STM, SECPM, AFM and Electrochemistry on Single Crystalline Surfaces

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Scanning Tunneling Microscope (STM) in Air and Under Electrochemical Conditions
1.2. Atomic force microscope (AFM)
1.3. Scanning Electrochemical Potential Microscopy (SECPM)
1.4. Single Crystals and Single Crystalline Supports
2. Results and Discussion
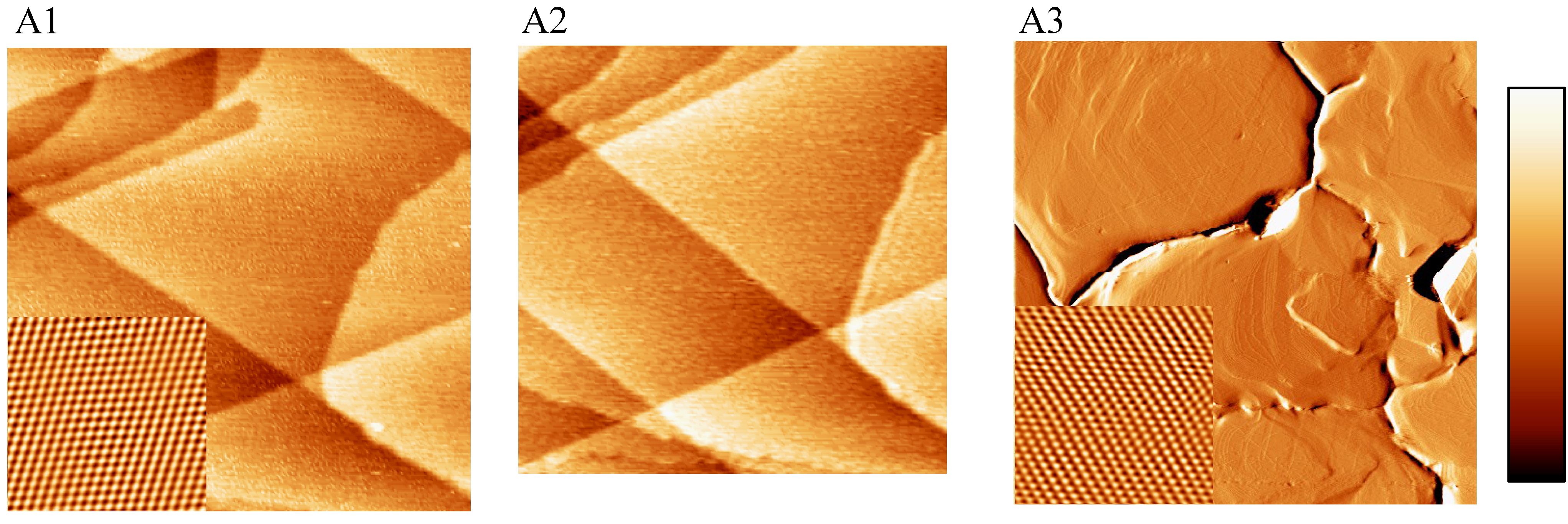
2.1. EC-STM, SECPM and AFM Imaging of the Au(111) Crystalline Surfaces
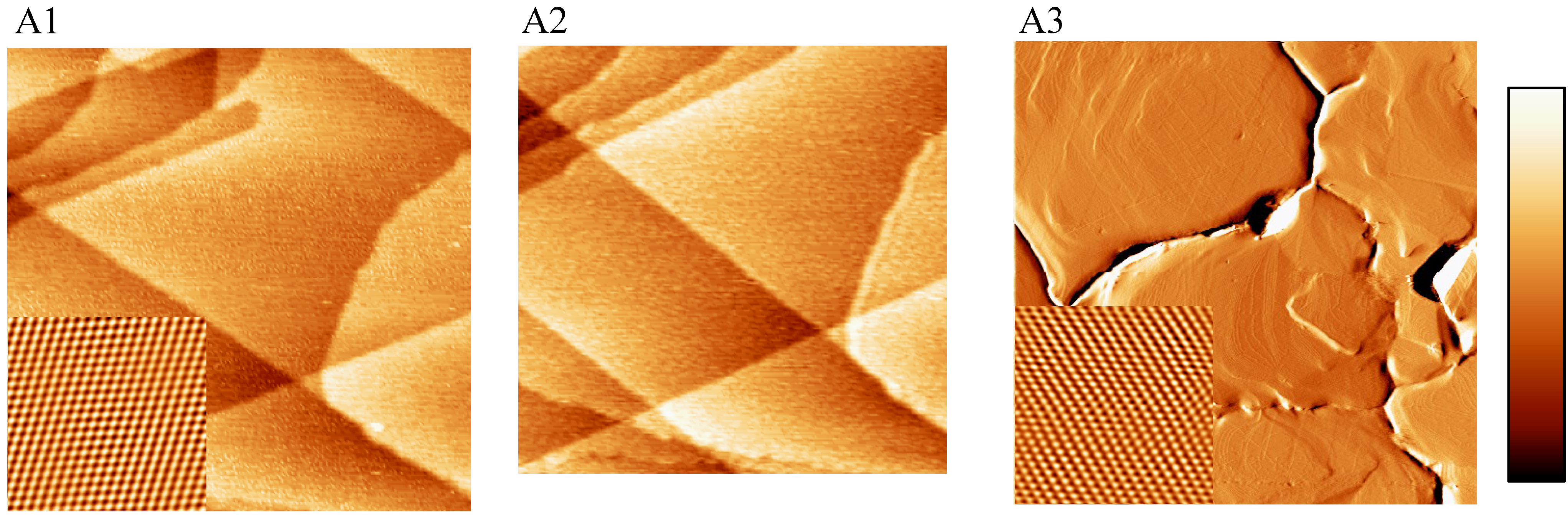
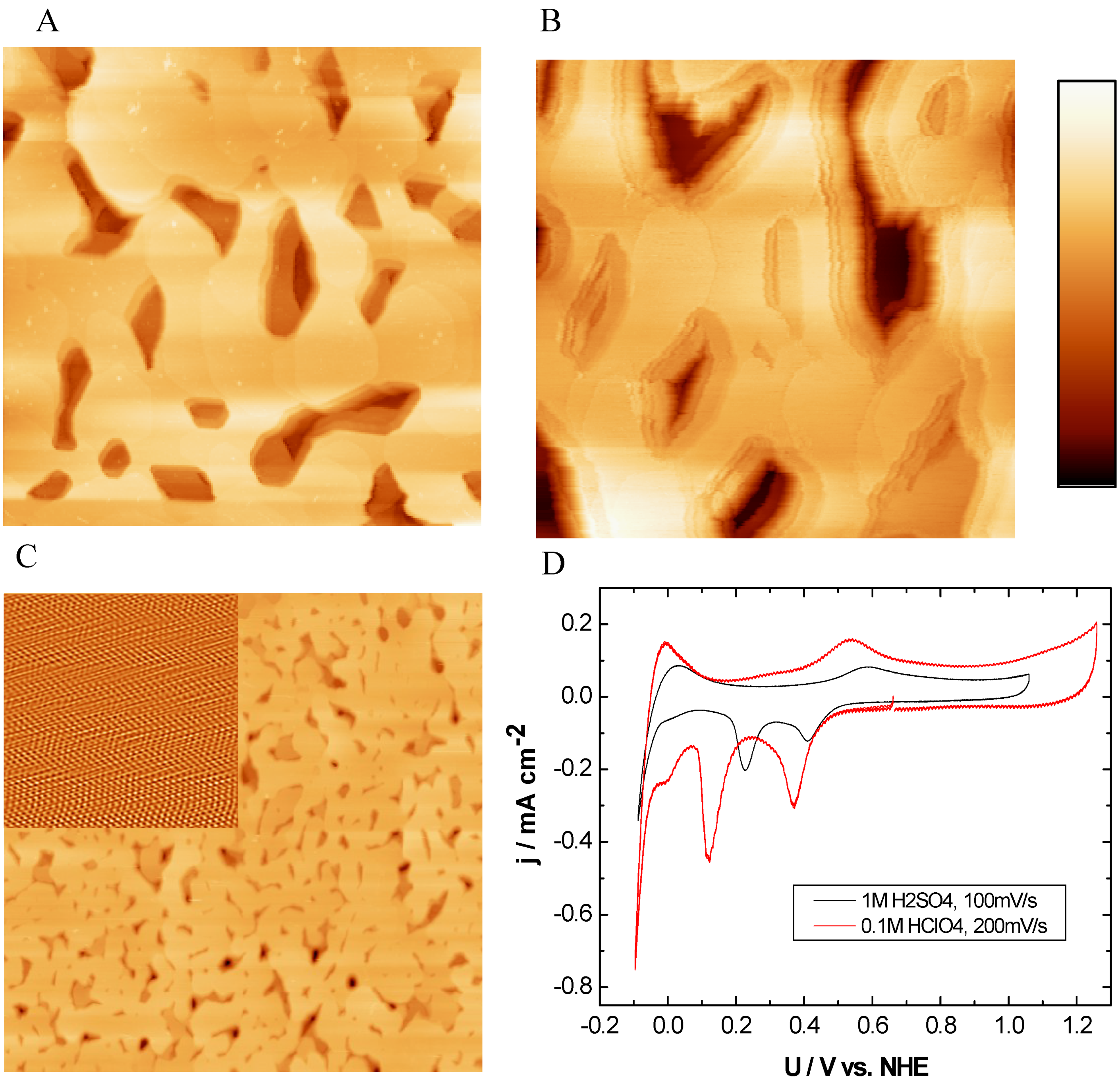
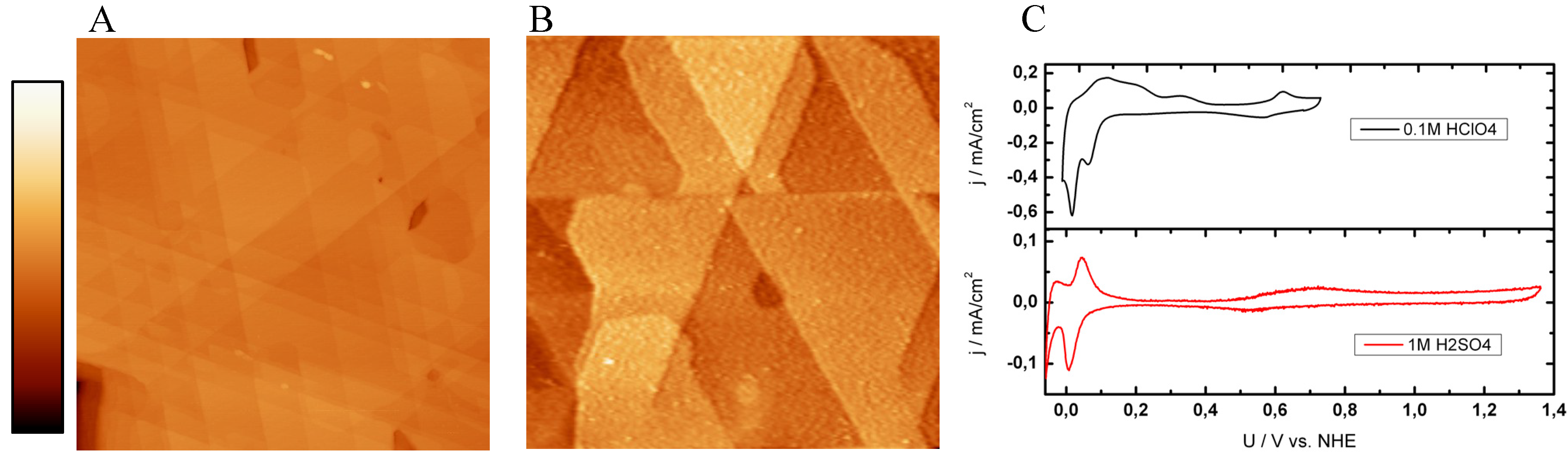
2.2. Single Crystalline Surfaces (Ru(0001), Rh(111), Ir(111), Ir(100))
2.2.1. Ru(0001)

2.2.2. Rh(111)

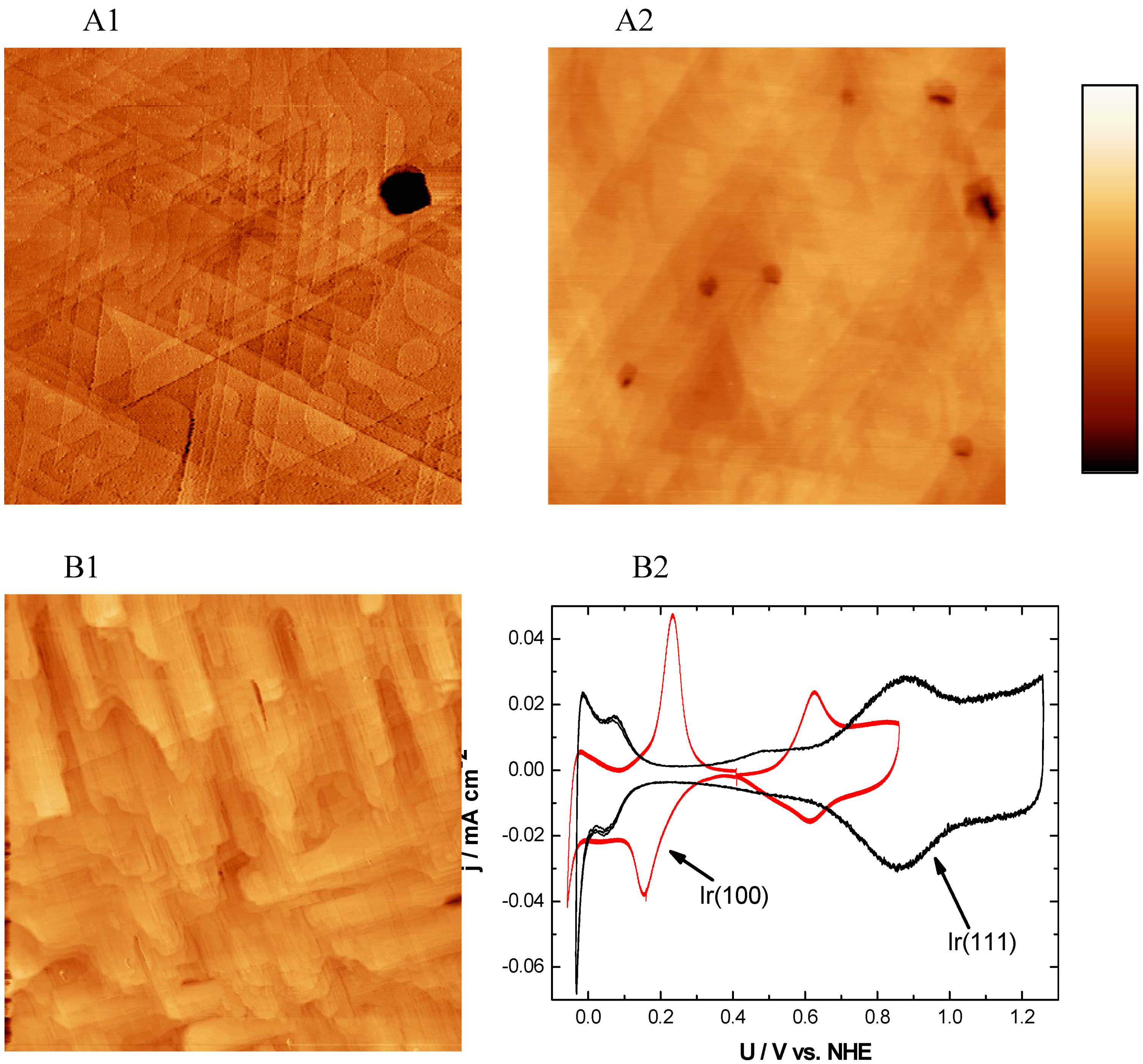
2.2.3. Ir(111) and Ir(100)
3. Experimental Section
3.1. Materials and Instrumentation
3.2. Sample Preparation
3.2.1. Au(111) electrodes
3.2.2. Ru(0001), Rh(111), Ir(111), Ir(100)
4. Conclusions
Acknowledgements
References and Notes
- Binnig, G.; Rohrer, H.; Gerber, C.; Weibel, E. Tunneling through a Controllable Vacuum Gap. Appl. Phys. Lett. 1982, 40, 178–180. [Google Scholar] [CrossRef]
- Binnig, G.; Rohrer, H.; Gerber, C.; Weibel, E. Surface Studies by Scanning Tunneling Microscopy. Phys. Rev. Lett. 1982, 49, 57–61. [Google Scholar] [CrossRef]
- Hansma, P.K.; Elings, V.B.; Marti, O.; Bracker, C.E. Scanning tunneling microscopy and atomic force microscopy: Application to biology and technology. Science 1988, 242, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Frommer, J. Scanning tunneling microscopy and atomic force microscopy in organic chemistry. Angew. Chem. Int. Ed. 1992, 31, 1298–1328. [Google Scholar] [CrossRef]
- Samori, P. Scanning probe microscopies beyond imaging. J. Mater. Chem. 2004, 14, 1353–1366. [Google Scholar] [CrossRef]
- Itaya, K. In situ scanning tunneling microscopy in electrolyte solutions. Prog. Surf. Sci. 1998, 58, 121–247. [Google Scholar] [CrossRef]
- Kolb, D.M.; Simeone, F.C. Electrochemical nanostructuring with an STM: A status report. Electrochim. Acta 2005, 50, 2989–2996. [Google Scholar] [CrossRef]
- Guntherodt, H.J.; Wiesendanger, R. Scanning Tunneling Microscopy Volume I–III; Springer: Berlin, Germany, 1993; Volume I–III. [Google Scholar]
