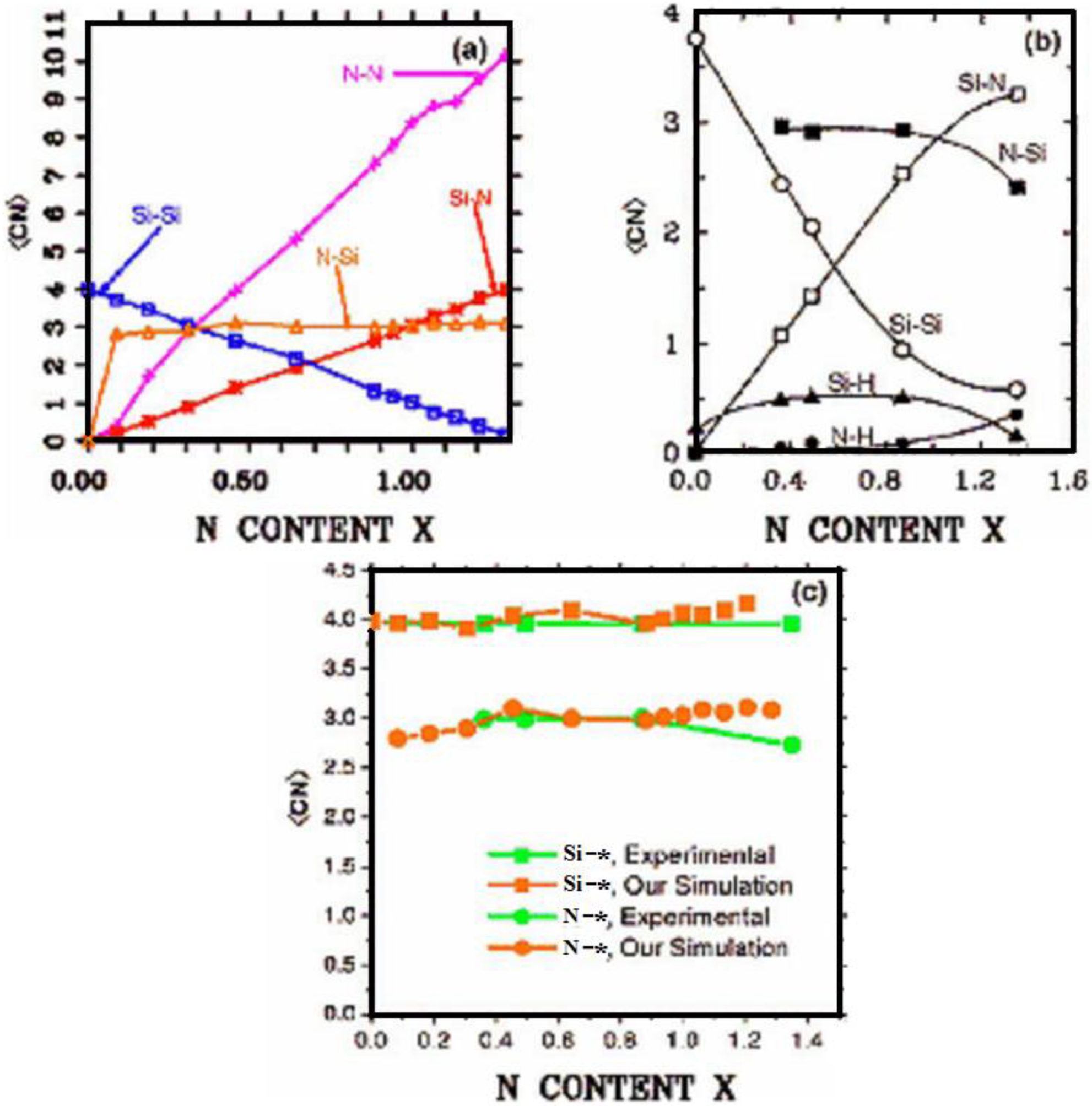
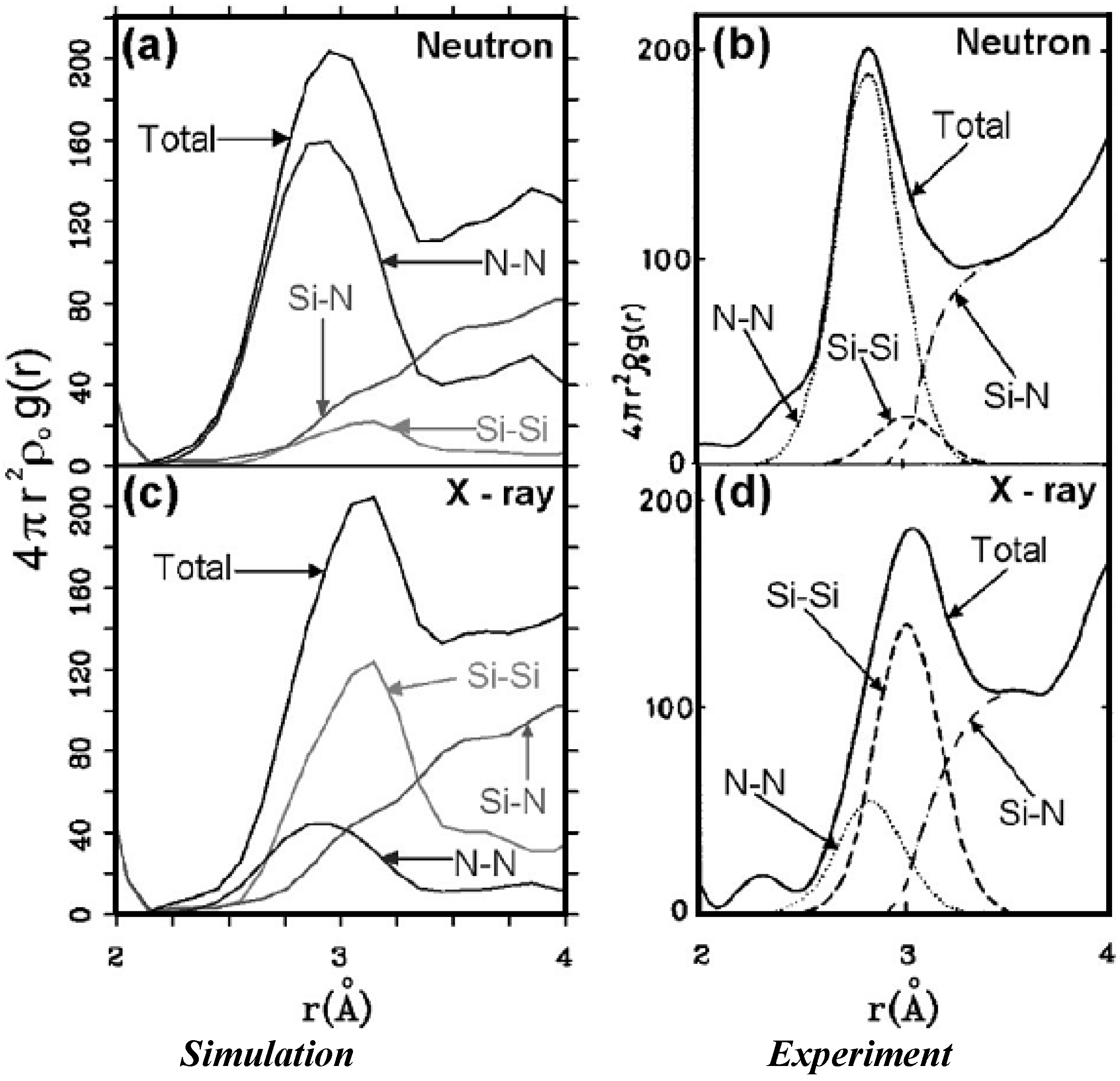
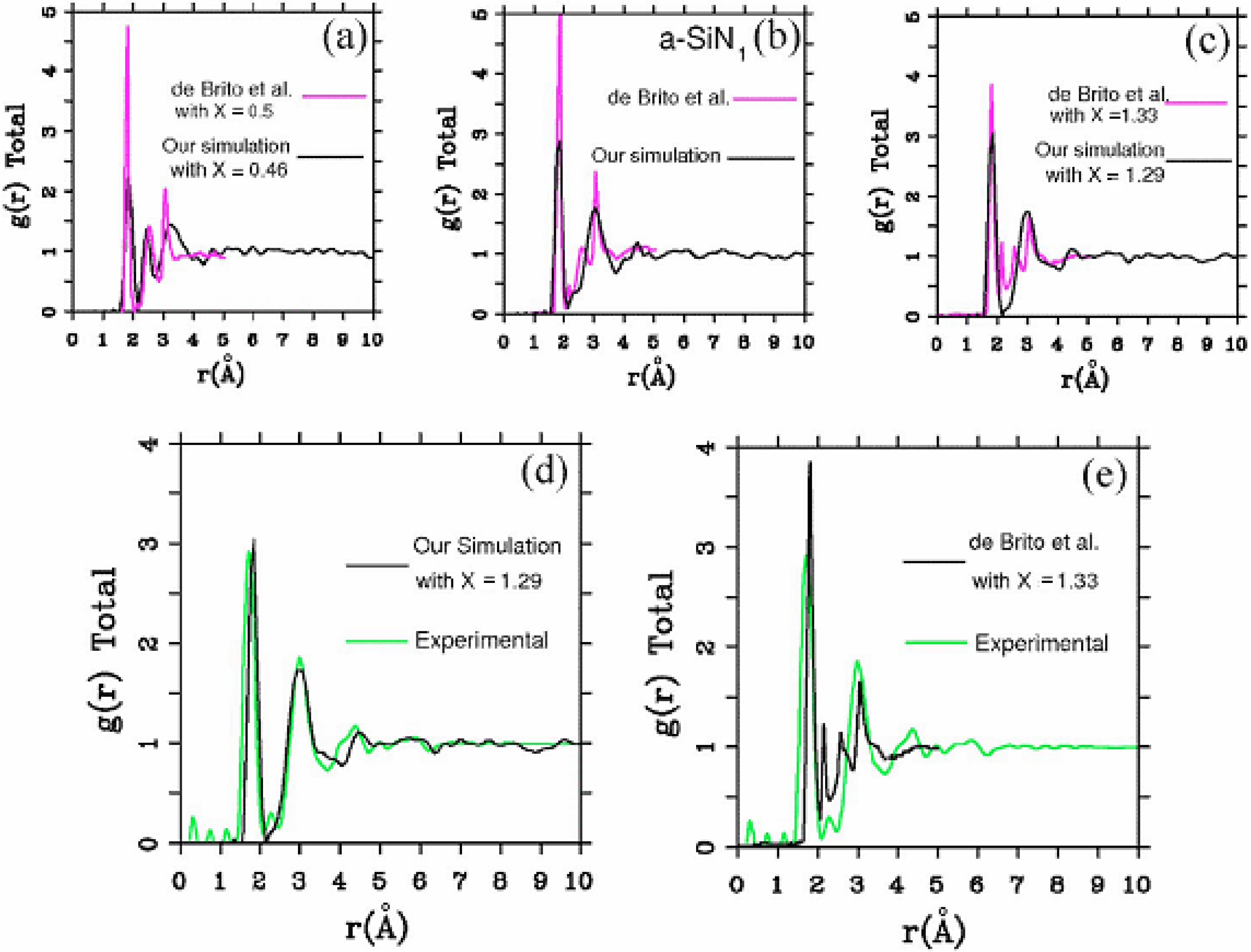
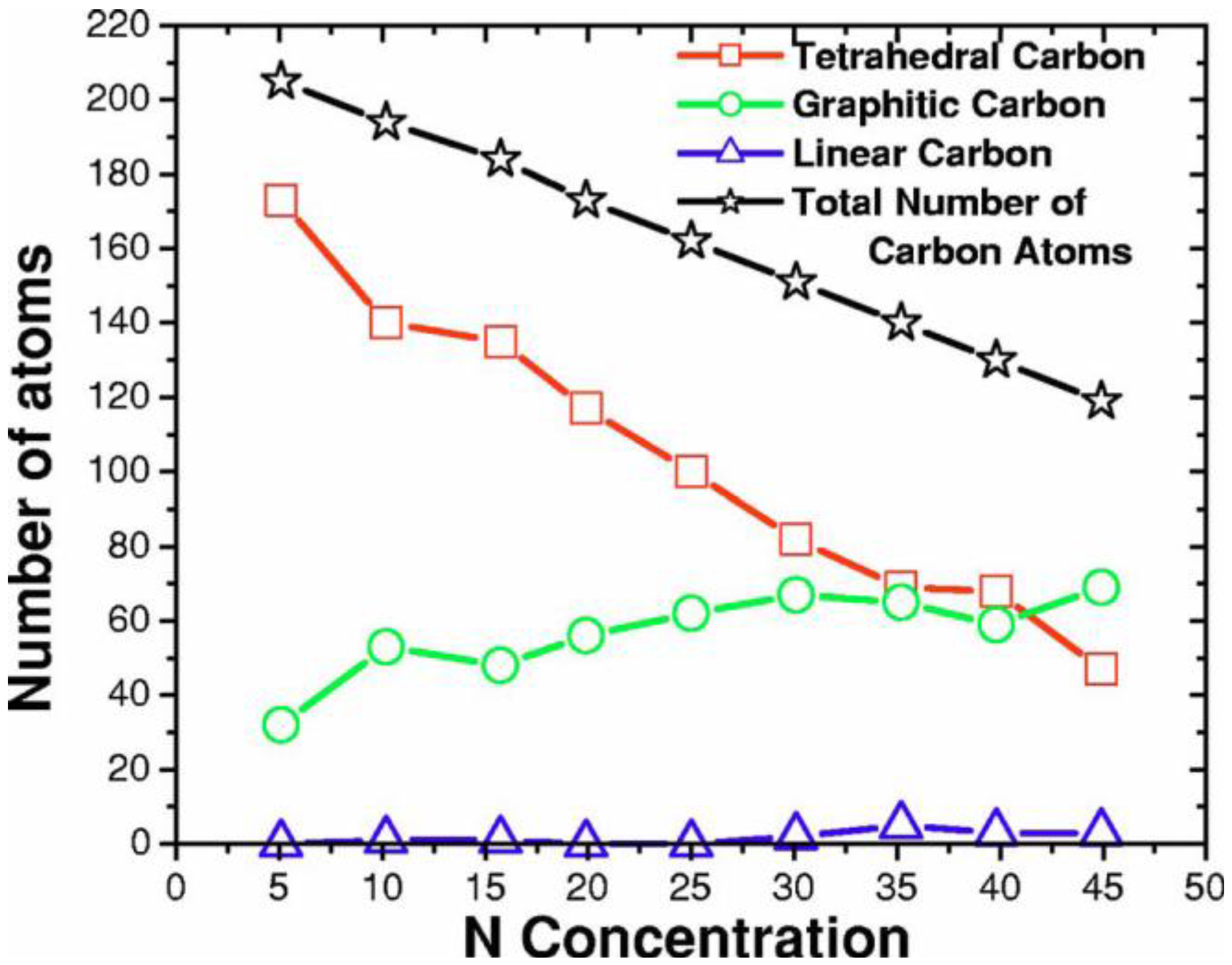
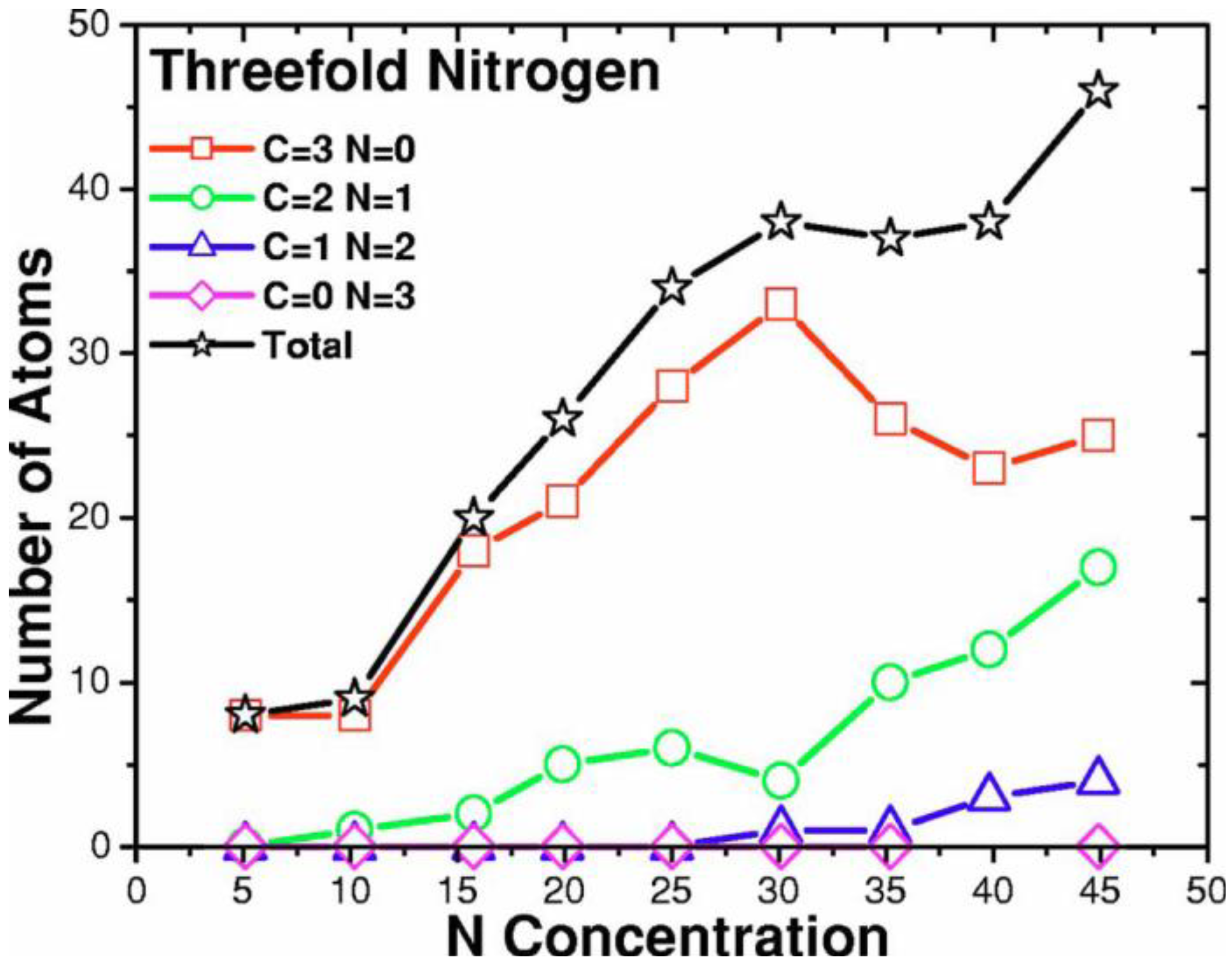
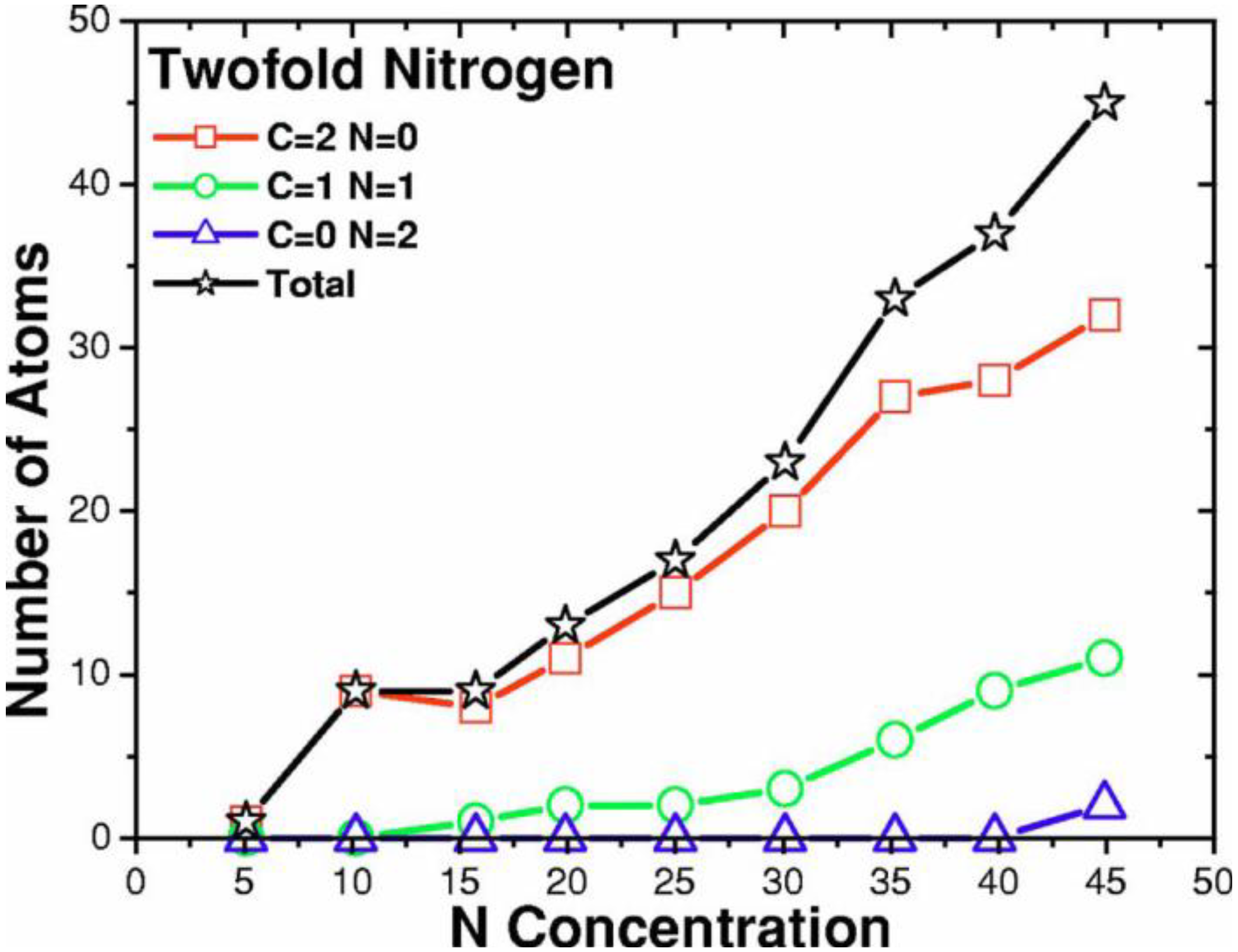
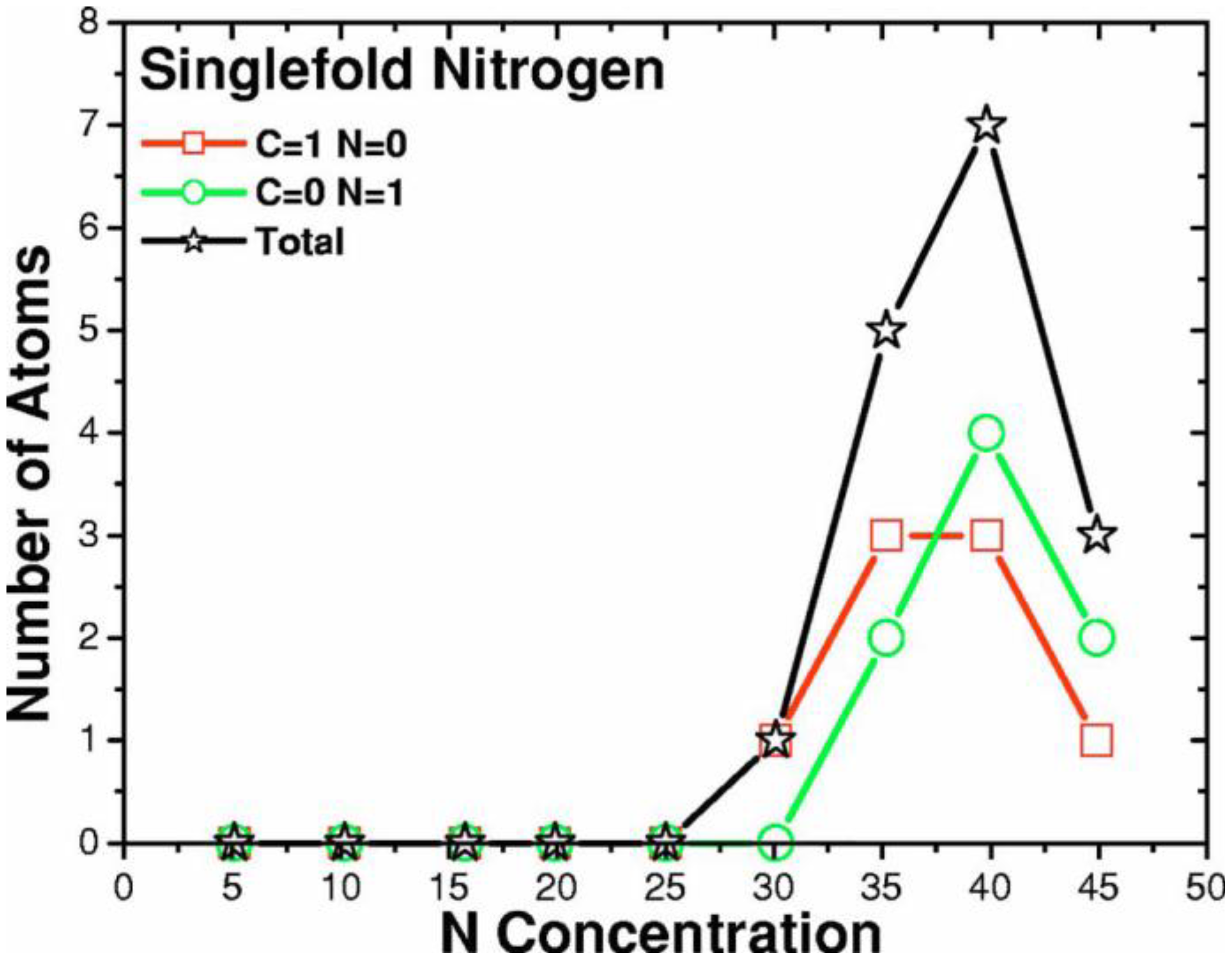
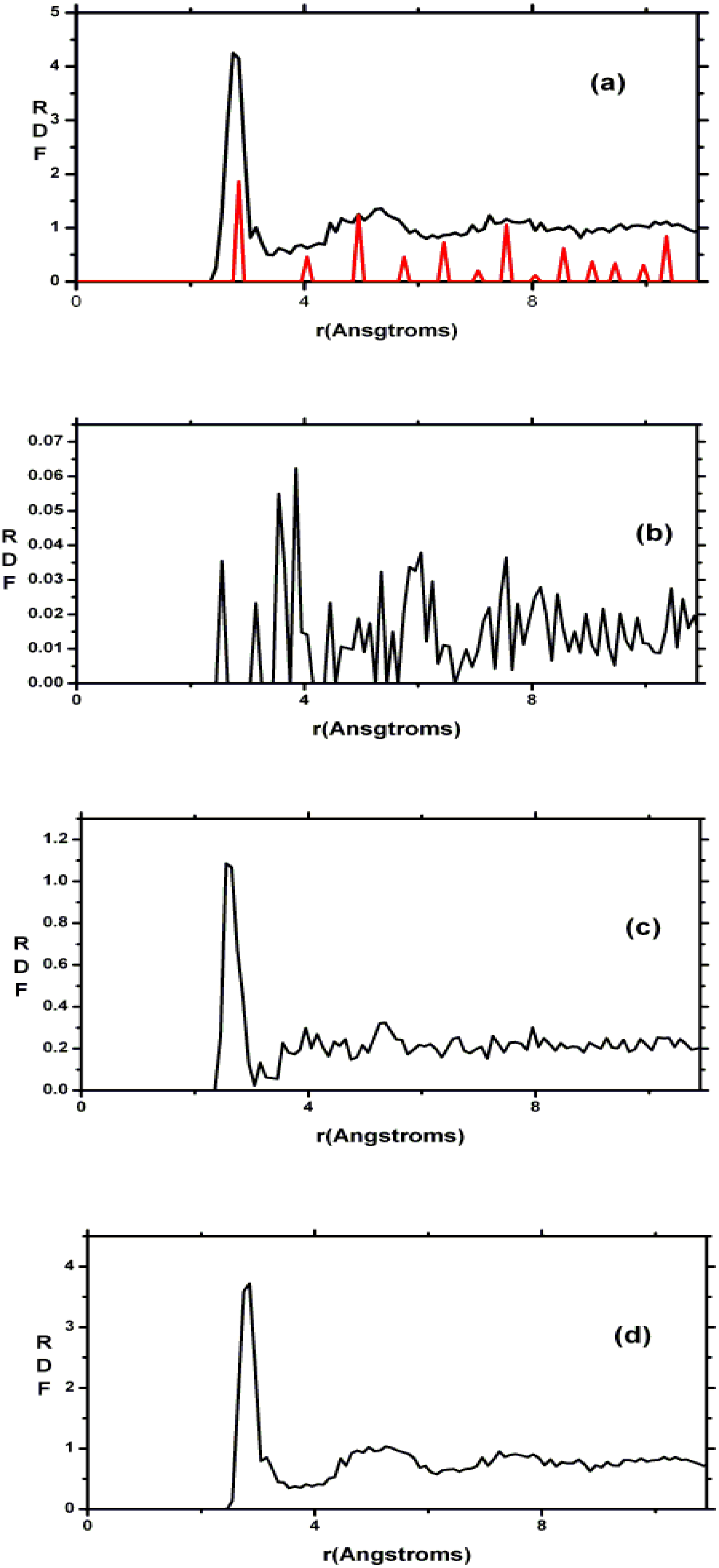
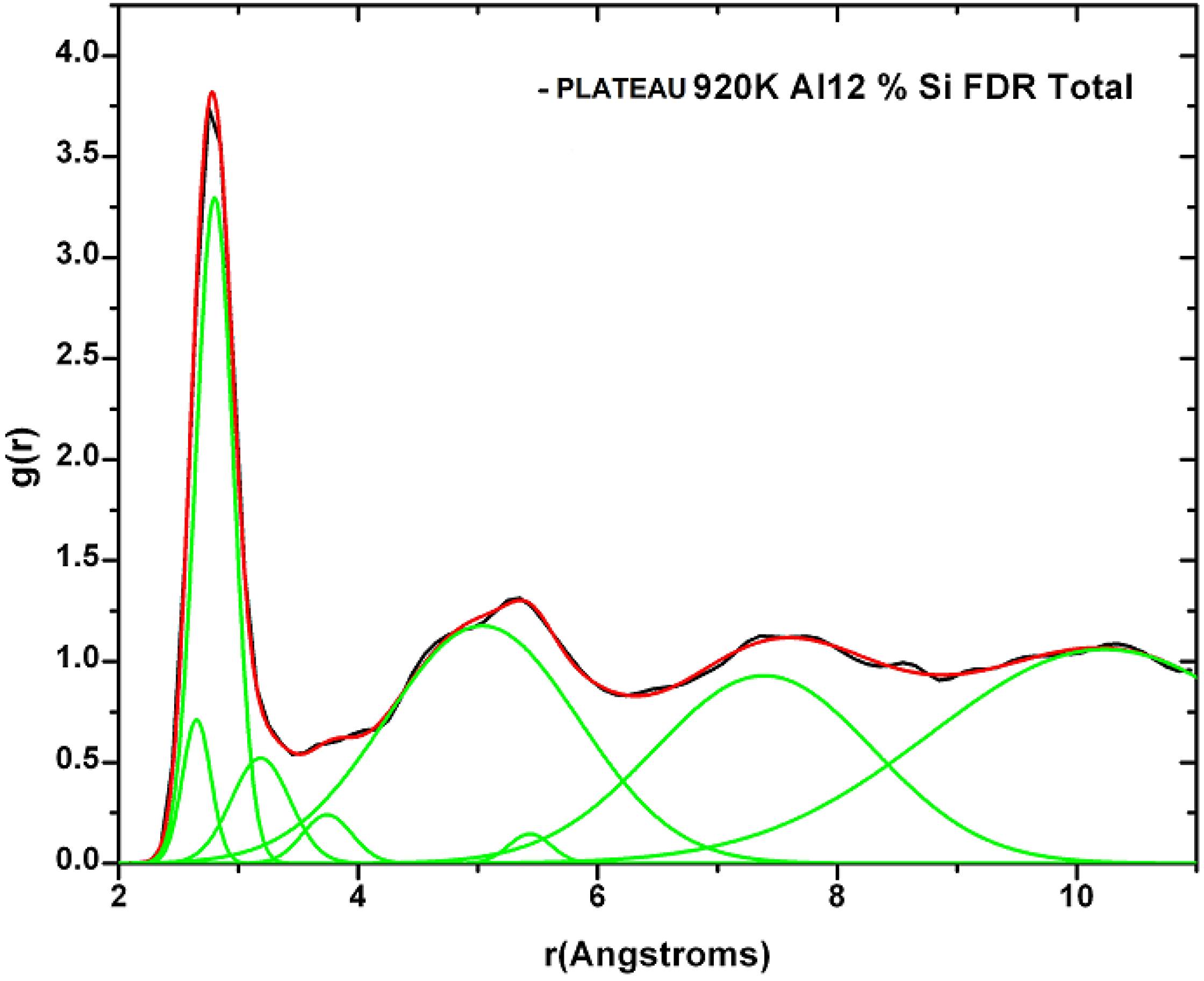
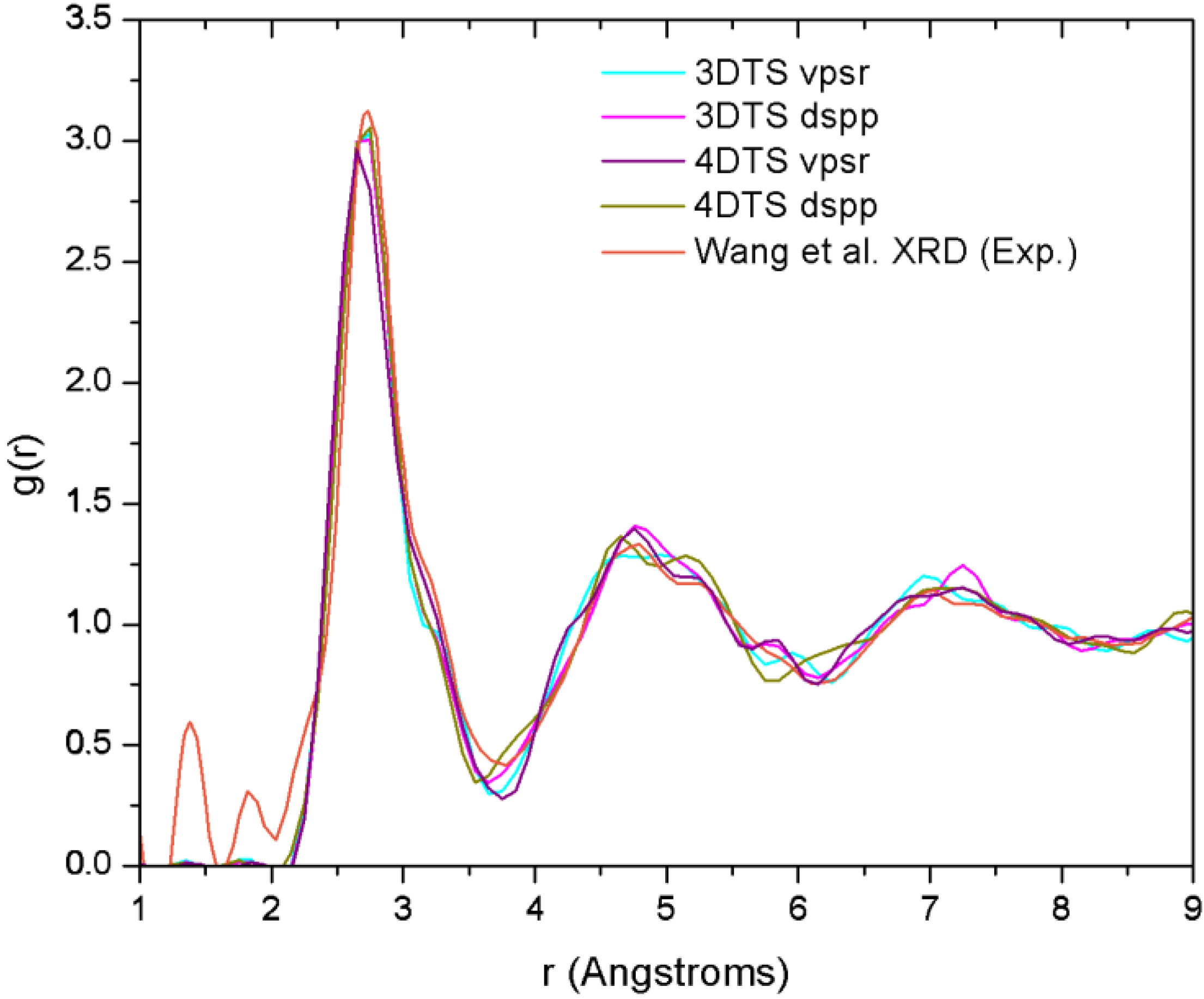
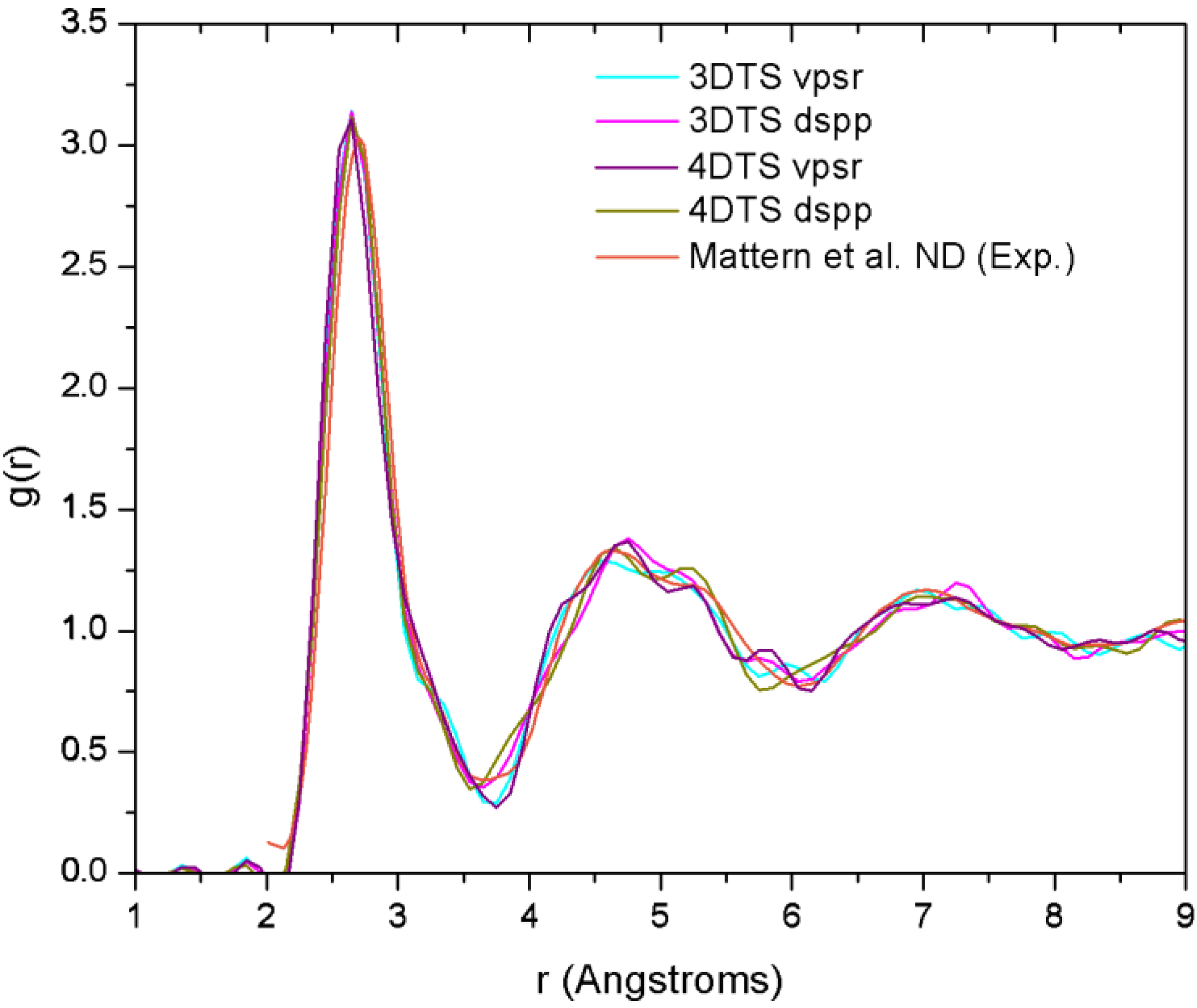
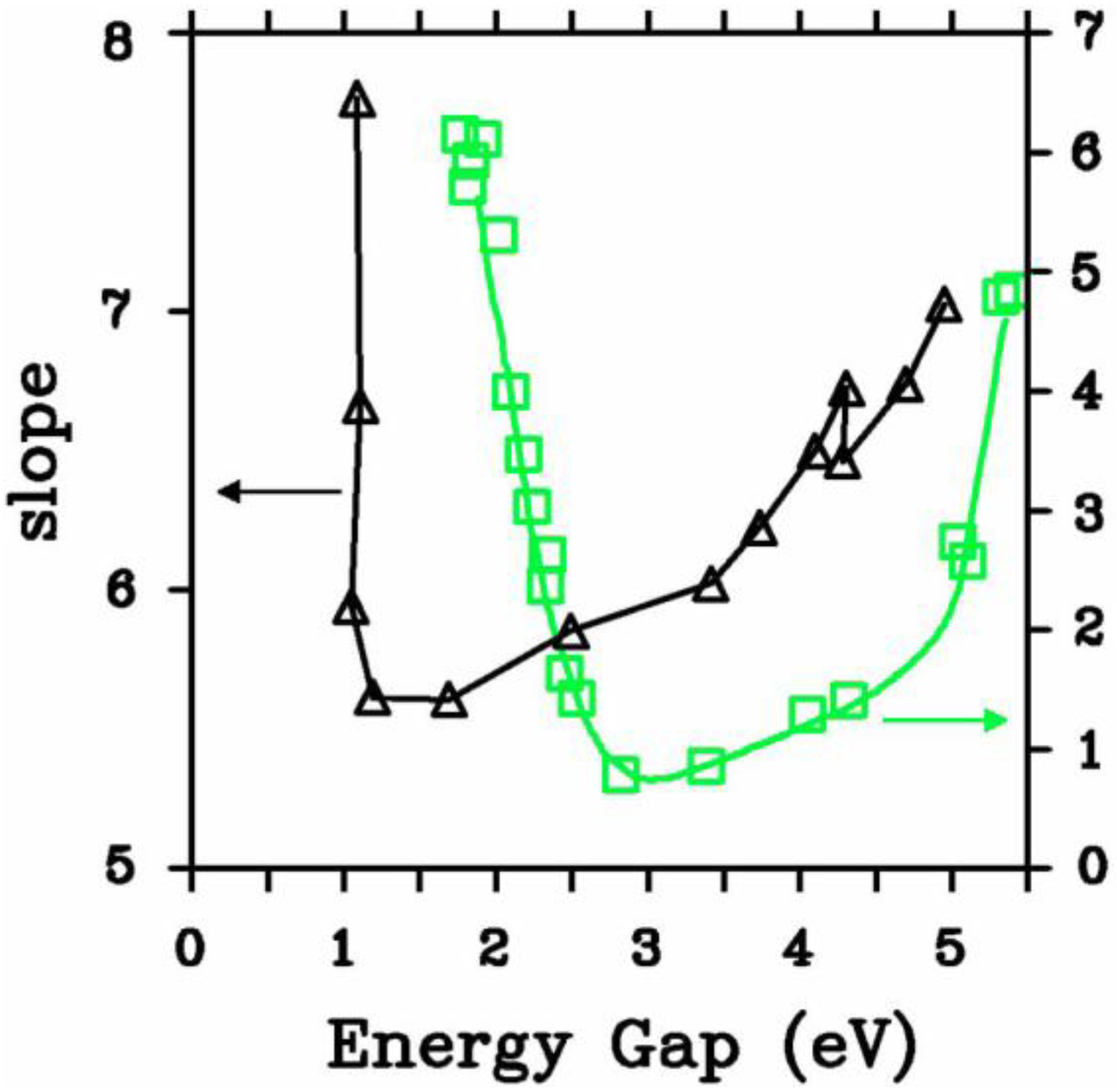
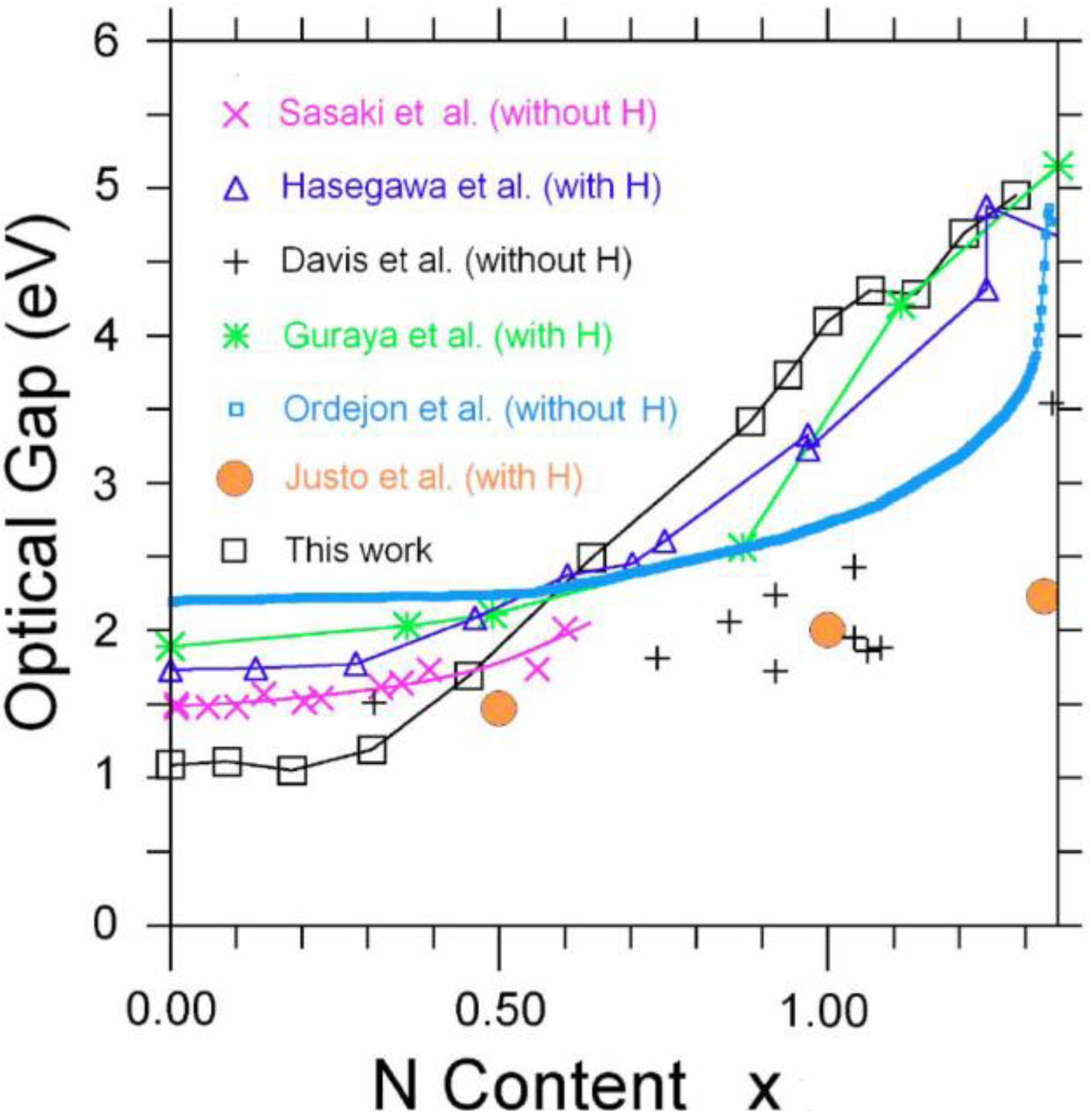
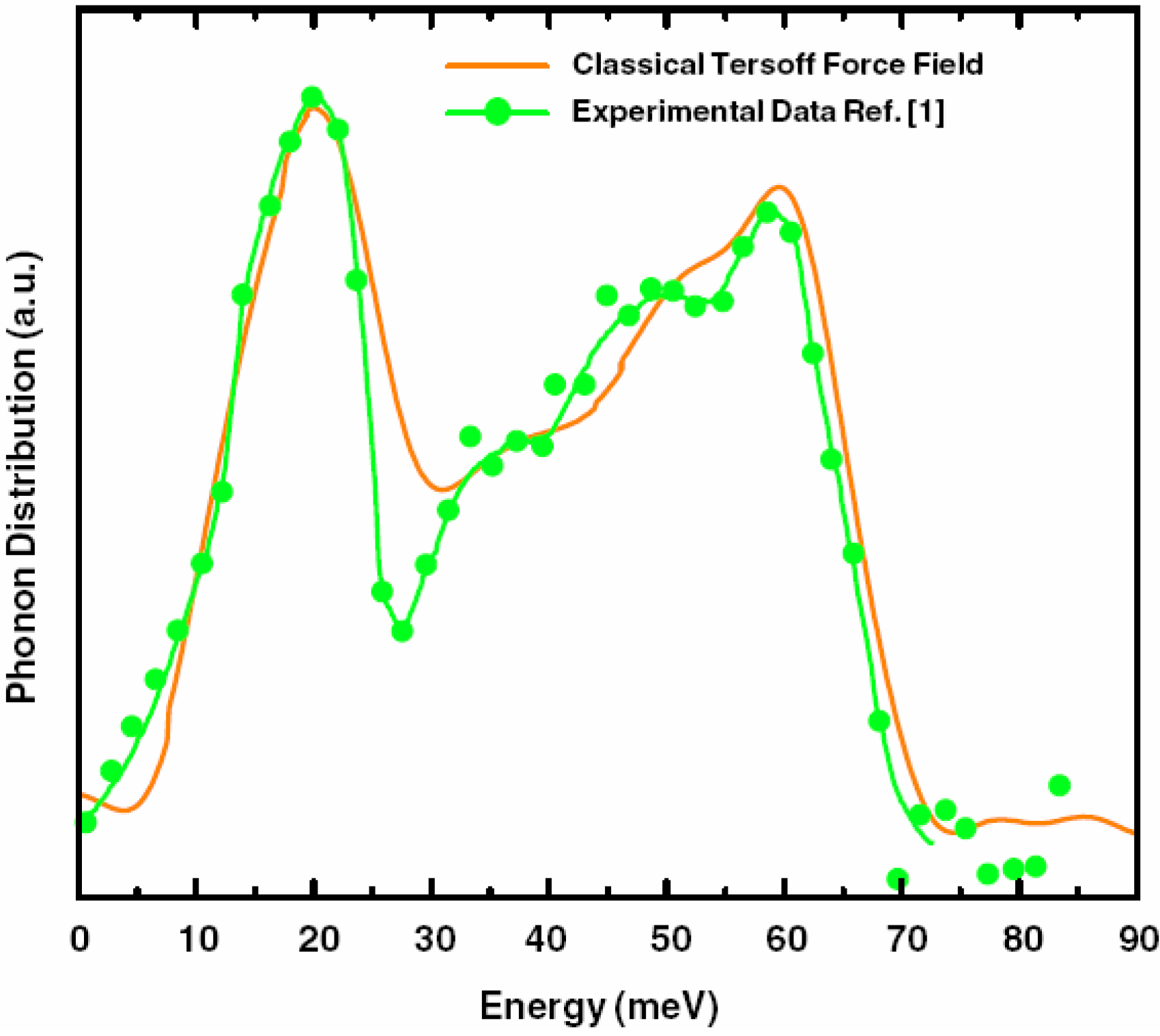
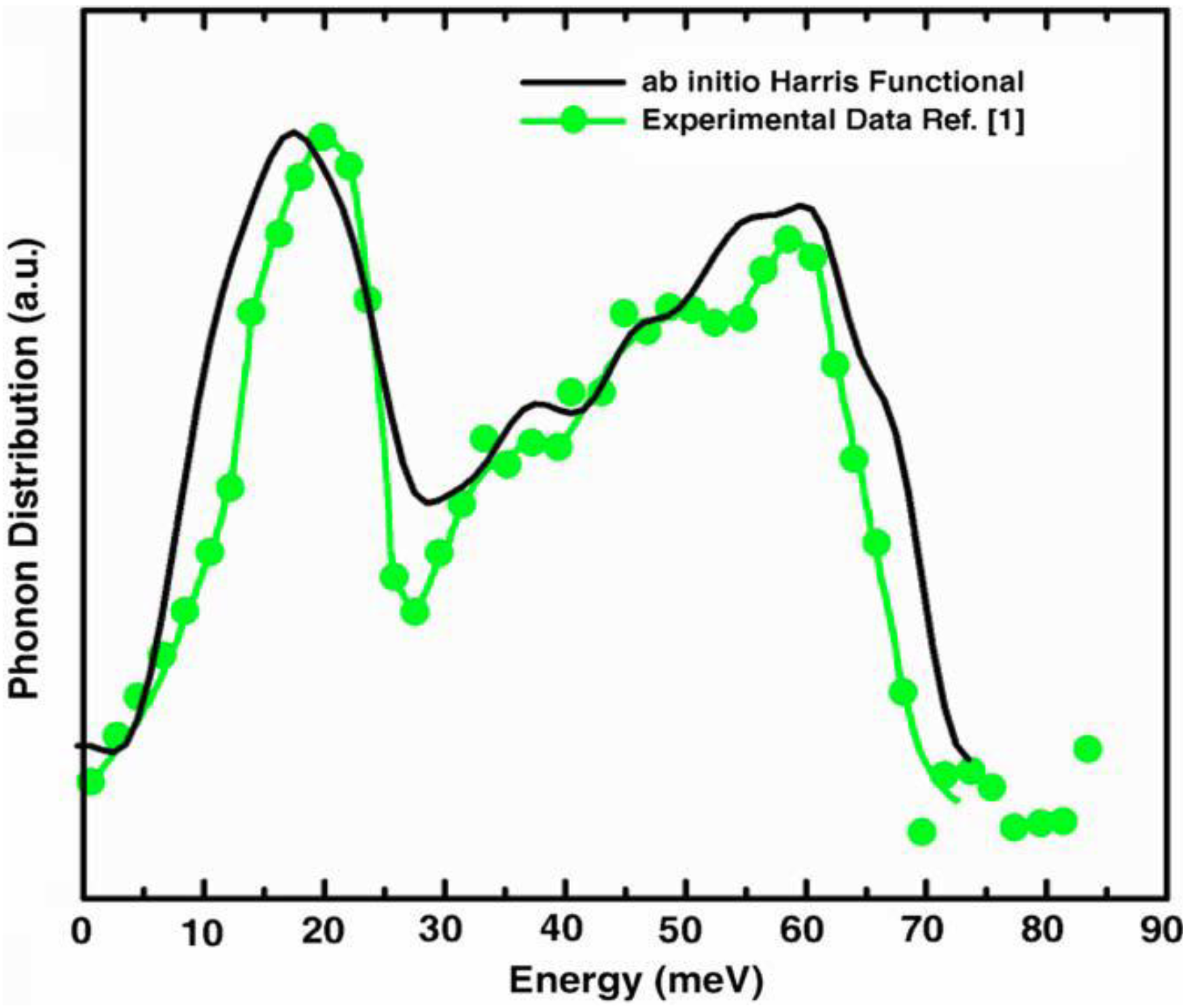
2. Antecedents [1]
The generation of amorphous topologies has a long history of approaches using a variety of force fields and different dynamical approximation methods. Since crystalline semiconductors provoked a revolution in electronics it is understandable that amorphous semiconductors were extensively produced and studied experimentally. This in turn provoked the development of theoretical models that contributed to the understanding of the atomic structure of these disordered materials. Amorphous metallic systems are on the other hand very difficult to produce in the laboratory since they tend to crystallize rapidly because the amorphous phases can be very unstable. In some materials, high cooling rates of ~10
6 K/s are needed to bypass crystallization and this restricts the thickness of the samples obtained to a few micrometers [
3]. The first attempts at producing metallic glasses were done by metal deposition on cold substrates which invariably led to thin film samples. Metallic glasses obtained by rapidly quenching melts were reported in 1960 by Klement
et al. who quickly cooled an Au–Si alloy from about 1,300 °C to room temperature leading to samples in the micrometer regime [
3].
Ab initio modeling of metallic systems has been widely used to study local structures in pure and alloyed liquid metals [
4], but its application to the generation of amorphous atomic topologies of solid metallic alloys has been very limited due perhaps to the small number of atoms that can be dealt with, despite their potential and necessary applicability to Bulk Metallic Glasses (BMG). Recent work reports the use of the Honeycutt-Anderson method to analyze amorphous alloys generated via computer simulations [
5].
For amorphous systems the attempts to generate reasonable atomic topologies can be classified via two extremes: (i) calculations based on
