Cell Type Influences Local Delivery of Biomolecules from a Bioinspired Apatite Drug Delivery System



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials Fabrication
2.1.1. bCaP Application with or without Antimycin A
2.1.2. PEM Bilayer Coating
2.1.3. FGF-2 Factor Absorption
2.2. Characterization
2.2.1. Cell Culture Assays
2.2.2. Evaluation of Cell Access Mechanism
2.3. Statistical Analysis
3. Results
3.1. Characterizations of bCaP-PEM Coating
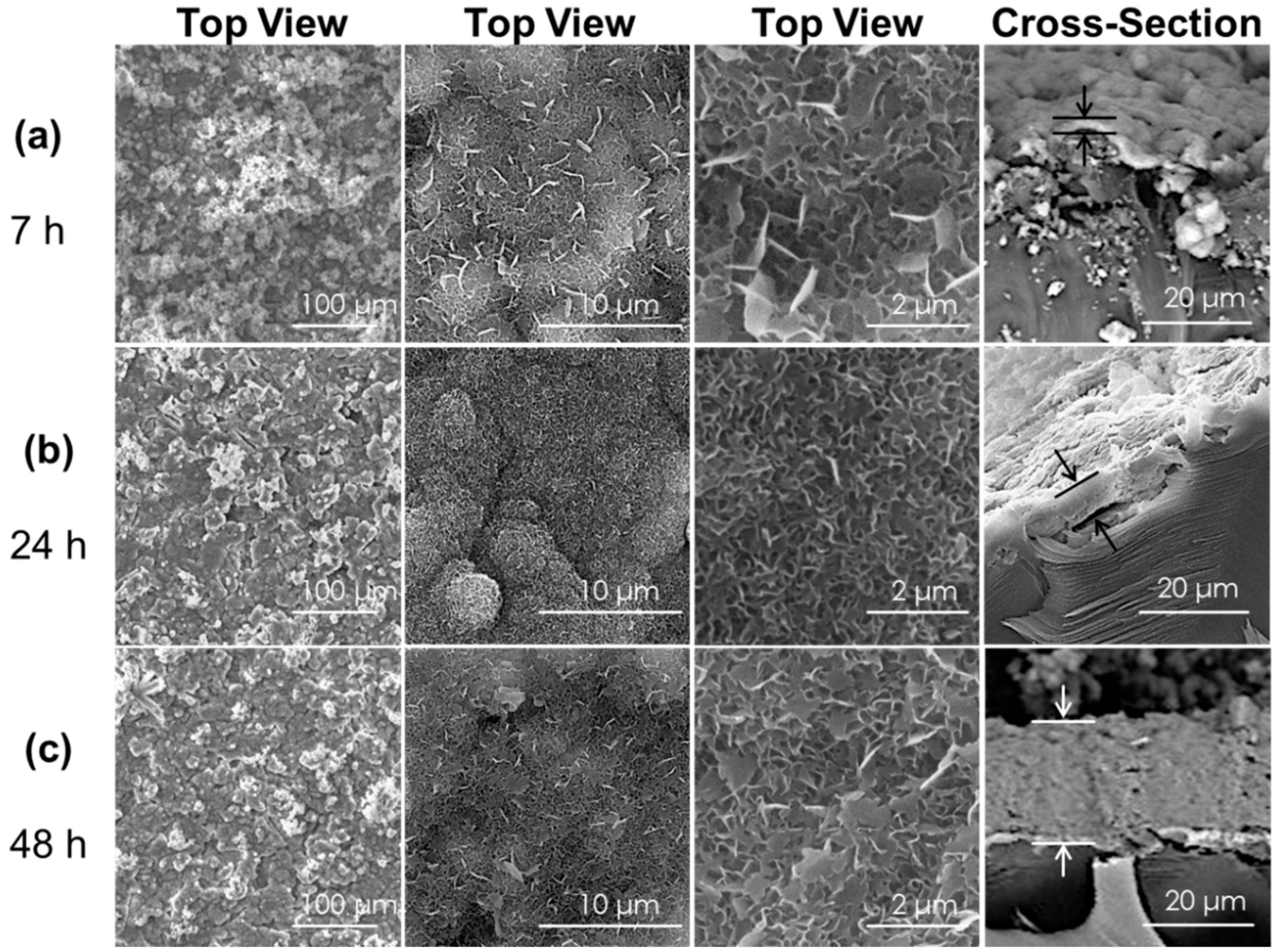
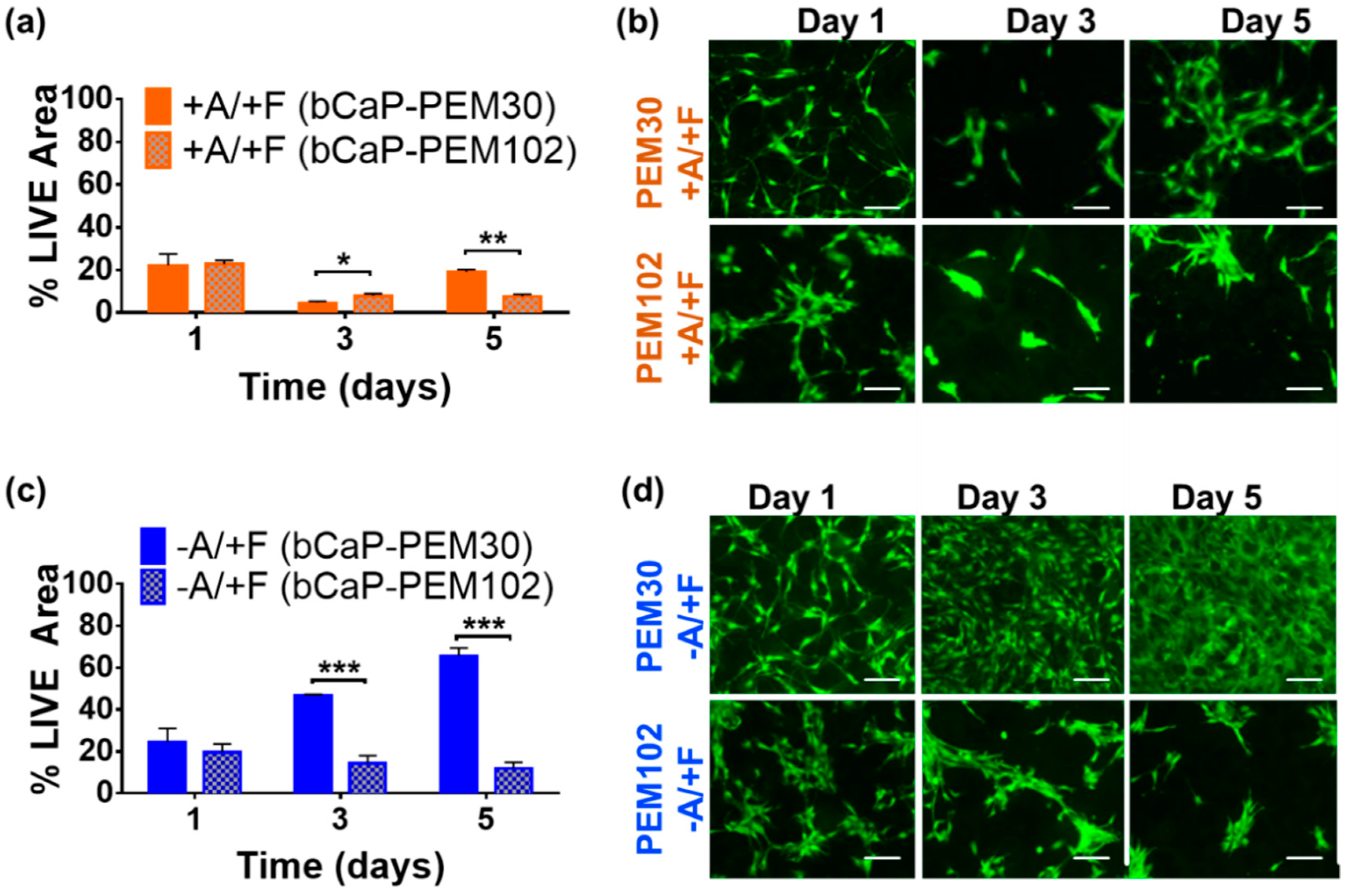
3.1.1. Changing bCaP Layer Thickness in the Presence of PEM
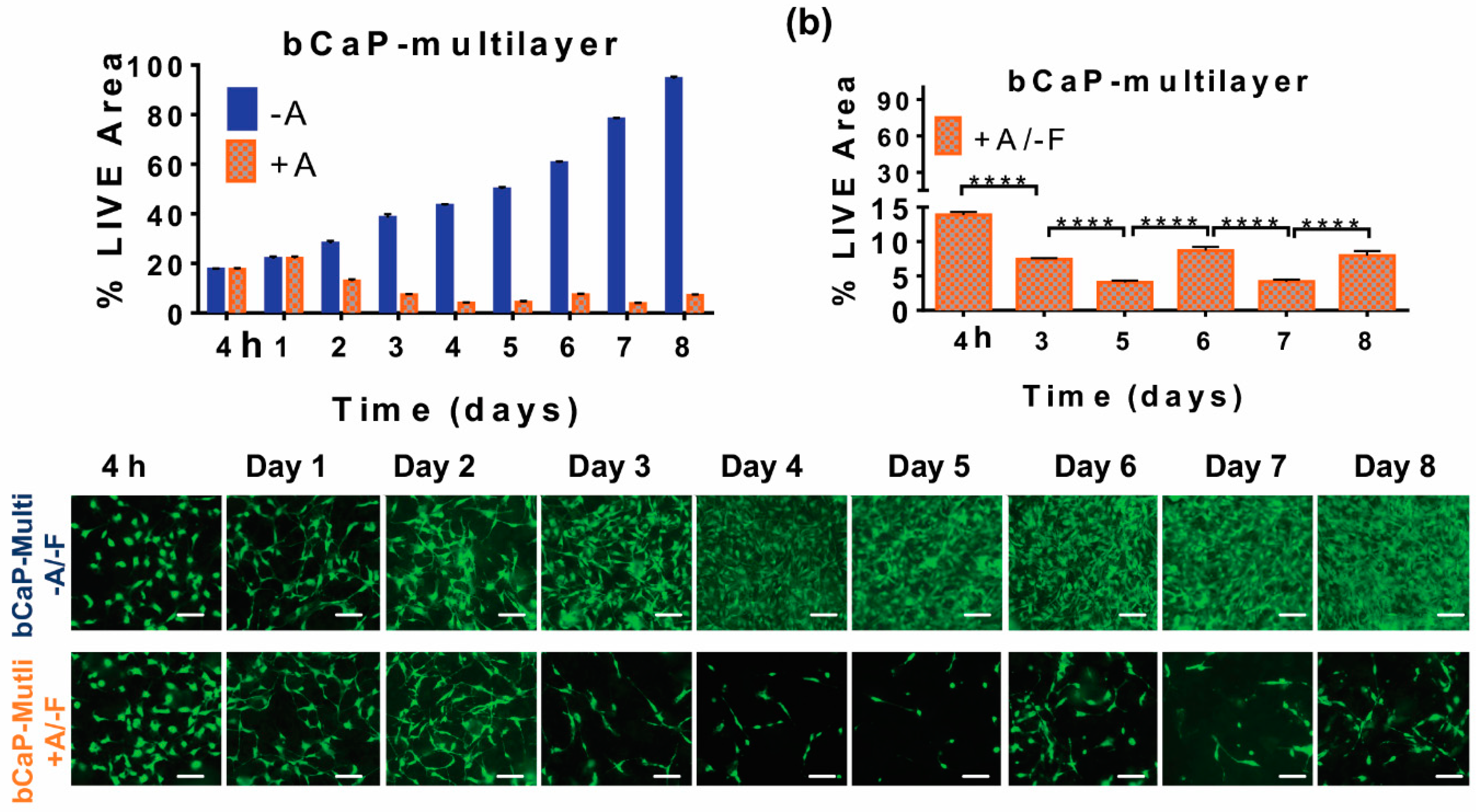
3.1.2. Increasing Number of PEM Bilayers
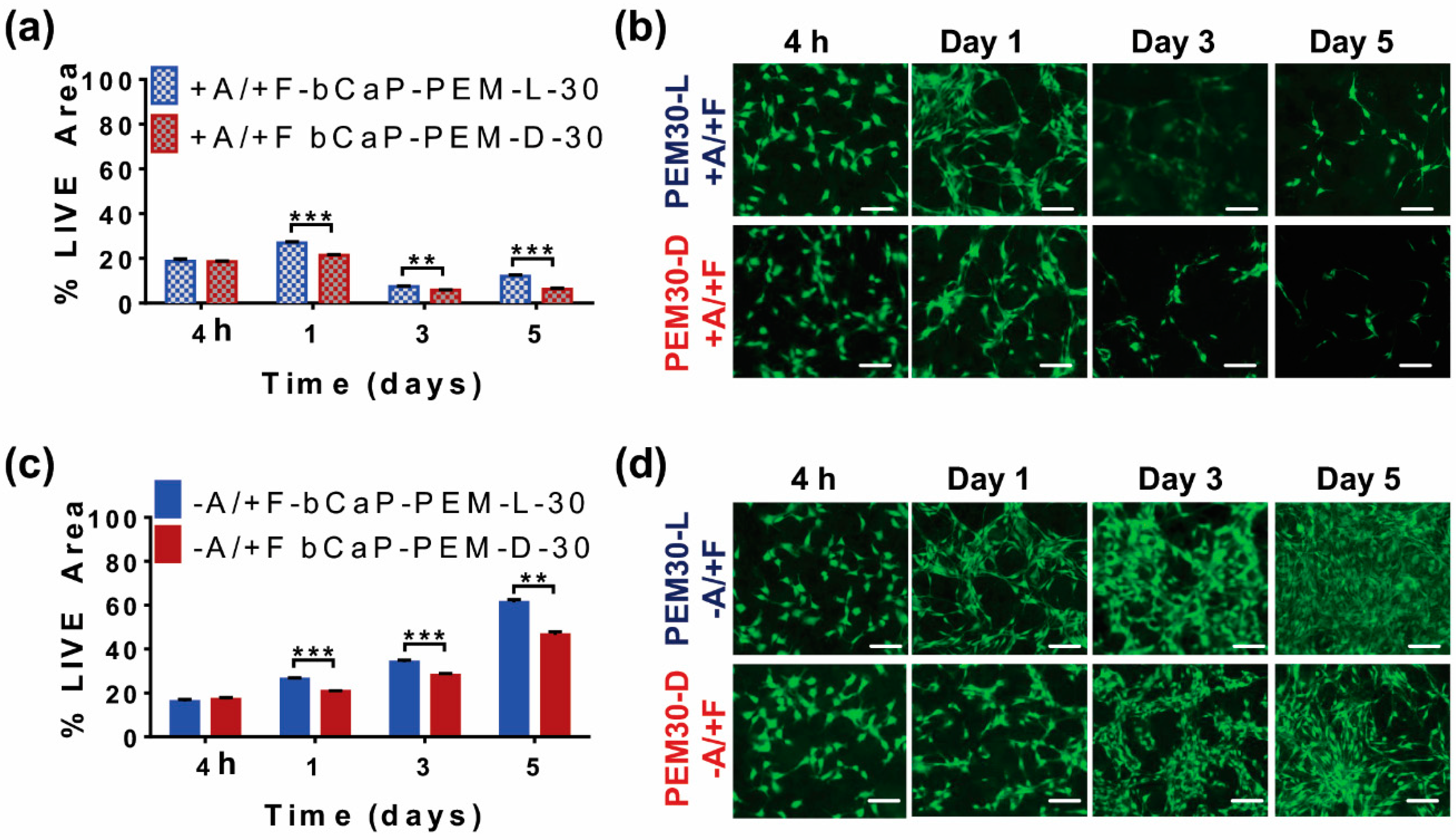
3.1.3. Use of D-Enantiomers: PEM-D
3.2. Characterization of the Coatings bCaP without PEM
3.2.1. Effects of Removing PEM Film
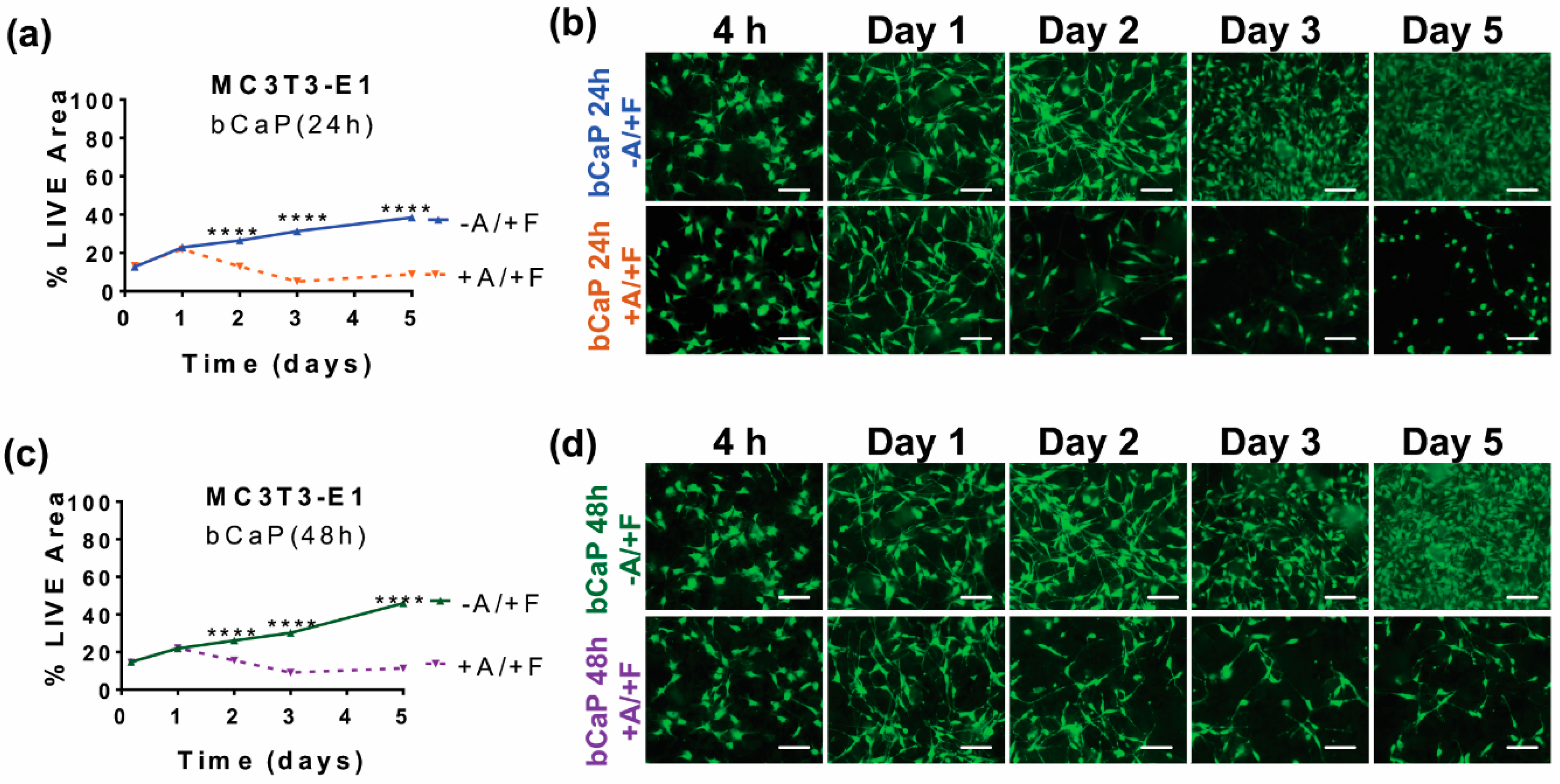
3.2.2. Pulsatile Delivery from bCaP
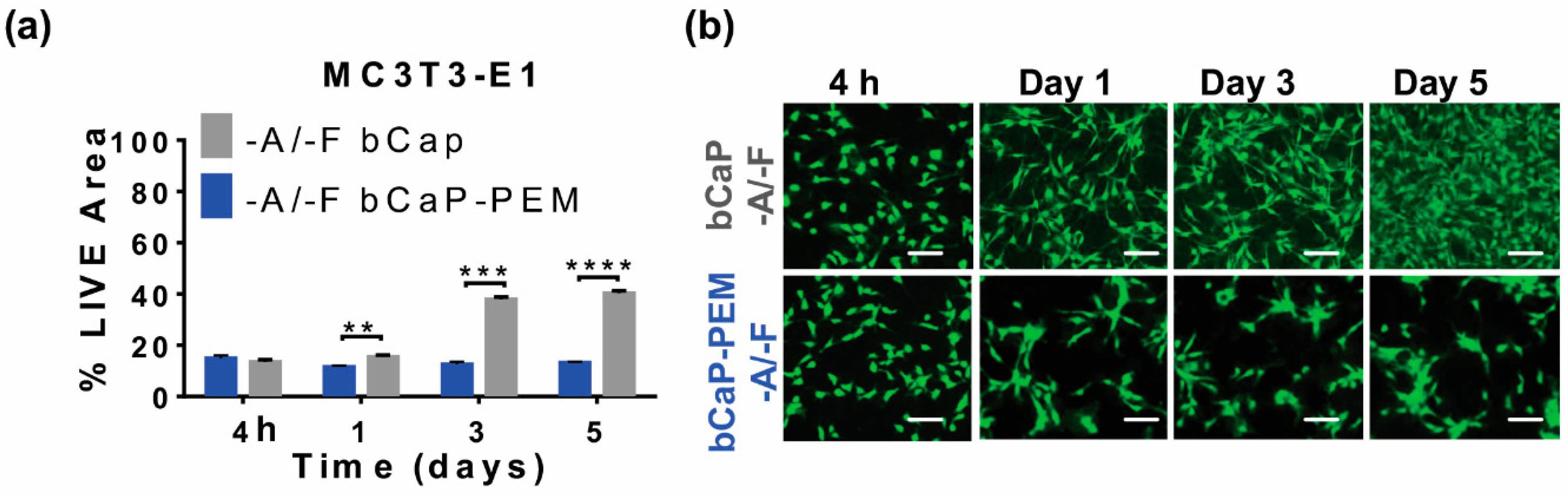
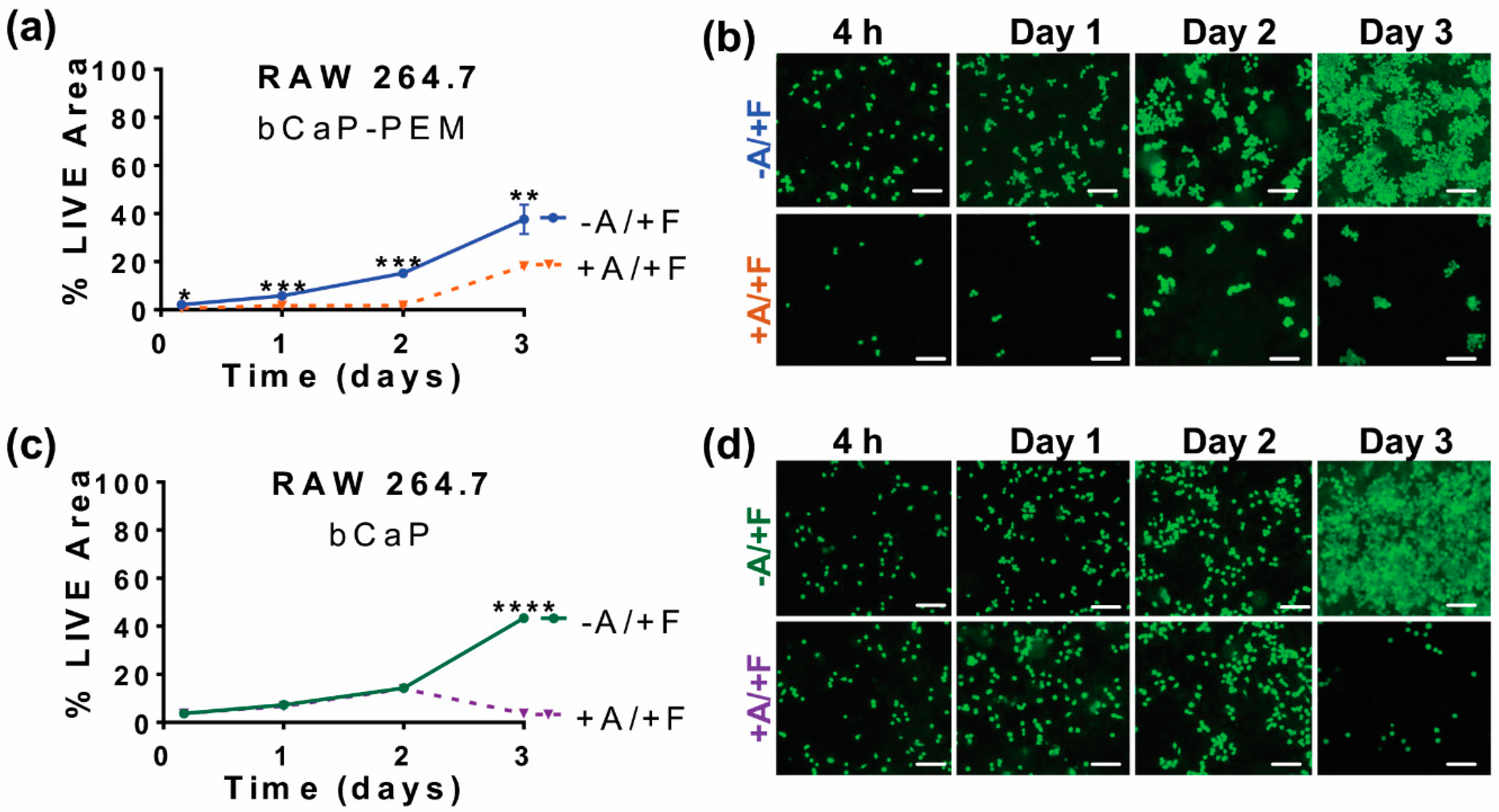
3.3. Studying Macrophages to Mimic Cells Present during Inflammation and Bone Remodeling
3.4. Cells Access Mechanism as Measured by SEM Imaging
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Appendix A




References
- Batoon, L.; Millard, S.M.; Raggatt, L.J.; Pettit, A.R. Osteomacs and bone regeneration. Curr. Osteoporos. Rep. 2017, 15, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Michalski, M.N.; McCauley, L.K. Macrophages and skeletal health. Pharmacol. Ther. 2017, 174, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.C.; Raggatt, L.J.; Alexander, K.A.; Pettit, A.R. Unraveling macrophage contributions to bone repair. BoneKEy Rep. 2013, 2, 373. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Dubon, M.J.; Yu, J.; Choi, S.; Park, K.S. Transforming growth factor beta induces bone marrow mesenchymal stem cell migration via noncanonical signals and N-cadherin. J. Cell. Physiol. 