Comparative Physicochemical and Electrochemical Characterization of the Structure and Composition of Thin Pd Binary and Ternary Codeposits with Pt, Ru, and Rh

 , and
, and 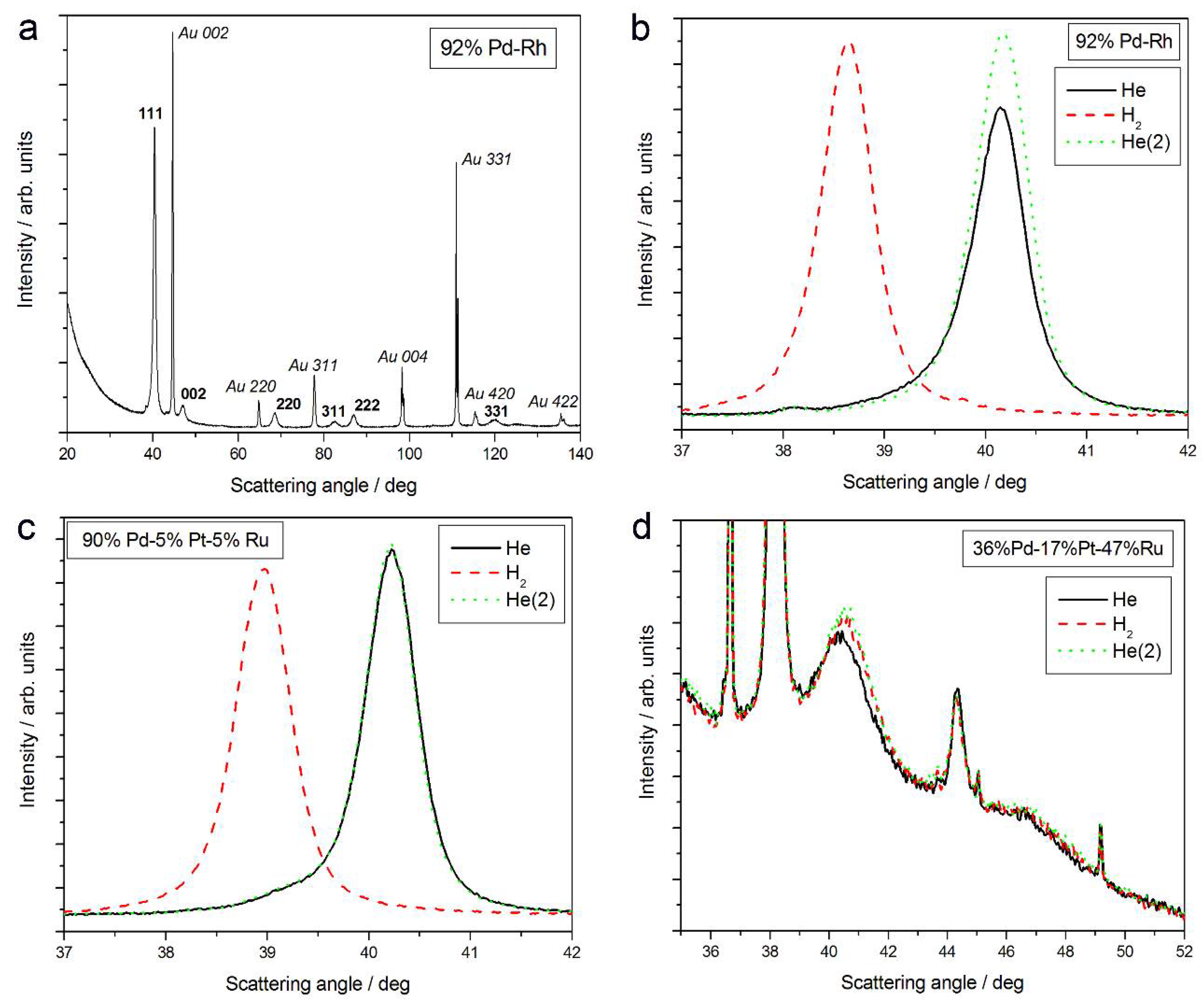{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
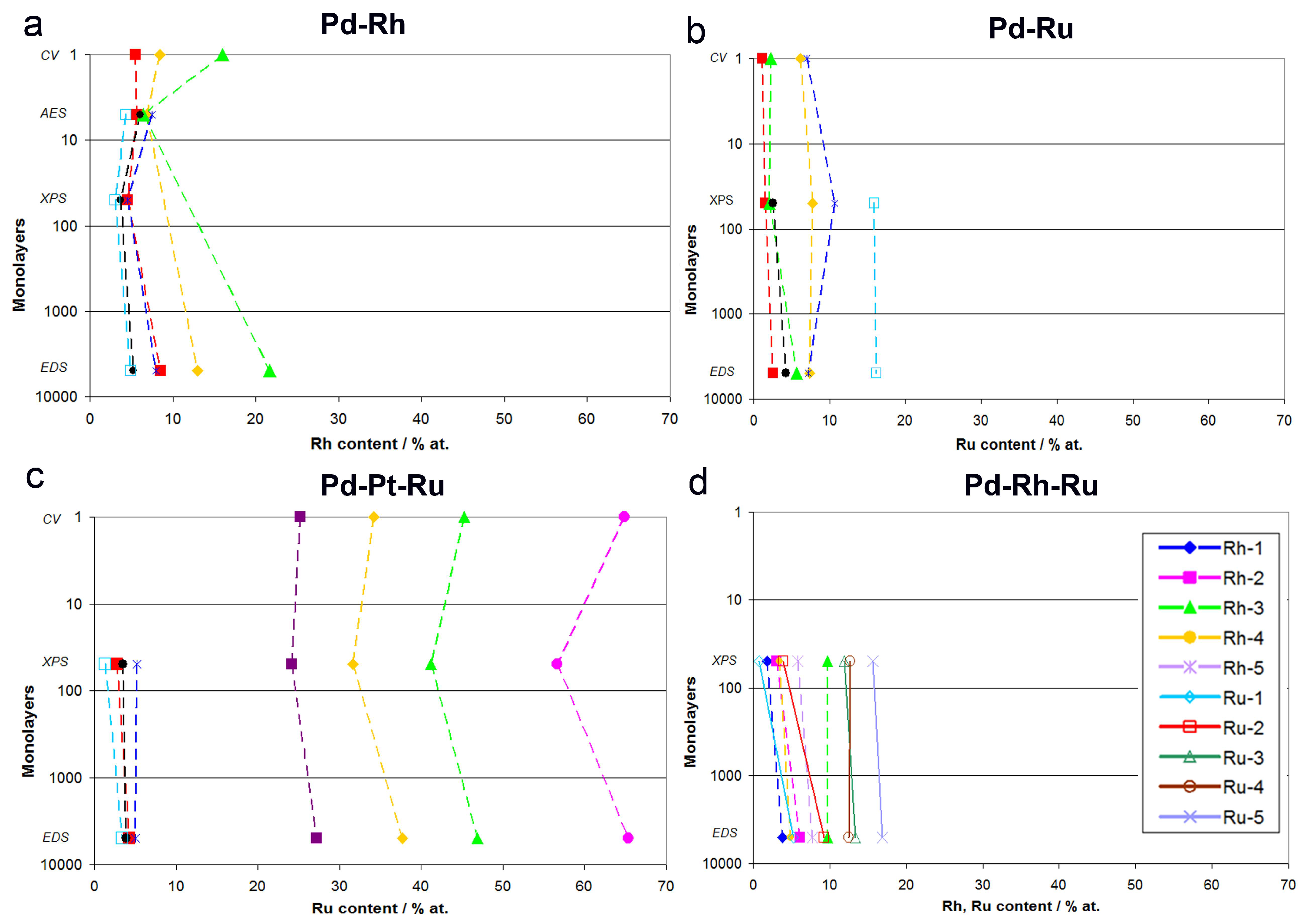
3.1. Structural Characterization
- (1)
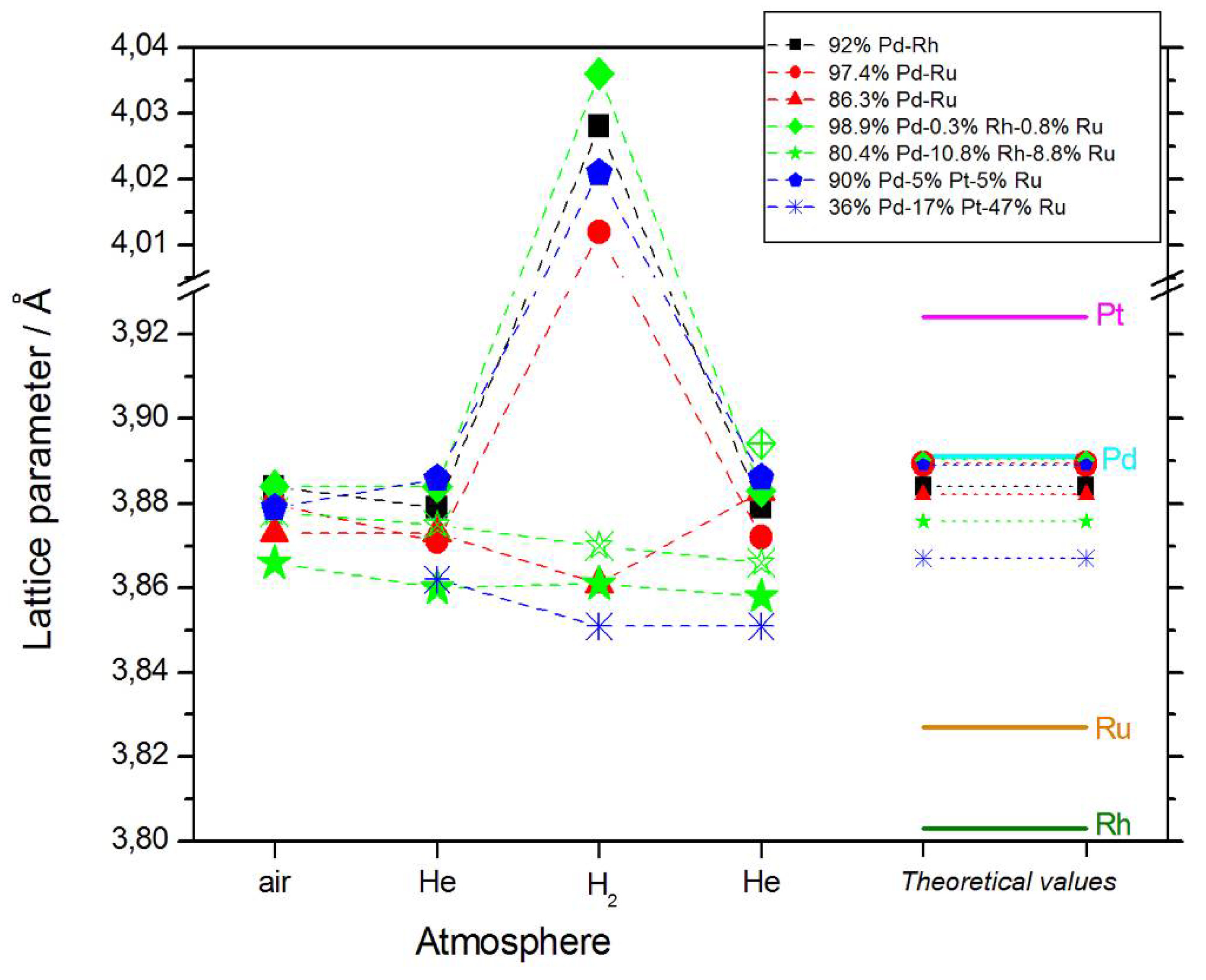
- In most cases, lattice parameters of fresh deposits are close to those predicted by Vegard’s law;
- (2)
- Under hydrogen atmosphere, the lattice parameter for Pd-rich samples considerably increases, while for the samples poorer in Pd, it almost does not alter, or even slightly decreases;
- (3)
- Lattice parameter in vacuum or in helium before or after exposure to hydrogen is often slightly smaller than that in air.
- (1)
- The good agreement with Vegard’s law suggests that the alloy phase is the only one present in the samples, i.e., that the deposits are highly homogeneous. However, the literature data show that such a conclusion should be drawn with care (see Januszewska et al. [65] and the references therein). In particular, there are evidences that even in homogenous systems, the deviations from Vegard’s law are observed, and vice versa, Vegard’s law can be very well fulfilled in the case of heterogeneous materials.