ad hoc classical, parameter-dependent potentials, constructed for the specific purpose of generating amorphous samples of certain materials; (ii) quantum methods, parameterized and
ab initio, that can deal from the outset with the thermalization processes used to generate the amorphous structures; with the interactions among electrical charges that lead to their structures and lead to their electronic and related properties, to understand their physical and chemical behavior. Much simulational work has been carried out both on pure elements and on alloyed systems [
1]. However, for the purposes of the present work we shall ignore some of the results we have obtained for pure elements and will concentrate on the description of amorphous semiconducting and metallic alloys.
For solid amorphous metallic systems the generation of disordered structures using first principles techniques is to our knowledge non-existent. There is some published work on the properties of liquid metallic systems: pure elements, alloys, and semiconductors, like Si and Ge, which are metallic in nature when in the liquid state. In what follows we report some of our unpublished results on the generation of amorphous metallic alloys in the solid state.
Since 1985 Car-Parrinello molecular dynamics [
6] and quenching from the melt of periodically-continued supercells have been extensively used to
ab initio generate amorphous structures of covalent semiconductors. Without doubt the pioneering work of Car and Parrinello has been a landmark in the development of the field, and has permeated most efforts up to the present. This technique of quenching from the melt is frequently used and in the literature is commonly known as the
melt-and-quench approach; it invariably generates a large number of bond defects, floating or dangling, in amorphous semiconductors when their liquid phases are metallic with larger coordination numbers. There is another common approach to the generation of amorphous substances in which ‘perfect’ random networks are constructed by hand, by switching bonds and adjusting plane and dihedral angles, where no bond defects are incorporated. The two procedures are opposite and only partially represent real amorphous materials. So it was necessary to search for a different thermal procedure that avoids the melting history of the first process and the ‘perfect’ construction of the second, hence the
undermelt-quench approach that we have developed [
1].