2018, 233, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Xian, L.; Wu, X.; Pang, L.; Lou, M.; Rosen, C.J.; Qiu, T.; Crane, J.; Frassica, F.; Zhang, L.; Rodriguez, J.P.; et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat. Med. 2012, 18, 1095–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, A.; Kousteni, S.; Bisikirska, B.; Shah, J.G.; Manavalan, J.S.; Recker, R.R.; Lappe, J.; Dempster, D.W.; Zhou, H.; McMahon, D.J.; et al. IGF-1 Receptor expression on circulating osteoblast progenitor cells predicts tissue-based bone formation rate and response to teriparatide in premenopausal women with idiopathic osteoporosis. J. Bone Miner. Res. 2017, 32, 1267–1273. [Google Scholar] [CrossRef] [PubMed]
- Lowery, J.W.; Pazin, D.; Intini, G.; Kokabu, S.; Chappuis, V.; Capelo, L.P.; Rosen, V. The role of BMP2 signaling in the skeleton. Crit. Rev. Eukaryot. Gene Expr. 2011, 21, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Fei, Y.; Xiao, L.; Doetschman, T.; Coffin, D.J.; Hurley, M.M. Fibroblast growth factor 2 stimulation of osteoblast differentiation and bone formation is mediated by modulation of the Wnt signaling pathway. J. Biol. Chem. 2011, 286, 40575–40583. [Google Scholar] [CrossRef] [PubMed]
- Montero, A.; Okada, Y.; Tomita, M.; Ito, M.; Tsurukami, H.; Nakamura, T.; Doetschman, T.; Coffin, J.D.; Hurley, M.M. Disruption of the fibroblast growth factor-2 gene results in decreased bone mass and bone formation. J. Clin. Investig. 2000, 105, 1085–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagayasu-Tanaka, T.; Nozaki, T.; Miki, K.; Sawada, K.; Kitamura, M.; Murakami, S. FGF-2 promotes initial osseointegration and enhances stability of implants with low primary stability. Clin. Oral Implants Res. 2017, 28, 291–297. [Google Scholar] [CrossRef] [PubMed]
- El Bialy, I.; Jiskoot, W.; Reza Nejadnik, M. Formulation, delivery and stability of bone morphogenetic proteins for effective bone regeneration. Pharm. Res. 2017, 34, 1152–1170. [Google Scholar] [CrossRef] [PubMed]