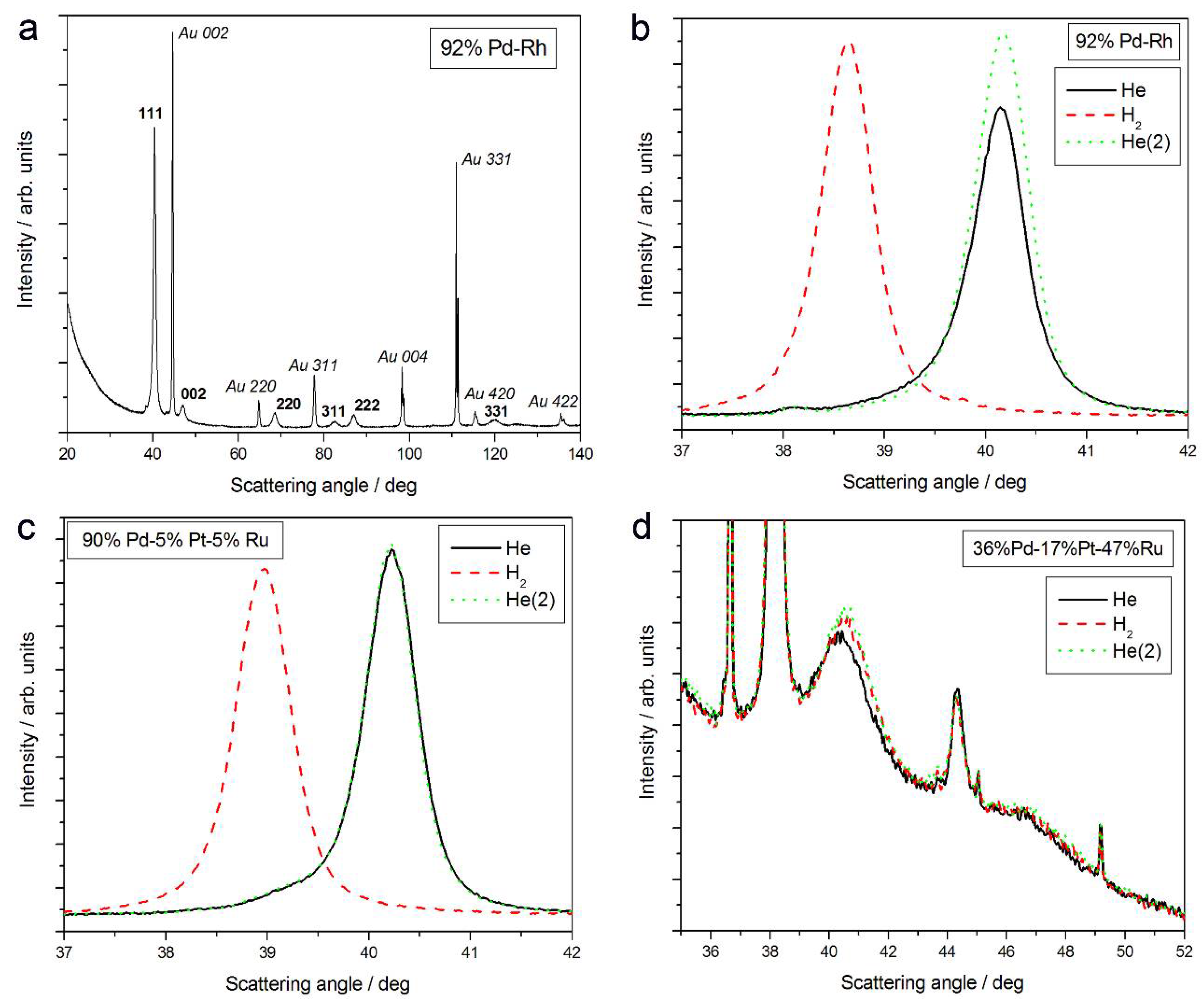
- (2)
- The magnitude of changes in lattice parameter under hydrogen atmosphere depends on the fact whether a given sample can absorb hydrogen or not. This is illustrated in Figure 1c,d, comparing XRD patterns for two Pd-Pt-Ru samples with different abilities to absorb hydrogen. While for a Pd-rich (ca. 95% Pd in the bulk) Pd-Pt-Ru sample, lattice parameter increases under hydrogen atmosphere and the diffraction lines are shifted negatively, in the case of the sample containing ca. 36% Pd in the bulk, no expansion of the crystal lattice occurs, as mirrored by no alterations of XRD spectrum. The constant position of signals in XRD spectra during the exposure to gaseous hydrogen is typical of those Pd-based deposits which contain too little Pd in the bulk for hydrogen absorption in the β-phase to occur [9]. In the presence of only negligible amounts of hydrogen absorbed in the α-phase, the lattice parameter increases very weakly in comparison to that of a non-hydrogenated material [31,32], and therefore its slight change is not detected by XRD. It should be added that lattice parameter for the hydride phase in the Pd-Rh alloys containing ca. 92 at % Pd in the bulk (4.028–4.043 Å) is even greater than that for Pd hydride (4.025 Å), which is consistent with the literature [31].
- (3)
- The small decrease in lattice parameter after pumping out the air and then after contact with hydrogen could indicate changes in the composition of the alloy phases. For instance, using Vegard’s law, one obtains the alloy bulk composition as ca. 92% Pd-Rh for the fresh alloy, and ca. 86% Pd-Rh after hydrogen absorption/desorption. The former value is in a very good agreement with atomic emission spectroscopy data. The small decrease in lattice parameter, converted via Vegard’s law into the apparent alloy enrichment with Rh, can be explained taking into account the following possibilities [64]: (i) An irreversible surface segregation accelerated by superabundant vacancy formation (SAV) [66] that may accompany hydrogen absorption, and (ii) an additional reduction of small amounts of Rh and/or Ru surface oxides (present on the surface after contact with the deposition bath) to the metallic form by atomic hydrogen taking part in the absorption process.
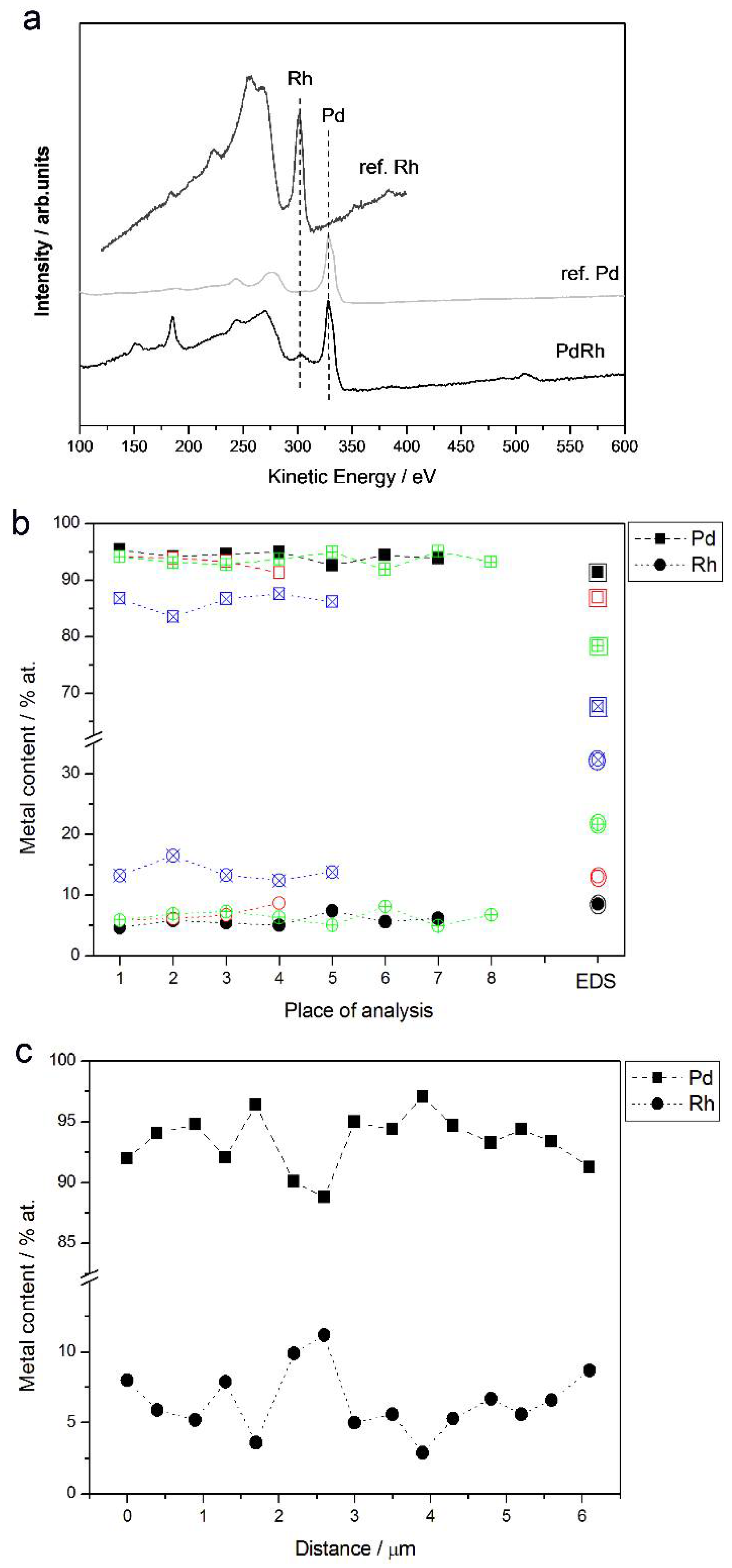
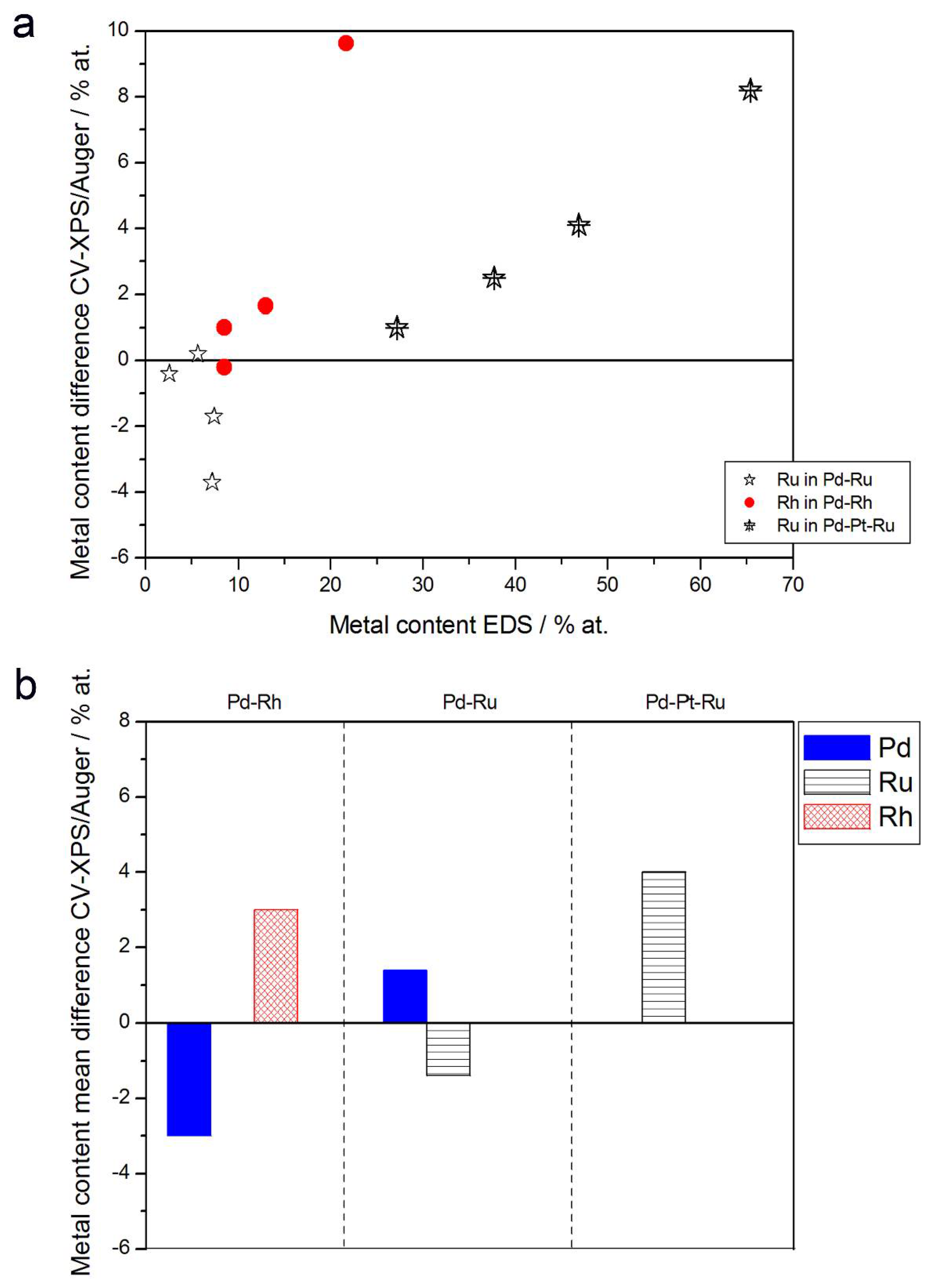
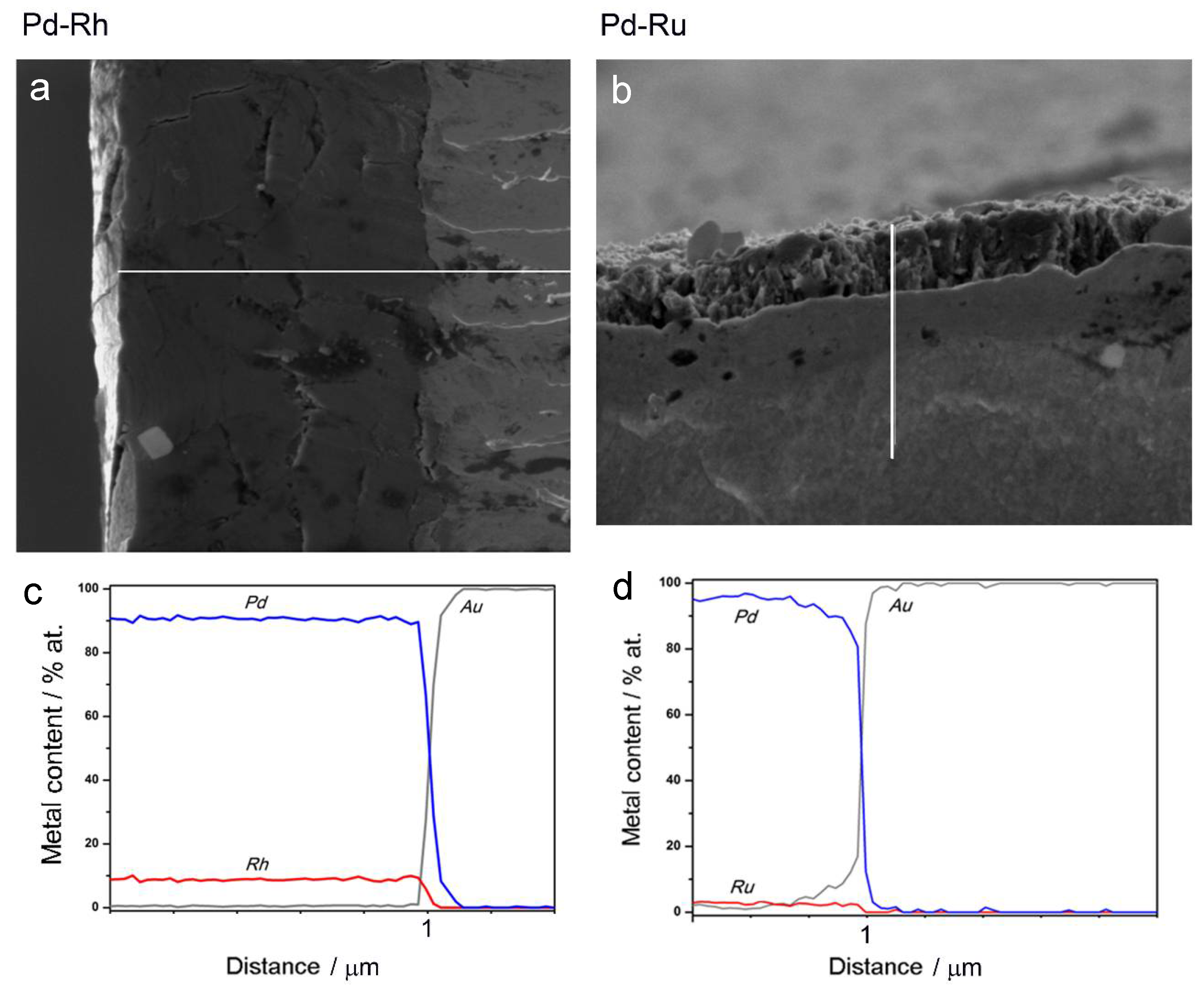
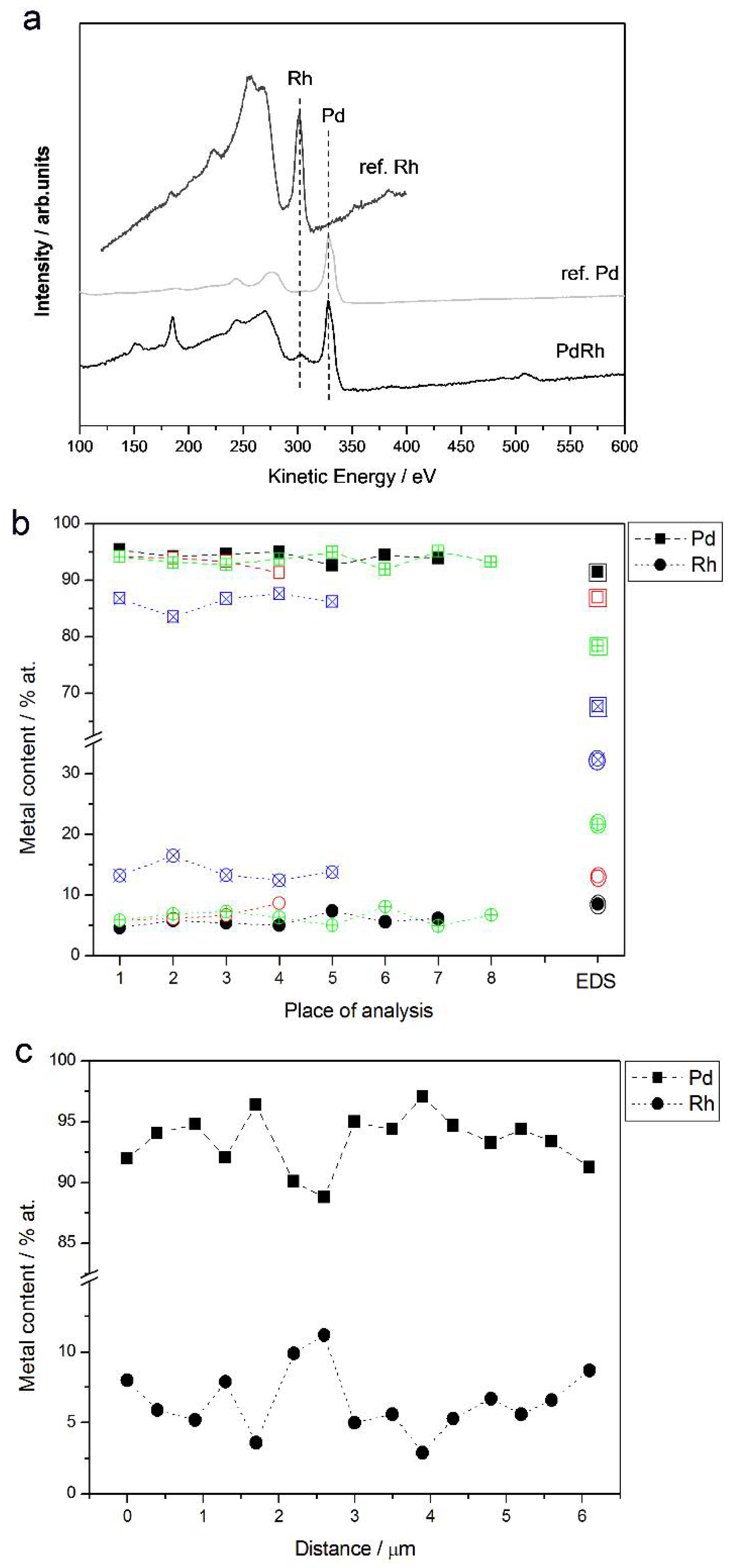
3.2. Characterization by Spectroscopic Techniques
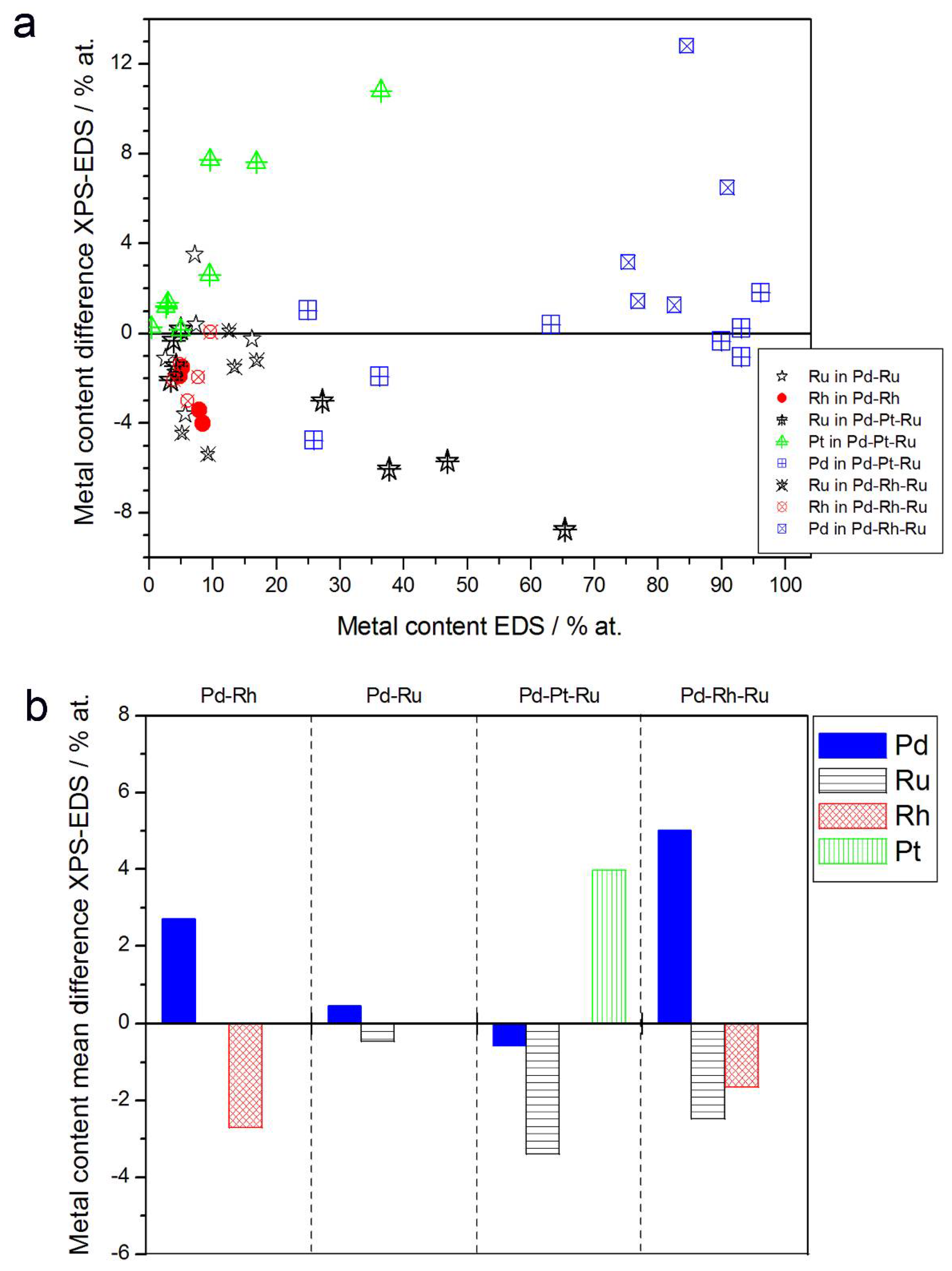
- (1)
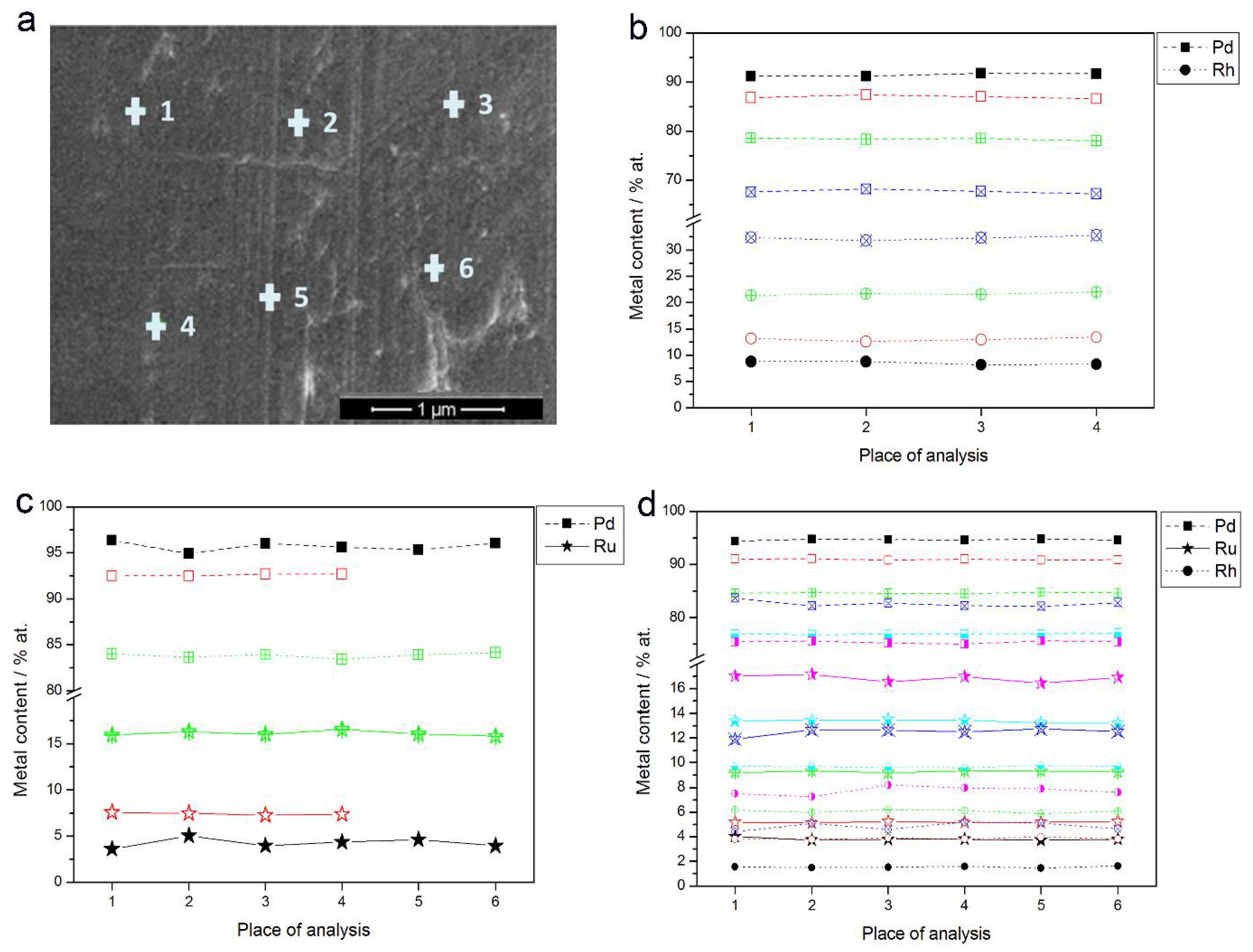
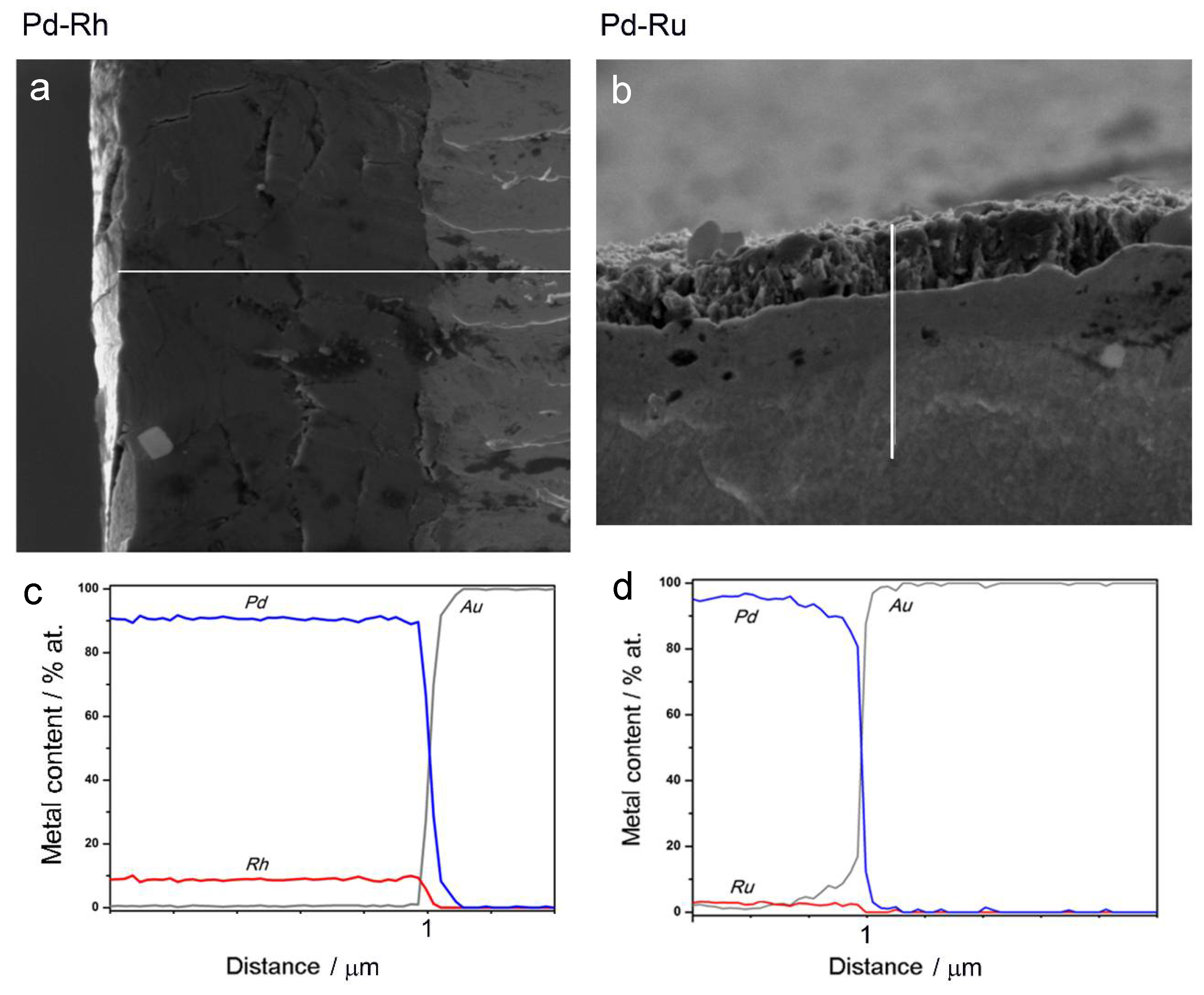
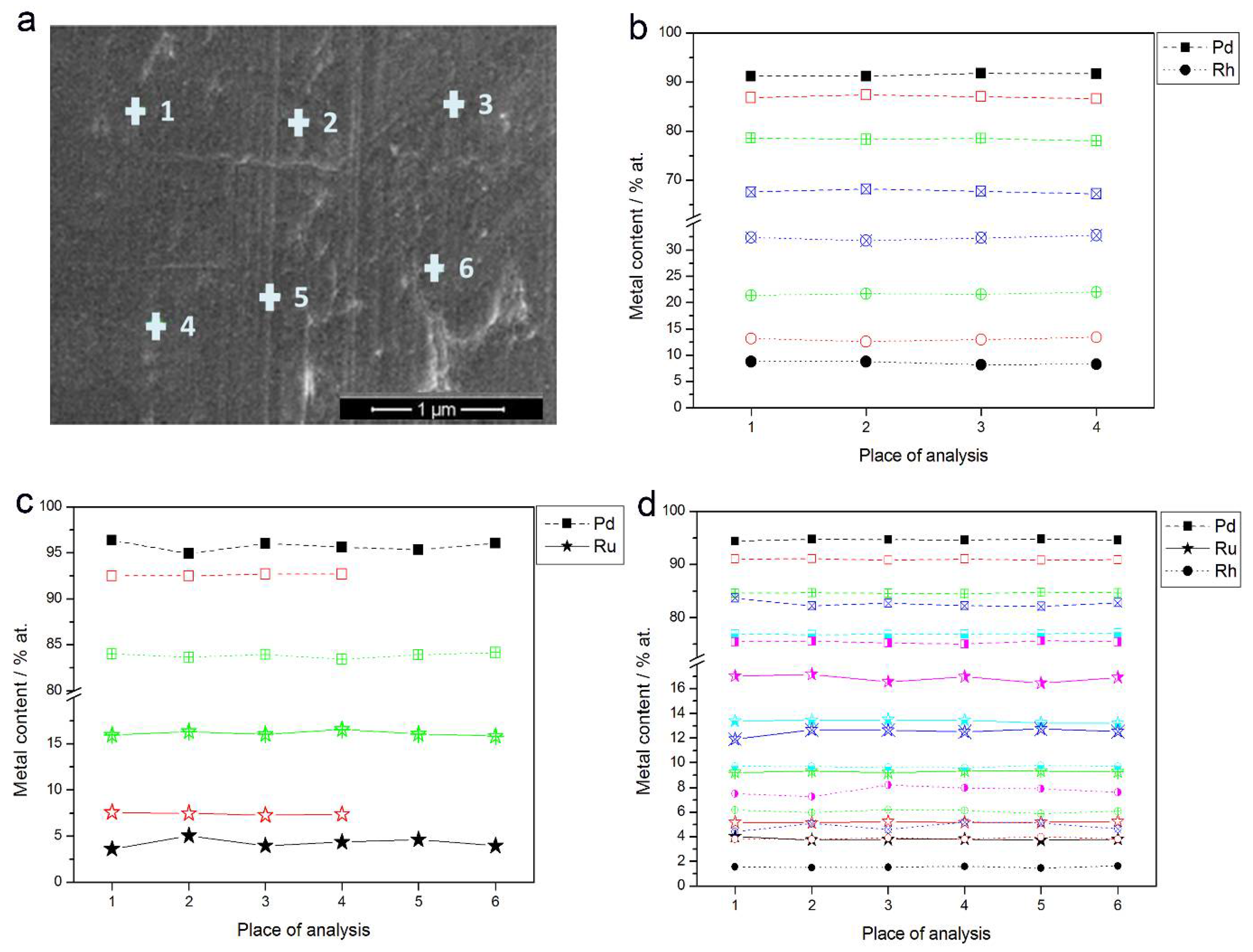
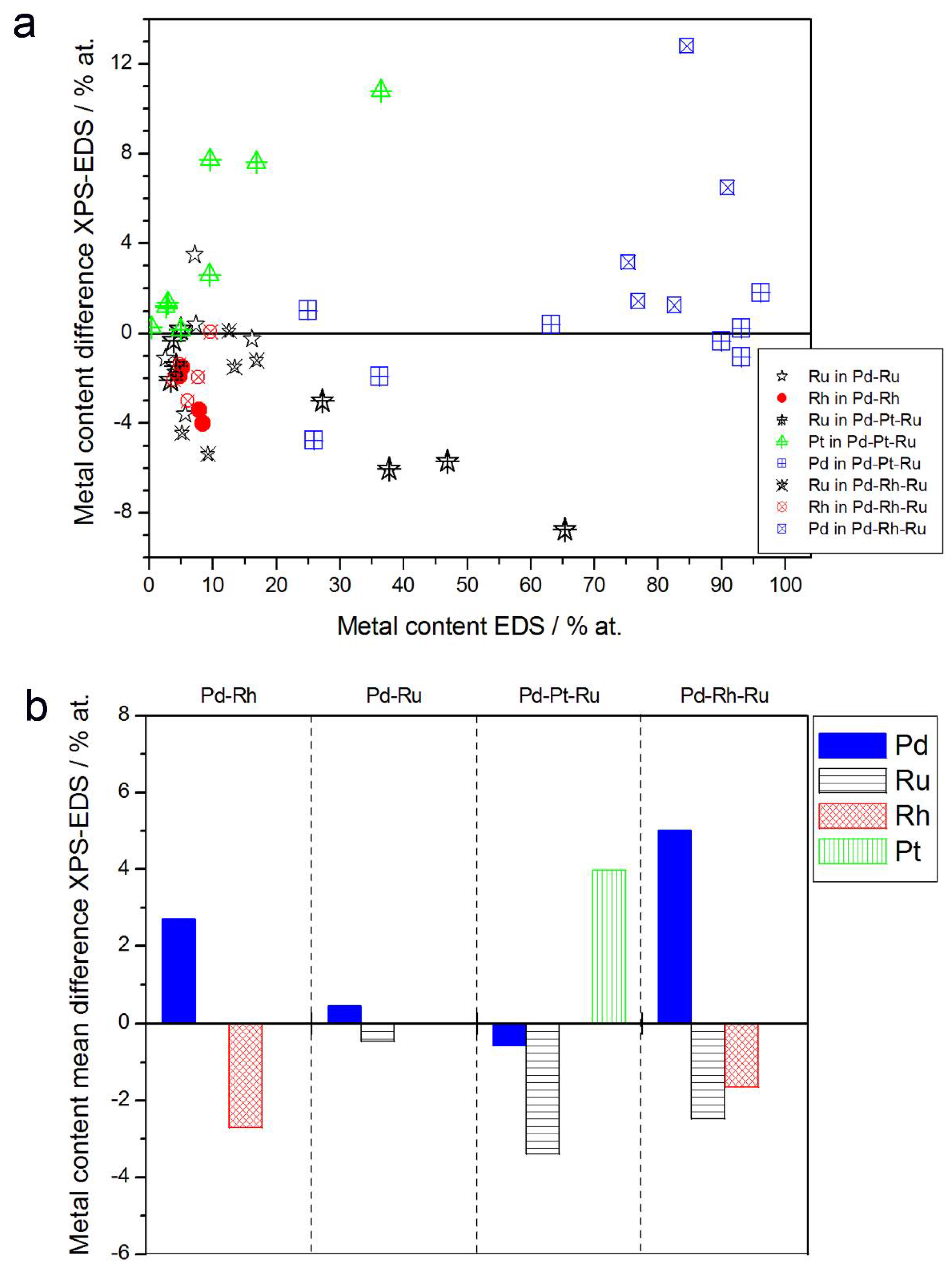
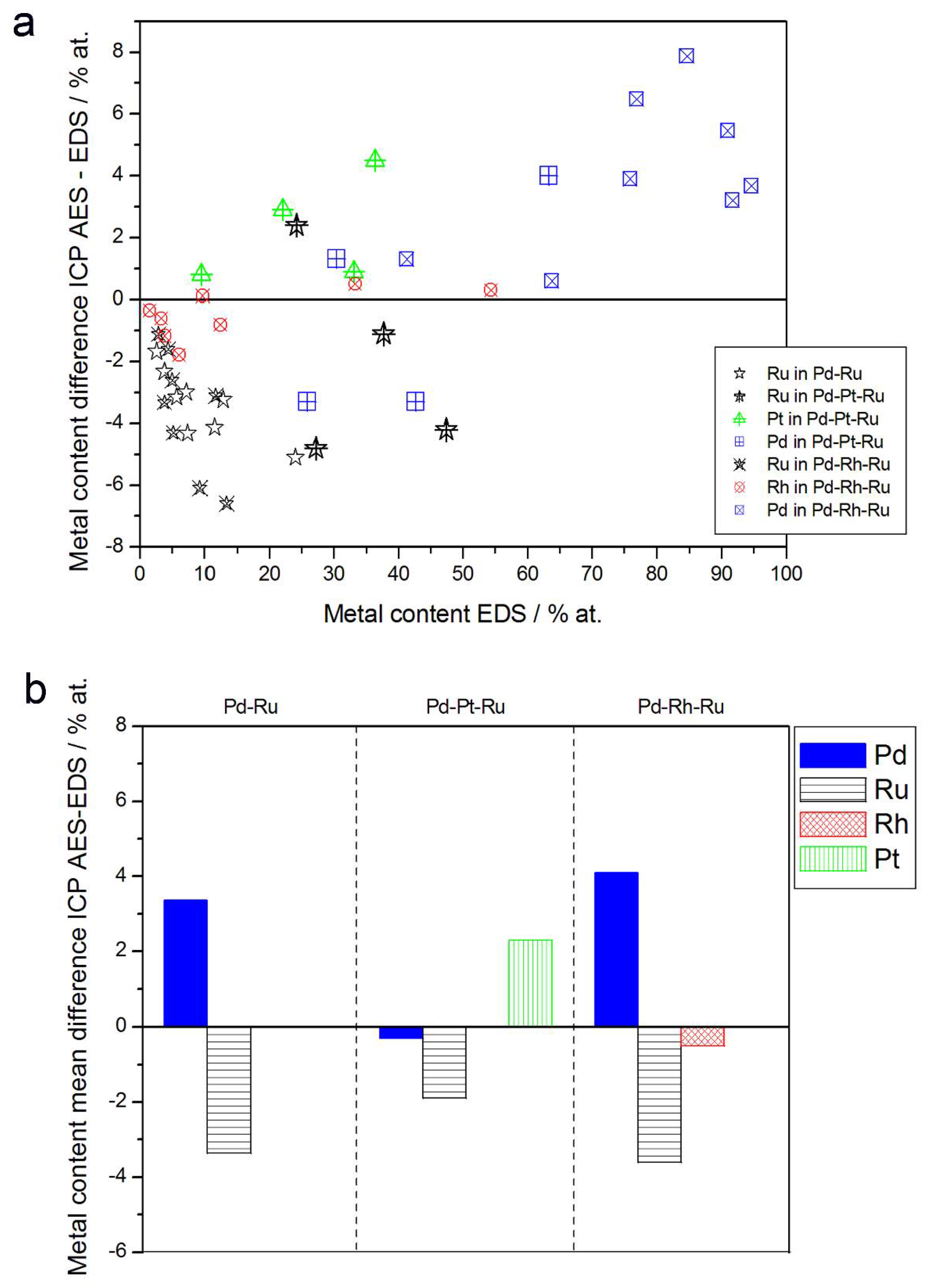
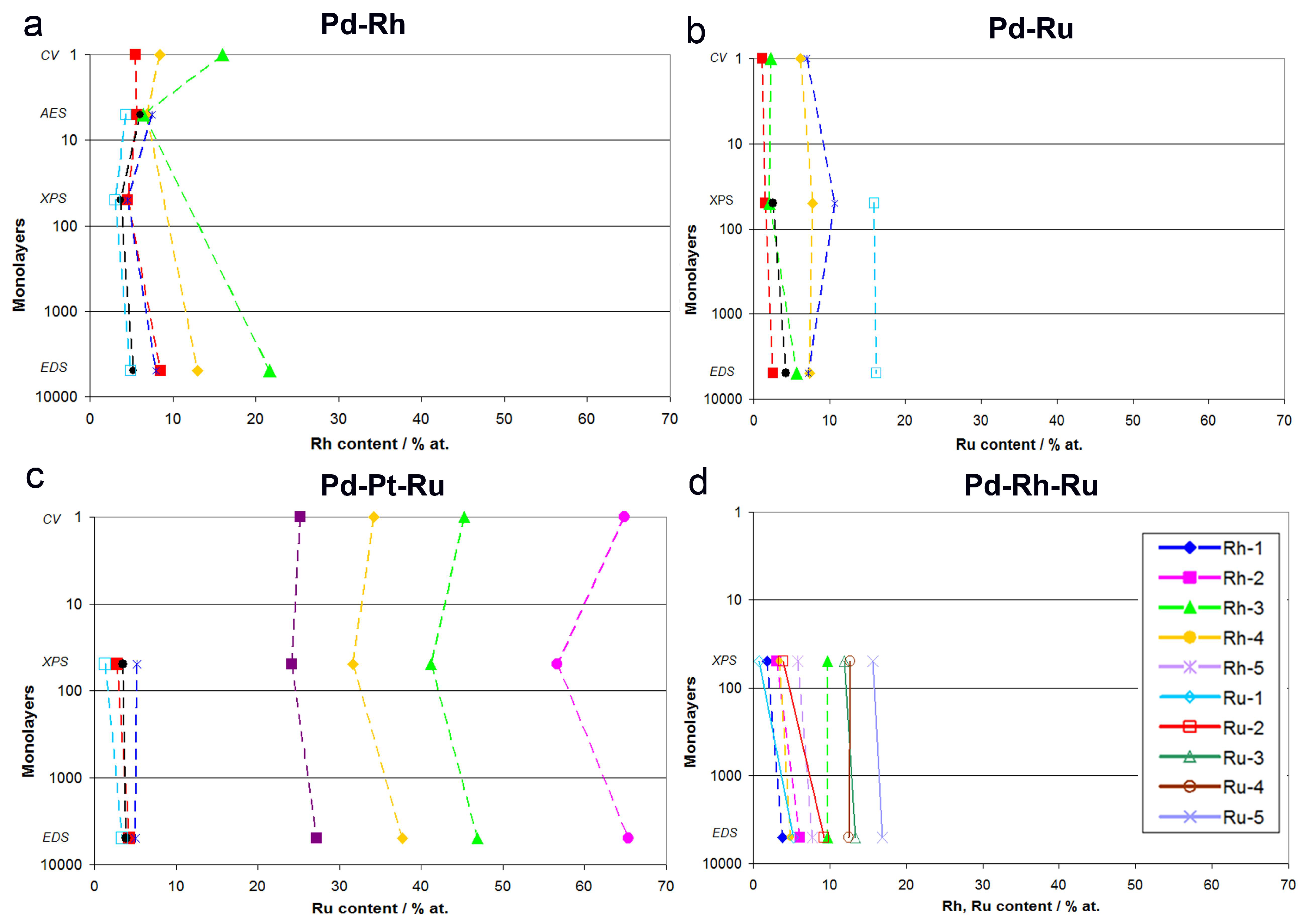
- In the case of Pd-rich (>85% Pd in the bulk) binary Pd-Ru deposits, it is difficult to find a distinct, unequivocal tendency in the relation between bulk and surface/subsurface content of the metals. For individual Pd-Ru samples, a rather large scatter of data is observed within ±4%, and the mean difference between the metal contents derived from XPS and EDS does not exceed ±0.5 at %;
- (2)