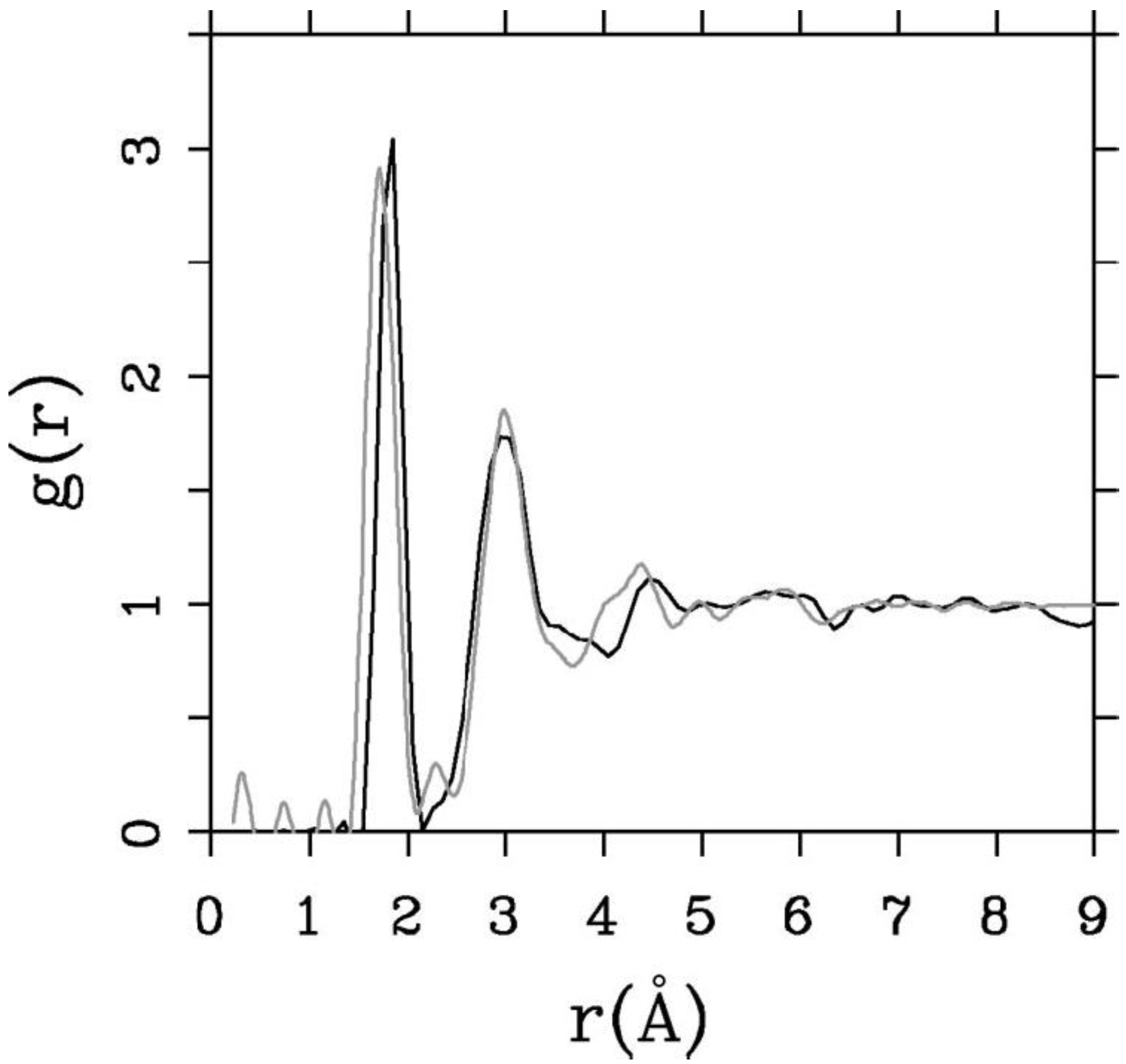
Car, Parrinello and collaborators applied their first-principles plane-wave molecular dynamics method (Car-Parrinello Molecular Dynamics, or CPMD) to C, Si, and Ge. Their simulations were done starting from the corresponding liquid phases and, after cooling them, radial distribution functions (RDFs) were calculated for the range 0 < r < l/2, where l is the length of the supercell edge used and generally includes the first two radial peaks. Even though the RDFs obtained reproduce reasonably well the first two peaks of the experimental results, the overall agreement with experiment varies from material to material. Furthermore, the procedure of quenching from the melt produces a large number of overcoordinated atoms since some of the liquid phases of these semiconductors are metallic; e.g., liquid silicon and liquid germanium have average coordination numbers between 6 and 7, and the quenching from the melt preserves some of this overcoordination. This excess of bond defects makes the electronic and/or optical gaps difficult to observe.
Chronologically, the first application of CPMD was to amorphous Si,
a-Si [
6] and then to liquid silicon [
7,
8] and most of the existing calculations stem from this original work [
7,
8,
9,
10,
11,
12]. Car and Parrinello performed this first
ab initio molecular dynamics (MD) study on an fcc periodic supercell with 54 atoms of silicon using their plane wave MD method. In their approach a non-local pseudopotential was used together with the parameterized local density approximation (LDA) form of Perdew and Zunger for the exchange-correlation effects [
7]. They obtained good agreement up to the second radial peak, with the experimental RDF of
a-Ge rescaled to simulate
a-Si, and argued that because of the size of their supercell, distances larger than 6 Å could not be studied. They pointed out that comparisons of simulated and experimentally determined atomic structures should be carried out with care in view of the large number of defects generated in the simulation. A typical simulation for
a-Si was started above the melting point at about 2,200 K, and the liquid was allowed to equilibrate for ≈ 0.7 ps before it was quenched down to ≈300 K at a cooling rate of ≈2 × 10
15 K/s. During the initial quenching the volume of the cell was gradually changed to 1080 Å
3, the crystalline value.
Since then several works have appeared that generate disordered structures using CPMD and the melt-and-quench procedure. We now present a brief résumé of the pertinent works for our purposes, introducing the nomenclature where generally a-AB refers to the family of amorphous alloys that have varying contents of the elements A and B, whereas a-AxB1−x or a-AyBz refer to a specific alloy. Sometimes we shall use the nomenclature a-ABx just to be consistent with the established identification procedure of having an amorphous alloy that has 1 part of element A for x parts of element B.
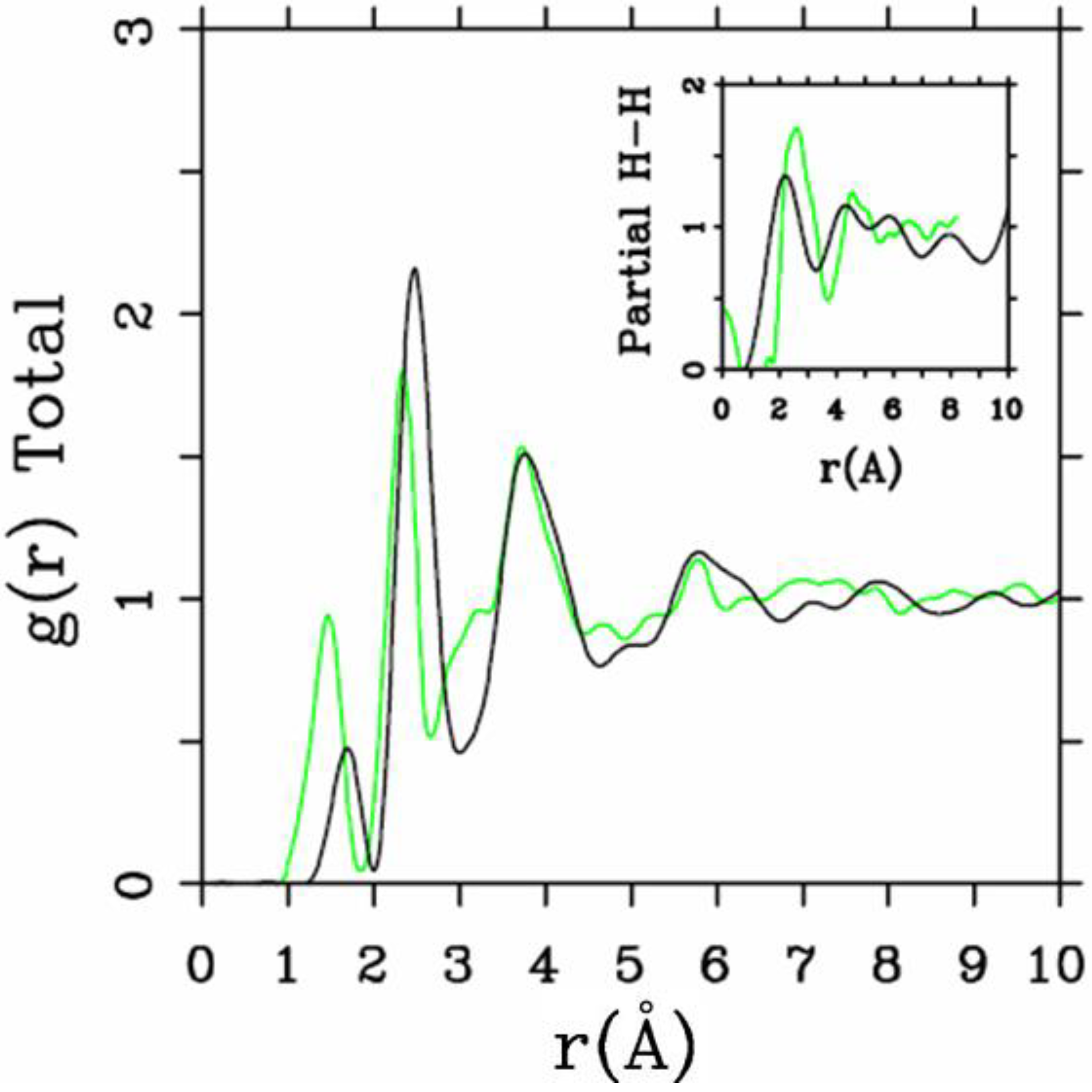
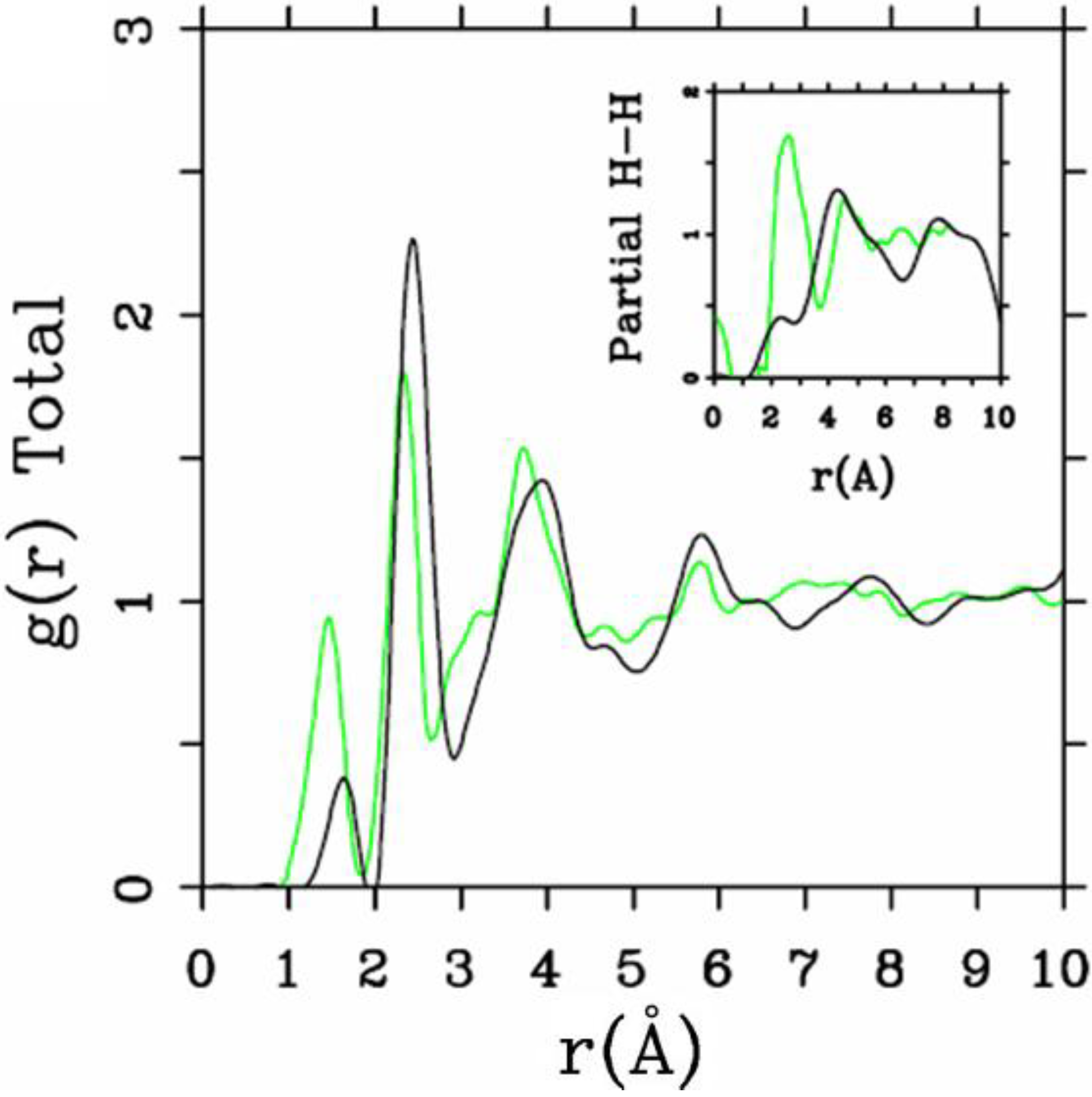
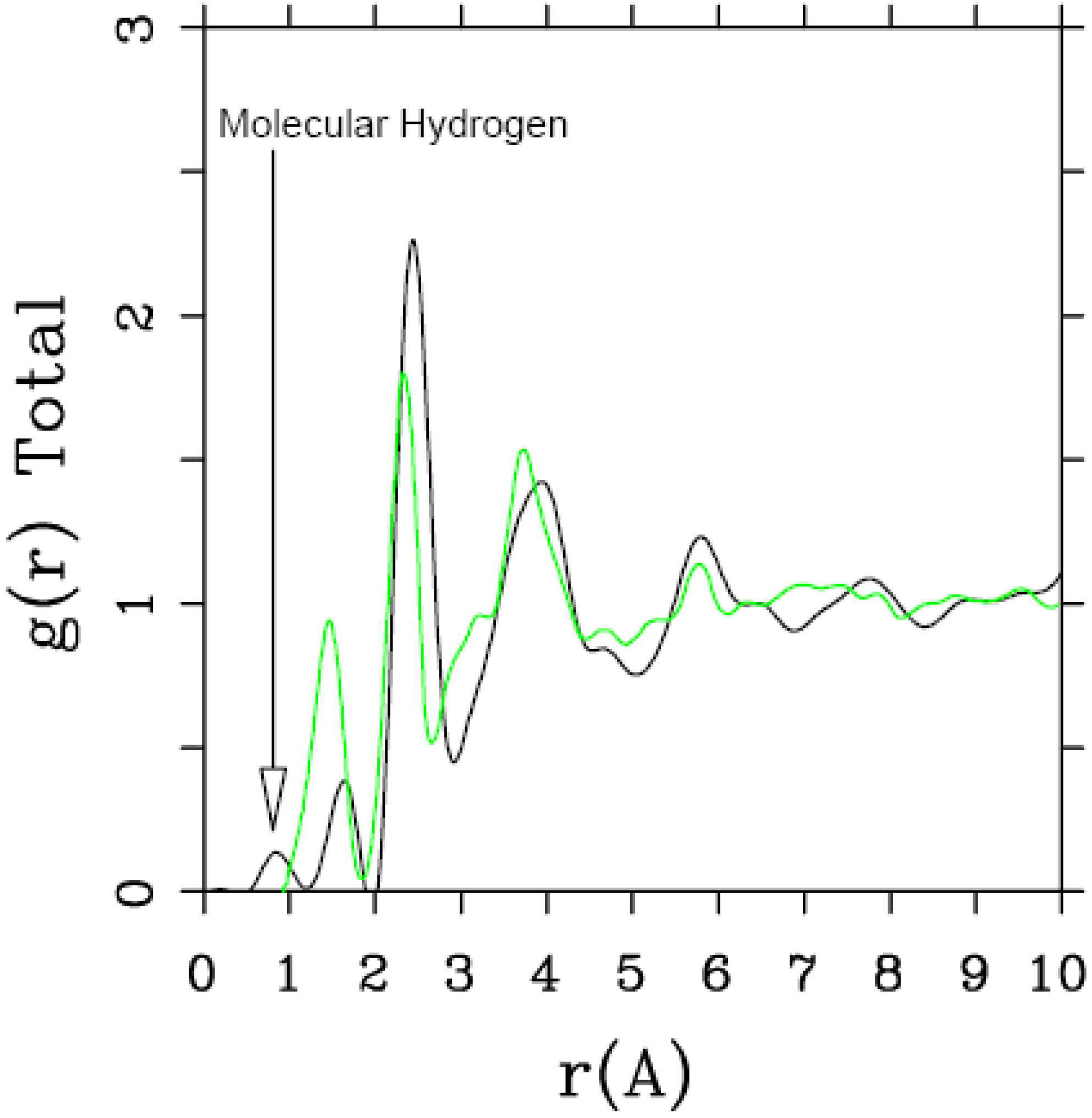
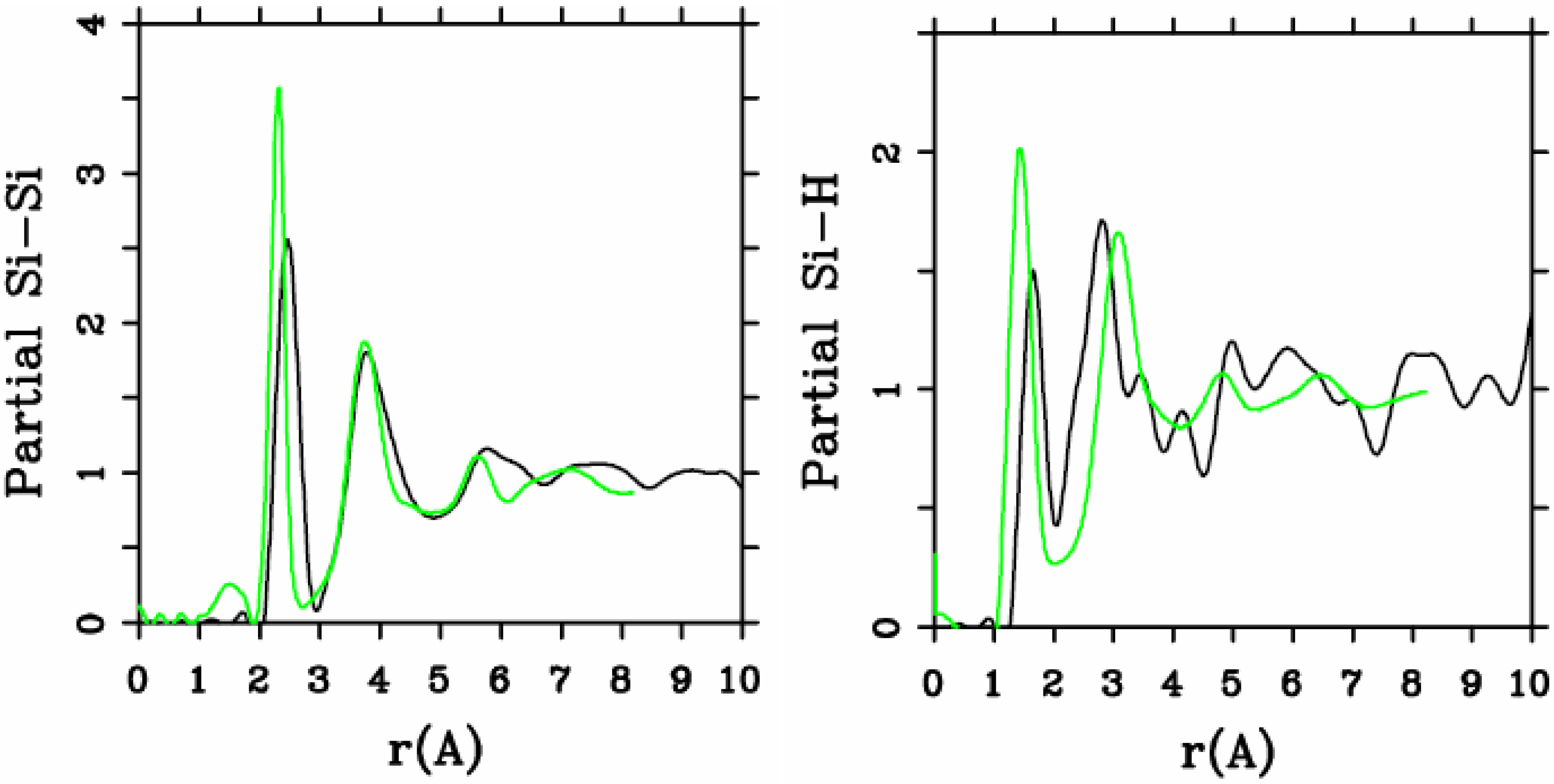
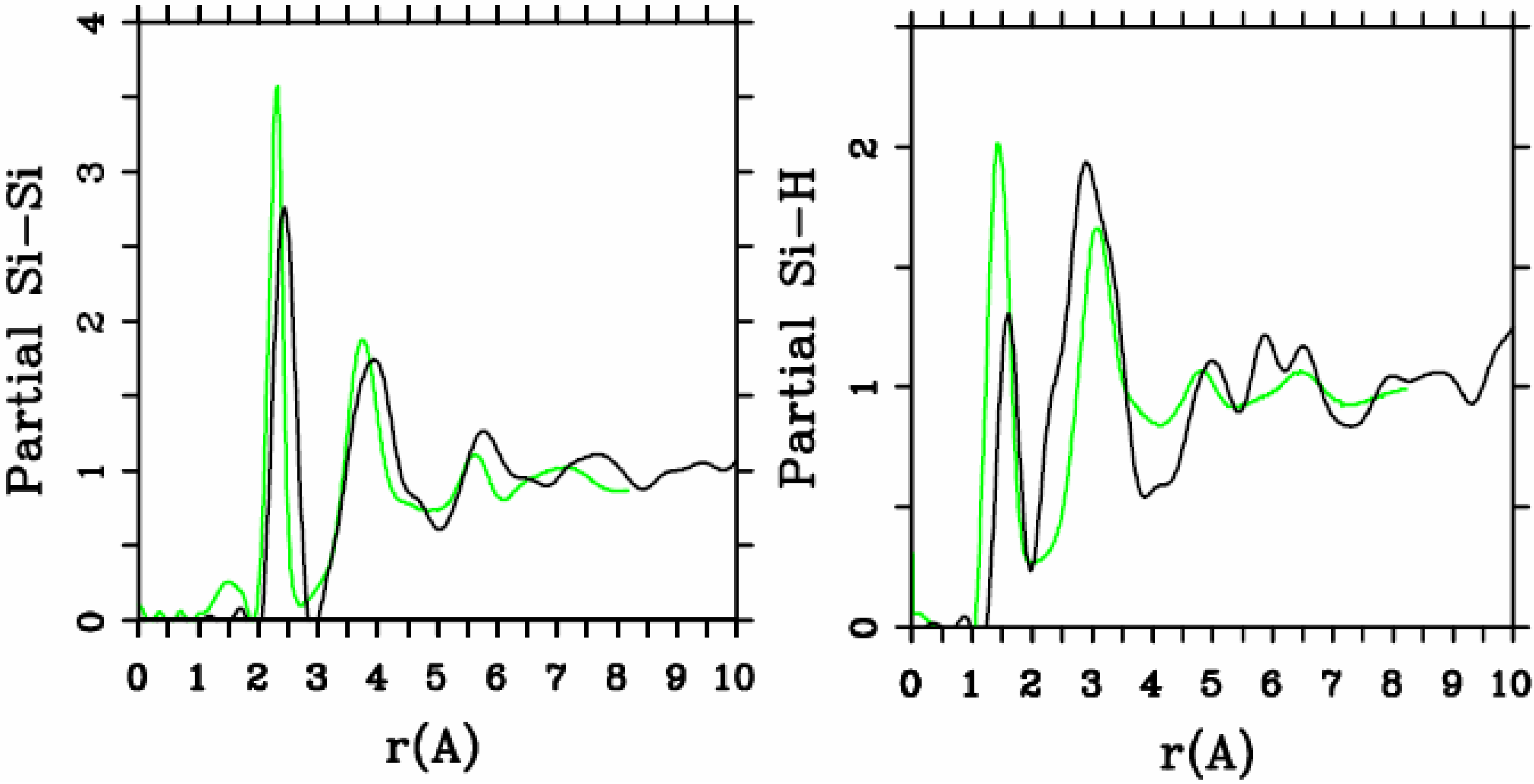
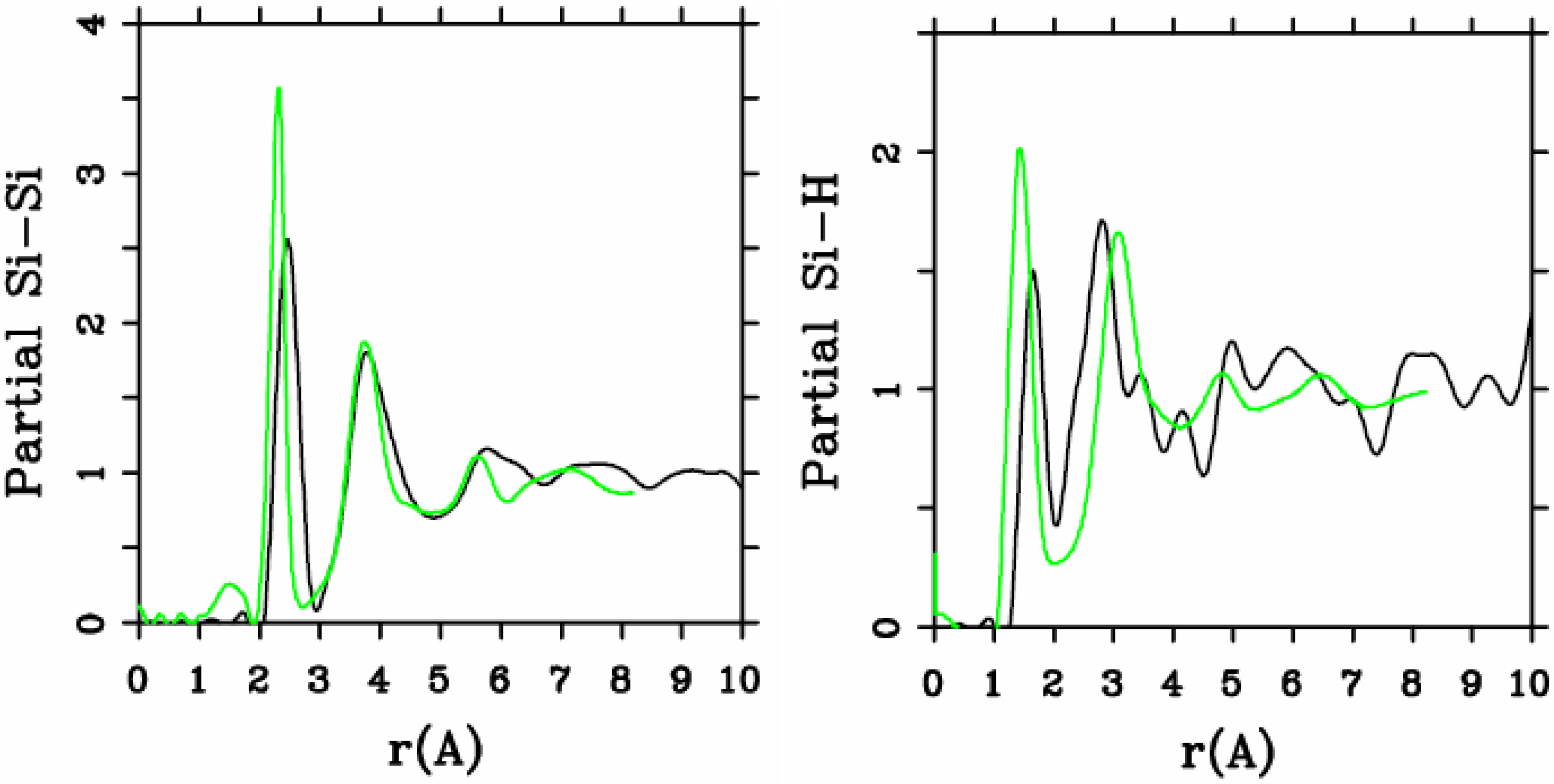
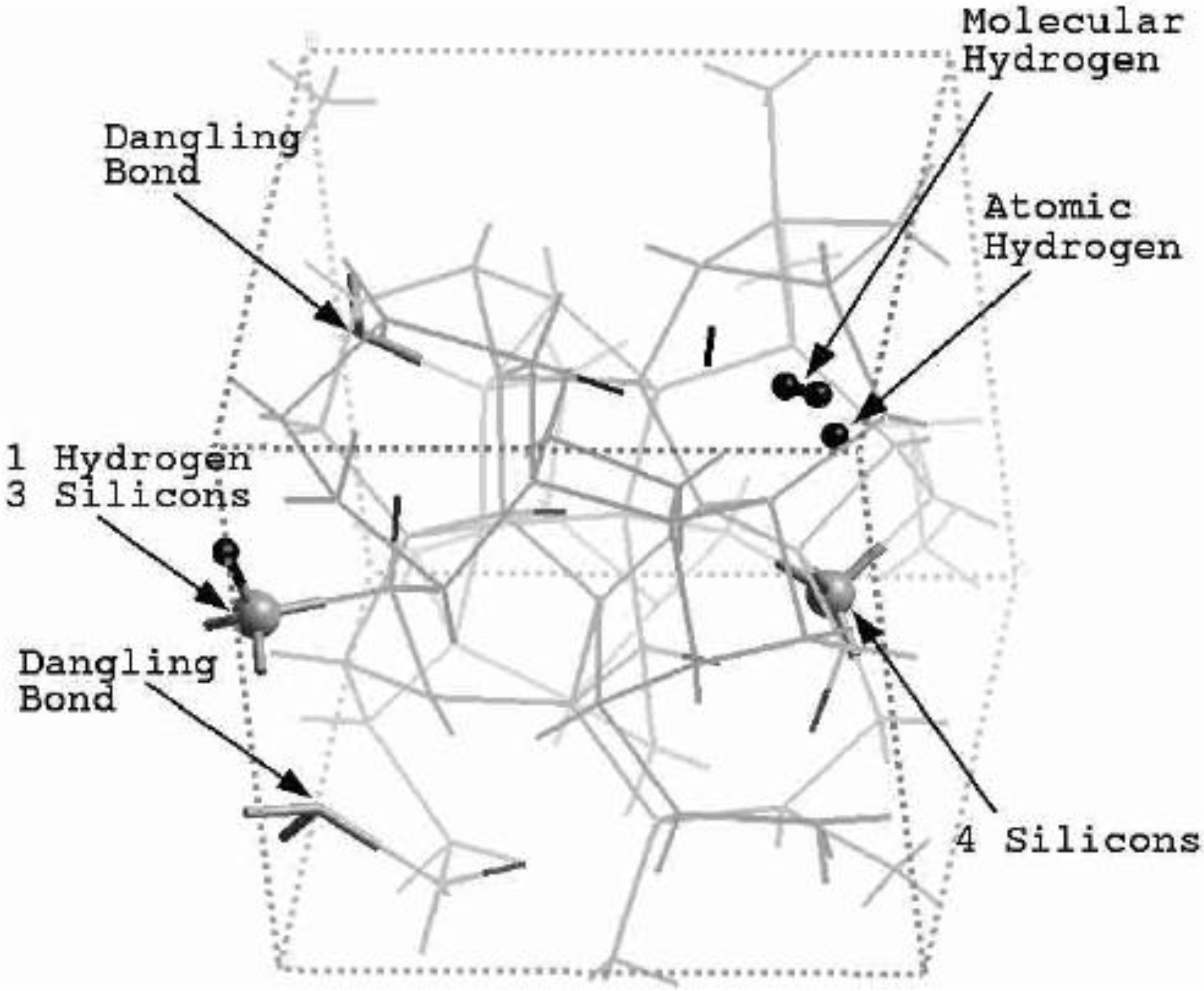
Let us begin with amorphous hydrogenated silicon. Simulating
a-SiH is a difficult task since there seems to be a strong dependence of its atomic topology on deposition conditions; also, the chemical reactivity of hydrogen is another factor that has to be addressed. Finally, the high mobility of H compared to the mobility of Si, and the role of its zero point energy have to be taken into account, indicating the necessity of a quantum mechanical
ab initio approach to this material. The corresponding CPMD work is due to Buda
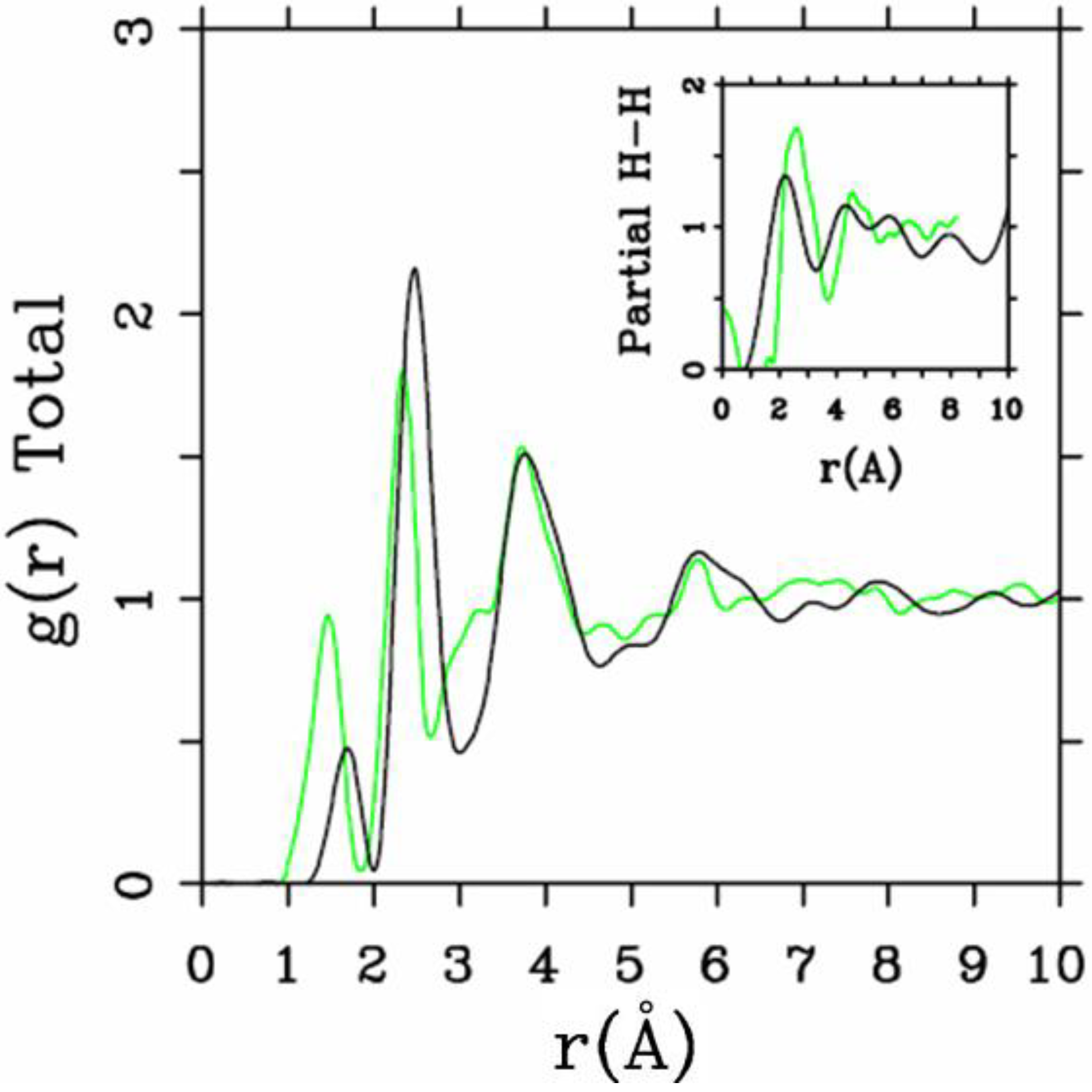
et al. where the plane-wave Car-Parrinello method was applied to an amorphous hydrogenated cubic cell of 64 silicons and 8 hydrogens (11% concentration) [
13]. These authors started out with a liquid material containing both silicon and hydrogen atoms which was rapidly quenched, maintaining a density equal to the value of the crystalline material. They report only partial distribution functions and the H-H RDF obtained in these simulations compares poorly to the existing neutron scattering experiments. We believe that the poor agreement is mainly due to the fact that the simulational supercells are melted before being solidified. Fedders and Drabold [
14] and Tuttle and Adams [
15] have also studied
a-SiH from first principles. Fedders and Drabold do not report any RDF, total or partial, whereas Tuttle and Adams report only the Si–Si and the Si-H RDFs of a cell of 242 atoms with 11% hydrogen. Tuttle and Adams generated their structures from a liquid at ≈1,800 K, which was then quenched to produce an amorphous sample at 300 K. They assumed that the mass of each hydrogen atom
equals the mass of each silicon atom leading to an unrealistic representation of the diffusion of hydrogen in the sample. This unrealistic assumption implies an unrealistic H-H RDF so perhaps that is why no H-H RDF is reported in their work. Furthermore, the high mobility of the hydrogen atoms relative to the mobility of the heavier silicons makes the handling of an adequate time step in the simulation more difficult.