- Bard, A.J.; Fan, F.R. Introductory lecture studies of the liquid-solid interface by scanning-Tunneling-microscopy and scanning electrochemical microscopy. Faraday Discuss. 1992, 94, 1–22. [Google Scholar] [CrossRef]
- Hansma, P.K.; Tersoff, J. Scanning tunneling microscopy. J. Appl. Phys. 1987, 61, R1–R23. [Google Scholar] [CrossRef]
- Sonnenfeld, R.; Hansma, P.K. Atomic-resolution microscopy in Water. Science 1986, 232, 211–213. [Google Scholar] [CrossRef] [PubMed]
- Lustenberger, P.; Rohrer, H.; Christoph, R.; Siegenthaler, H. Scanning tunneling microscopy at potential controlled electrode surfaces in electrolytic environment. J. Electroanal. Chem. 1988, 243, 225–235. [Google Scholar] [CrossRef]
- Itaya, K.; Tomita, E. Scanning tunneling microscope for electrochemistry—A new concept for the In situ scanning tunneling microscope in electrolyte solutions. Surf. Sci. 1988, 201, L507–L512. [Google Scholar] [CrossRef]
- Binnig, G.; Garcia, N.; Rohrer, H.; Soler, J.M.; Flores, F. Electron-metal-surface interaction potential with vacuum tunneling: Observation of the image force. Phys. Rev. B 1984, 30. [Google Scholar] [CrossRef]
- Engelmann, G.E.; Ziegler, J.C.; Kolb, D.M. Electrochemical fabrication of large arrays of metal nanoclusters. Surf. Sci. 1998, 401, L420–L424. [Google Scholar] [CrossRef]
- Binnig, G.; Quate, C.F.; Gerber, C. Atomic Force Microscope. Phys. Rev. Lett. 1986, 56, 930–933. [Google Scholar] [CrossRef] [PubMed]
- Meyer, G.; Amer, N.M. Novel optical approach to atomic force microscopy. Appl. Phys. Lett. 1988, 53, 1045–1047. [Google Scholar] [CrossRef]
- Giessibl, F.J. Advances in atomic force microscopy. Rev. Mod. Phys. 2003, 75, 949–983. [Google Scholar] [CrossRef]
- Garcia, R.; Perez, R. Dynamic atomic force microscoy methods. Surf. Sci. Rep. 2002, 47, 197–301. [Google Scholar] [CrossRef]
- Hofer, W.A.; Foster, A.S.; Shluger, A.L. Theories of scanning probe microscopes at the atomic scale. Rev. Mod. Phys. 2003, 75, 1287–1331. [Google Scholar] [CrossRef]