- In case of Pd-rich (>90% Pd in the bulk) binary Pd-Rh deposits, XPS detected ca. 1.5–4% less Rh than EDS (a difference mean value ca. −2.7 at %);
- (3)
- A systematic excess of Pd content in XPS vs. EDS data (a difference mean value ca. +5 at %) at the expense of Rh and Ru is observed in the case of Pd-rich (>80% in the bulk) Pd-Rh-Ru deposits;
- (4)
- Ru content determined from XPS data is smaller than that obtained by EDS for Pd-Pt-Ru and Pd-Rh-Ru electrodes. For individual Pd-Pt-Ru samples, the depletion with Ru visible by XPS becomes greater with increasing Ru bulk content, reaching ca. −8.7 at % for a sample containing 65% Ru in the bulk. The mean differences between both sets of data are ca. −3.7 at % (and almost −6 at % for Pd-Pt-Ru samples containing more than 25% Ru and less than 65% Pd in the bulk) and −2.5 at %, respectively;
- (5)
- In the case of Pd-Pt-Ru samples, much more Pt is indicated by XPS than by EDS, i.e., by up to +11 at % individually and ca. +4 at % on the average (and even +6 at % for Pd-Pt-Ru samples containing less than 65 at % Pd in the bulk).
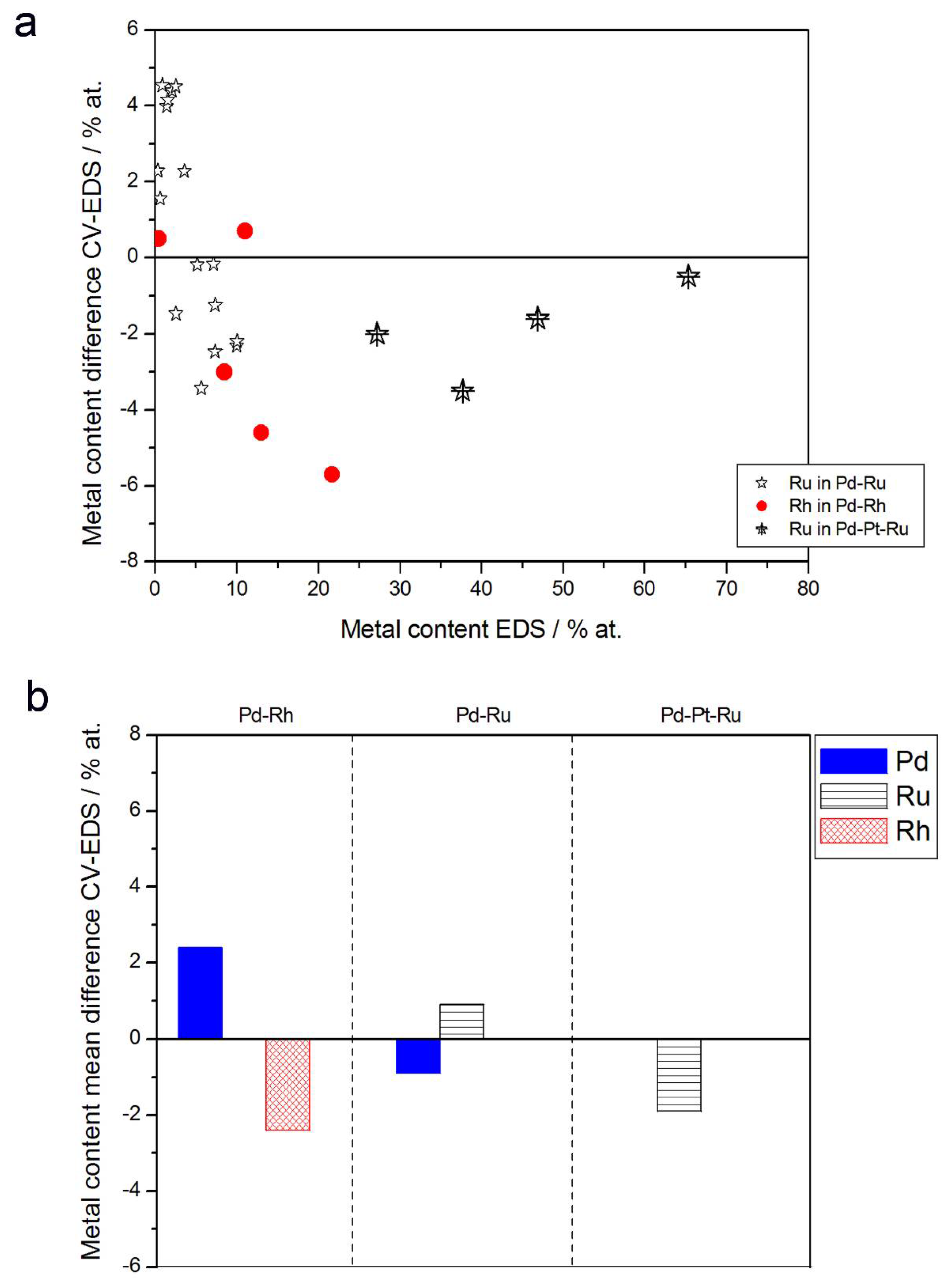
3.3. Electrochemical Characterization
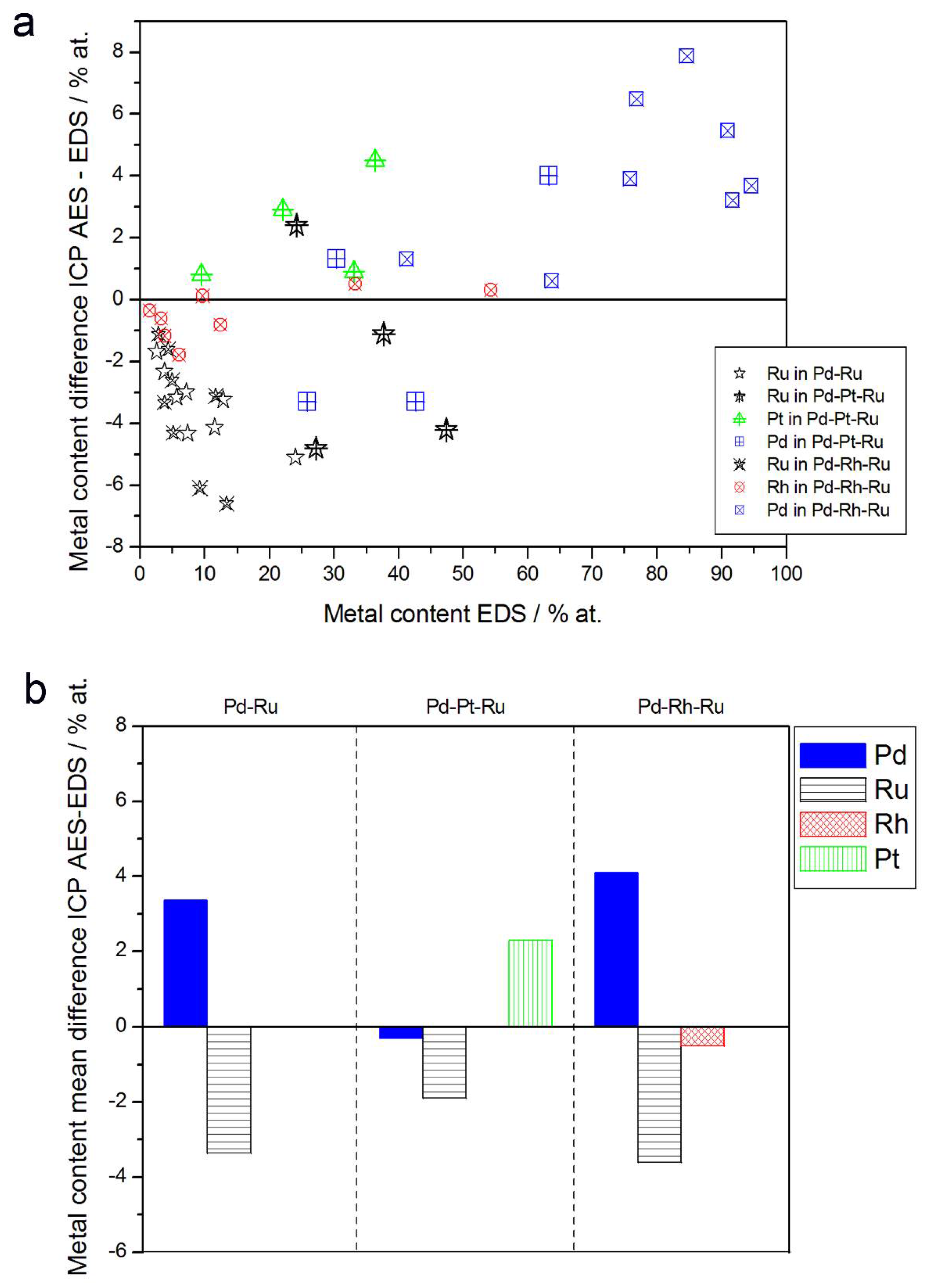
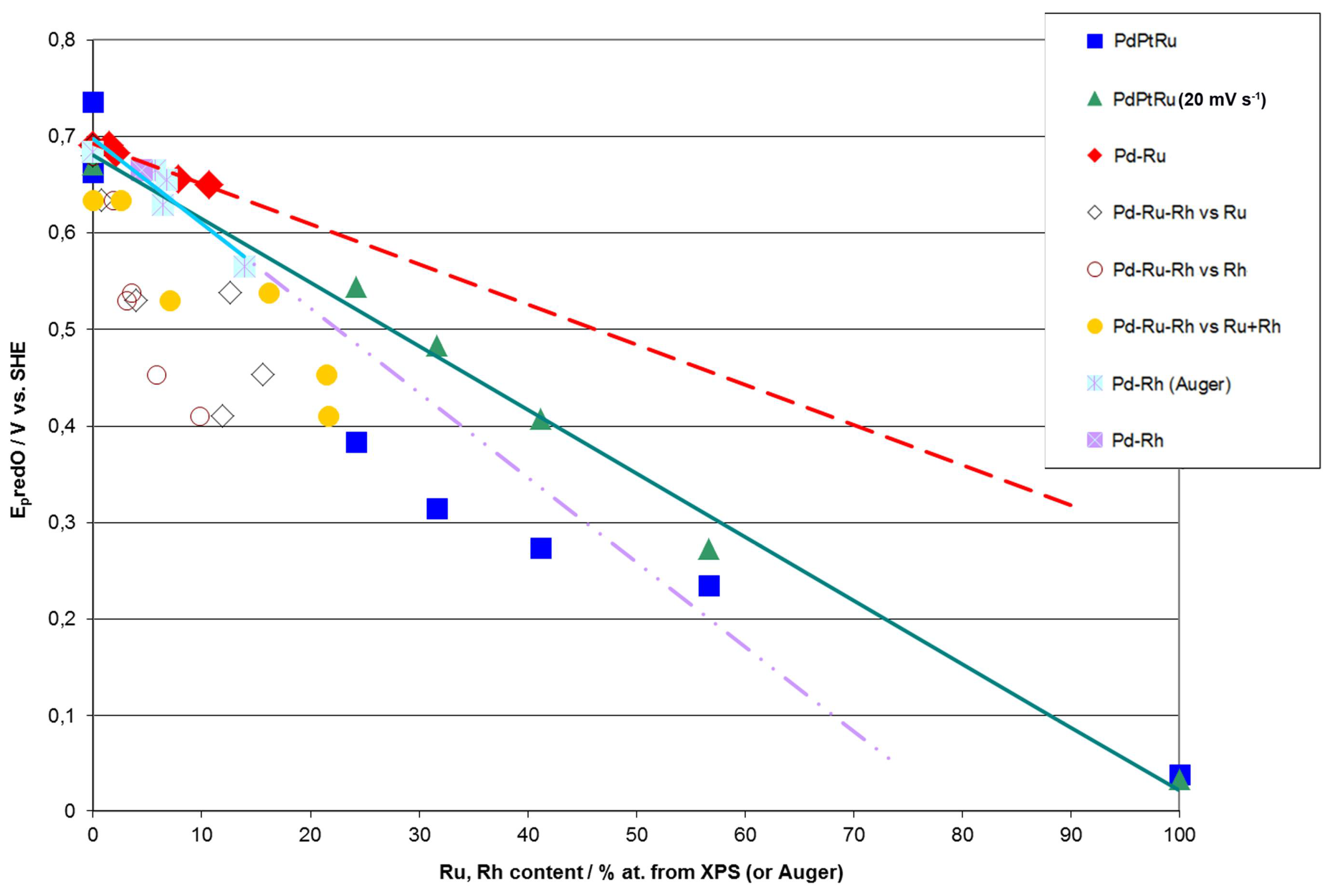
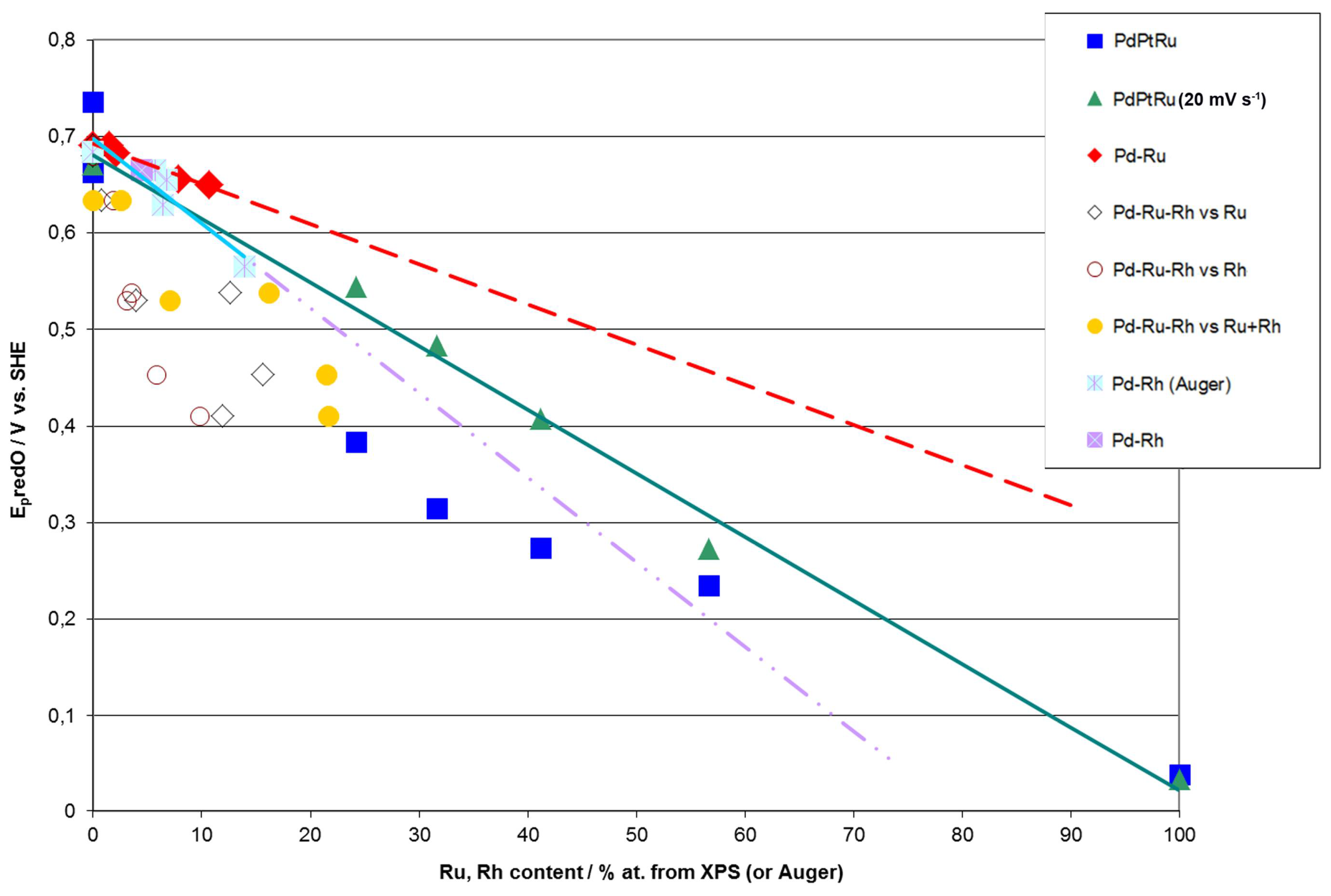
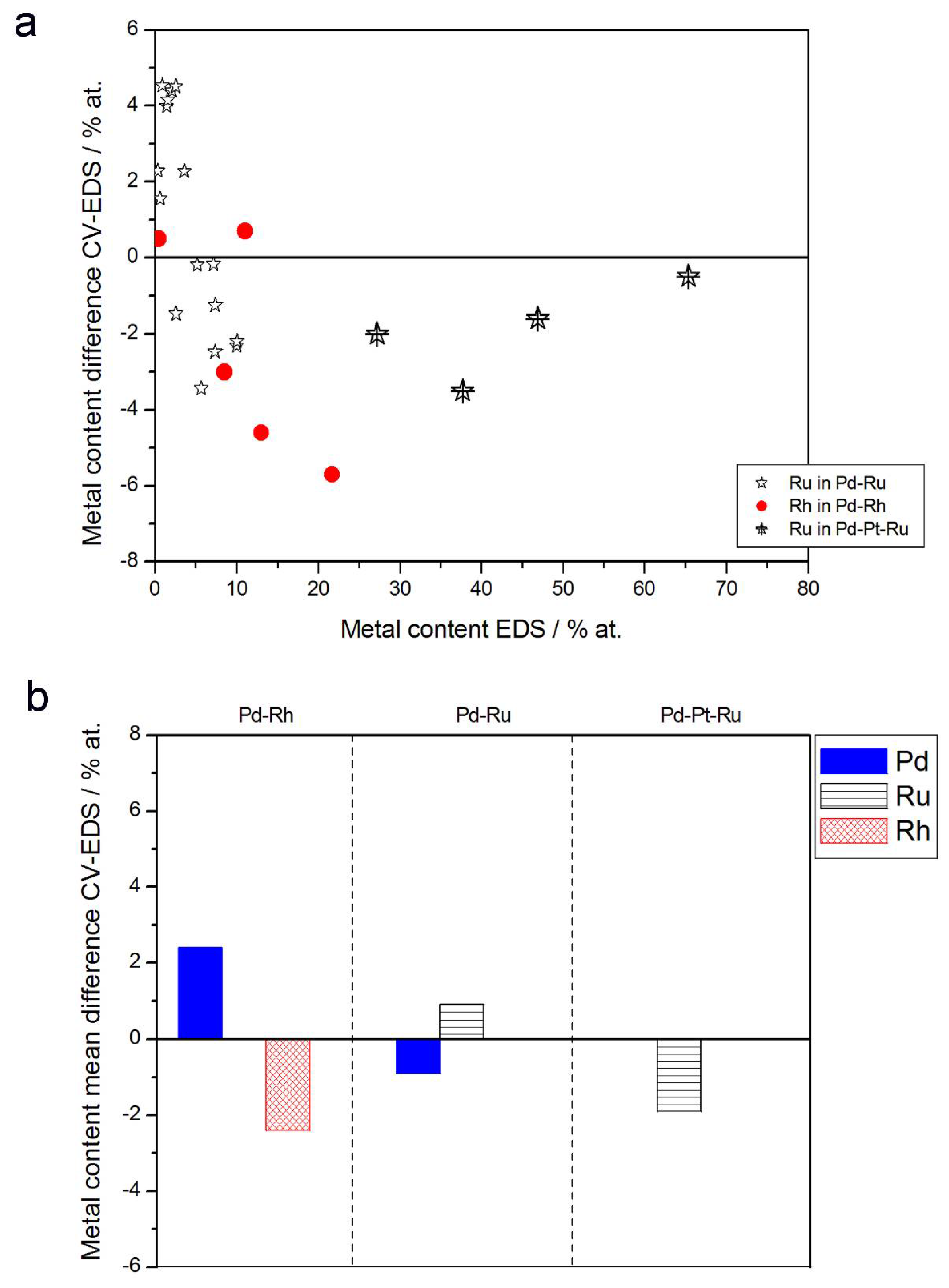
- (1)
- Rh content determined by EDS is usually higher than those derived from XPS or AES measurements, and at the same time Rh content originating from AES data is higher than that from XPS. For Pd-Rh samples in the composition range examined in this study, Rh concentration determined by CV is similar to or lower than that obtained by EDS (differences up to −5.7 at % for the individual samples, and ca. −2.4 at % on average). This is in line with our earlier data on the electrochemistry of the Pd-Rh system [79,80]. However, according to the results of the experiments performed with Pd-Rh electrodes in a much wider composition range, for Pd-Rh electrodeposits containing more than ca. 20% Rh in the bulk, the surface becomes more and more enriched with Rh [79,80];
- (2)
- For individual Pd-Ru samples, the differences in Ru contents determined by CV and EDS vary within the range ±4 at %, but the mean differences do not exceed +1 at %;
- (3)
- For Pd-Pt-Ru electrodes, the analysis of CV curves gives a systematically smaller Ru content with respect to EDS data (to −3.5 at % for the individual samples and ca. −2 at % on average);
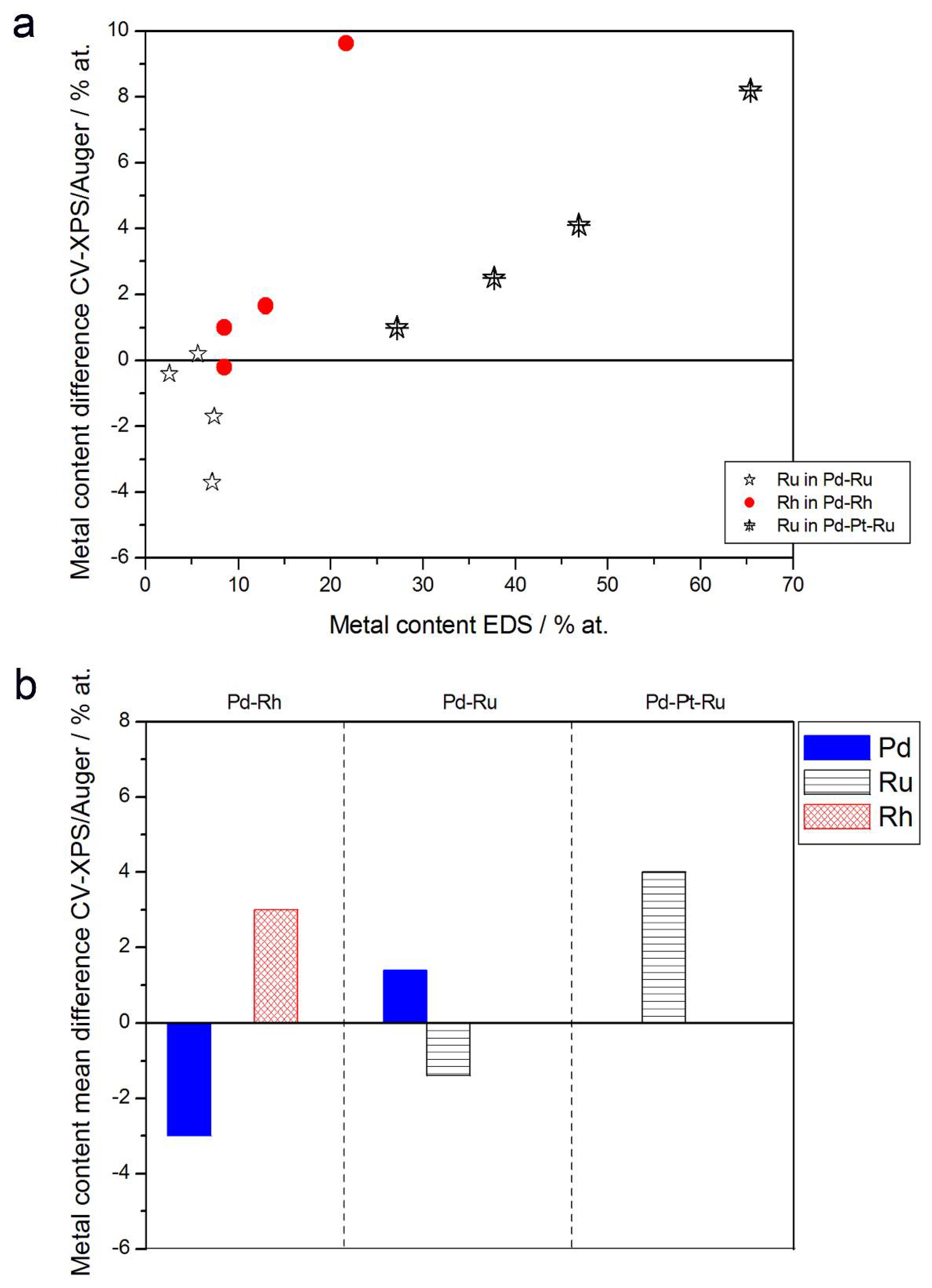
- (4)
- The differences in Ru or Rh contents determined by CV and XPS/AES are opposite to CV vs. EDS-derived values. In particular, for Pd-Pt-Ru alloys with increasing bulk Ru content (>25%), more Ru (by up to +8 at % individually and ca. +4 at % on average) is detected by CV than by XPS, while for the same samples, CV data are closer to EDS data. A similar situation occurs for CV vs. AES data for Pd-rich Pd-Rh alloys, while for Pd-rich Pd-Ru alloys CV analysis gives a smaller Ru content (to −4 at % individually and ca. −1.5 at % on average) than that based on XPS analysis.
3.4. Comparison of Data Obtained by Various Analytical Techniques
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Appleby, A.J.; Garche, J. Encyclopedia of Electrochemical Power Sources; Elsevier: Amsterdam, The Netherlands, 2009. [Google Scholar]