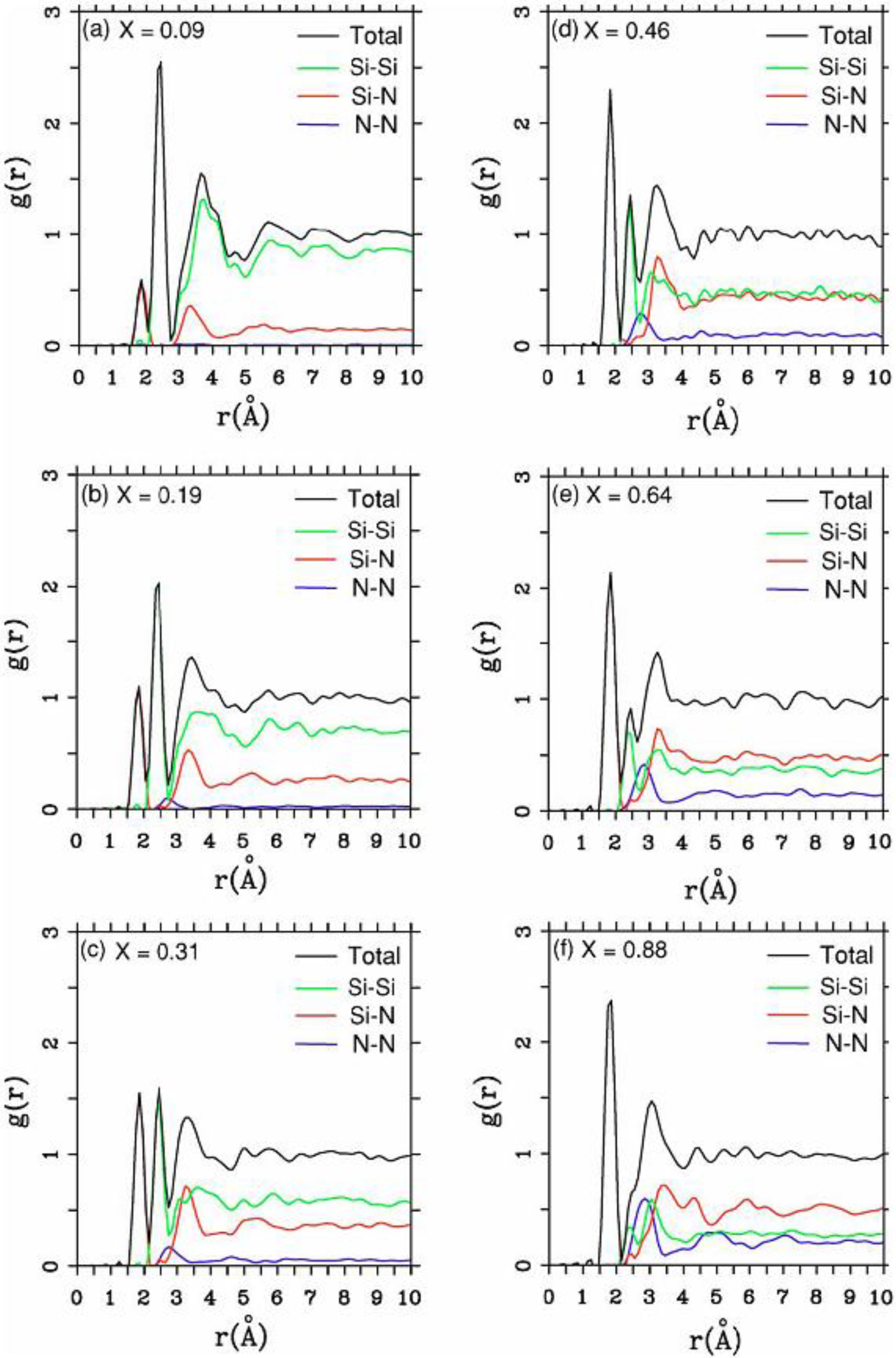
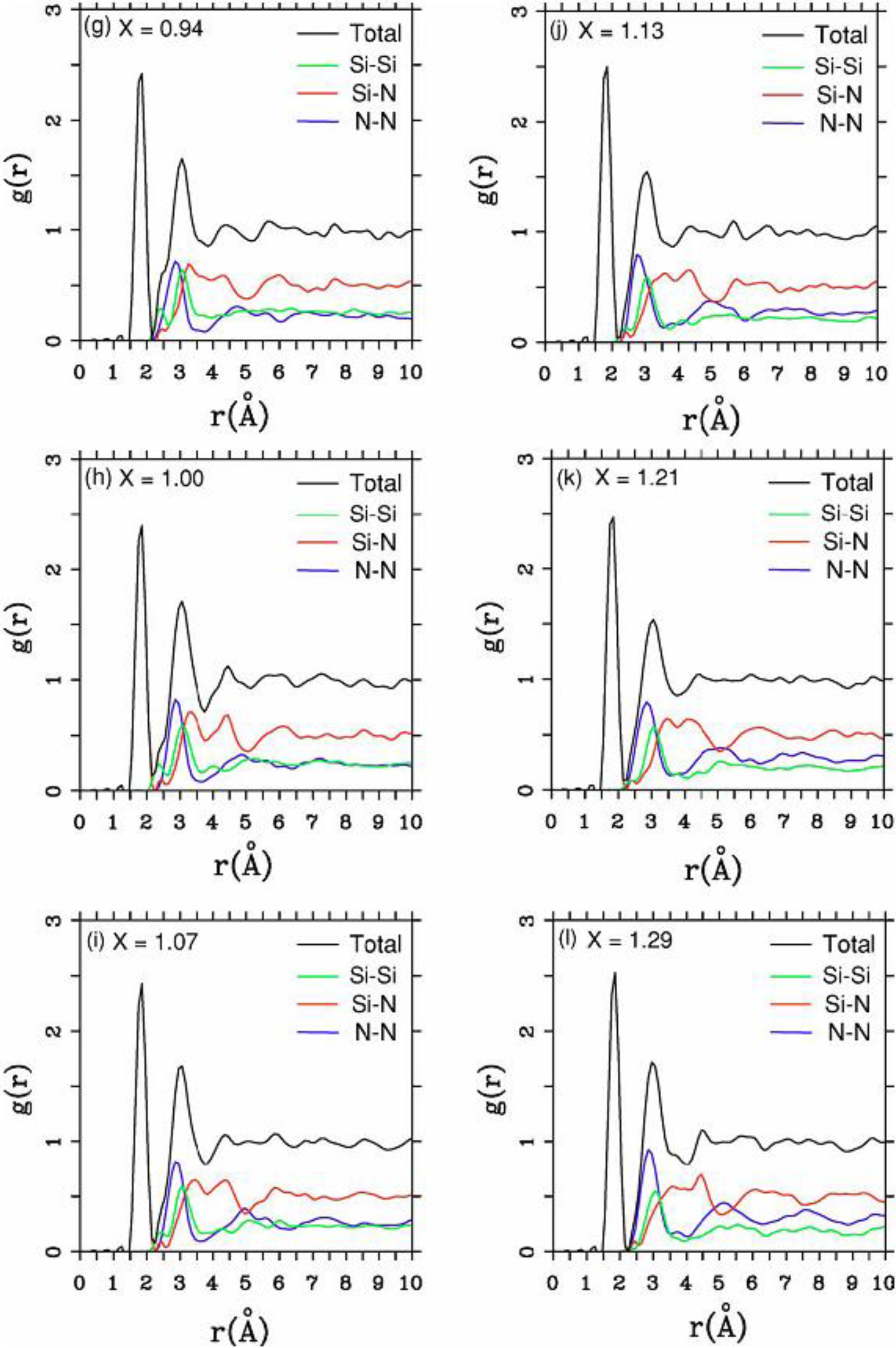
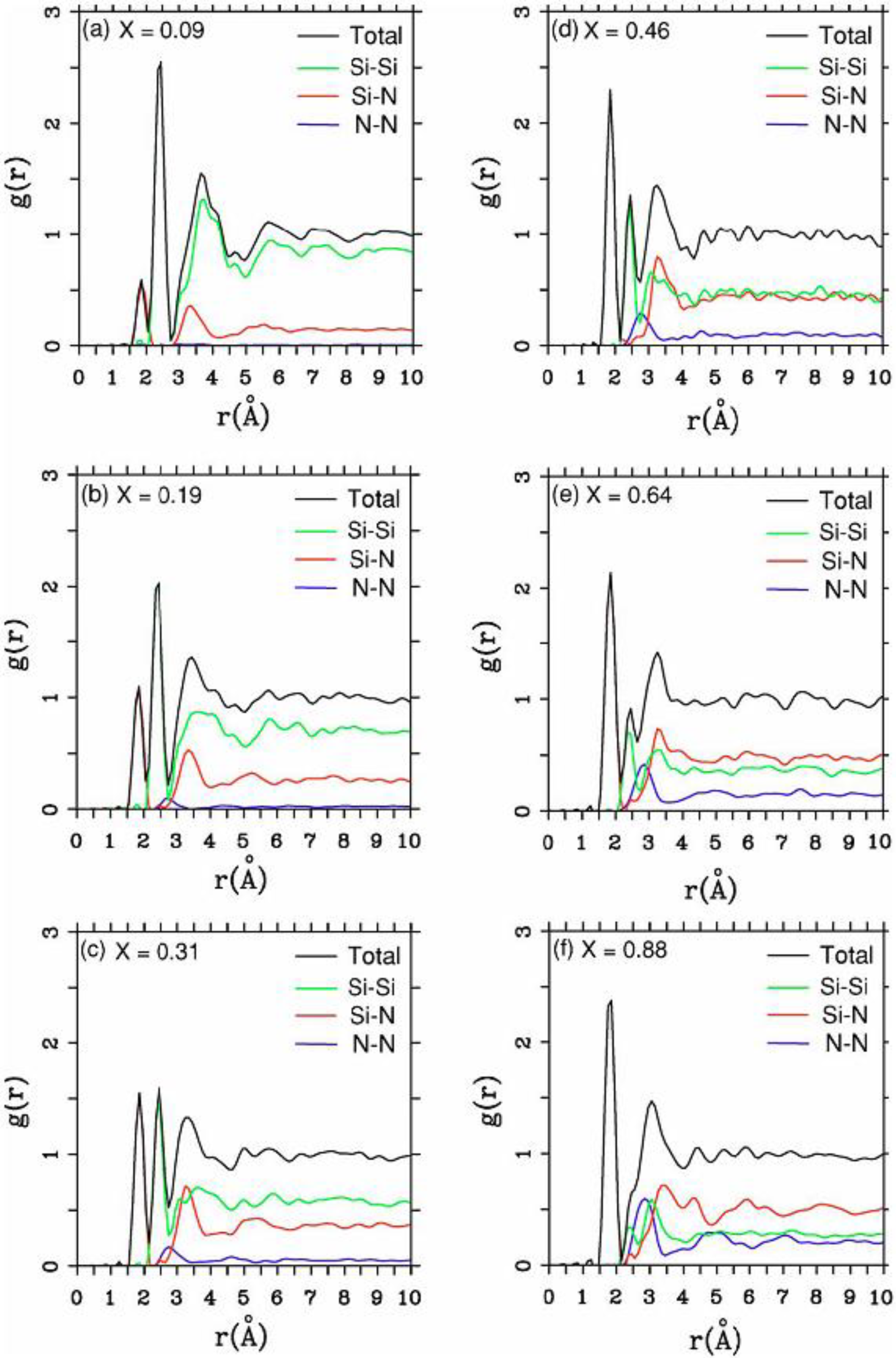
There were no CPMD-based calculations for
a-SiN before the publication of the results of our group in 2002, results that shall be presented in
Section 4 and
Section 5. As far as we know
a-SiN alloys have not been the subject of any other type of quantum molecular dynamics simulations up to the present, and therefore our work was the first, and so far it is the only
ab initio study of this material.
For
a-CN, there are some
ab initio studies using the CPMD approach [
16,
17]. McKenzie and coworkers use random networks that were generated by the
melt-and-quench method on a crystalline 64-atom supercell. They studied the electronic density of states to investigate the probable doping mechanism of carbon by nitrogen. Only three different densities were considered, each for two concentrations, influenced by the experimental work of Walters
et al. [
18]. The two concentrations were C
62N
2 and C
56N
8, and the three densities were 2.45, 2.95 and 3.20 g/cm
3. An additional simulation was carried out for a density of 2.7 g/cm
3 and a concentration of C
60N
4. They claimed that, contrary to the experimentally found substitutional doping, their results did not indicate that for low concentrations nitrogen behaves as a dopant in amorphous carbon.
The first CPMD simulation concerning the structure of amorphous silicon carbide,
a-Si
0.5C
0.5, was reported in 1992 by Finocchi
et al. [
19]. They performed CPMD on two different samples, one with 27 C and 27 Si atoms randomly distributed throughout the diamond crystalline positions, and the other with 32 C and 32 Si atoms randomly distributed throughout the rock-salt crystalline structure positions. Both samples have a density of 3.1 g/cm
3. The authors used a
melt-and-quench procedure where the samples were heated up to a temperature of approximately 4,000 K, then they were equilibrated during a 1 ps interval and cooled down to approximately 500 K. The authors affirmed that the two samples had very similar structural properties, thus they concluded that the simulations were not dependent on the initial structures. The electronic properties of these samples were calculated at the Γ point, despite the small number of atoms and the small size of the supercells. In this work the authors reported the total and partial RDFs, and the electronic density of states (eDOS). The authors observed that the RDFs had a peak around 1.5 Å due to C–C bonds and 1.9 Å due to Si-C bonds, so the first coordination shell was formed by two different bond types, also 40–45% of the bonds present in the sample were C–C homonuclear bonds. Two conclusions were drawn in the analysis of the electronic structure of the amorphous samples: the first was that the material is a semiconductor; the second was that the ionicity gap observed in crystalline silicon carbide,
c-SiC, located at about −11 eV with respect to the Fermi level disappears.
The second CPMD work was also done by Finocchi
et al. and had the objective of studying the local atomic environment of
a-Si
0.5C
0.5 [
20]. Here the authors generated the amorphous structure of SiC using a technique very similar to the previous work [
19]. In this paper they could not establish a sample structure either chemically ordered or completely random. The general remark was that a detailed analysis of each atomic species is of vital importance in order to understand its physical properties.
In order to explore the local atomic environment of
a-(SiC)H, Finocchi and Galli in 1994 worked out a CPMD amorphization of a simple cubic supercell of 3.18 g/cm
3 which was made up of 32 C, 32 Si and 12 H atoms [
21]. The
a-(SiC)H sample was produced by a rapid quenching from the melt, at ~4,000 K, to 500 K, using the same procedure as in Reference 19. The authors reported the total and partial distribution functions, and the coordination numbers, but since we have not studied this system we shall not dwell on it.