- Woo, D.H.; Yoo, J.S.; Park, S.M.; Jeon, I.C.; Kang, H. Direct probing into the electrochemical interface using a novel potential probe: Au(111) electrode/NaBF4 solution interface. Bull. Korean Chem. Soc. 2004, 25, 577–580. [Google Scholar] [CrossRef]
- Hurth, C.; Li, C.Z.; Bard, A.J. Direct probing of electrical double layers by scanning electrochemical potential microscopy. J. Phys. Chem. C 2007, 111, 4620–4627. [Google Scholar] [CrossRef]
- Kim, Y.T.; Bard, A.J. Imaging and etching of self-assembled N-Octadecanethiol layers on gold with the scanning tunneling Microscope. Langmuir 1992, 8, 1096–1102. [Google Scholar] [CrossRef]
- Chang, H.P.; Bard, A.J. Scanning tunneling microscopy studies of Carbon Oxygen reactions on highly oriented pyrolytic-graphite. J. Am. Chem. Soc. 1991, 113, 5588–5596. [Google Scholar] [CrossRef]
- Suggs, D.W.; Bard, A.J. Scanning tunneling microscopic study with atomic-resolution of the dissolution of Cu(100) electrodes in aqueous chloride media. J. Phys. Chem. 1995, 99, 8349–8355. [Google Scholar] [CrossRef]
- Corbella, C.; Pascual, E.; Oncins, G.; Canal, C.; Andujar, J.L.; Bertran, E. Composition and morphology of metal-containing diamond-like carbon films obtained by reactive magnetron sputtering. Thin Solid Films 2005, 482, 293–298. [Google Scholar] [CrossRef]
- Baier, C.; Stimming, U. Imaging single enzyme molecules under In situ conditions. Angew. Chem. Int. Ed. 2009, 48, 5542–5544. [Google Scholar] [CrossRef]
- Ohshima, H. Diffuse double layer interaction between two parallel plates with constant surface charge density in an electrolyte solution. Colloid Polym. Sci. 1974, 252, 158–164. [Google Scholar] [CrossRef]
- Sader, J.E.; Lenhoff, A.M. Electrical double-layer interaction between heterogeneously charged colloidal particles: A superposition formulation. J. Colloid Interface Sci. 1998, 201, 233–243. [Google Scholar] [CrossRef]
- Sader, J.E.; Chan, D.Y.C. Electrical double-layer interaction between charged particles near surfaces an in confined geometries. J. Colloid Interface Sci. 1999, 218, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Hamou, R.F.; Biedermann, P.U.; Rohwerder, M.; Blumenau, A.T. FEM Simulation of the scanning electrochemical potential microscopy (SECPM). In Excerpt from the Proceedings of the COMSOL Conference 2008, Hannover, Germany, 2009.