- Kakati, N.; Maiti, J.; Hee Lee, S.; Hyun Jee, S.; Viswanathan, B.; Yoon, Y.S. Anode catalysts for Direct methanol fuel cells in acidic media: Do we have any alternative for Pt or Pt–Ru? Chem. Rev. 2014, 114, 12397–12429. [Google Scholar] [CrossRef] [PubMed]
- Piela, P.; Czerwiński, A. Przegląd technologii ogniw paliwowych. Cz. 1. Zasada działania i możliwości. Przem. Chem. 2006, 85, 13–18. [Google Scholar]
- Brouzgou, A.S.; Song, Q.; Tsiakaras, P. Low and non-platinum electrocatalysts for PEMFCs: Current status, challenges and prospects. Appl. Catal. B Environ. 2012, 127, 371–388. [Google Scholar] [CrossRef]
- Tiwari, J.; Tiwari, R.; Singh, G.; Kim, K.S. Recent progress in the development of anode and cathode catalysts for direct methanol fuel cells. Nano Energy 2013, 2, 553–578. [Google Scholar] [CrossRef]
- Antolini, E. Structural parameters of supported fuel cell catalysts: The effect of particle size, inter-particle distance and metal loading on catalytic activity and fuel cell performance. Appl. Catal. B Environ. 2016, 181, 298–313. [Google Scholar] [CrossRef]
- Piela, P.; Czerwiński, A. Przegląd technologii ogniw paliwowych. Cz. 2. Typy ogniw paliwowych. Przem. Chem. 2006, 85, 164–170. [Google Scholar]
- Petrii, O.A. Pt-Ru electrocatalysts for fuel cells: A representative review. J. Solid State Electrochem. 2008, 12, 609–642. [Google Scholar] [CrossRef]
- Lewis, F.A. The Palladium/Hydrogen System; Academic Press: New York, NY, USA, 1967. [Google Scholar]
- Kleperis, J.; Wójcik, G.; Czerwiński, A.; Skowroński, J.; Kopczyk, M.; Bełtowska-Brzezińska, M. Electrochemical behavior of metal hydrides. J. Solid State Electrochem. 2001, 5, 229–249. [Google Scholar] [CrossRef]
- Wójcik, G.; Kopczyk, M.; Drulis, H.; Bełtowska-Brzezińska, M. Multi-component metallic systems absorbing hydrogen for rechargeable batteries. Wiad. Chem. 1995, 49, 285. [Google Scholar]
- Crabtree, G.W.; Dresselhaus, M.S.; Buchanan, M.V. The hydrogen economy. Phys. Today 2004, 57, 39–44. [Google Scholar] [CrossRef]
- Schlapbach, L.; Züttel, A. Hydrogen-storage materials for mobile applications. Nature 2001, 414, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Kijeński, J. Dlaczego Wodór? Przem. Chem. 2005, 84, 799. [Google Scholar]
- Conway, B.E. Electrochemical Capacitors. Scientific Fundamentals and Technological Applications; Kluwer Academia: Norwell, MA, USA; Plenum Publishers: New York, NY, USA, 1999. [Google Scholar]
- Tokarz, W.; Piela, P.; Czerwiński, A. Analysis of the influence of rhodium addition to platinum on its activity towords methanol electrooxidation by EIS. J. Solid State Electrochem. 2010, 14, 515–521. [Google Scholar] [CrossRef]
- Tokarz, W.; Piela, P.; Czerwiński, A. Electro-oxidation of methanol on Pt-Rh alloys. Electrochim. Acta 2007, 52, 5565–5573. [Google Scholar] [CrossRef]
- Siwek, H.; Tokarz, W.; Piela, P.; Czerwiński, A. Electrochemical behavior of CO, CO2 and methanol adsorption products formed on Pt–Rh alloys of various surface compositions. J. Power Sources 2008, 181, 24–30. [Google Scholar] [CrossRef]
- Waszczuk, P.; Solla-Gull, J.; Kim, H.-S.; Tong, Y.Y.; Montiel, V.; Aldaz, A.; Wieckowski, A. Methanol Electrooxidation on Platinum/Ruthenium Nanoparticle Catalysts. J. Catal. 2001, 203, 1–6. [Google Scholar] [CrossRef]
