, and Al2O3. The threshold of the hydride spectrum was intermediate between those of Al and Al2O3. The first-principles calculation reasonably reproduced the shape and chemical shift of the experimental spectra.
- (2)
- Theoretical chemical bonding data obtained by Mulliken population analysis of the calculated electronic structures revealed that the ionic bonding and covalent bonding strengths of the hydride were intermediate between those of Al and Al2O3.
- (3)
- The theoretical Al-L2,3 spectra and chemical bonding between the four Al compounds (Al, AlH3, Al2O3, and AlF3) exhibited systematic trends between the chemical bonding and their spectral features (fine peak structure and chemical shift). These trends are expected to be useful for further TEM-EELS nanoscale analysis of the chemical bonding changes that occur during the dehydrogenation of AlH3 hydrides.
Acknowledgments
References
- Sandrock, G.; Reilly, J.; Graetz, J.; Zhou, W.M.; Johnson, J.; Wegrzyn, J. Accelerated thermal decomposition of AlH3 for hydrogen-fueled vehicles. J. Appl. Phys. A 2005, 80, 687–690. [Google Scholar] [CrossRef]
- Graetz, J.; Reilly, J.J. Decomposition Kinetics of the AlH3 Polymorphs. J. Phys. Chem. B 2005, 109, 22181–22185. [Google Scholar] [CrossRef] [PubMed]
- Orimo, S.; Nakamori, Y.; Kato, T.; Brown, C.; Jensen, C.M. Intrinsic and mechanically modified thermal stabilities of α-, β- and α-aluminum trihydrides AlH3. Appl. Phys. A Mater. 2006, 83, 5–8. [Google Scholar] [CrossRef]
- Ke, X.; Kuwabara, A.; Tanaka, I. Cubic and orthorhombic structures of aluminum hydride AlH3 predicted by a first-principles study. Phys. Rev. B 2005, 71, 184107:1–184107:7. [Google Scholar]
- Pickard, C.J.; Needs, R.J. Metallization of aluminum hydride at high pressures: A first-principles study. Phys. Rev. B 2007, 76, 144114:1–144114:5. [Google Scholar] [CrossRef]
- Graetz, J.; Chaudhuri, S.; Lee, Y.; Vogt, T.; Muckerman, J.T.; Reilly, J.J. Pressure-induced structural and electronic changes in α-AlH3. Phys. Rev. B 2006, 74, 214114:1–214114:7. [Google Scholar] [CrossRef]
- Graetz, J.; Reilly, J.J. Thermodynamics of the α, β and γ polymorphs of AlH3. J. Alloys Comp. 2006, 424, 262–265. [Google Scholar] [CrossRef]
- Brinks, H.W.; Langley, W.; Jensen, C.M; Graetz, J.; Reilly, J.J.; Hauback, B.C. Synthesis and crystal structure of β-AlD3. J. Alloys Compounds 2006, 424, 262–265. [Google Scholar] [CrossRef]
- Yartys, V.A.; Denys, R.V.; Maehlen, J.P.; Frommen, C.; Fichtner, M.; Bulychev, B.M.; Emerich, H. Double-bridge bonding of aluminium and hydrogen in the crystal structure of γ-AlH3. Inorg. Chem. 2007, 46, 1051–1055. [Google Scholar] [CrossRef] [PubMed]
- Vajeeston, P.; Ravindran, P.; Fjellvåg, H. Novel high pressure phases of β-alh3: A density-functional study. Chem. Mater. 2008, 20, 5997–6002. [Google Scholar] [CrossRef]
- Takeda, Y.; Saitoh, Y.; Saitoh, H.; Machida, A.; Aoki, K.; Yamagami, H.; Muro, T.; Kato, Y.; Kinoshita, T. Electronic structure of aluminium trihydride studied using soft X-ray emission and absorption spectroscopy. Phys. Rev. B 2011, 84, 153102:1–153102:4. [Google Scholar] [CrossRef]
- Ikeda, K.; Muto, S.; Tatsumi, K.; Menjo, M.; Kato, S.; Bielman, M.; Züttel, A.; Jensen, C.M.; Orimo, S. Dehydriding reaction of AlH3: In situ microscopic observations combined with thermal and surface analyses. Nanotechnology 2009, 20, 204004:1–204004:4. [Google Scholar] [CrossRef]