- Noh, S.S.; Bhang, S.H.; La, W.G.; Lee, S.; Shin, J.Y.; Ma, Y.J.; Jang, H.K.; Kang, S.; Jin, M.; Park, J.; et al. A dual delivery of substance P and bone morphogenetic protein-2 for mesenchymal stem cell recruitment and bone regeneration. Tissue Eng. Part A 2015, 21, 1275–1287. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.J.; Modes, J.E.; Flanagan, C.L.; Krebsbach, P.H.; Edwards, S.P.; Hollister, S.J. Dual delivery of EPO and BMP2 from a novel modular poly-varepsilon-caprolactone construct to increase the bone bormation in prefabricated bone flaps. Tissue Eng. Part C Methods 2015, 21, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Sharmin, F.; McDermott, C.; Lieberman, J.; Sanjay, A.; Khan, Y. Dual growth factor delivery from biofunctionalized allografts: Sequential VEGF and BMP-2 release to stimulate allograft remodeling. J. Orthop. Res. 2017, 35, 1086–1095. [Google Scholar] [CrossRef] [PubMed]
- Shu, X.; Feng, J.; Feng, J.; Huang, X.; Li, L.; Shi, Q. Combined delivery of bone morphogenetic protein-2 and insulin-like growth factor-1 from nano-poly(gamma-glutamic acid)/beta-tricalcium phosphate-based calcium phosphate cement and its effect on bone regeneration in vitro. J. Biomater. Appl. 2017, 32, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.J.; Han, Z.W.; Duan, K.; Mu, Y.D.; Weng, J. Multilayered pore-closed PLGA microsphere delivering OGP and BMP-2 in sequential release patterns for the facilitation of BMSCs osteogenic differentiation. J. Biomed. Mater. Res. A 2018, 106, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Facca, S.; Ferrand, A.; Mendoza-Palomares, C.; Perrin-Schmitt, F.; Netter, P.; Mainard, D.; Liverneaux, P.; Benkirane-Jessel, N. Bone formation induced by growth factors embedded into the nanostructured particles. J. Biomed. Nanotechnol. 2011, 7, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Begam, H.; Nandi, S.K.; Kundu, B.; Chanda, A. Strategies for delivering bone morphogenetic protein for bone healing. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 70, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Gronowicz, G.; Jacobs, E.; Peng, T.; Zhu, L.; Hurley, M.; Kuhn, L.T. Calvarial bone regeneration is enhanced by sequential delivery of FGF-2 and BMP-2 from Layer-by-Layer coatings with a biomimetic calcium phosphate barrier layer. Tissue Eng. Part A 2017, 23, 1490–1501. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, E.E.; Gronowicz, G.; Hurley, M.M.; Kuhn, L.T. Biomimetic calcium phosphate/polyelectrolyte multilayer coatings for sequential delivery of multiple biological factors. J. Biomed. Mater. Res. A 2017, 105, 1500–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulmedais, F.; Frisch, B.; Etienne, O.; Lavalle, P.; Picart, C.; Ogier, J.; Voegel, J.C.; Schaaf, P.; Egles, C. Polyelectrolyte multilayer films with pegylated polypeptides as a new type of anti-microbial protection for biomaterials. Biomaterials 2004, 25, 2003–2011. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Shah, N.J.; Drake, A.C.; DeMuth, P.C.; Lee, J.B.; Chen, J.; Hammond, P.T. Graphene multilayers as gates for multi-week sequential release of proteins from surfaces. ACS Nano 2012, 6, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Diamanti, E.; Muzzio, N.; Gregurec, D.; Irigoyen, J.; Pasquale, M.; Azzaroni, O.; Brinkmann, M.; Moya, S.E. Impact of thermal annealing on wettability and antifouling characteristics of alginate poly-l-lysine polyelectrolyte multilayer films. Colloids Surf. B Biointerfaces 2016, 145, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.J.; Kang, H.; Kim, Y.H.; Han, S.; Yoo, P.J. Layer-by-layer assembly of polyelectrolyte multilayers in three-dimensional inverse opal structured templates. ACS Appl. Mater. Interfaces 2012, 4, 2107–2115. [Google Scholar] [CrossRef] [PubMed]