- Yépez, O.; Pickup, G. Electrochemical Oxidation of Methanol at Hydrogen-Loaded PdPtRu-Coated Pd Electrodes. Electrochem. Solid State Lett. 2005, 8, 35–38. [Google Scholar] [CrossRef]
- Hamnett, A. Mechanism and electrocatalysis in the direct methanol fuel cell. Catal. Today 1997, 38, 445–457. [Google Scholar] [CrossRef]
- Léger, J.-M. Mechanistic aspects of methanol oxidation on platinum-based electrocatalysts. J. Appl. Electrochem. 2001, 31, 767–771. [Google Scholar] [CrossRef]
- Conway, B.E. Electrochemical oxide film formation at noble metals as a surface-chemical process. Prog. Surf. Sci. 1995, 49, 331–452. [Google Scholar] [CrossRef]
- Jerkiewicz, G.; Vatankhah, G.; Lessard, J.; Soriaga, M.P.; Park, Y.-S. Surface-oxide growth at platinum electrodes in aqueous sulfuric acid reexamination of its mechanism through combined cyclic-voltammetry, electrochemical quartz-crystal nanobalance, and AES measurements. Electrochim. Acta 2004, 49, 1451–1459. [Google Scholar]
- Stamenkovic, V.; Mun, B.S.; Mayrhofer, K.J.J.; Ross, P.N.; Markovic, N.M.; Rossmeisl, J.; Greeley, J.; Norskov, J.K. Changing the activity of electrocatalysts for oxygen reduction by tuning the surface electronic structure. Angew. Chem. Int. Ed. 2006, 45, 2897–2901. [Google Scholar] [CrossRef] [PubMed]
- Fiçicilar, B.; Bayrakçeken, A.; Eroğlu, İ. Effect of Pd loading in Pd-Pt bimetallic catalysts doped into hollow core mesoporous shell carbon on performance of proton exchange membrane fuel cells. J. Power Sources 2009, 193, 17–23. [Google Scholar] [CrossRef]
- Watanabe, M.; Uchida, H.; Ikeda, N. Electrochemical quartz crystal microbalance study of copper ad-atoms on gold and platinum electrodes Part I. Adsorption of anions in sulfuric acid. J. Electroanal. Chem. 1995, 380, 255–260. [Google Scholar] [CrossRef]
- Gojković, S.L.; Vidaković, T.R.; Durović, D.R. Kinetic study of methanol oxidation on carbon-supported PtRu electrocatalyst. Electrochim. Acta 2003, 48, 3607–3614. [Google Scholar] [CrossRef]
- Waszczuk, P.; Lu, G.-Q.; Wieckowski, A.; Lu, C.; Rise, C.; Masel, R.I. UHV and electrochemical studies of CO and methanol adsorbed at platinum/ruthenium surfaces, and reference to fuel cell catalysis. Electrochim. Acta 2002, 47, 3637–3652. [Google Scholar] [CrossRef]
- Flanagan, T.B.; Sakamoto, Y. Hydrogen in Disordered and Ordered Palladium Alloys. Platin. Met. Rev. 1993, 37, 26–37. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Baba, K.; Flanagan, T.B. The Effect of Alloying of Palladium on the Hydrogen-Palladium Miscibility Gap. Z. Phys. Chem. 1988, 158, 223–236. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Yuwasa, K.; Hirayama, K. X-ray investigation of the absorption of hydrogen by several palladium and nickel solid solution alloys. J. Less Common Met. 1982, 88, 115–124. [Google Scholar] [CrossRef]
- Sakamoto, Y.; Chen, F.L.; Ura, M.; Flanagan, T.B. Thermodynamic Properties for Solution of Hydrogen in Palladium-Based Binary Alloys. Ber. Bunsenges. Phys. Chem. 1995, 99, 807–820. [Google Scholar] [CrossRef]
- Wicke, E.; Frölich, K. Electronic and Elastic Effects in the Phase Diagrams of Binary Pd Alloy Hydrides*. Z. Phys. Chem. 1989, 163, 35–40. [Google Scholar] [CrossRef]