- Hamou, R.F.; Biedermann, P.U.; Erbe, A.; Rohwerder, M. Numerical simulation of probing the electric double layer by scanning electrochemical potential microscopy. Electrochim. Acta 2010, 55, 5210–5222. [Google Scholar] [CrossRef]
- Clavilier, J.; Faure, R.; Guinet, G.; Durand, R. Preparation of mono-crystalline Pt microelectrodes and electrochemical study of the plane surfaces cut in the direction of the (111) and (110) planes. J. Electroanal. Chem. 1980, 107, 205–209. [Google Scholar] [CrossRef]
- Soriaga, M.P. Ultra-High vacuum techniques in the study of single-crystal electrode surfaces. Prog. Surf. Sci. 1992, 39, 325–443. [Google Scholar] [CrossRef]
- Hubbard, A.T. Electrochemistry at well-characterized Surfaces. Chem. Rev. 1988, 88, 633–656. [Google Scholar] [CrossRef]
- Kibler, L.A. Preparation and Characterization of Noble Metal Single Crystal Electrode Surfaces; International Society of Electrochemistry: Lausanne, Switzerland, 2003. [Google Scholar]
- Chen, T.L.; Li, X.M.; Zhang, S.; Zhang, X. Comparative study of epitaxial growth of Pt and Ir electrode films grown on MgO-buffered Si(100) by PLD. Appl. Phys. A 2005, 80, 73–76. [Google Scholar] [CrossRef]
- Akai, D.; Hirabayashi, K.; Yokawa, M.; Sawada, K.; Ishida, M. Epitaxial growth of Pt(001) thin films on Si substrates using an epitaxial gamma-Al2O3(001) buffer layer. J. Cryst. Growth 2004, 264, 463–467. [Google Scholar] [CrossRef]
- Gsell, S.; Fischer, M.; Brescia, R.; Schreck, M.; Huber, P.; Bayer, F.; Stritzker, B.; Schlom, D.G. Reduction of mosaic spread using iridium interlayers: A route to improved oxide heteroepitaxy on silicon. Appl. Phys. Lett. 2007, 91. [Google Scholar] [CrossRef]
- Gsell, S.; Bauer, T.; Goldfuss, J.; Schreck, M.; Stritzker, B. A route to diamond wafers by epitaxial deposition on silicon via iridium/yttria-stabilized zirconia buffer layers. Appl. Phys. Lett. 2004, 84, 4541–4543. [Google Scholar] [CrossRef]
- Kibler, L.A. Hydrogen electrocatalysis. ChemPhysChem 2006, 7, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Wolfschmidt, H.; Bußar, R.; Stimming, U. Charge transfer reactions at nanostructured Au(111) surfaces: Influence of the substrate material on electrocatalytic activity. J. Phys.: Condens. Matter 2008, 20. [Google Scholar] [CrossRef]