- Salvi, C.; Lyu, X.; Peterson, A.M. Effect of assembly pH on polyelectrolyte multilayer surface properties and BMP-2 release. Biomacromolecules 2016, 17, 1949–1958. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.M.; Caridade, S.G.; Costa, R.R.; Alves, N.M.; Groth, T.; Picart, C.; Reis, R.L.; Mano, J.F. pH Responsiveness of multilayered films and membranes made of polysaccharides. Langmuir ACS J. Surf. Colloids 2015, 31, 11318–11328. [Google Scholar] [CrossRef] [PubMed]
- Crouzier, T.; Sailhan, F.; Becquart, P.; Guillot, R.; Logeart-Avramoglou, D.; Picart, C. The performance of BMP-2 loaded TCP/HAP porous ceramics with a polyelectrolyte multilayer film coating. Biomaterials 2011, 32, 7543–7554. [Google Scholar] [CrossRef] [PubMed]
- Guzman, E.; Mateos-Maroto, A.; Ruano, M.; Ortega, F.; Rubio, R.G. Layer-by-Layer polyelectrolyte assemblies for encapsulation and release of active compounds. Adv. Colloid Interface Sci. 2017, 249, 290–307. [Google Scholar] [CrossRef] [PubMed]
- Picart, C.; Mutterer, J.; Richert, L.; Luo, Y.; Prestwich, G.D.; Schaaf, P.; Voegel, J.C.; Lavalle, P. Molecular basis for the explanation of the exponential growth of polyelectrolyte multilayers. Proc. Natl. Acad. Sci. USA 2002, 99, 12531–12535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsell, R.; Einhorn, T.A. The biology of fracture healing. Injury 2011, 42, 551–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claes, L.; Recknagel, S.; Ignatius, A. Fracture healing under healthy and inflammatory conditions. Nat. Rev. Rheumatol. 2012, 8, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Sokic, S.; Christenson, M.C.; Larson, J.C.; Appel, A.A.; Brey, E.M.; Papavasiliou, G. Evaluation of MMP substrate concentration and specificity for neovascularization of hydrogel scaffolds. Biomater. Sci. 2014, 2, 1343–1354. [Google Scholar] [CrossRef] [PubMed]