- Burch, R. Theoretical aspects of the absorption of hydrogen by palladium and its alloys. Part 1—A reassessment and comparison of the various proton models. Trans. Faraday Soc. 1970, 66, 736–748. [Google Scholar] [CrossRef]
- Khanra, B.C.; Bertolini, J.C.; Rousset, J.L. Effect of surface segregation on the catalytic activity of alloys: CO hydrogenation on Pd–Ni(111) surface. J. Mol. Catal. A. 1998, 129, 233. [Google Scholar] [CrossRef]
- Kuijers, F.J.; Tieman, B.M.; Ponec, V. The surface composition of platinum-palladium alloys determined by AES. Surf. Sci. 1978, 75, 657–680. [Google Scholar] [CrossRef]
- Gossner, K.; Mizera, E. The adsorption and electrochemical oxidation of CO on gold + palladium alloys 3. J. Electroanal. Chem. 1979, 98, 37–48. [Google Scholar]
- Kadirgan, F.; Beden, B.; Leger, J.-M.; Lamy, C. Synergistic effect in the electrocatalytic oxidation of methanol on platinum+palladium alloy electrodes. J. Electroanal. Chem. 1981, 125, 89–103. [Google Scholar] [CrossRef]
- Garcia, A.C.; Paganin, V.A.; Ticianelli, E.A. CO tolerance of PdPt/C and PdPtRu/C anodes for PEMFC. Electrochim. Acta 2008, 53, 4309–4315. [Google Scholar] [CrossRef]
- He, C.; Kunz, H.R.; Fenton, J.M. Evaluation of Platinum-Based Catalysts for Methanol Electro-oxidation in Phosphoric Acid Electrolyte. J. Electrochem. Soc. 1997, 144, 970–979. [Google Scholar] [CrossRef]
- Jiang, J.; Kucernak, A. Electrodeposition of highly alloyed quaternary PtPdRuOs catalyst with highly ordered nanostructure. Electrochem. Commun. 2009, 11, 1005–1008. [Google Scholar] [CrossRef]
- Plieth, W. Electrochemical alloy deposition: New properties by formation of intermetallic compounds. Surf. Coat. Technol. 2003, 169–170, 96–99. [Google Scholar] [CrossRef]
- Plieth, W. Kinetic models for alloy and semiconductor electrodeposition. Electrochim. Acta 2007, 53, 245–249. [Google Scholar] [CrossRef]
- Georgiev, G.S.; Georgieva, V.T.; Plieth, W. Markov chain model of electrochemical alloy deposition. Electrochim. Acta 2005, 51, 870–876. [Google Scholar] [CrossRef]
- Bockris, J.O.M.; Reddy, A.K. Modern Electrochemistry; Rosseta Edition: New York, NY, USA, 1973. [Google Scholar]
- Brenner, A. Electrodeposition of Alloys; Academic Press: New York, NY, USA; London, UK, 1963. [Google Scholar]
- Baumgartner, M.E.; Raub, C.J. The Electrodeposition of Platinum and Platinum Alloys. Platin. Met. Rev. 1988, 32, 188–197. [Google Scholar]
- Rao, C.R.K.; Trivedi, D.C. Chemical and electrochemical depositions of platinum group metals and their applications. Coord. Chem. Rev. 2005, 249, 613–631. [Google Scholar] [CrossRef]
- Yang, B.; Lu, Q.; Wang, Y.; Zhang, L.; Lu, J.; Liu, P. Simple and Low-Cost Preparation Method for Highly Dispersed PtRu/C Catalysts. Chem. Mater. 2003, 15, 3552–3557. [Google Scholar] [CrossRef]
- Chekin, F.; Bagheri, S.; Bee, S.; Hamid, A. Synthesis of Pt doped TiO2 nanoparticles: Characterization and application for electrocatalytic oxidation of l-methionine. Sens. Actuators B 2013, 177, 898–903. [Google Scholar] [CrossRef]
- Hu, L.; Boateng, K.A.; Hill, J.M. Sol-gel synthesis of Pt/Al2O3 catalysts: Effect of Pt precursor and calcination procedure on Pt dispersion. J. Mol. Catal. A Chem. 2006, 259, 51–60. [Google Scholar] [CrossRef]