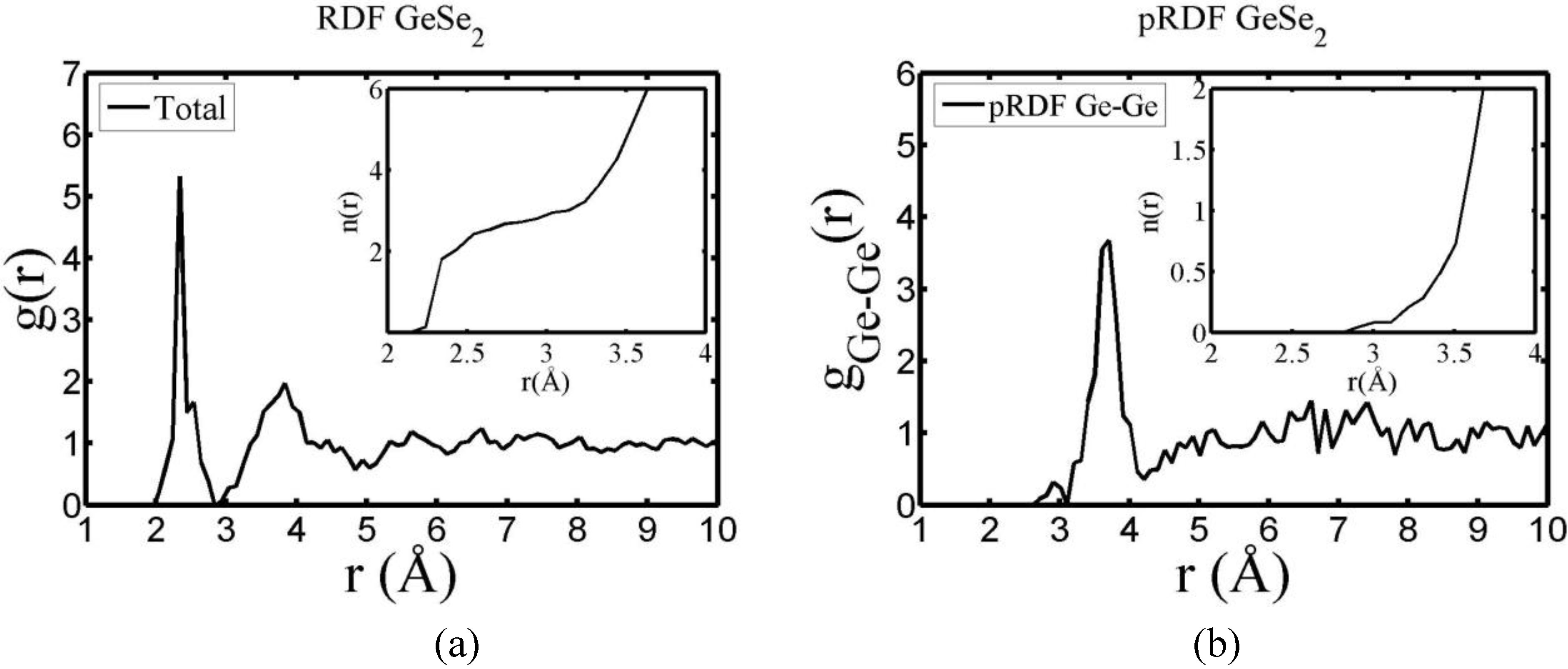
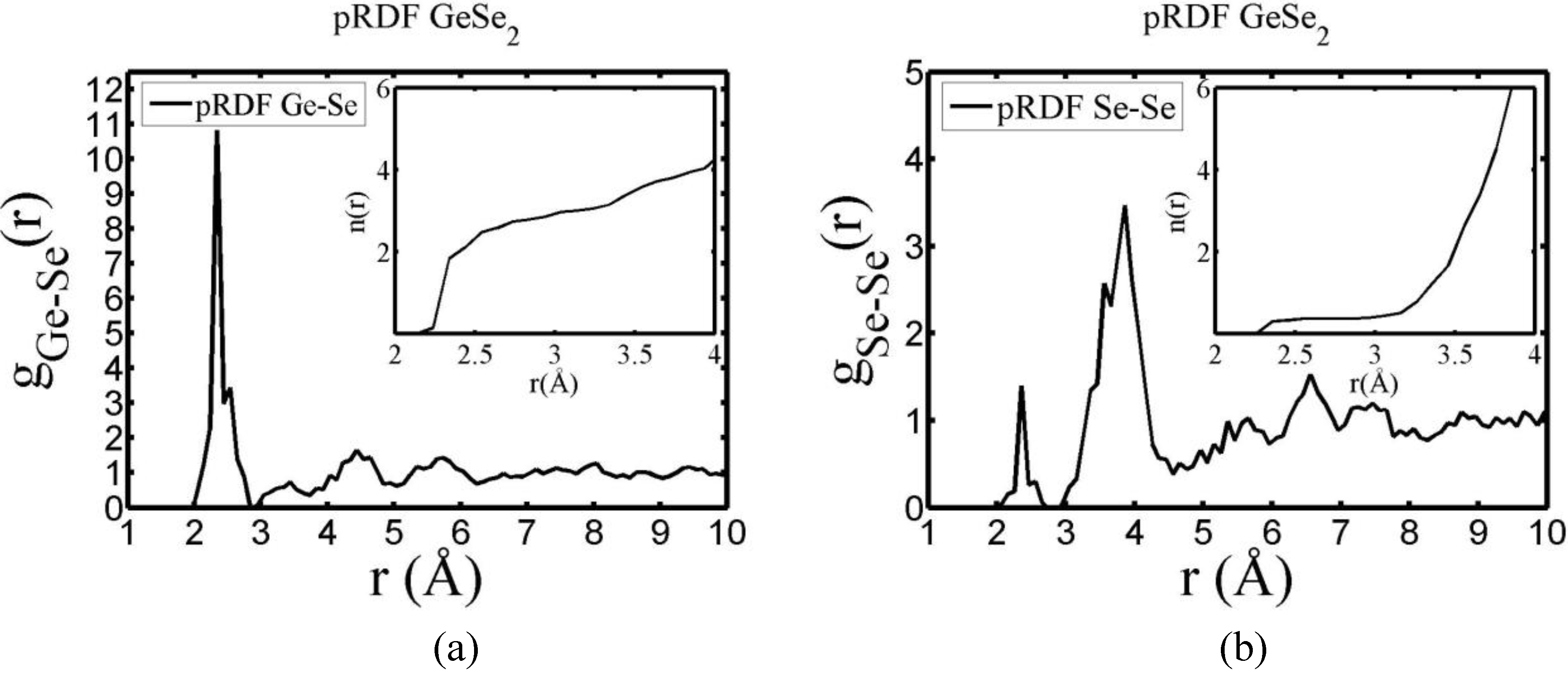
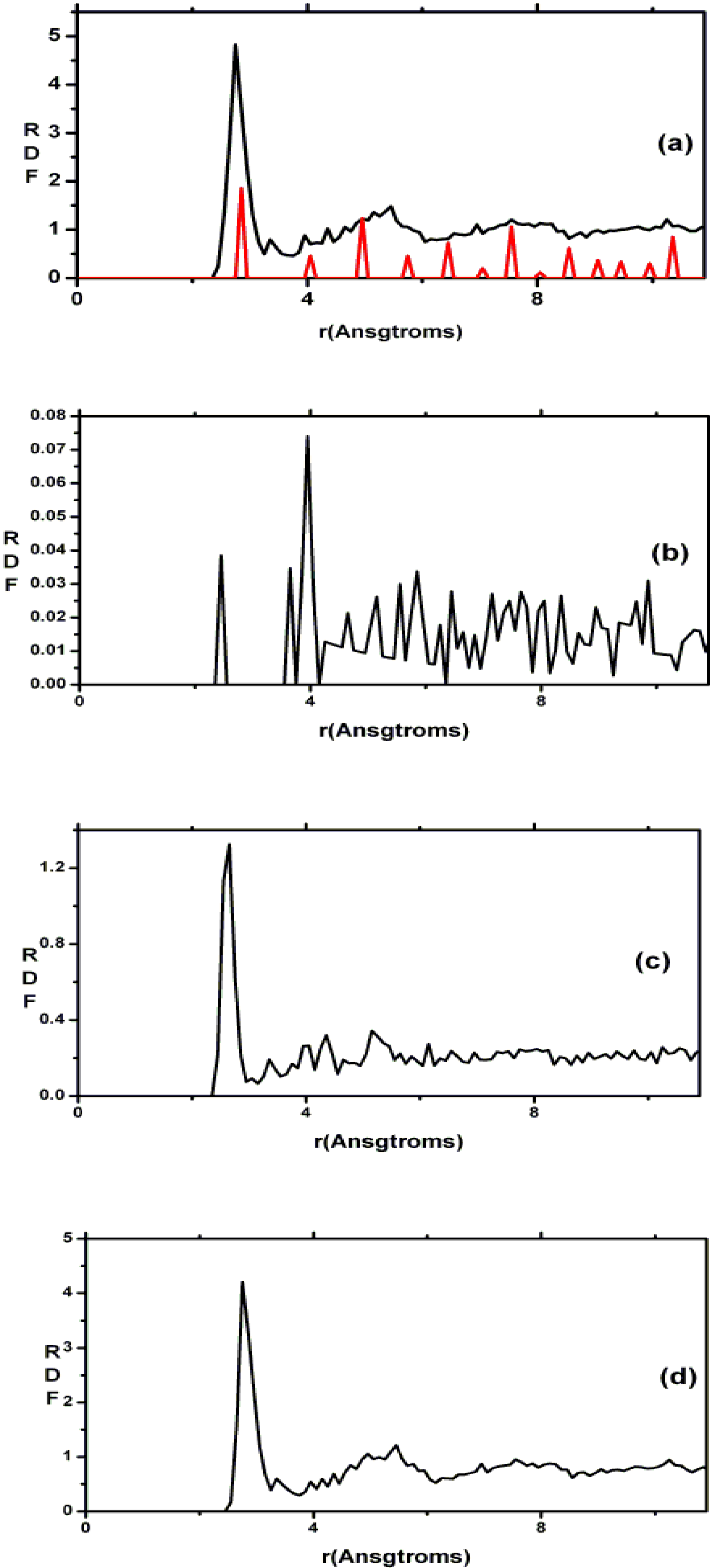
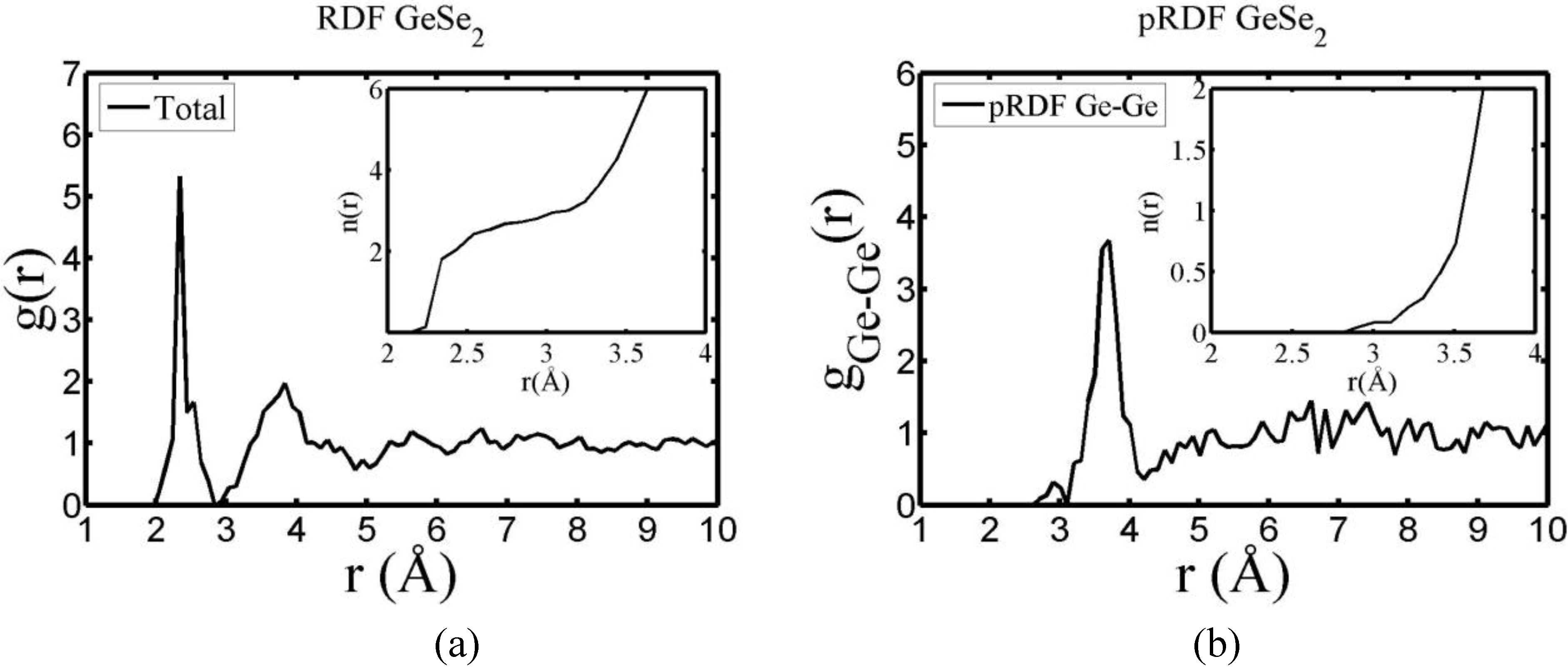
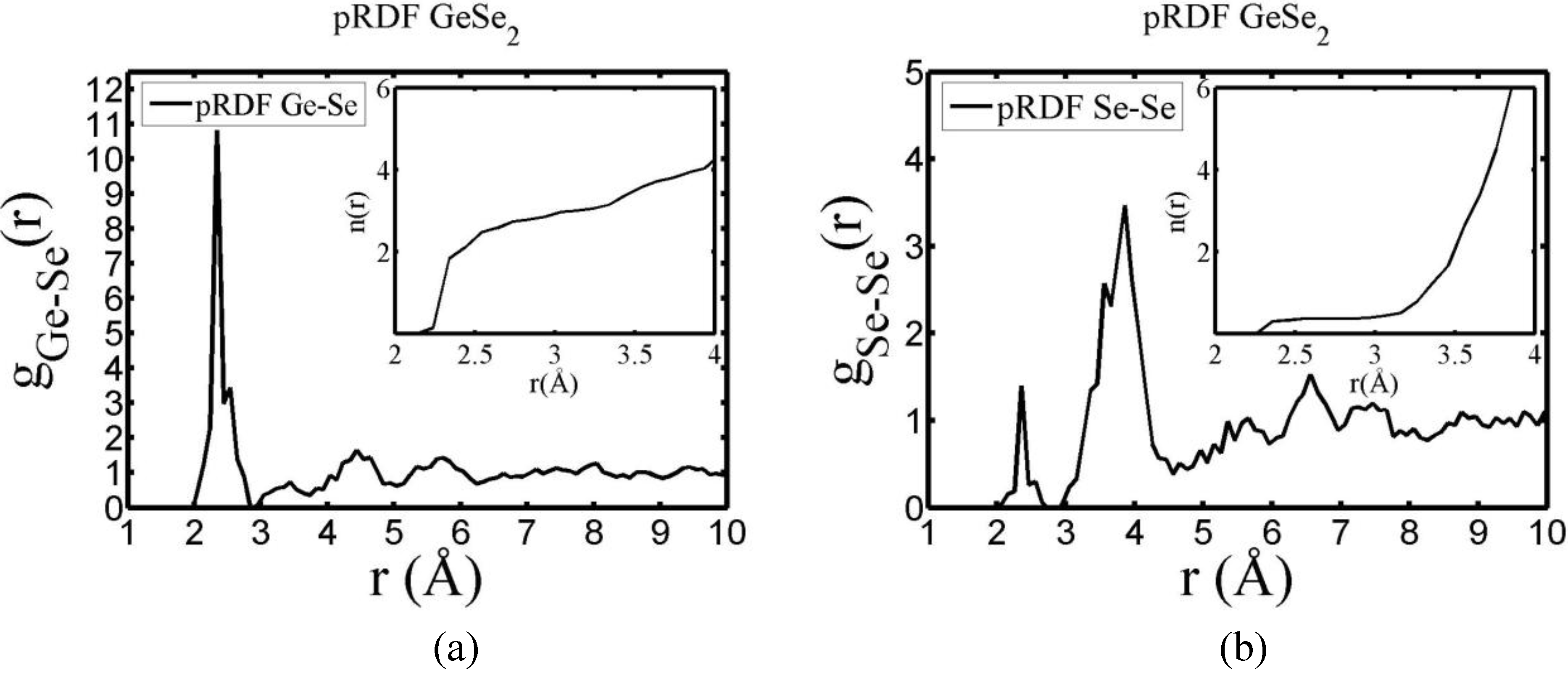
There are a few CPMD studies on amorphous GeSe
2. Recently, Massobrio and Pasquarello generated amorphous networks by cooling a 120-atom liquid supercell [
22,
23]. They used 40 Ge and 80 Se and a periodic cubic supercell with an edge length of 15.16 Å and a density of 4.38 g/cm
3. There are
ab initio works based on the Harris density-functional method developed by Sankey and coworkers [
24]. In the first work, Cappelletti
et al. studied the vibrations in amorphous GeSe
2 using a diamond-like supercell of 63 atoms, 20 Ge and 43 Se [
25]. They applied the
melt-and-quench process to their model and did some removing and adding of atoms to end up with exact stoichiometry: 21 Ge and 42 Se, and a resulting density of 4.20 g/cm
3. They compared their vibrational density of states (vDOS) results with the experimental one. In the second work on
a-GeSe
2, Cobb
et al. report a 216-atom supercell [
26]. In this
ab initio study they reported topological, vibrational and electronic properties that will be compared with our results later on.
For completeness, and to the best of our knowledge, there is no CPMD work reported on
a-InSe, but tight-binding calculations were performed by Kohary and collaborators [
27]. They amorphized In-Se with different densities and numbers of atoms per supercell, depending on the basis set: 64 and 124 atoms for DNP and SN basis sets. These cells were made amorphous by the
melt-and-quench procedure. The positions of the first peak were in the range of 2.60 and 2.76 Å. They concluded that there were no Se-Se homopolar bonds for
a-In
0.5Se
0.5.
No CPMD works on aluminum-based alloys were found, except for a calculation of Alemany
et al. where the Kohn-Sham
ab initio molecular dynamics is applied to study liquid aluminum near the melting point [
28]. Our own work, which shall be presented at a later stage, is based on the Lin-Harris MD, LHMD, and is not self consistent as the CPMD.
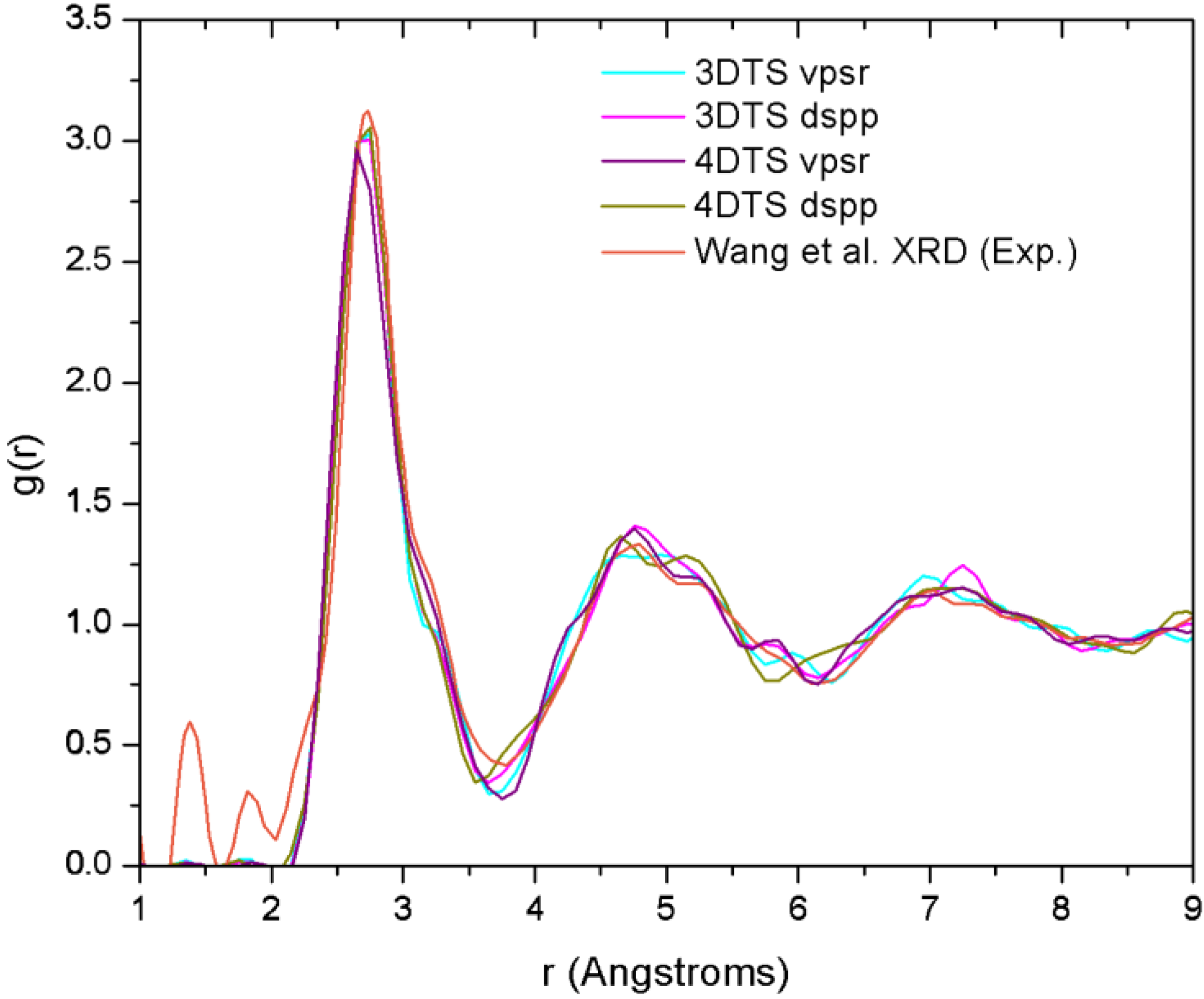
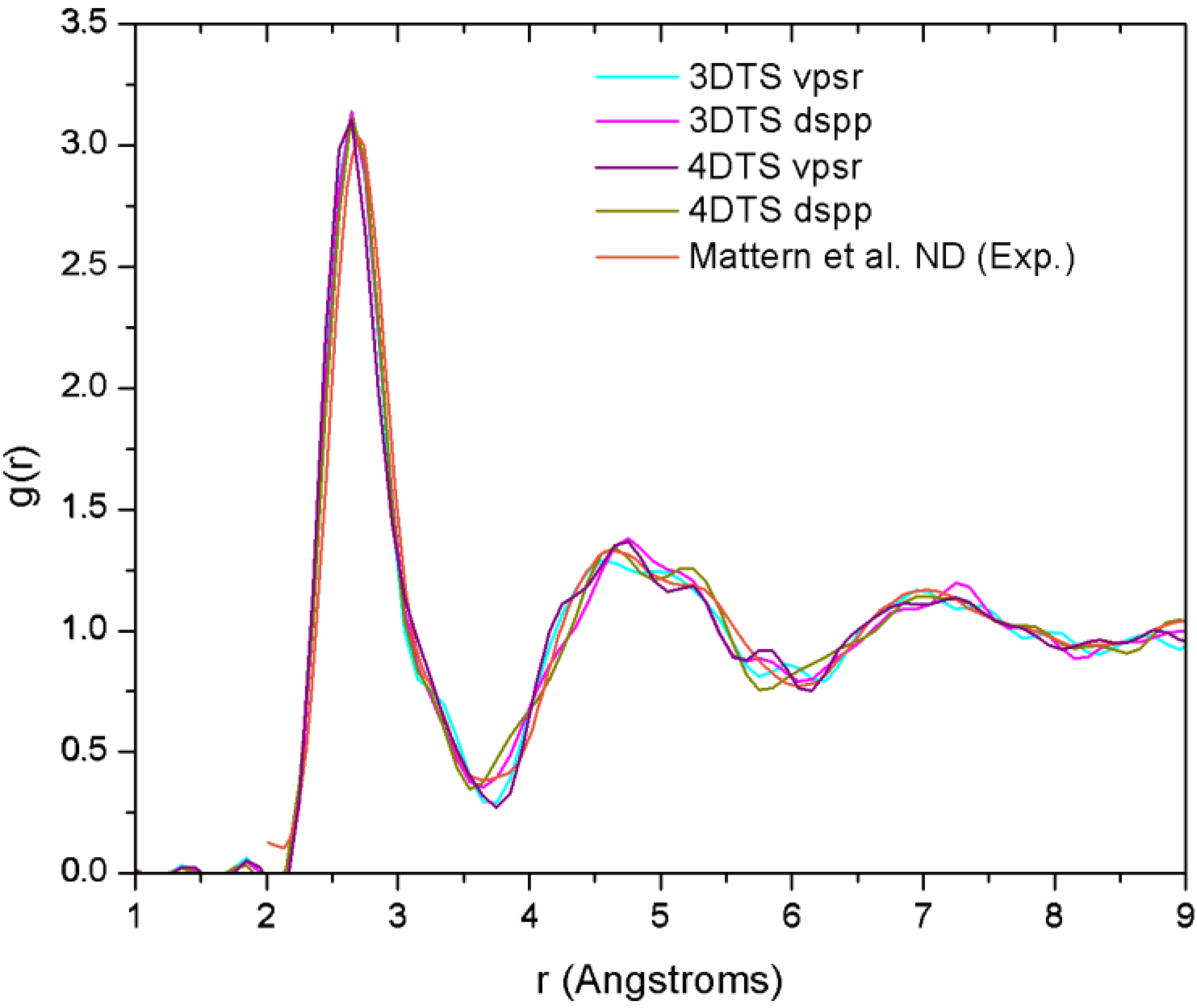
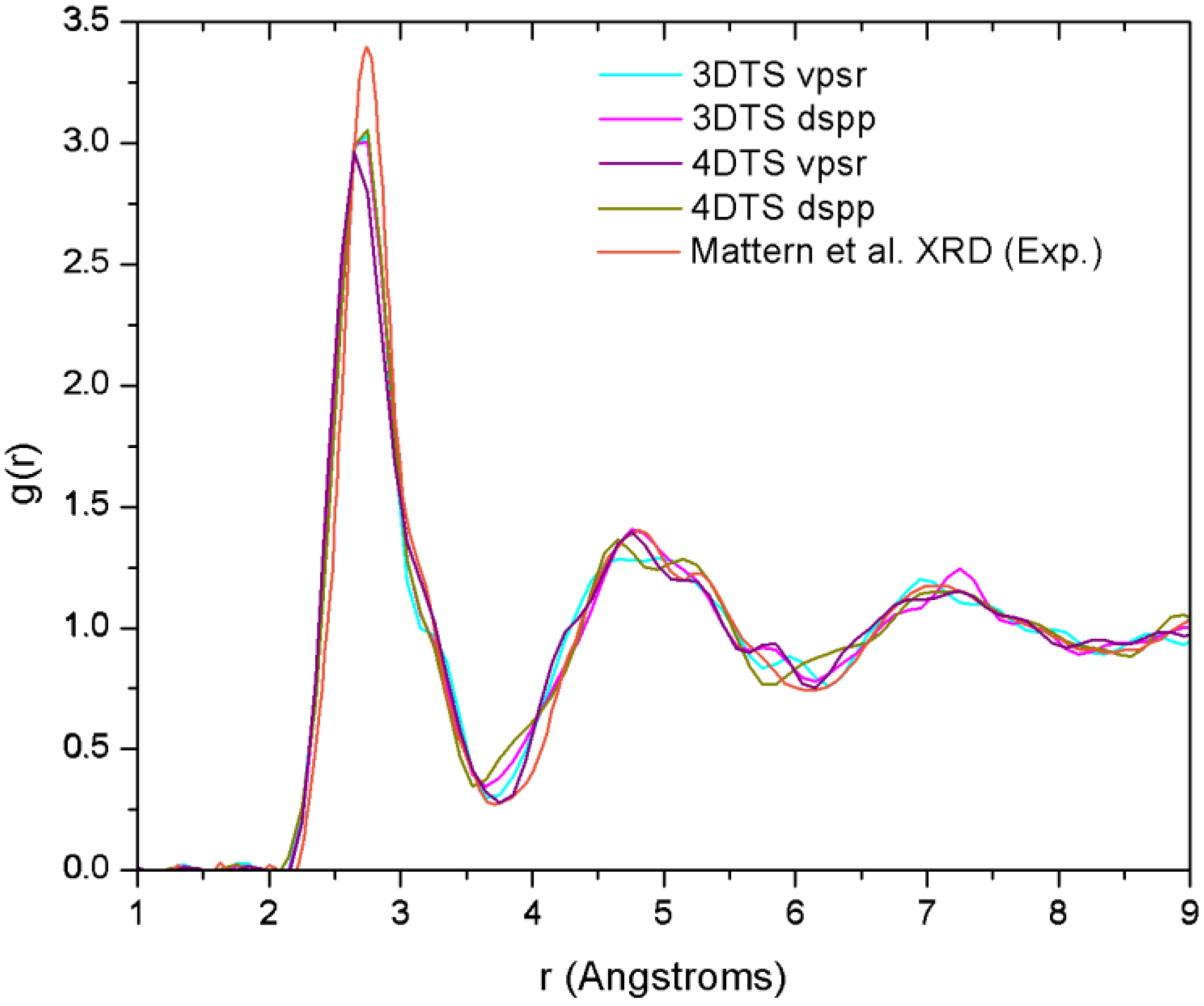
Finally, theoretical studies via simulational modeling exist for the glassy system CuZr,
g-CuZr, but not for the
amorphous CuZr,
a-CuZr. Wang
et al. performed some
ab initio molecular dynamics (AIMD) and reverse Monte Carlo studies (RMC) on a sample with the same concentration they had used for their X-ray diffraction (XRD) analysis [
29]. By comparing the AIMD with the XRD results, and RMC with extended X-ray absorption fine structure (EXAFS), they obtained the 3D structure of the samples from which they established the short range ordering. Likewise, Mattern
et al. carried out an RMC study to resolve the partial radial distribution functions (pRDFs) and consequently the coordination number [
30]. In addition to this, Jakse and Pasturel have reported an AIMD study for the Cu
64Zr
36 alloy [
31,
32]. They obtained a coordination number closer to the one found by Mattern and co-workers,
i.e., 13.1. It is noteworthy that these computational works have in common the use of plane waves as basis sets and a thermal procedure which leads to obtaining the metallic glass cooling from the melt. Also, neither the AIMD studies (except for the work of Jakse and Pasturel) nor the values reported experimentally establish clearly the method they used to compute the coordination numbers.
Another work on the
g-CuZr system, worth mentioning although it is not
ab initio, is the one done by Sun and coworkers where the Finnis-Sinclair potential was used [
33]. A complete study of the temperature effects on the structural evolutions and diffusivity of this alloy was conducted. In particular, the pair distribution functions and common-neighbor analysis were used to investigate the structural variations. Also, the mean square displacement and the self part of the van Hove function were calculated to evaluate the relaxation and transport properties. Finally, the critical temperature
Tc, a predicted glass transition temperature for Cu
60Zr
40 glass former, is calculated to be 1,008 K. An interesting challenge is to see what the results would be when using an
ab initio approach to this problem.
It should be clear that, before our incursion in the field, the ab initio amorphization of crystalline supercells was essentially based on the melt-and-quench procedure that had been in use in the literature since both classical and quantum computer simulations began to appear. It should also be clear, as mentioned before, that because the group IV semiconductors are metallic in the liquid state, melting them leads to the appearance of extra bonds in the atomic coordination of the liquid state so that when the supercells are quenched, some of this overcoordination is carried over into the disordered solid phase. The result is that the samples so generated are not fully representative of the experimental samples obtained by techniques other than melting and quenching.
Our contribution to the field of amorphous semiconductors is having devised a new method: the
undermelt-quench approach, which does not melt the crystalline supercells but heats them to just
below the melting temperature, avoiding the liquid state and consequently the overcoordination that occurs in this phase compared to the coordination of the crystalline or amorphous solid phases. Clearly, the code used was also a determining factor (F
ast S
tructure S
imulated A
nnealing by Molecular Simulations, Inc.) since it was devised for finding in a rapid manner the minimum energy structures of atomic aggregates after having disordered them
stochastically. Because of this, our results are more representative of the experimental ones and agree better with them. In
Section 3 we present this method, and its variants. Using our approach, in
Section 4 we review results that we obtained, and already published, for the following amorphous systems: SiH, SiN, CN, new results are reported for silicon carbide, for the chalcogenide binary alloy
a-GeSe
2, for aluminum-based alloys,
a-AlN,
a-AlSi, and for
a-CuZr. In
Section 5 we present some physical properties calculated for some of these amorphous systems. In particular, we report: the electronic density of states of the hydrogenated amorphous silicon samples; the optical gaps of the amorphous silicon nitrogen alloys using an approach
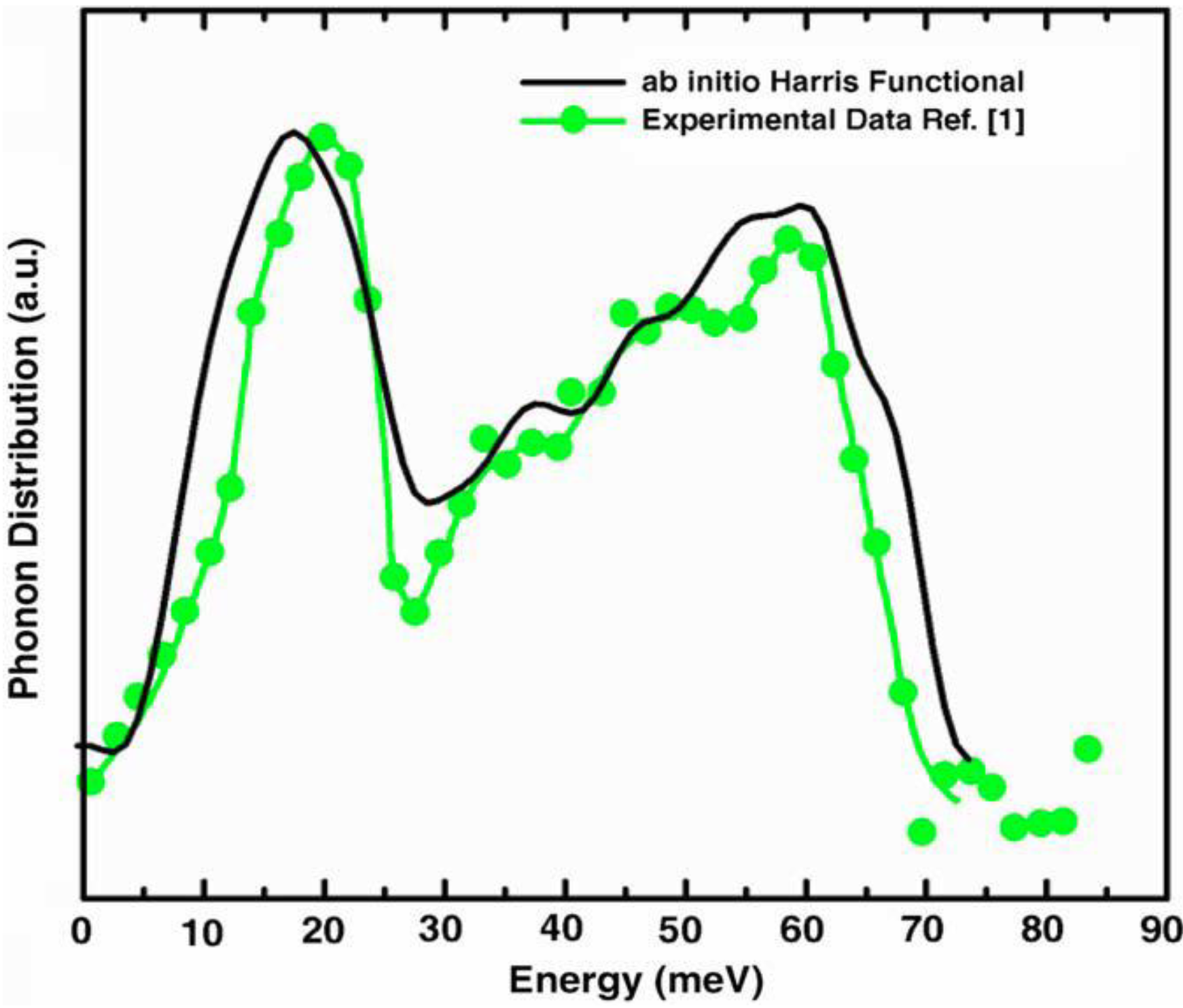
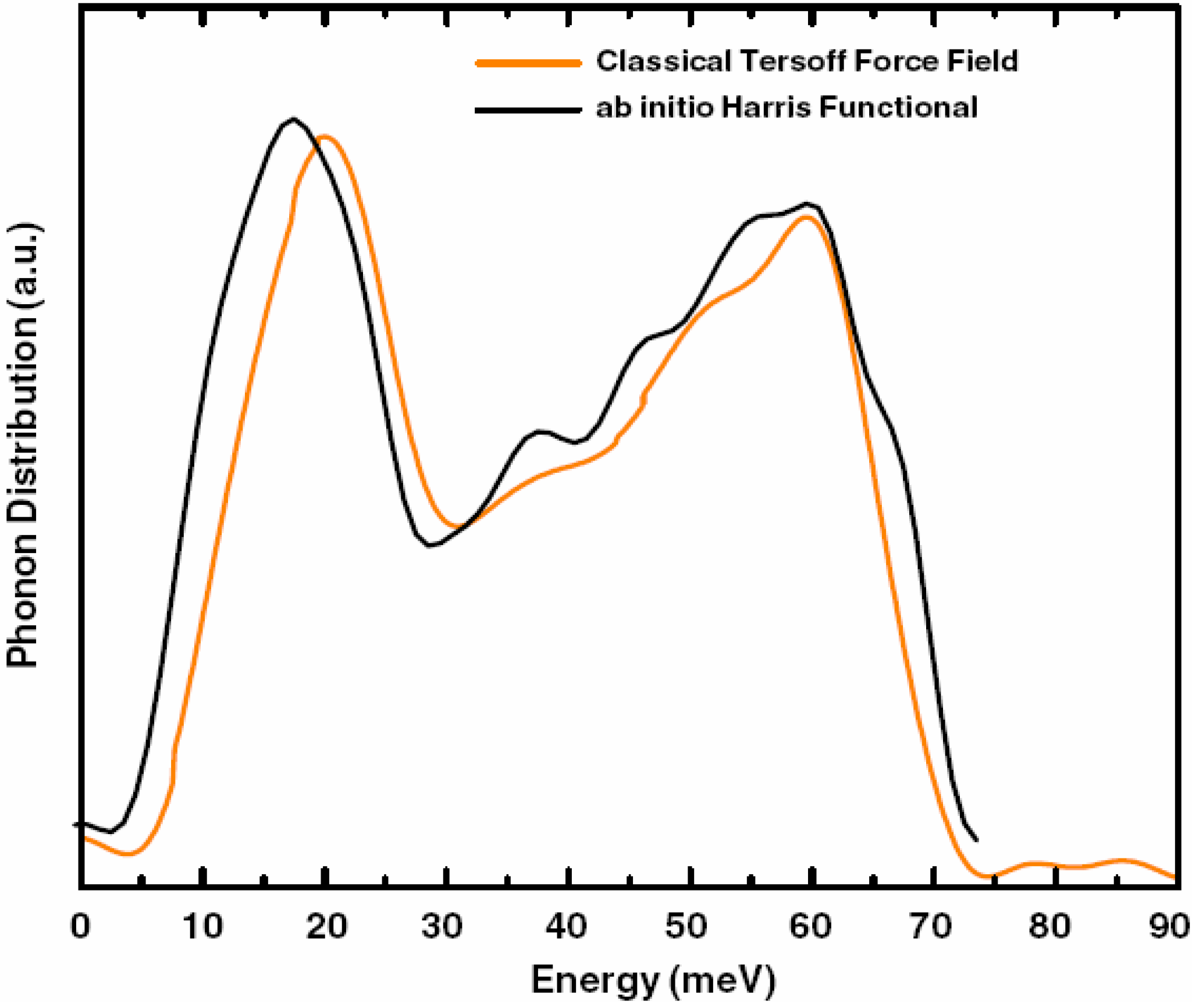
a la Tauc devised by us and the vDOS using a large, 216-atom amorphous silicon sample that lead to results in remarkable agreement with experiment.