- Hunziker, E.B.; Jovanovic, J.; Horner, A.; Keel, M.J.; Lippuner, K.; Shintani, N. Optimisation of BMP-2 dosage for the osseointegration of porous titanium implants in an ovine model. Eur. Cell Mater. 2016, 32, 241–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Wu, G.; Zheng, Y.; Wismeijer, D.; Everts, V.; Liu, Y. Cell-mediated BMP-2 release from a novel dual-drug delivery system promotes bone formation. Clin. Oral Implants Res. 2014, 25, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.J.; Liu, Y.; Advincula, M.C.; Gronowicz, G.; Habibovic, P.; Kuhn, L.T. Fabrication and characterization of hydroxyapatite-coated polystyrene disks for use in osteoprogenitor cell culture. J. Biomater. Sci. Polym. Ed. 2010, 21, 1371–1387. [Google Scholar] [CrossRef] [PubMed]
- Barrere, F.; Layrolle, P.; Van Blitterswijk, C.A.; De Groot, K. Biomimetic coatings on titanium: A crystal growth study of octacalcium phosphate. J. Mater. Sci. Mater. Med. 2001, 12, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Habibovic, P.; Barrere, F.; Van Blitterswijk, C.A.; de Groot, K.; Layrolle, P. Biomimetic hydroxyapatite coating on metal implants. J. Am. Ceram. Soc. 2002, 85, 517–522. [Google Scholar] [CrossRef]
- Barrere, F.; van Blitterswijk, C.A.; de Groot, K.; Layrolle, P. Influence of ionic strength and carbonate on the Ca-P coating formation from SBFx5 solution. Biomaterials 2002, 23, 1921–1930. [Google Scholar] [CrossRef]
- Allen, A.M.; Graham, A. Mitochondrial function is involved in regulation of cholesterol efflux to apolipoprotein (apo)A-I from murine RAW 264.7 macrophages. Lipids Health Dis. 2012, 11, 169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Facca, S.; Cortez, C.; Mendoza-Palomares, C.; Messadeq, N.; Dierich, A.; Johnston, A.P.; Mainard, D.; Voegel, J.C.; Caruso, F.; Benkirane-Jessel, N. Active multilayered capsules for in vivo bone formation. Proc. Natl. Acad. Sci. USA 2010, 107, 3406–3411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkirane-Jessel, N.; Lavalle, P.; HüBSCH, E.; Holl, V.; Senger, B.; Haïkel, Y.; Voegel, J.C.; Ogier, J.; Schaaf, P. Short-time tuning of the biological activity of functionalized polyelectrolyte multilayers. Adv. Funct. Mater. 2005, 15, 648–654. [Google Scholar] [CrossRef]