- Wolfschmidt, H.; Weingarth, D.; Stimming, U. Enhanced reactivity for hydrogen reactions at Pt nanoislands on Au(111). ChemPhysChem 2010, 11, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Brülle, T.; Stimming, U. Platinum nanostructured HOPG—Preparation, characterization and reactivity. J. Electroanal. Chem. 2009, 636, 10–17. [Google Scholar] [CrossRef]
- Lima, F.H.B.; Zhang, J.; Shao, M.H.; Sasaki, K.; Vukmirovic, M.B.; Ticianelli, E.A.; Adzic, R.R. Catalytic activity-d-band center correlation for the O-2 reduction reaction on platinum in alkaline solutions. J. Phys. Chem. C 2007, 111, 404–410. [Google Scholar] [CrossRef]
- Greeley, J.; Nørskov, J.K.; Kibler, L.A.; El-Aziz, A.M.; Kolb, D.M. Hydrogen evolution over bimetallic systems: Understanding the trends. ChemPhysChem 2006, 7, 1032–1035. [Google Scholar] [CrossRef] [PubMed]
- Greeley, J.; Nørskov, J.K. Large-scale, density functional theory-based screening of alloys for hydrogen evolution. Surf. Sci. 2007, 601, 1590–1598. [Google Scholar] [CrossRef]
- Norskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the exchange current for hydrogen evolution. J. Electrochem. Soc. 2005, 152, J23–J26. [Google Scholar] [CrossRef]
- Roudgar, A.; Gross, A. Local reactivity of metal overlayers: Density functional theory calculations of Pd on Au. Phys. Rev. B 2003, 67. [Google Scholar] [CrossRef]
- Santos, E.; Schmickler, W. Electronic interactions decreasing the activation barrier for the hydrogen electro-oxidation reaction. Electrochim. Acta 2008, 53, 6149–6156. [Google Scholar] [CrossRef]
- Santos, E.; Quaino, P.; Soldano, G.; Schmickler, W. Some properties of electrochemical nanostructures. J. Chem. Sci. 2009, 121, 575–577. [Google Scholar] [CrossRef]
- Baldauf, M.; Kolb, D.M. A Hydrogen Adsorption and Absorption Study with Ultrathin Pd Overlayers on Au(111) and Au(100). Electrochim. Acta 1993, 38, 2145–2153. [Google Scholar] [CrossRef]
- Hernandez, F.; Baltruschat, H. Electrochemical characterization of gold stepped surfaces modified with Pd. Langmuir 2006, 22, 4877–4884. [Google Scholar] [CrossRef] [PubMed]