Section 6 contains general conclusions.
It remains an issue as to how generally and how accurately an ab initio method that uses a relatively small supercell can describe the properties of amorphous materials both semiconducting and metallic. The present work addresses this issue and presents new alternative methods to computer generate disordered atomic topologies of alloys that agree very well with experiment, when available, as long as the defects present are not in the mesoscopic regime and as long as the properties studied are mainly dependent on the short and middle range order of the material.
3. The Undermelt-Quench Approach [1], Its Variants, the Method and the Bonding Criterion
The new amorphizing thermal procedure described here, was developed by one of the authors (AAV) while spending a sabbatical year (1997) at Molecular Simulations Inc, MSI, (now known as Accelrys, Inc.), in San Diego, CA, USA, since their code F
ast S
tructure S
imulated A
nnealing (F
ast for short) [
34] seemed appropriate to generate bulk amorphous structures. The code was developed by John Harris and collaborators to fast find the structure of atom aggregates, but the periodic boundary conditions incorporated, and the initial randomness in the atomic velocity distribution gave it a broader applicability [
35].
The
ab initio methods attempt to answer questions from first principles and are generally applicable without adjustment of parameters. Since these methods are very demanding on computer resources they are presently limited to handling a relatively small number of atoms;
i.e., to relatively small supercells. There is also a notable amount of ‘approximations’ that depend on the particular method used, or on the type of wave functions, or whether full core or pseudopotentials are incorporated in the calculations, or the ‘parameters’ that appear when using approximations for the exchange-correlation interaction, to mention those that occur most commonly and that determine the quality of the approximation. Within the first principles methods one has the option of using a recursive self-consistent approach [
36,
37] like the one used in CPMD, or using a linear combination of atomic orbitals, LCAO, non self-consistent approach employing a functional like the one developed by Harris [
38] and implemented in the LHMD. In general the self-consistent method is more computer-demanding than the non self-consistent one.
F
ast, the code that we used at the beginning is, in short, a density functional code based on the Harris functional, which generates energies and forces faster than traditional Kohn-Sham functional methods, since the code is not self-consistent. However, it is not always possible to use the Harris functional since, until recently, it was unable to handle partially filled d-band materials. New developments that shall be mentioned later on seem to remedy this situation. In all our calculations we have used the LDA with the parameterization of Vosko, Wilk and Nusair (VWN) [
39]. Some of our calculations are performed on an all electron basis, some use pseudopotentials, particularly when heavy atoms are considered. The valence orbitals are described either viaminimal or standard basis sets of ‘finite-range’ atomic orbitals with a cutoff radius chosen as a compromise between computational cost and accuracy, and in general are different for the various materials. F
ast, and later developments of the Code, can handle three types of basis sets: (i) minimal, consisting of the atomic orbitals occupied in the neutral atom;
sp-valence type; (ii) standard, broadly equivalent to a Double Numeric basis set, DN and (iii) enhanced, broadly equivalent to a Double Numeric set together with Polarization functions, DNP. The computation scales with a high power of the cutoff radius, because the time-limiting factor is the number of three-center integrals that have to be carried out [
40,
41]. The linear combination of atomic orbitals utilized makes the minimum energy atomic structures generated very close to experiment. The interatomic distances fall within 1% of the experimental values for a large variety of small molecules [
34,
35]. F
ast uses optimization techniques through a force generator to allow simulated annealing/molecular-dynamics studies with quantum force calculations. The forces are calculated using rigorous formal derivatives of the expression for the energy in the Harris functional, as discussed by Lin and Harris [
42]. Three-center integrals were performed using the weight-function method of Delley [
40,
41] with correction for the dependence of the mesh on the nuclear coordinates. This is the essence of F
ast, the code developed by Harris
et al. for MSI.
Fast initially disrupts the atomic aggregates stochastically, then heats them using molecular dynamics to foster the rearrangement of their atomic constituents and finally cools them to what would be the structure of minimum energy, at least locally. Rather than using this code to find the minimum-energy atomic structure of a cell, we use it to generate random structures from an originally crystalline supercell with periodic boundary conditions.