- Alyousef, Y.M.; Datta, M.K.; Kadakia, K.S.; Yao, S.C.; Kumta, P.N. Sol–gel synthesis of Pt-Ru-Os-Ir based anode electro-catalysts for direct methanol fuel cells. J. Alloys Compd. 2010, 506, 698–702. [Google Scholar] [CrossRef]
- Chen, M.; Xing, Y. Polymer-Mediated Synthesis of Highly Dispersed Pt Nanoparticles on Carbon Black. Langmuir 2005, 2, 9334–9338. [Google Scholar] [CrossRef] [PubMed]
- Hakamada, M.; Mabuchi, M. Preparation of Nanoporous Palladium by Dealloying: Anodic Polarization Behaviors of Pd-M (M=Fe, Co, Ni) Alloys. Mater. Trans. 2009, 50, 431–435. [Google Scholar] [CrossRef]
- Peng, Z.M.; You, H.J.; Yang, H. An Electrochemical Approach to PtAg Alloy Nanostructures Rich in Pt at the Surface. Adv. Funct. Mater. 2010, 20, 3734–3741. [Google Scholar] [CrossRef]
- Srivastava, R.; Mani, P.; Hahn, N.; Strasser, P. Efficient Oxygen Reduction Fuel Cell Electrocatalysis on Voltammetrically Dealloyed Pt–Cu–Co Nanoparticles. Angew. Chem. Int. Ed. 2007, 46, 8988–8991. [Google Scholar] [CrossRef] [PubMed]
- Soszko, M.; Dłubak, J.; Czerwiński, A. Quartz crystal microbalance study of palladium alloys. Part 1: Electrodeposition of Pt–Pd–Ru alloys. J. Electroanal. Chem. 2014, 729, 27–33. [Google Scholar] [CrossRef]
- Paulus, U.A.; Schmidt, T.J.; Gasteiger, H.A.; Behm, R.J. Oxygen reduction on a high-surface area Pt/Vulcan carbon catalyst: A thin-film rotating ring-disk electrode study. J. Electroanal. Chem. 2001, 495, 134–145. [Google Scholar] [CrossRef]
- Schmidt, T.J.; Gasteiger, H.A.; Stäb, G.D.; Urban, P.M.; Kolb, D.M.; Behm, R.J. Characterization of high-surface-area electrocatalysts using a rotating disk electrode configuration. J. Electrochem. Soc. 1998, 145, 2354–2358. [Google Scholar] [CrossRef]
- Clewley, J.D.; Lynch, J.F.; Flanagan, T.B. Thermodynamics of hydrogen dissolved in palladium/rhodium and palladium/platinum alloys. J. Chem. Soc. Faraday Trans. I 1977, 73, 494–504. [Google Scholar] [CrossRef]
- Thiébaut, S.; Bigot, A.; Achard, J.C.; Limacher, B.; Leroy, D.; Percheron-Guégan, A. Structural and thermodynamic properties of the deuterium-palladium solid solutions systems: D2-[Pd(Pt), Pd(Rh), Pd(Pt, Rh)]. J. Alloys Compd. 1995, 231, 440–447. [Google Scholar] [CrossRef]
- Moysan, I.; Paul-Boncour, V.; Thiébaut, S.; Sciora, E.; Fournier, J.M.; Cortes, R.; Bourgeois, S.; Percheron-Guégan, A. Pd–Pt alloys: Correlation between electronic structure and hydrogenation properties. J. Alloys Compd. 2001, 322, 14–20. [Google Scholar] [CrossRef]
- Kaszkur, Z. A Report from Commissioned Analyses Performed at Laboratory of X-ray Powder Diffraction and Spectrometry; Polish Academy of Sciences: Warsaw, Poland, 2016. [Google Scholar]
- Januszewska, A.; Dercz, G.; Lewer, A.; Jurczakowski, R. Spontaneous Chemical Ordering in Bimetallic Nanoparticles. J. Phys. Chem. C 2015, 119, 19817–19825. [Google Scholar] [CrossRef]
- Zhang, C.; Alavi, A. First-Principles Study of Superabundant Vacancy Formation in Metal Hydrides. J. Am. Chem. Soc. 2005, 127, 9808–9817. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.N.; Bharadwaj, S.R.; Dharwadkar, S.R. The Pd-Ru system (palladium-ruthenium). J. Phase Equilib. 1993, 14, 638–642. [Google Scholar] [CrossRef]
- Rudnitskii, A.A.; Polyakova, R.S. The Palladium-Ruthenium system. Russ. J. Inorg. Chem. 1959, 4, 631–636. [Google Scholar]