- Kibler, L.A.; El-Aziz, A.M.; Kolb, D.M. Electrochemical behaviour of pseudomorphic overlayers: Pd on Au(111). J. Mol. Catal. A: Chem. 2003, 199, 57–63. [Google Scholar] [CrossRef]
- Meier, J.; Schiotz, J.; Liu, P.; Norskov, J.K.; Stimming, U. Nano-scale effects in electrochemistry. Chem. Phys. Lett. 2004, 390, 440–444. [Google Scholar] [CrossRef]
- Pandelov, S.; Stimming, U. Reactivity of monolayers and nano-islands of palladium on Au(111) with respect to proton reduction. Electrochim. Acta 2007, 52, 5548–5555. [Google Scholar] [CrossRef]
- Pluntke, Y.; Kibler, L.A.; Kolb, D.M. Unique activity of Pd monomers: Hydrogen evolution at AuPd(111) surface alloys. Phys. Chem. Chem. Phys. 2008, 10, 3684–3688. [Google Scholar] [CrossRef] [PubMed]
- Crown, A.; Johnston, C.; Wieckowski, A. Growth of ruthenium islands on Pt(hkl) electrodes obtained via repetitive spontaneous deposition. Surf. Sci. 2002, 506, L268–L274. [Google Scholar] [CrossRef]
- Ferrin, P.; Mavrikakis, M. Structure Sensitivity of Methanol Electrooxidation on Transition Metals. J. Am. Chem. Soc. 2009, 131, 14381–14389. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.L.; Vukmirovic, M.B.; Xu, Y.; Mavrikakis, M.; Adzic, R.R. Controlling the catalytic activity of platinum-monolayer electrocatalysts for oxygen reduction with different substrates. Angew. Chem. Int. Ed. 2005, 44, 2132–2135. [Google Scholar] [CrossRef]
- Schnyder, B.; Alliata, D.; Kötz, R.; Siegenthaler, H. Electrochemical intercalation of perchlorate ions in HOPG: An SFM/LFM and XPS study. Appl. Surf. Sci. 2001, 173, 221–232. [Google Scholar] [CrossRef]
- Soler, J.M.; Baro, A.M.; Garcia, N.; Rohrer, H. Interatomic Forces in Scanning Tunneling Microscopy—Giant Corrugations of the Graphite Surface. Phys. Rev. Lett. 1986, 57, 444–447. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Durkan, C.; Chandrasekhar, N. Tailoring the Local Interaction between Graphene Layers in Graphite at the Atomic Scale and Above Using Scanning Tunneling Microscopy. ACS Nano 2009, 3, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Zeh, R.; Wolfschmidt, H.; Baier, C.; Stimming, U. in preparation.