- Fischer, D.; Li, Y.; Ahlemeyer, B.; Krieglstein, J.; Kissel, T. In vitro cytotoxicity testing of polycations: Influence of polymer structure on cell viability and hemolysis. Biomaterials 2003, 24, 1121–1131. [Google Scholar] [CrossRef]
- Arnold, L.J., Jr.; Dagan, A.; Gutheil, J.; Kaplan, N.O. Antineoplastic activity of poly(l-lysine) with some ascites tumor cells. Proc. Natl. Acad. Sci. USA 1979, 76, 3246–3250. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Thompson, D.L.; Hellenbrand, D.J.; Lee, J.S.; Madura, C.J.; Wesley, M.G.; Dillon, N.J.; Sharma, T.; Enright, C.J.; Murphy, W.L. Sustained release of neurotrophin-3 via calcium phosphate-coated sutures promotes axonal regeneration after spinal cord injury. J. Neurosci. Res. 2016, 94, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Lu, Y.; Baer, G.S.; Markel, M.D.; Murphy, W.L. Controllable protein delivery from coated surgical sutures. J. Mater. Chem. 2010, 20, 8894–8903. [Google Scholar] [CrossRef]
- Jongpaiboonkit, L.; Franklin-Ford, T.; Murphy, W.L. Mineral-coated polymer microspheres for controlled protein binding and release. Adv. Mater. 2009, 21, 1960–1963. [Google Scholar] [CrossRef]
- Leuschner, F.; Dutta, P.; Gorbatov, R.; Novobrantseva, T.I.; Donahoe, J.S.; Courties, G.; Lee, K.M.; Kim, J.I.; Markmann, J.F.; Marinelli, B.; et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat. Biotechnol. 2011, 29, 1005–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, B.N.; Londono, R.; Tottey, S.; Zhang, L.; Kukla, K.A.; Wolf, M.T.; Daly, K.A.; Reing, J.E.; Badylak, S.F. Macrophage phenotype as a predictor of constructive remodeling following the implantation of biologically derived surgical mesh materials. Acta Biomater. 2012, 8, 978–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiller, K.L.; Anfang, R.R.; Spiller, K.J.; Ng, J.; Nakazawa, K.R.; Daulton, J.W.; Vunjak-Novakovic, G. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials 2014, 35, 4477–4488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richert, L.; Schneider, A.; Vautier, D.; Vodouhe, C.; Jessel, N.; Payan, E.; Schaaf, P.; Voegel, J.C.; Picart, C. Imaging cell interactions with native and crosslinked polyelectrolyte multilayers. Cell Biochem. Biophys. 2006, 44, 273–285. [Google Scholar] [CrossRef]
- Sheikh, Z.; Abdallah, M.N.; Hanafi, A.A.; Misbahuddin, S.; Rashid, H.; Glogauer, M. Mechanisms of in vivo degradation and resorption of calcium phosphate based biomaterials. Materials 2015, 8, 7913–7925. [Google Scholar] [CrossRef] [PubMed]
- Dar, H.Y.; Azam, Z.; Anupam, R.; Mondal, R.K.; Srivastava, R.K. Osteoimmunology: The nexus between bone and immune system. Front. Biosci. 2018, 23, 464–492. [Google Scholar]
- Okamoto, K.; Nakashima, T.; Shinohara, M.; Negishi-Koga, T.; Komatsu, N.; Terashima, A.; Sawa, S.; Nitta, T.; Takayanagi, H. Osteoimmunology: The conceptual framework unifying the immune and skeletal systems. Physiol. Rev. 2017, 97, 1295–1349. [Google Scholar] [CrossRef] [PubMed]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alhamdi, J.; Jacobs, E.; Gronowicz, G.; Benkirane-Jessel, N.; Hurley, M.; Kuhn, L. Cell Type Influences Local Delivery of Biomolecules from a Bioinspired Apatite Drug Delivery System. Materials 2018, 11, 1703. https://doi.org/10.3390/ma11091703
Alhamdi J, Jacobs E, Gronowicz G, Benkirane-Jessel N, Hurley M, Kuhn L. Cell Type Influences Local Delivery of Biomolecules from a Bioinspired Apatite Drug Delivery System. Materials. 2018; 11(9):1703. https://doi.org/10.3390/ma11091703
Chicago/Turabian StyleAlhamdi, Jumana, Emily Jacobs, Gloria Gronowicz, Nadia Benkirane-Jessel, Marja Hurley, and Liisa Kuhn. 2018. "Cell Type Influences Local Delivery of Biomolecules from a Bioinspired Apatite Drug Delivery System" Materials 11, no. 9: 1703. https://doi.org/10.3390/ma11091703