The difference between our approach and previous techniques resides in the heating procedure and in the code used. It is clear from previous work that quenching from a melt, or from partially melted samples, generates structures that only partially resemble the local arrangement of atoms in the amorphous material; therefore, we took a different route. A corresponding crystalline supercell with the chosen number of atoms and the same density as the amorphous phase is subjected to the following process. The supercell is heated from 300 K to
just below the corresponding melting temperature (the liquidus temperature in binary systems) in 100 steps, with a time step three to four times the default value. It is then immediately cooled down to 0 K in the necessary number of steps required by the cooling rate which is the same in magnitude as the heating rate. This is the proper amorphizing stage. Physical masses of the atoms are used throughout and this allows realistic atomic diffusive processes to occur in the system, and lets them move within each periodic supercell. Once this first stage is complete, several variants have evolved. In the original process each supercell is subjected to six annealing cycles at a temperature dictated by experiment, with intermediate quenching processes, in order to release the stresses generated. Finally, a geometry optimization is carried out. Other variants have been developed to describe specific systems. If the atoms are heavy, like in the chalcogenides or the BMG-like systems, the stress releasing cycles are reduced to one, or none, to minimize computational times but the structure optimization process stays. Finally, a plateau is sometimes introduced at constant temperature either below the melting point or above the melting point, depending on the material to be disordered and the process desired,
i.e., this constant temperature is the value at which the system is maintained long enough to foster the amorphizing or the liquefying procedure. Then the system is cooled rapidly, usually in one step, or maintained at this temperature. The original, amorphizing process will generically be referred to as the
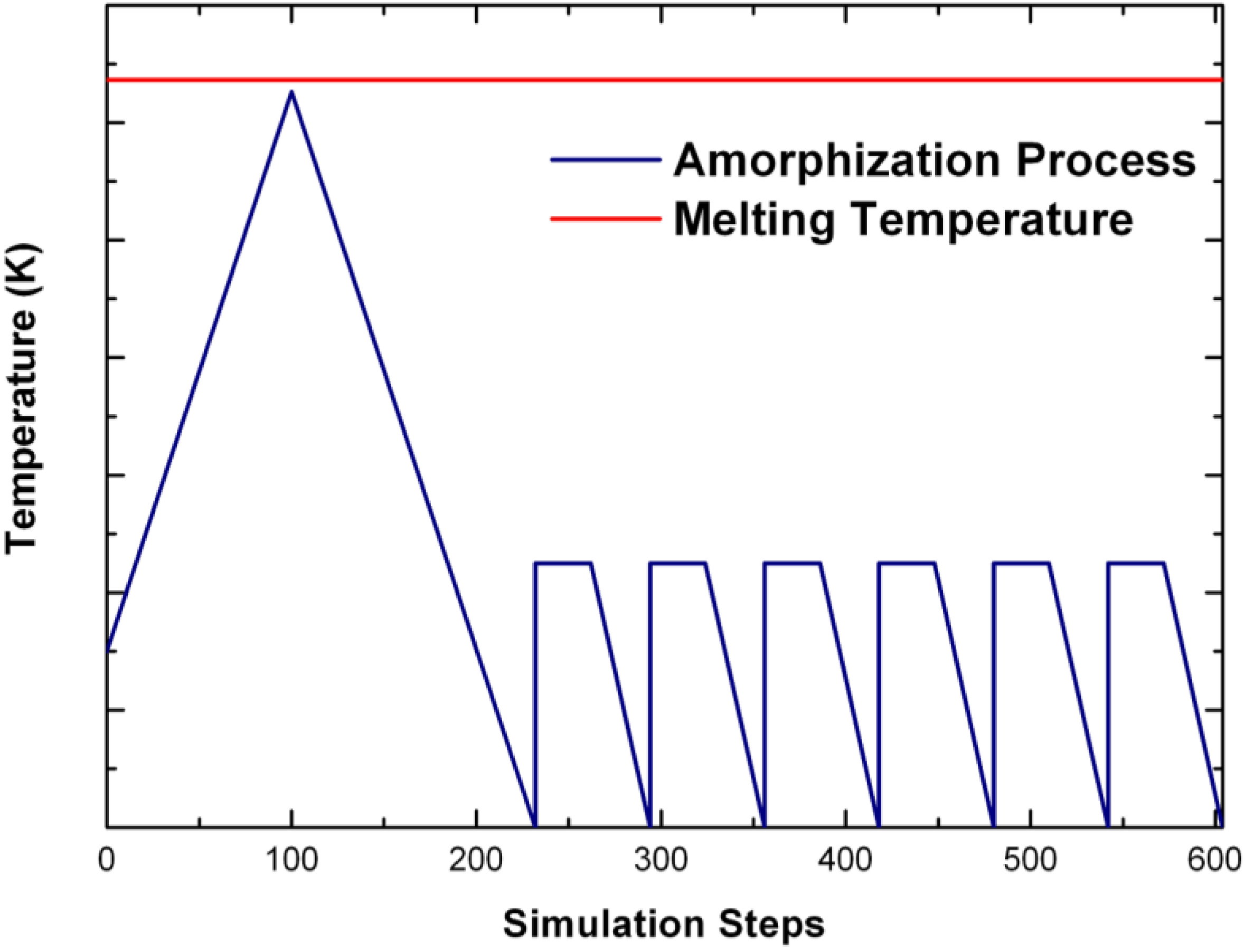
undermelt-quench approach with its variants. The original process is shown schematically in
Figure 1.
Figure 1.
Schematic representation of the original undermelt-quench approach. The first process (triangular) is the randomizing part; the rest are the stress-relieving cycles. At the end a geometry optimization is carried out. The temperatures considered are specific to each material. For variants of this process see text.
Figure 1.
Schematic representation of the original undermelt-quench approach. The first process (triangular) is the randomizing part; the rest are the stress-relieving cycles. At the end a geometry optimization is carried out. The temperatures considered are specific to each material. For variants of this process see text.
Since F
ast initially disrupts the atomic aggregates randomly, the probability of returning to a crystalline structure after the initial heating and cooling (amorphizing) cycle is nil for semiconductors. One might think that our approach may be dependent on the initial crystalline structures used, but we have demonstrated that the RDFs generated starting with diamond-like low density carbon structures are practically indistinguishable from those obtained from initial hexagonal or rhombohedral structures [
43,
44].
In the present work the amorphizing procedure was performed self-consistently for CuZr, since the Harris functional lacks the tools to deal with partially filled d-band metals. However, recent developments indicate the possibility of generalizing the Harris functional to deal with these metals [
45]. Sometimes energy calculations were carried out using both F
ast and the full Kohn-Sham DFT approach [
36,
37] implemented in the
ab initio DM
ol3 commercial code [
40,
41,
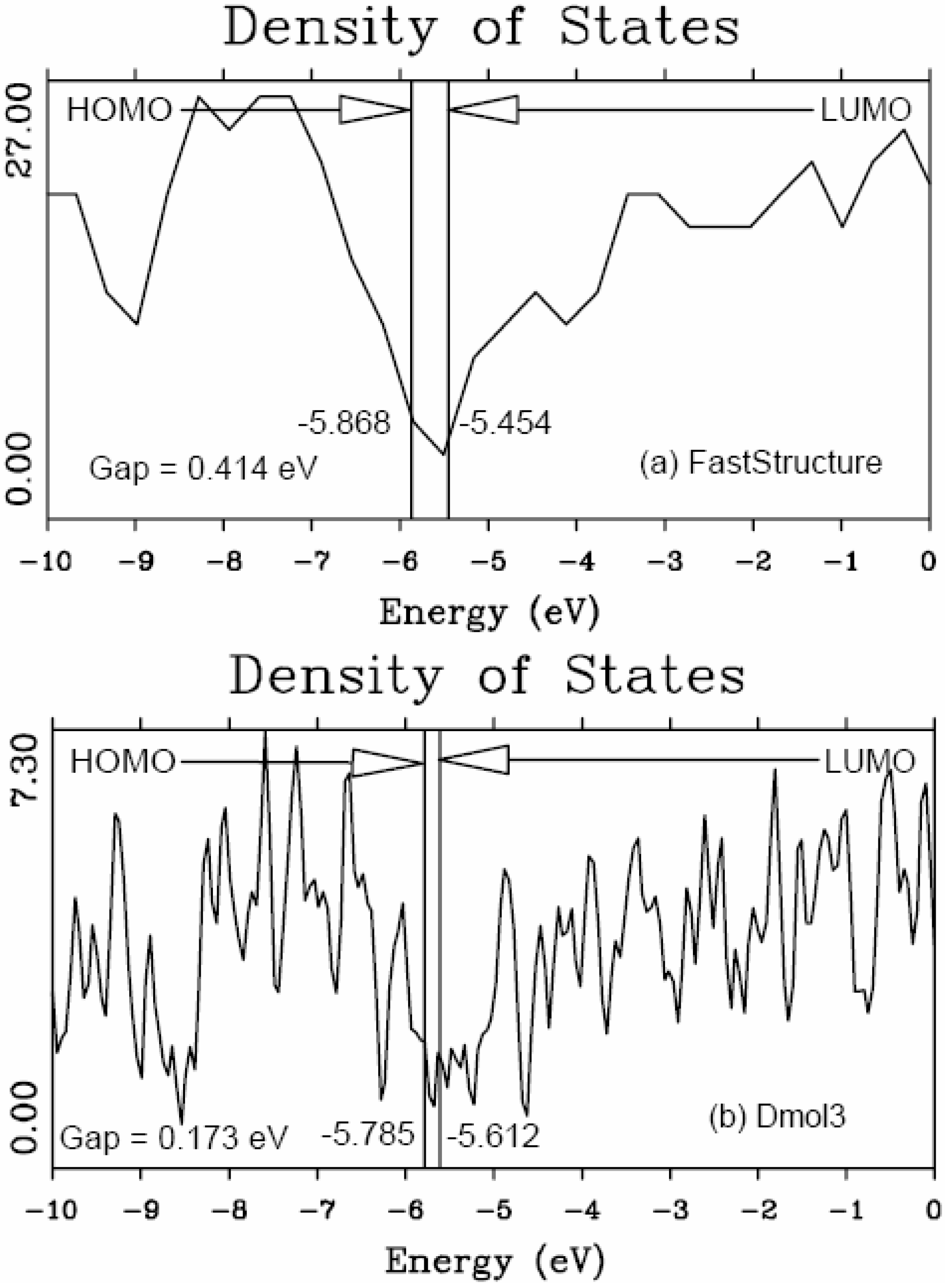
46] to obtain the eDOS curves of the final amorphous atomic structures.
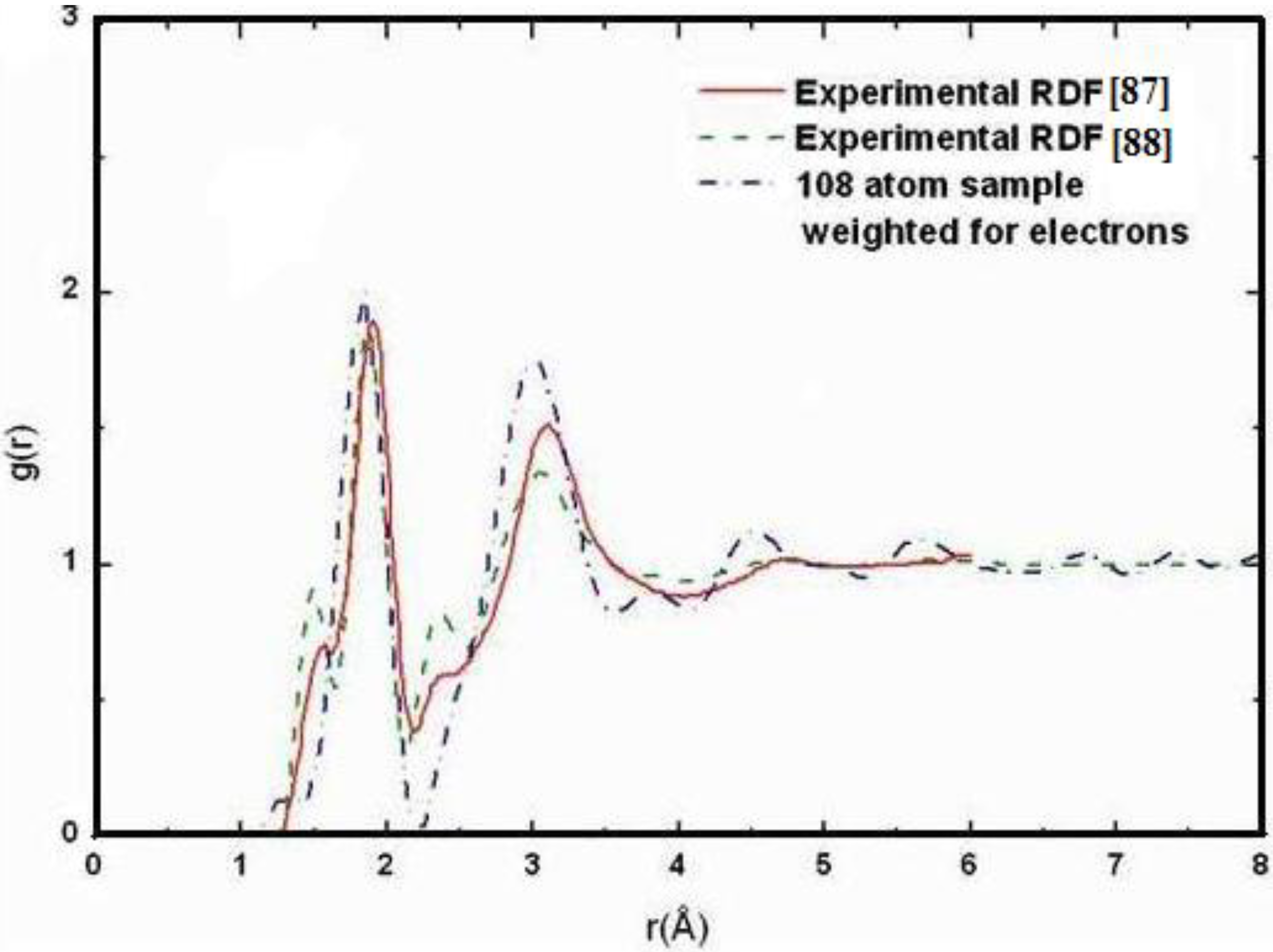
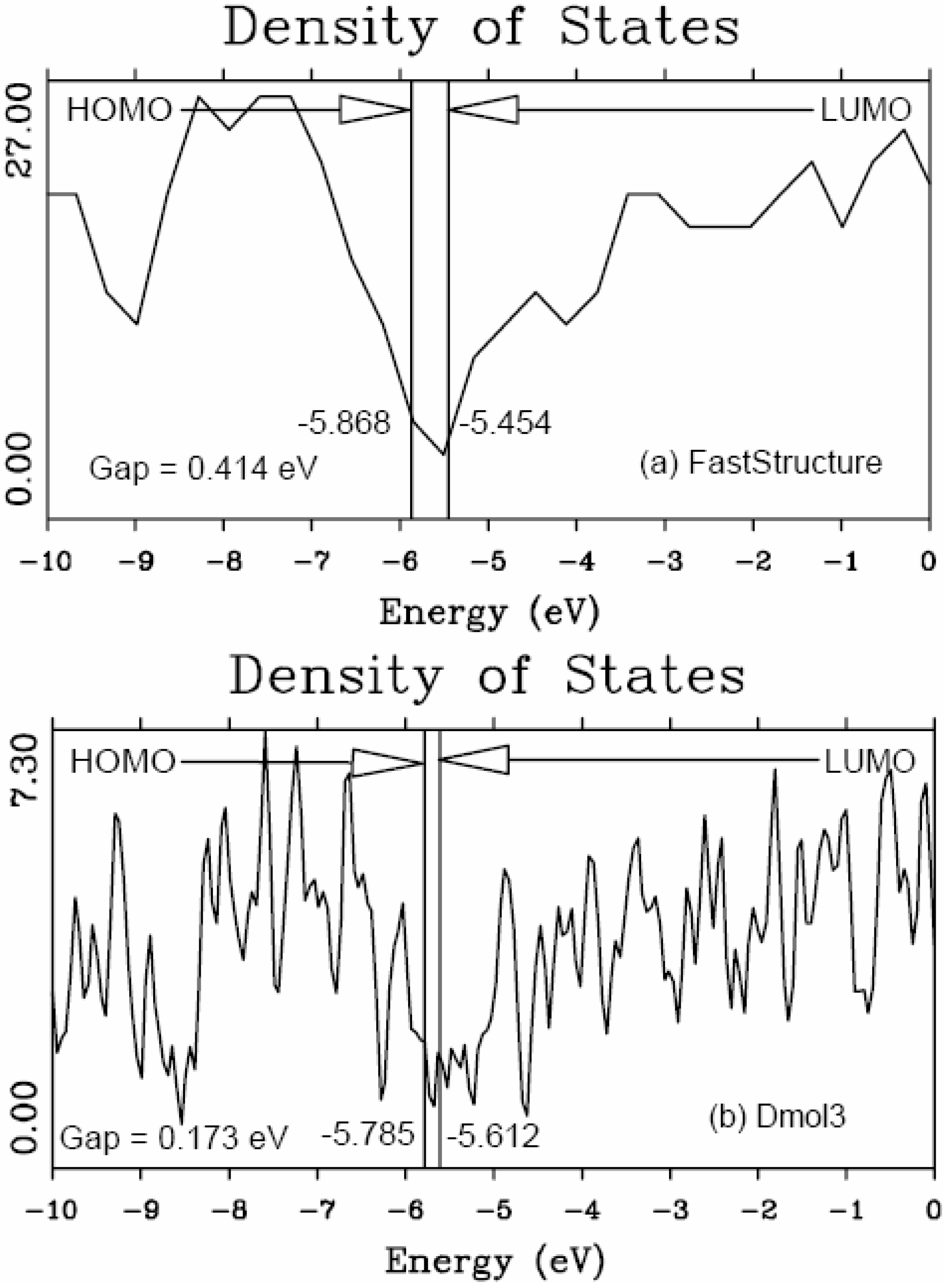
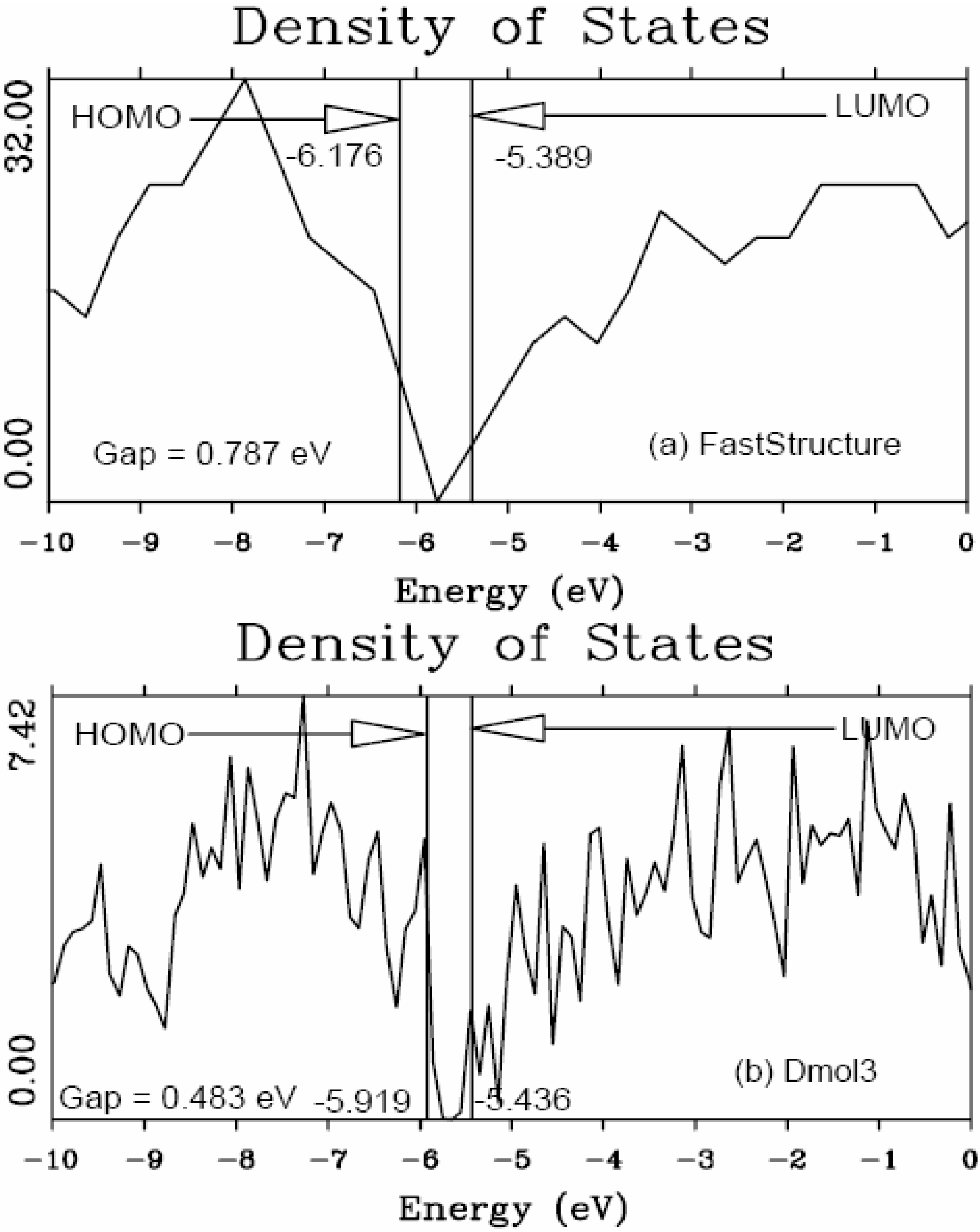
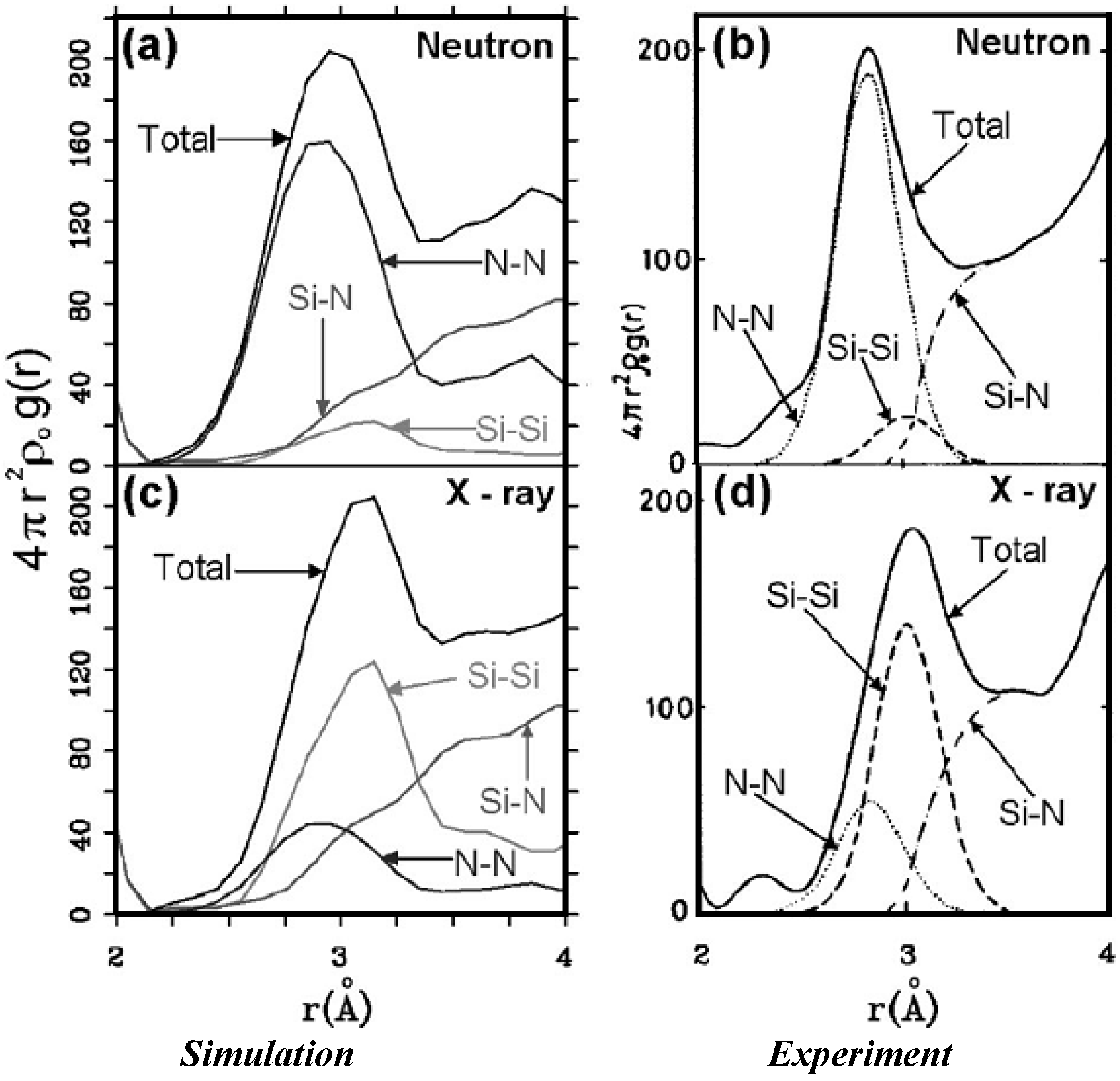
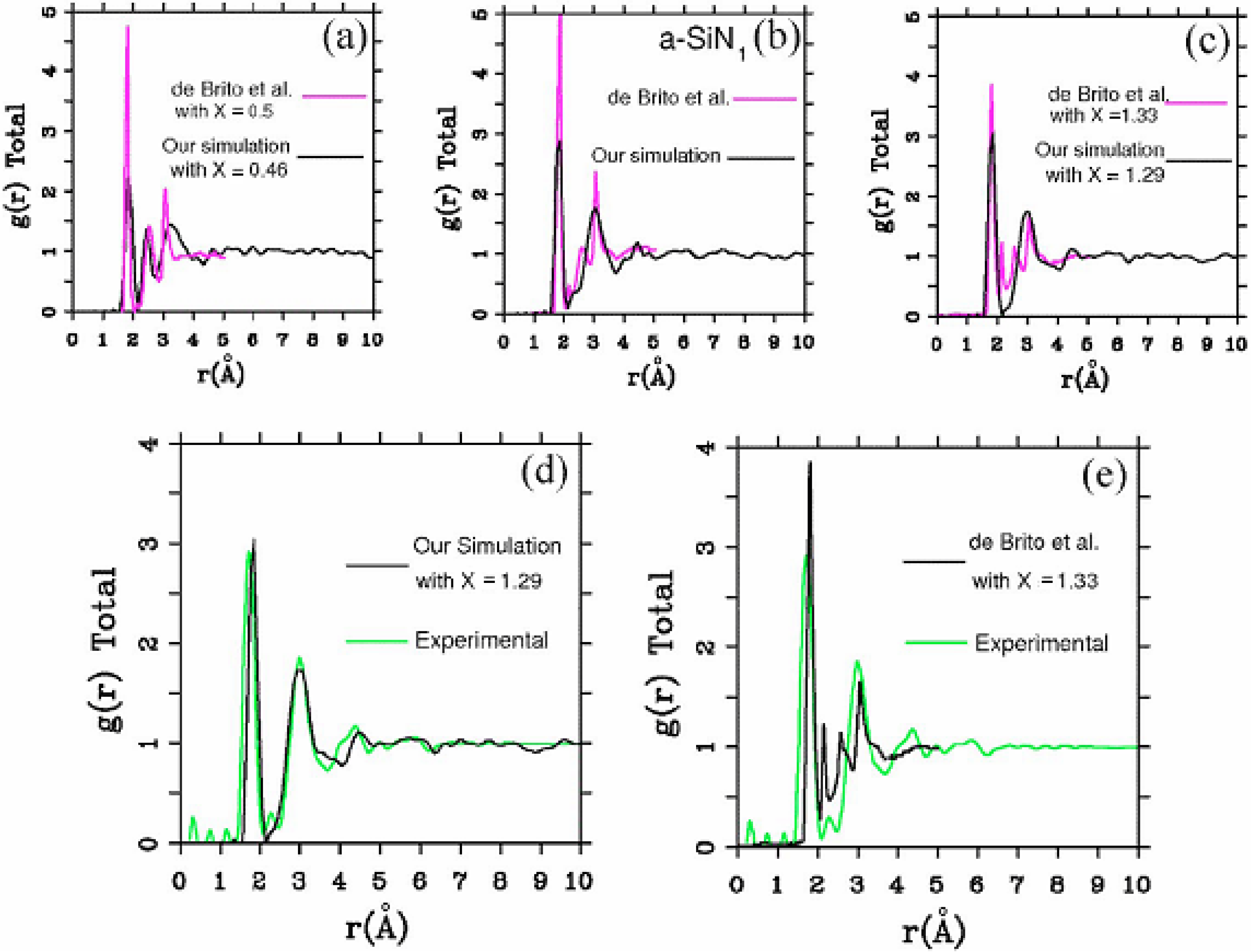
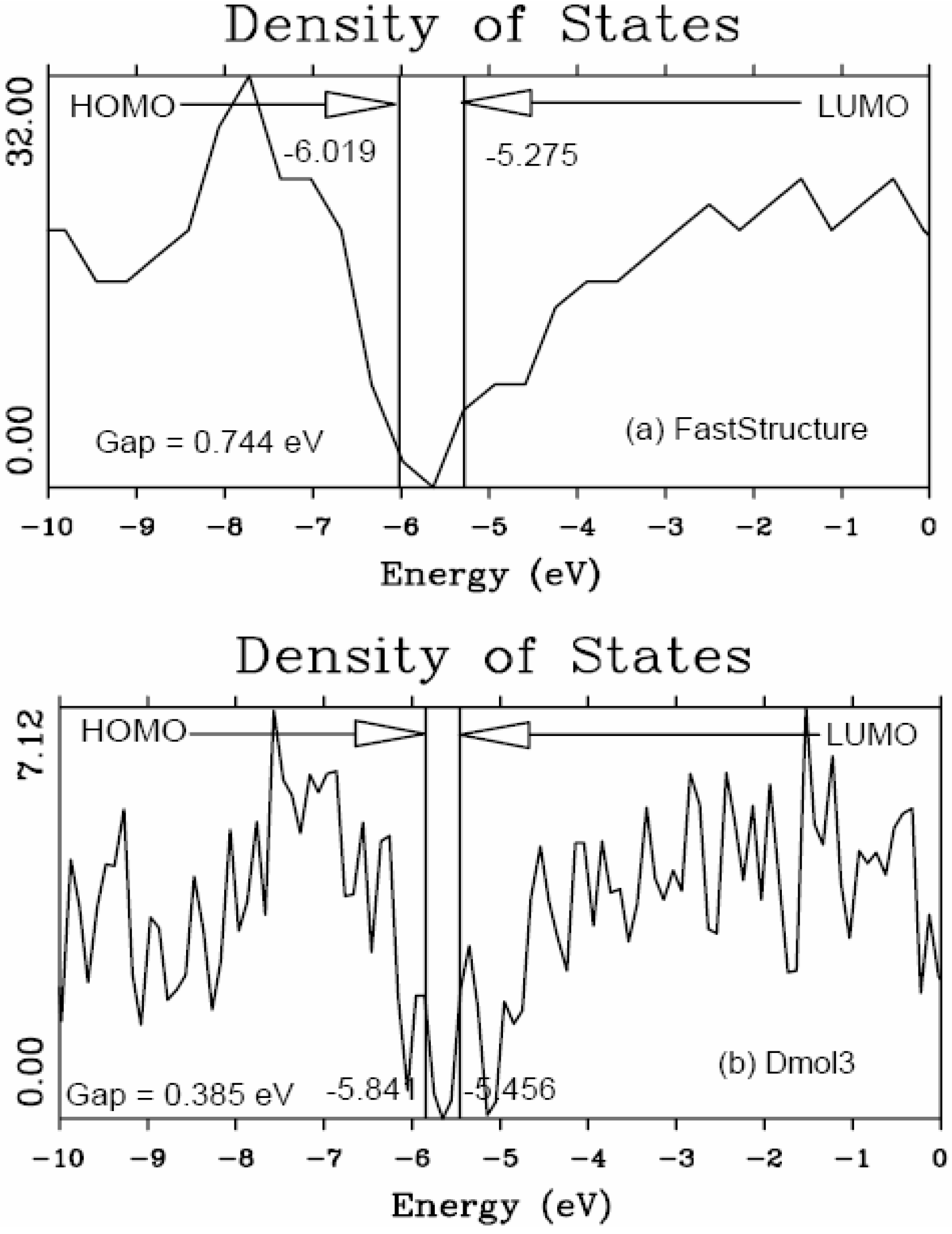
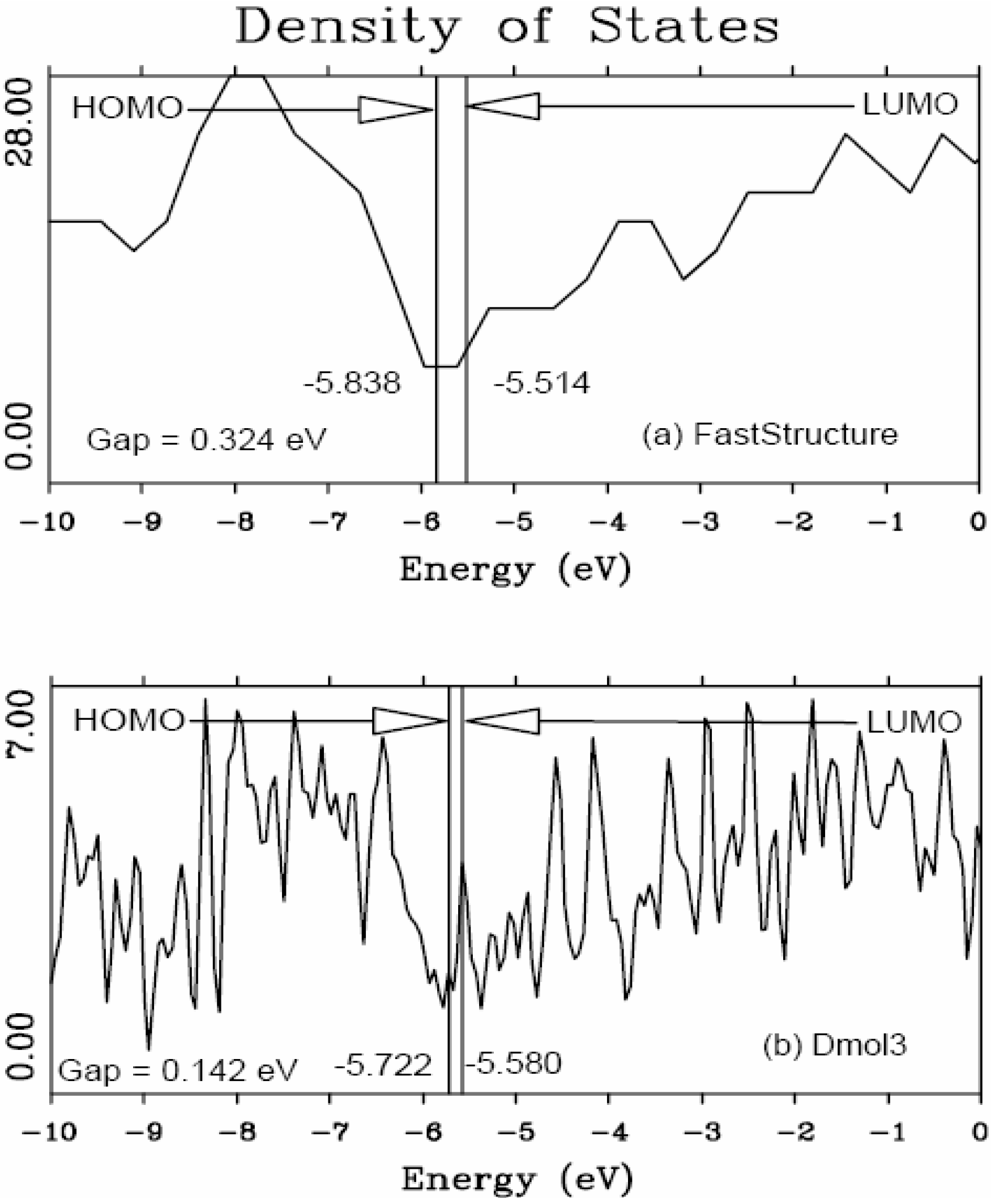
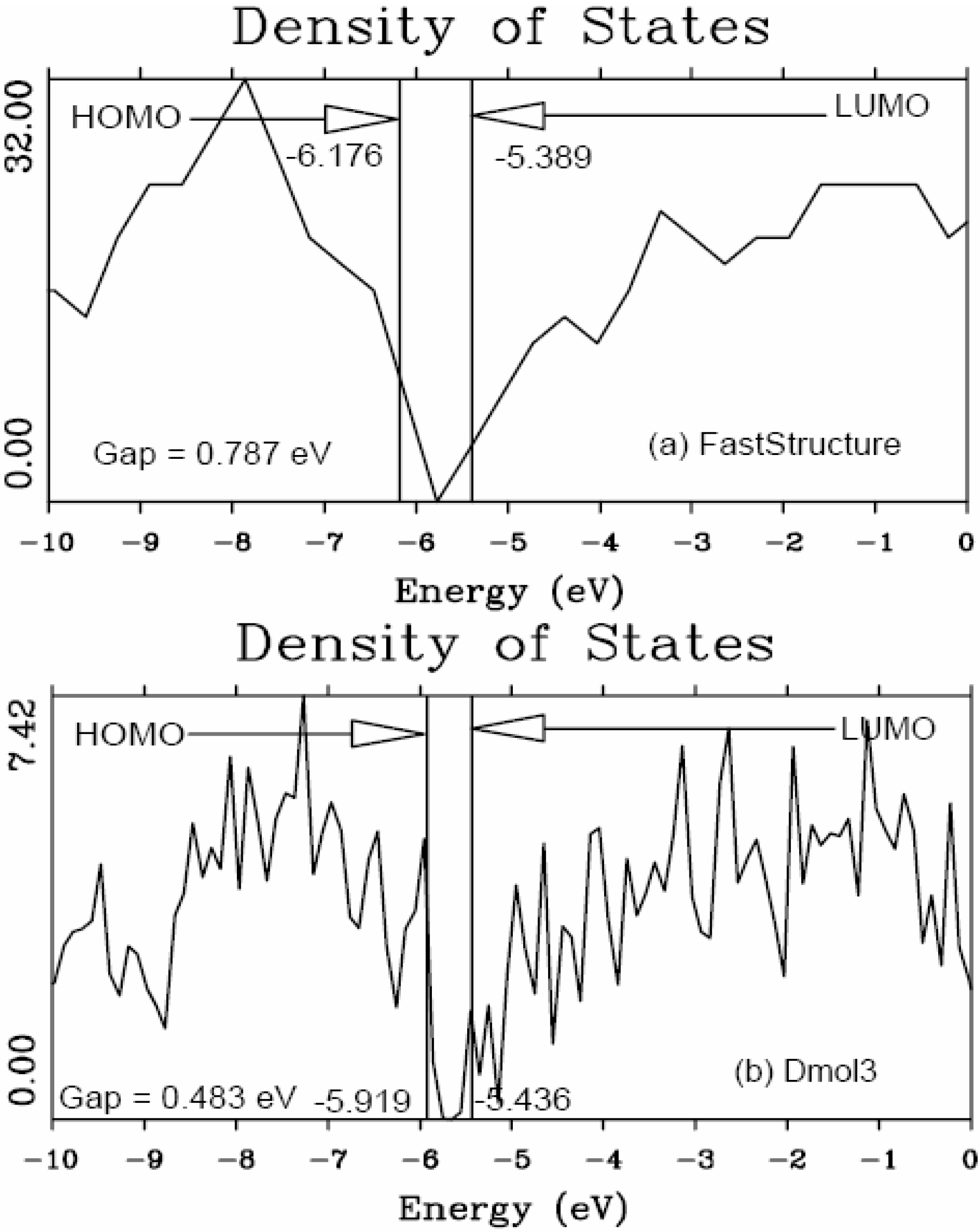
It should be clear from the outset that our computational processes do not pretend to mimic the production of such materials, but only to generate random networks, using ab initio techniques, that lead to RDFs that agree with experiment; this agreement allows us to study their topological, electronic, optical and vibrational properties.
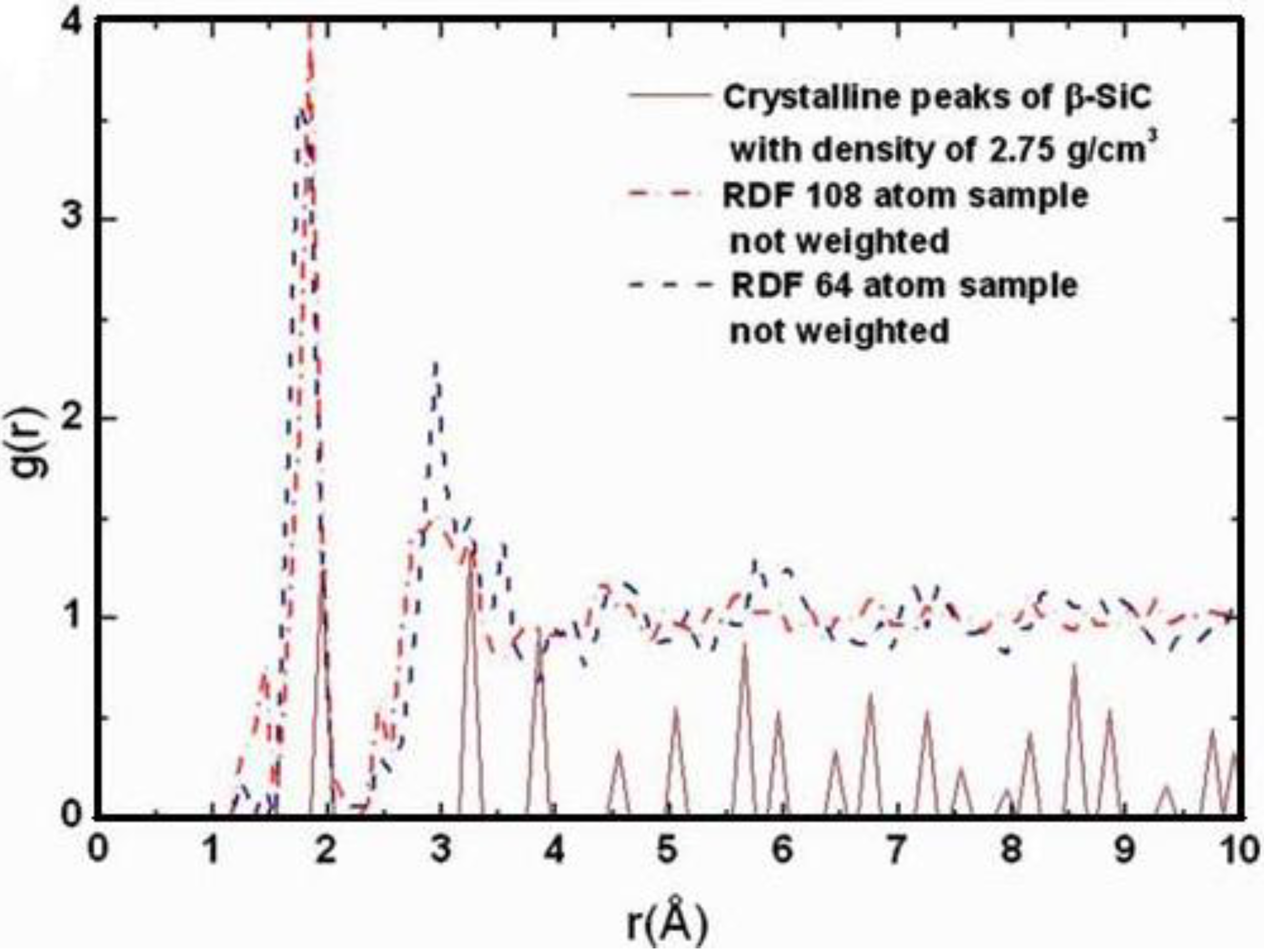
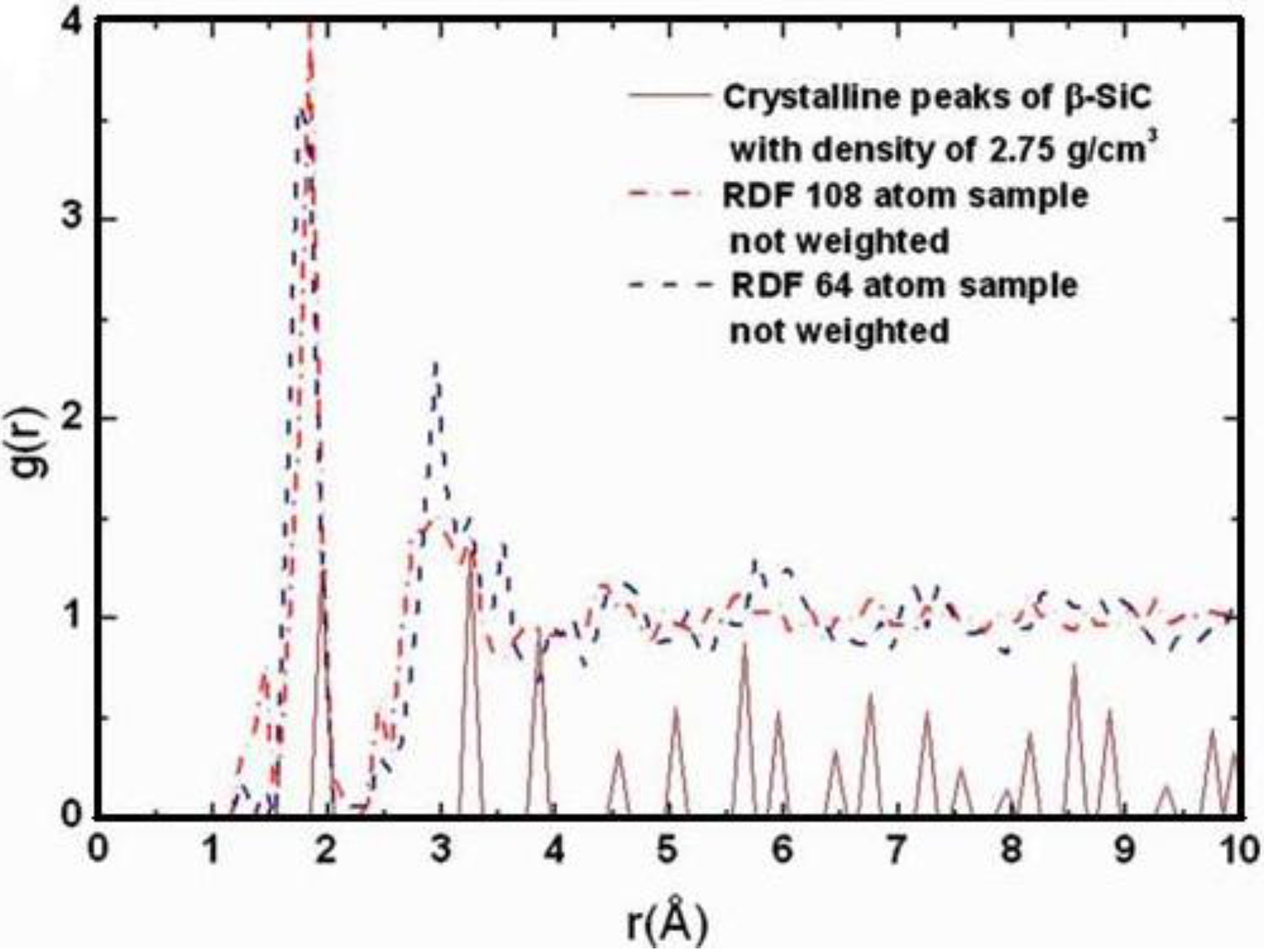
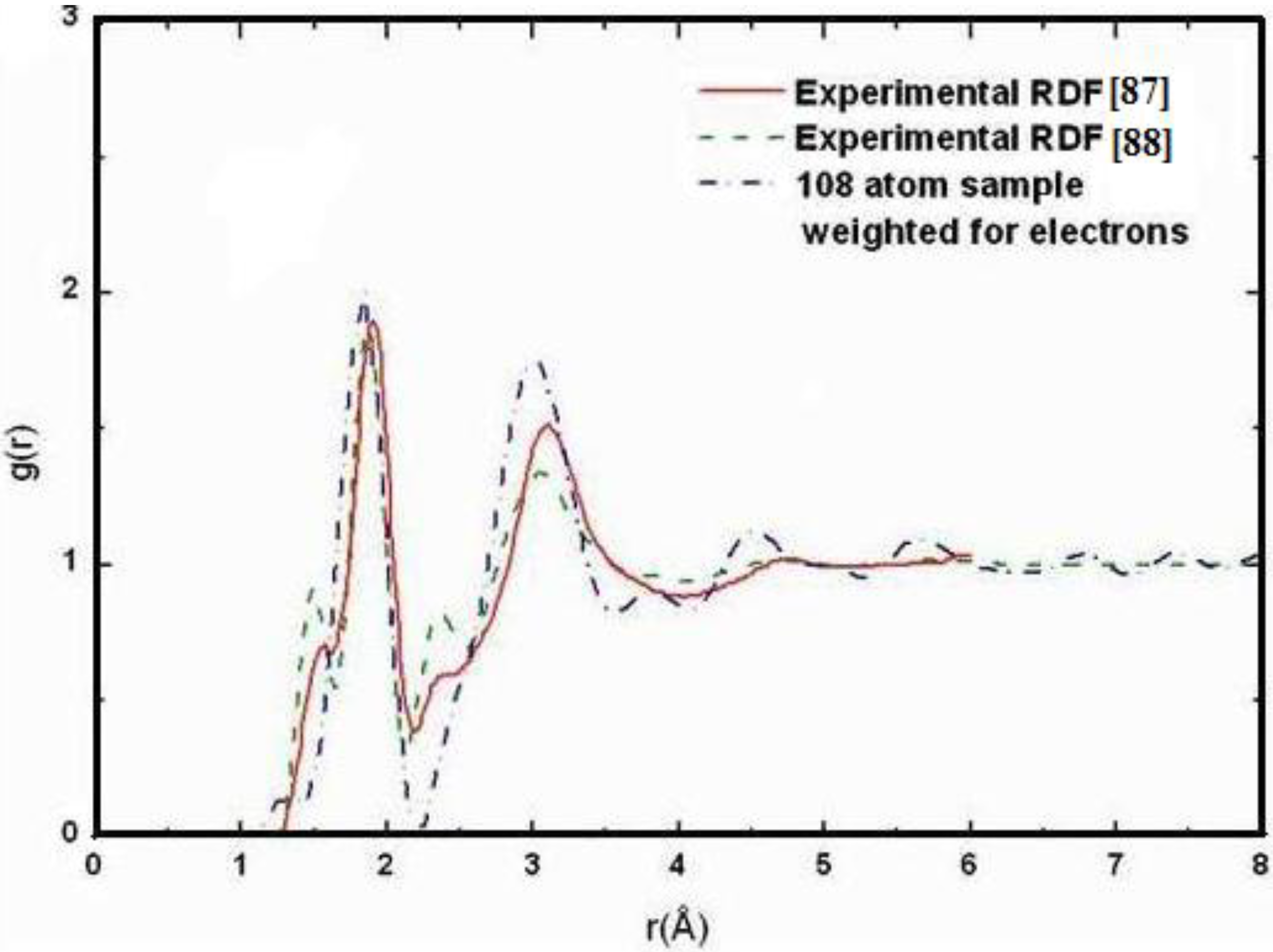
Also, since we use at least 108 atoms almost everywhere, we have Fourier-smoothed the RDFs to have adequate curves to allow comparison with experiment. The number of atoms used is low and this leads to statistical fluctuations that are not representative of the bulk.