- Cline, K.K.; McDermott, M.T.; McCreery, R.L. Anomalously Slow Electron Transfer at Ordered Graphite Electrodes: Influence of Electronic Factors and Reactive Sites. J. Phys. Chem. 1994, 98, 5314–5319. [Google Scholar] [CrossRef]
- Randin, J.P.; Yeager, E. Differential capacitance study on the basal plane of stress-annealed pyrolytic graphite. J. Electroanal. Chem. 1972, 36, 257–276. [Google Scholar] [CrossRef]
- Rice, R.J.; McCreery, R.L. Quantitative Relationship between Electron-Transfer Rate and Surface Microstructure of Laser-Modified Graphite-Electrodes. Anal. Chem. 1989, 61, 1637–1641. [Google Scholar] [CrossRef]
- Ray, K.; McCreery, R.L. Spatially Resolved Raman Spectroscopy of Carbon Electrode Surfaces: Observations of Structural and Chemical Heterogeneity. Anal. Chem. 1997, 69, 4680–4687. [Google Scholar] [CrossRef]
- Esplandiu, M.J.; Hagenstrom, H.; Kolb, D.M. Functionalized self-assembled alkanethiol monolayers on Au(111) electrodes: 1. Surface structure and electrochemistry. Langmuir 2001, 17, 828–838. [Google Scholar] [CrossRef]
- Kopitzki, K. Einführung in die Festkörperphysik; Teubner Studienbücher: Physik: Didderse, Deutschland, 1993. [Google Scholar]
- Grant, J.T.; Haas, T.W. A study of Ru(0001) and Rh(111) surfaces using LEED and Auger Electron Spectroscopy. Surf. Sci. 1970, 21, 76–85. [Google Scholar] [CrossRef]
- Stampfl, C.; Scheffler, M. Theoretical study of O adlayers on Ru(0001). Phys. Rev. B 1996, 54, 2868. [Google Scholar] [CrossRef]
- Stampfl, C.; Schwegmann, S.; Over, H.; Scheffler, M.; Ertl, G. Structure and stability of a high-coverage (1x1) oxygen phase on Ru(0001). Phys. Rev. Lett. 1996, 77, 3371–3374. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.F.; Zei, M.S.; Kim, Y.D.; Over, H.; Ertl, G. Electrochemical versus gas-phase oxidation of Ru single-crystal surfaces. J. Phys. Chem. B 2000, 104, 6040–6048. [Google Scholar] [CrossRef]
- Marinkovic, N.S.; Wang, J.X.; Zajonz, H.; Adzic, R.R. Adsorption of bisulfate on the Ru(0001) single crystal electrode surface. J. Electroanal. Chem. 2001, 500, 388–394. [Google Scholar] [CrossRef]
- El-Aziz, A.M.; Kibler, L.A. New information about the electrochemical behaviour of Ru(0001) in perchloric acid solutions. Electrochem. Commun. 2002, 4, 866–870. [Google Scholar] [CrossRef]
- Inoue, H.; Wang, J.X.; Sasaki, K.; Adzic, R.R. Electrocatalysis of H2 oxidation on Ru(0001) and Ru(10-10) single crystal surfaces. J. Electroanal. Chem. 2003, 554–555, 77–85. [Google Scholar] [CrossRef]
- El-Aziz, A.M.; Kibler, L.A. New information about the electrochemical behaviour of Ru(0 0 0 1) in perchloric acid solutions. Electrochem. Commun. 2002, 4, 866–870. [Google Scholar] [CrossRef]
- Mavrikakis, M.; Rempel, J.; Greeley, J.; Hansen, L.B.; Norskov, J.K. Atomic and molecular adsorption on Rh(111). J. Chem. Phys. 2002, 117, 6737–6744. [Google Scholar] [CrossRef]
- CRC Handbook of Chemistry and Physics, 76th ed.; CRC Press: New York, NY, USA, 1996.