- Muto, S.; Tatsumi, K.; Ikeda, K.; Orimo, S. Dehydriding process of α-AlH3 observed by transmission electron microscopy and electron energy-loss spectroscopy. J. Appl. Phys. 2009, 105, 123514:1–123514:4. [Google Scholar] [CrossRef]
- Keast, V.J.; Scott, A.J.; Kappers, M.J.; Foxon, C.T.; Humphyreys, C.J. Electronic structure of GaN and InxGa1−xN measured with electron energy-loss spectroscopy. Phys. Rev. B 2002, 66, 125319:1–125319:7. [Google Scholar] [CrossRef]
- Mauchamp, V.; Boucher, F.; Ouvrard, G.; Moreau, P. Ab initio simulation of the electron energy-loss near-edge structures at the Li K edge in Li, Li2O, and LiMn2O4. Phys. Rev. B 2006, 74, 115106:1–115106:4. [Google Scholar] [CrossRef]
- Olovsson, W.; Tanaka, I.; Mizoguchi, T.; Radtke, G.; Puschnig, P.; Ambrosch-Draxl, C. Al L2,3 edge X-ray absorption spectra in III–V semiconductors: Many-body perturbation theory in comparison with experiment. Phys. Rev. B 2011, 83, 195206:1–195206:8. [Google Scholar] [CrossRef]
- Filippetti, K.; Spaldin, N.A. Self-interaction-corrected pseudopotential scheme for magnetic and strongly-correlated systems. Phys. Rev. B 2003, 67, 125109:1–125109:15. [Google Scholar] [CrossRef]
- Muto, S.; Puetter, R.C.; Tatsumi, K. Spectral restoration and energy resolution improvement of electron energy-loss spectra by Pixon reconstruction: I. Principle and test examples. J. Electron Microsc. 2006, 55, 215–223. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11181. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Ching, W.Y. Theoretical studies of the electronic properties of ceramic materials. J. Am. Ceram. Soc. 1990, 73, 3135–3160. [Google Scholar] [CrossRef]
- Mo, S.D.; Ching, W.Y. Ab initio calculation of the core-hole effect in the electron energy-loss near-edge structure. Phys. Rev. B 2000, 62, 7901–7907. [Google Scholar] [CrossRef]
- Mogi, M.; Yamamoto, T.; Mizoguchi, T.; Tatasumi, K.; Yoshioka, S.; Kameyama, S.; Tanaka, I.; Adachi, H. Theoretical investigation of Al K-edge X-ray absorption spectra of Al, AlN and Al2O3. Mater. Trans. 2004, 45, 2031–2034. [Google Scholar]
- Mulliken, R.S. Electronic population analysis on LCAO-MO molecular wave functions. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Mizoguchi, T.; Tatsumi, K.; Tanaka, I. Peak assignments of ELNES and XANES using overlap population diagrams. Ultramicroscopy 2006, 106, 1120–1128. [Google Scholar] [CrossRef] [PubMed]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tatsumi, K.; Muto, S.; Ikeda, K.; Orimo, S.-I. Chemical Bonding of AlH3 Hydride by Al-L2,3 Electron Energy-Loss Spectra and First-Principles Calculations. Materials 2012, 5, 566-574. https://doi.org/10.3390/ma5040566
Tatsumi K, Muto S, Ikeda K, Orimo S-I. Chemical Bonding of AlH3 Hydride by Al-L2,3 Electron Energy-Loss Spectra and First-Principles Calculations. Materials. 2012; 5(4):566-574. https://doi.org/10.3390/ma5040566
Chicago/Turabian StyleTatsumi, Kazuyoshi, Shunsuke Muto, Kazutaka Ikeda, and Shin-Ichi Orimo. 2012. "Chemical Bonding of AlH3 Hydride by Al-L2,3 Electron Energy-Loss Spectra and First-Principles Calculations" Materials 5, no. 4: 566-574. https://doi.org/10.3390/ma5040566