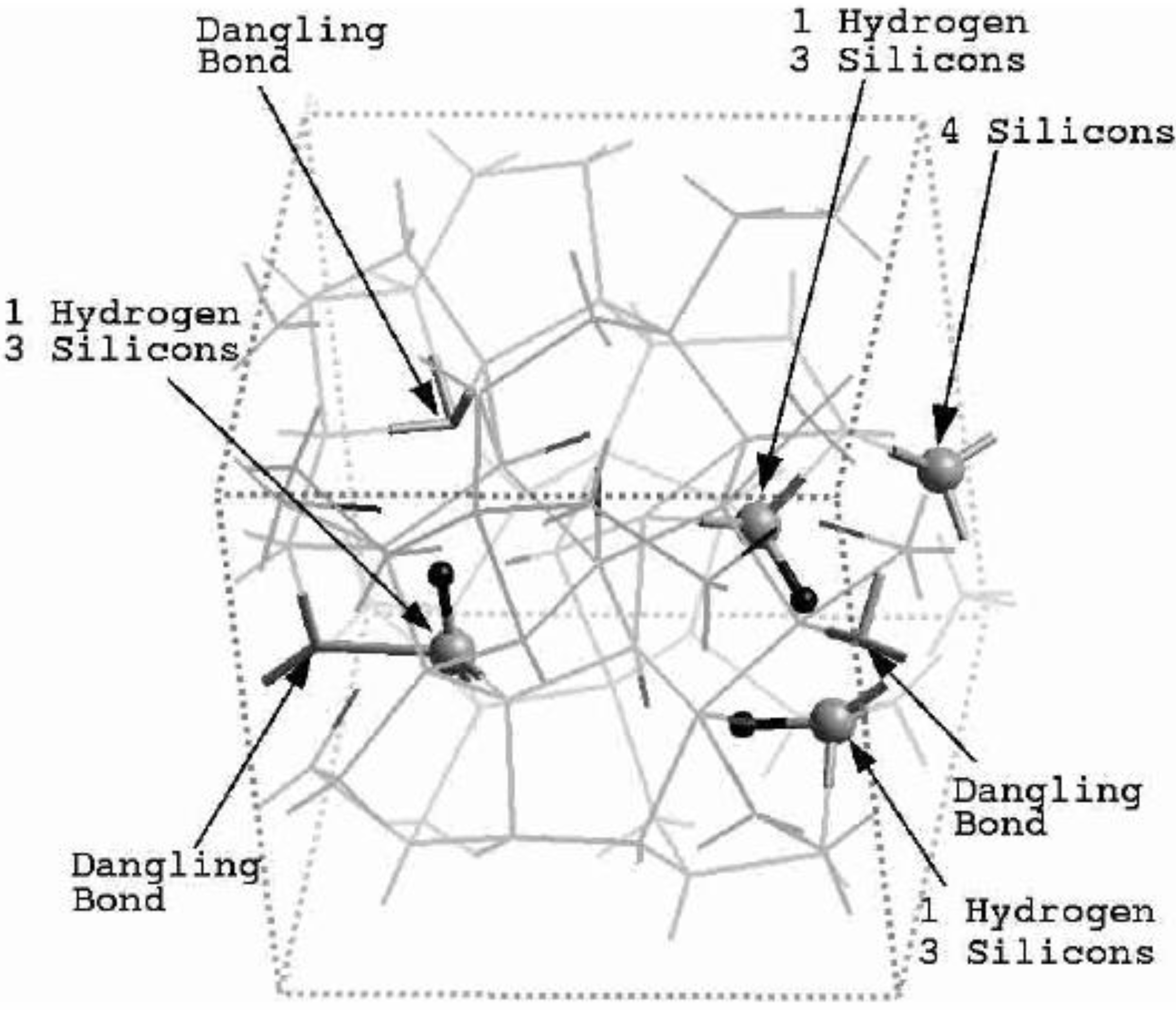
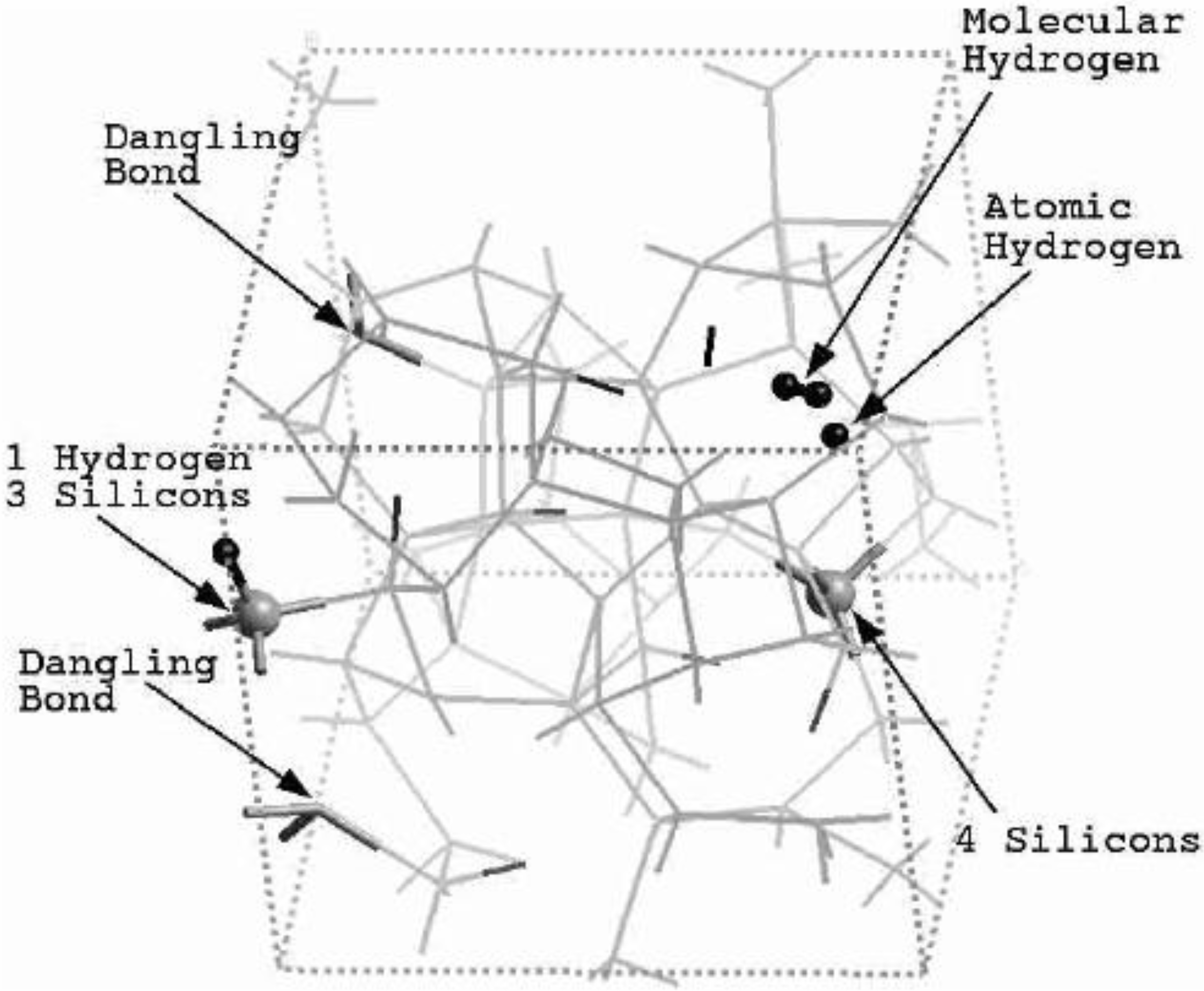
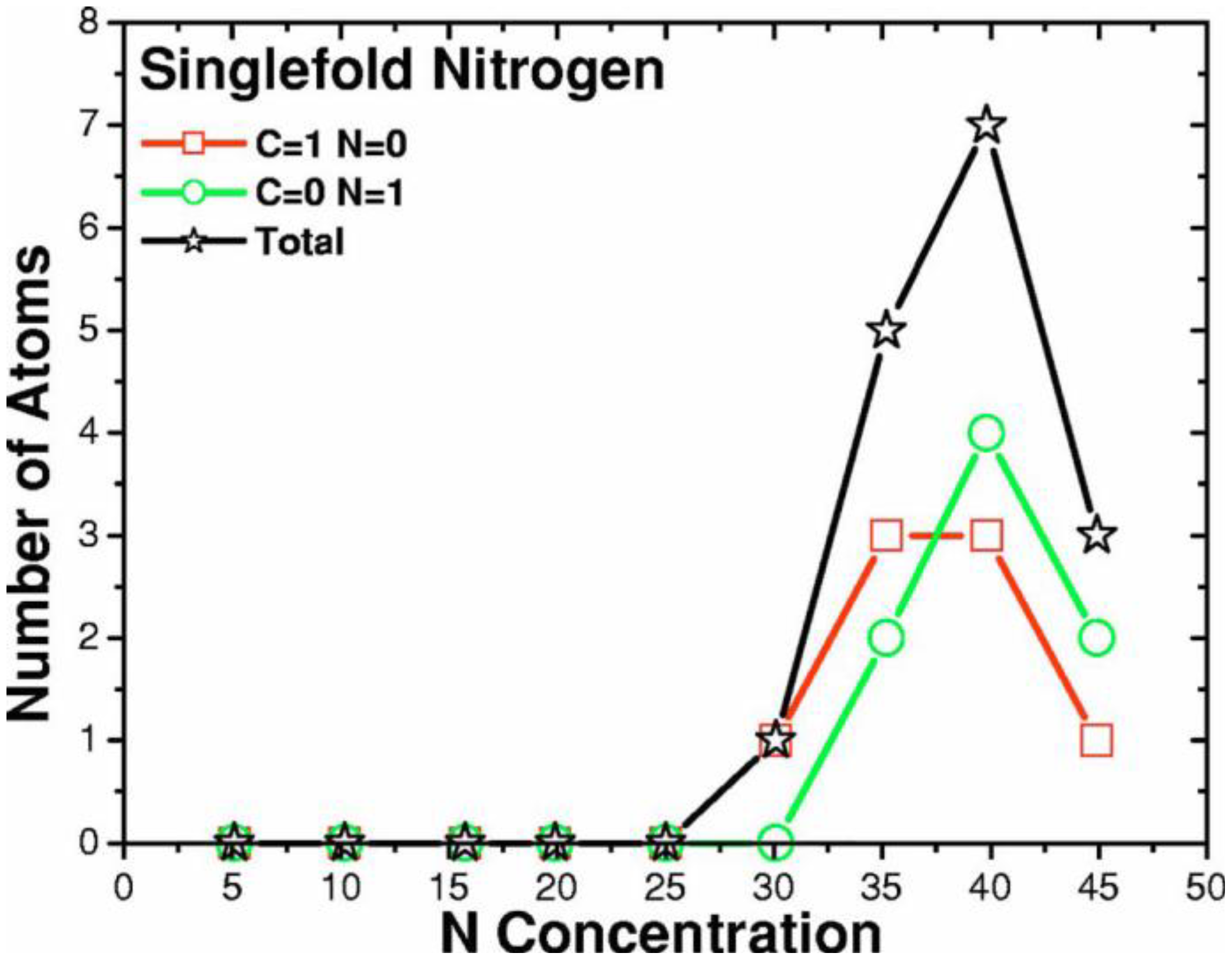
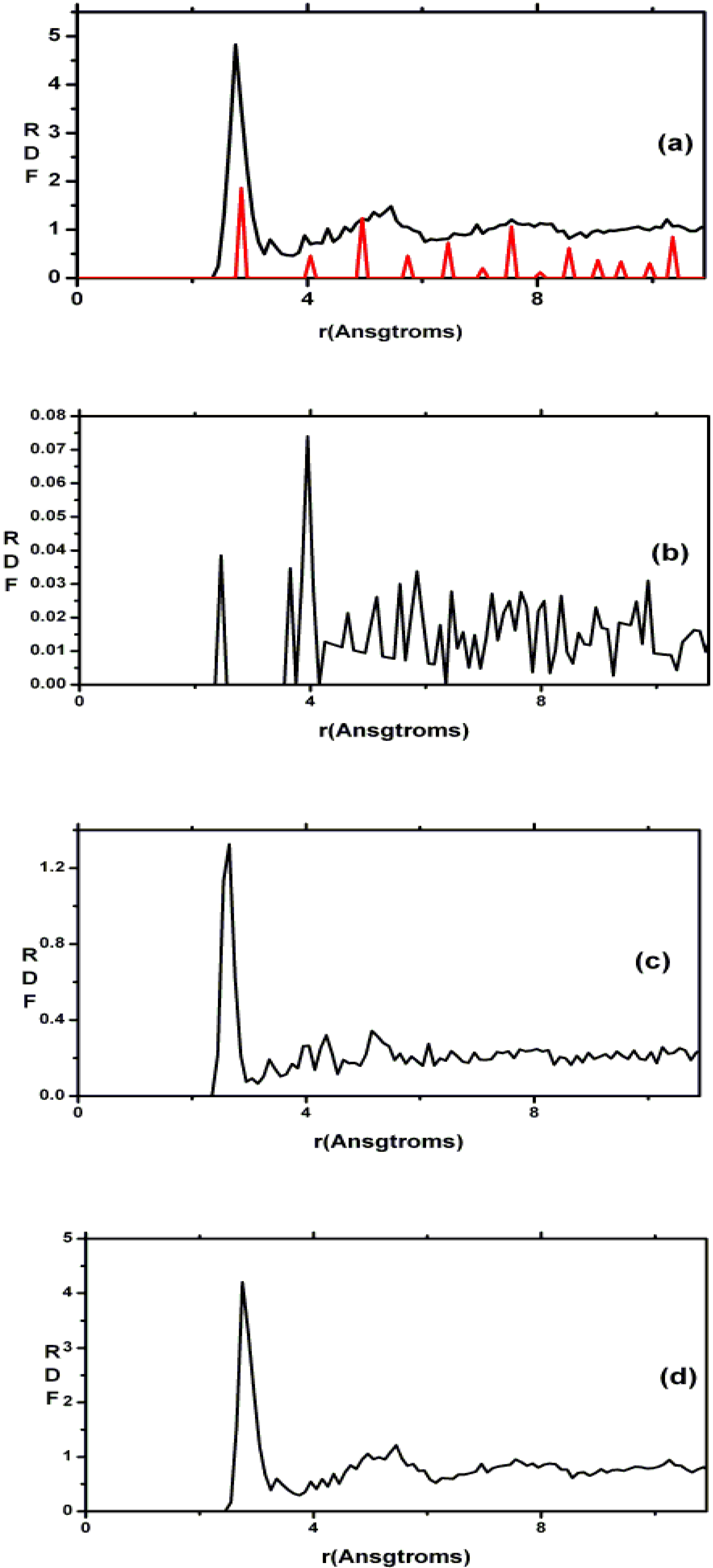
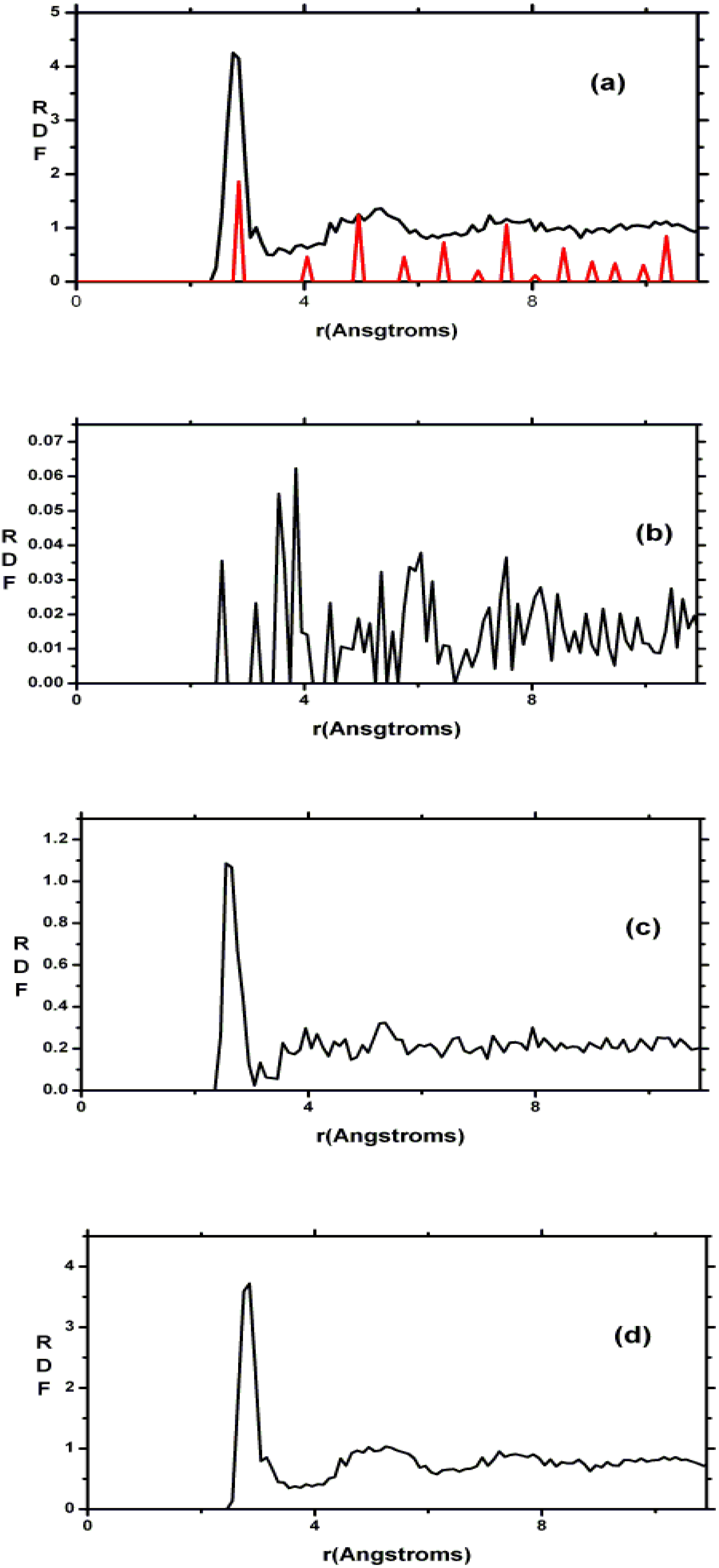
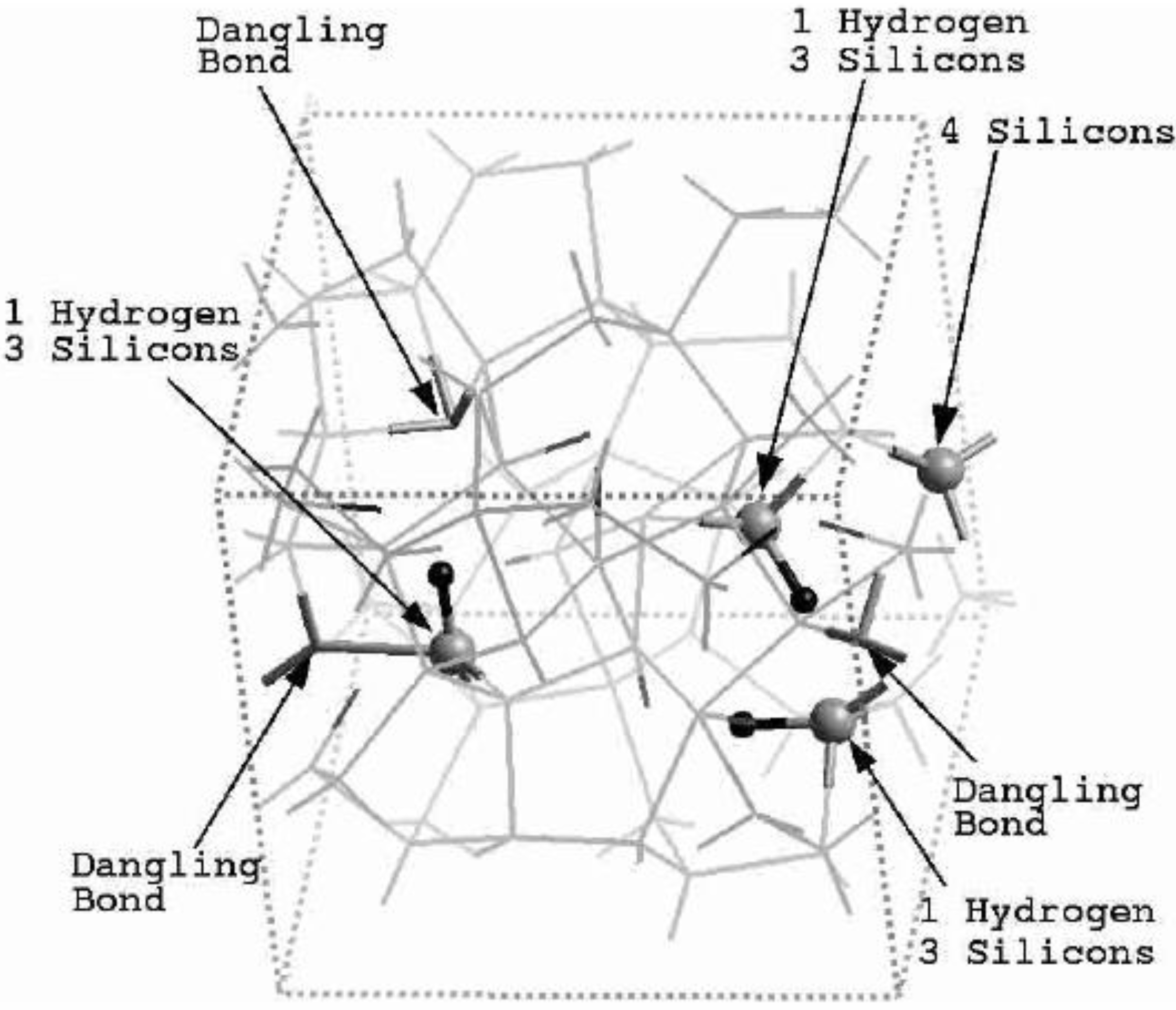
Finally, we would like to mention the criterion that we have used to determine the ‘extension of a bond’. Bonds, being electrical in nature, have in principle an infinite range, but in practice they have a finite range, so how is one to define when two atoms are bonded? This is a difficult problem in amorphous materials and some authors have carried out extensive searches for possible molecular (cluster) structures of a given element, silicon for example, to infer a probable bond length [
47]. Others opted for the use of localized wave functions, like the Wannier-type, to get an estimate of the bond lengths [
16]. One can also look at the charge distribution between atoms and set a limit below which the bonding is declared nonexistent. We decided to use throughout this work a geometric approach to the bonding problem. We believe that the structure of radial distribution functions is a manifest way to determine the
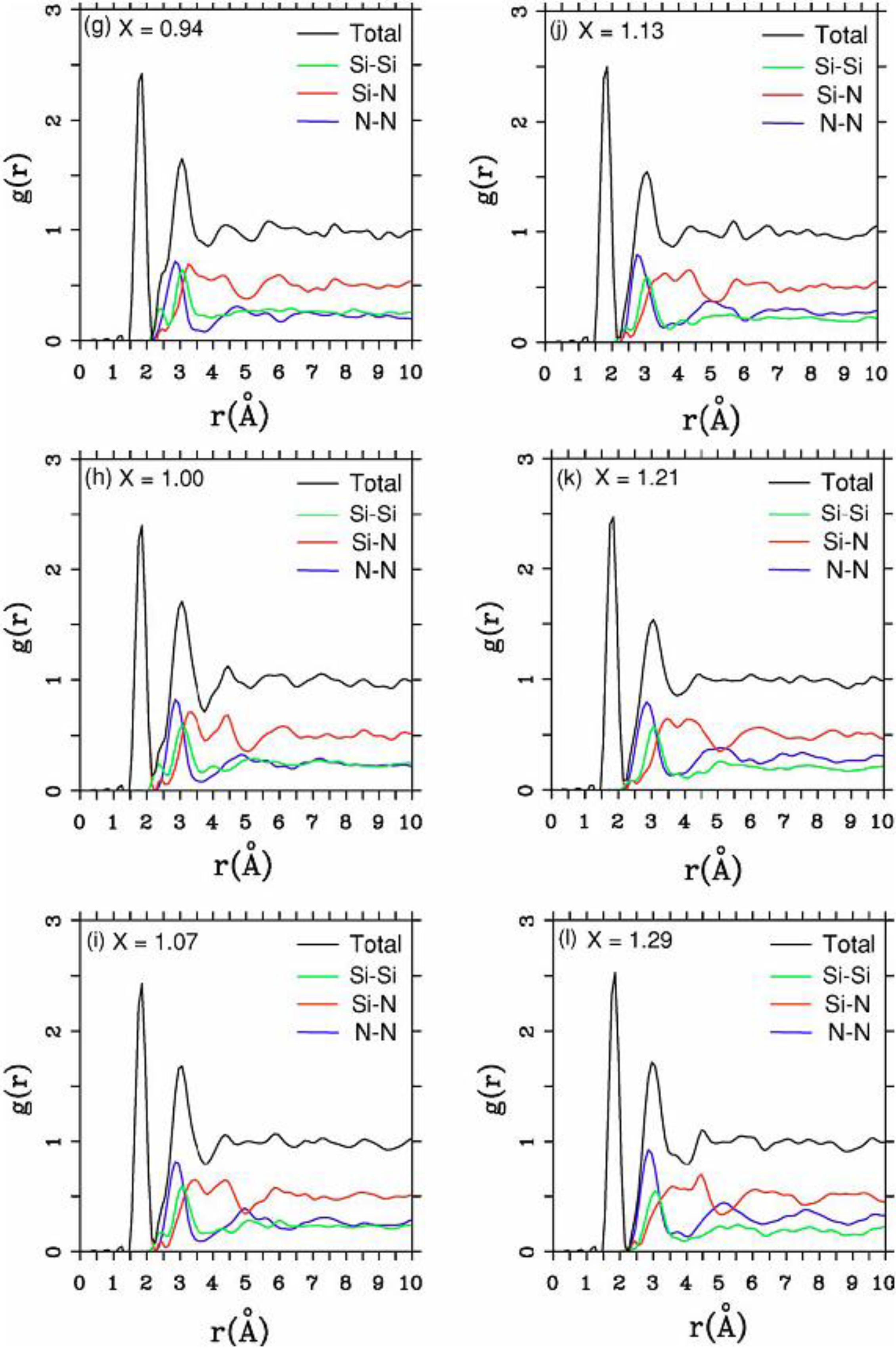
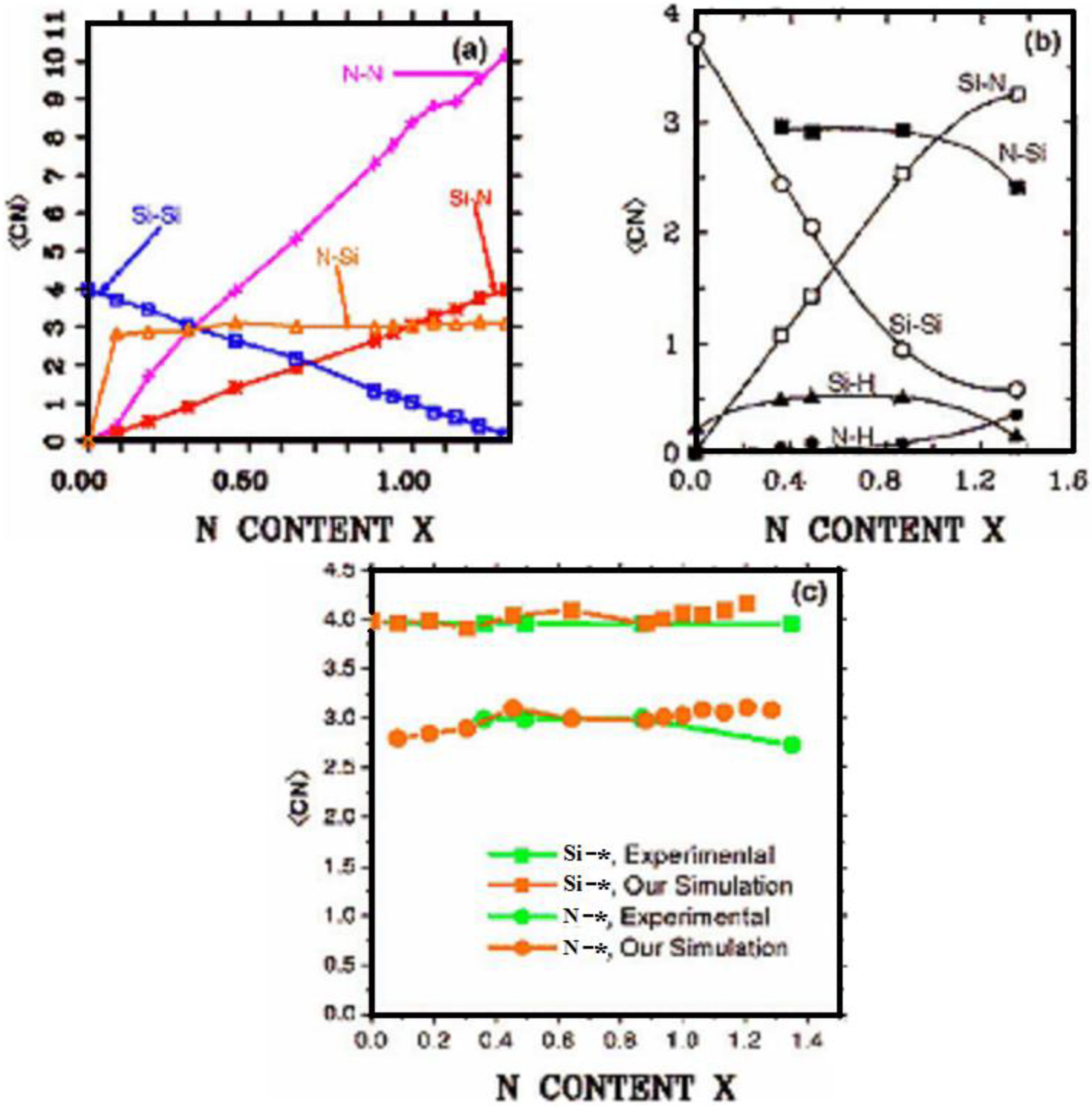
maximum bond length, especially when there is a clear zero minimum between the first and the second peaks which is the case for most elemental, monatomic, amorphous semiconductors. When amorphous alloys are considered, a way to determine the bond lengths among the diverse species is by looking at the minimum between the first and second peaks of the corresponding pRDFs; the maximum bond lengths are then set equal to the position of these minima. Using this approach we determine the extension of the bond, and by integrating the area under the corresponding peak of the adequate RDF we can also calculate the number of neighbors, although with this procedure it is difficult to determine the multiplicity of the bonds (single, double or triple) in
a-semiconductors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}