- Tanaka, K.; Sasahara, A. Interfacial Electrochmistry; Wieckowski, A., Ed.; Dekker, M.: New York, NY, USA, 1999; p. 493. [Google Scholar]
- Sung, Y.E.; Thomas, S.; Wieckowski, A. Characterization of the Rh(111) Electrode by Ceels, Aes, Leed, and Voltammetry—Adsorption of (Bi)Sulfate, Perchlorate, and Carbon-Monoxide. J. Phys. Chem. 1995, 99, 13513–13521. [Google Scholar] [CrossRef]
- Wan, L.J.; Yau, S.L.; Swain, G.M.; Itaya, K. In-Situ Scanning-Tunneling-Microscopy of Well-Ordered Rh(111) Electrodes. J. Electroanal. Chem. 1995, 381, 105–111. [Google Scholar] [CrossRef]
- Wan, L.J.; Yau, S.L.; Itaya, K. Atomic-Structure of Adsorbed Sulfate on Rh(111) in Sulfuric-Acid-Solution. J. Phys. Chem. 1995, 99, 9507–9513. [Google Scholar] [CrossRef]
- Funtikov, A.M.; Stimming, U.; Vogel, R. Anion adsorption from sulfuric acid solutions on Pt(111) single crystal electrodes. J. Electroanal. Chem. 1997, 428, 147–153. [Google Scholar] [CrossRef]
- Funtikov, A.M.; Linke, U.; Stimming, U.; Vogel, R. An In-Situ Stm Study of Anion Adsorption on Pt(111) from Sulfuric-Acid-Solutions. Surf. Sci. 1995, 324, L343–L348. [Google Scholar] [CrossRef]
- Xu, Q.; Linke, U.; Bujak, R.; Wandlowski, T. Preparation and electrochemical characterization of low-index rhodium single crystal electrodes in sulfuric acid. Electrochim. Acta 2009, 54, 5509–5521. [Google Scholar] [CrossRef]
- Clavilier, J.; Wasberg, M.; Petit, M.; Klein, L.H. Detailed Analysis of the Voltammetry of Rh(111) in Perchloric-Acid Solution. J. Electroanal. Chem. 1994, 374, 123–131. [Google Scholar] [CrossRef]
- Gomez, R.; Weaver, M.J. Reduction of nitrous oxide on iridium single-crystal electrodes. Langmuir 2002, 18, 4426–4432. [Google Scholar] [CrossRef]
- Pajkossy, T.; Kibler, L.A.; Kolb, D.M. Voltammetry and impedance measurements of Ir(111) electrodes in aqueous solutions. J. Electroanal. Chem 2005, 582, 69–75. [Google Scholar] [CrossRef]
- Pajkossy, T.; Kibler, L.A.; Kolb, D.M. Voltammetry and impedance measurements of Ir(100) electrodes in aqueous solutions. J. Electroanal. Chem 2007, 600, 113–118. [Google Scholar] [CrossRef]
- Motoo, S.; Furuya, N. Hydrogen and Oxygen-Adsorption on Ir(111), Ir(100) and Ir(110) Planes. J. Electroanal. Chem. 1984, 167, 309–315. [Google Scholar] [CrossRef]
- Horcas, I.; Fernandez, R.; Gomez-Rodriguez, J.M.; Colchero, J.; Gomez-Herrero, J.; Baro, A.M. WSXM: A software for scanning probe microscopy and a tool for nanotechnology. Rev. Sci. Instrum. 2007, 78. [Google Scholar] [CrossRef] [PubMed]
- Gsell, S.; Fischer, M.; Schreck, M.; Stritzker, B. Epitaxial films of metals from the platinum group (Ir, Rh, Pt and Ru) on YSZ-buffered Si(111). J. Cryst. Growth 2009, 311, 3731–3736. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wolfschmidt, H.; Baier, C.; Gsell, S.; Fischer, M.; Schreck, M.; Stimming, U. STM, SECPM, AFM and Electrochemistry on Single Crystalline Surfaces. Materials 2010, 3, 4196-4213. https://doi.org/10.3390/ma3084196
Wolfschmidt H, Baier C, Gsell S, Fischer M, Schreck M, Stimming U. STM, SECPM, AFM and Electrochemistry on Single Crystalline Surfaces. Materials. 2010; 3(8):4196-4213. https://doi.org/10.3390/ma3084196
Chicago/Turabian StyleWolfschmidt, Holger, Claudia Baier, Stefan Gsell, Martin Fischer, Matthias Schreck, and Ulrich Stimming. 2010. "STM, SECPM, AFM and Electrochemistry on Single Crystalline Surfaces" Materials 3, no. 8: 4196-4213. https://doi.org/10.3390/ma3084196