- Pisarek, M.; Łukaszewski, M.; Winiarek, P.; Kędzierzawski, P.; Janik-Czachor, M. Selective hydrogenation of isopropane on Ni-Al. Alloy modified with Cr. Mater. Chem. Phys. 2009, 114, 774–779. [Google Scholar] [CrossRef]
- Janik-Czachor, M.; Pisarek, M. Modern Aspects of Electrochemistry; Pyun, S.-I., Lee, J.-W., Eds.; Springer: Dordrecht, The Netherlands; Heidelberg, Germany; London, UK, 2009; Volume 46, pp. 175–230. [Google Scholar]
- Chastain, J.; King, R.C., Jr. (Eds.) Handbook of X-Ray Photoelectron Spectroscopy; Physical Electronics Inc.: Chanhassen, MN, USA, 1995. [Google Scholar]
- Batirev, I.G.; Leiro, J.A. Surface segregation of PdRh, PdNi and PdCo alloys. J. Electron Spectrosc. Relat. Phenom. 1995, 71, 79–86. [Google Scholar] [CrossRef]
- Leiro, J.A.; Heinonen, M.H.; Batirev, I.G. Surface segregation and core-level shift of a Pd-Rh alloy studied by XPS. Appl. Surf. Sci. 1995, 90, 515–521. [Google Scholar] [CrossRef]
- Hedberg, C.L. (Ed.) Handbook of AES; Physical Electronics Inc.: Chanhassen, MN, USA, 1995. [Google Scholar]
- Donten, M. Mikrosonda elektronowa (EDS). Analityka: Nauka i Praktyka 2002, 1, 4–8. (In Polish) [Google Scholar]
- Żurowski, A.; Łukaszewski, M.; Czerwiński, A. Electrosorption of hydrogen into palladium–rhodium alloys. Electrochim. Acta 2006, 51, 3112–3117. [Google Scholar] [CrossRef]
- Rand, D.A.J.; Woods, R. Determination of the surface composition of smooth noble metal alloys by cyclic voltammetry. J. Electroanal. Chem. 1972, 36, 57–69. [Google Scholar] [CrossRef]
- Hubkowska, K.; Łukaszewski, M.; Koss, U.; Czerwiński, A. Characterization and electrochemical behavior of Pd-rich Pd-Ru alloys. Electrochim. Acta 2014, 132, 214–222. [Google Scholar] [CrossRef]
- Łukaszewski, M.; Czerwiński, A. Electrochemical preparation and characterization of thin deposits of Pd-noble metal alloys. Thin Solid Films 2010, 518, 3680–3689. [Google Scholar] [CrossRef]
- Łukaszewski, M.; Czerwiński, A. Electrochemical behavior of Pd–Rh alloys. J. Solid State Electrochem. 2007, 11, 339–349. [Google Scholar] [CrossRef]
- Czerwiński, A.; Kiersztyn, I.; Grdeń, M.; Czapla, J. The study of hydrogen sorption in palladium limited volume electrodes (Pd-LVE): I. Acidic solutions. J. Electroanal. Chem. 1999, 471, 190–195. [Google Scholar] [CrossRef]
- Czerwiński, A.; Kiersztyn, I.; Grdeń, M. The study of hydrogen sorption in palladium limited volume electrodes (Pd-LVE): Part II. Basic solutions. J. Electroanal. Chem. 2000, 492, 128–136. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hubkowska, K.; Łukaszewski, M.; Soszko, M.; Koss, U.; Hamankiewicz, B.; Czerwiński, A. Comparative Physicochemical and Electrochemical Characterization of the Structure and Composition of Thin Pd Binary and Ternary Codeposits with Pt, Ru, and Rh. Materials 2018, 11, 798. https://doi.org/10.3390/ma11050798
Hubkowska K, Łukaszewski M, Soszko M, Koss U, Hamankiewicz B, Czerwiński A. Comparative Physicochemical and Electrochemical Characterization of the Structure and Composition of Thin Pd Binary and Ternary Codeposits with Pt, Ru, and Rh. Materials. 2018; 11(5):798. https://doi.org/10.3390/ma11050798
Chicago/Turabian StyleHubkowska, Katarzyna, Mariusz Łukaszewski, Michał Soszko, Urszula Koss, Bartosz Hamankiewicz, and Andrzej Czerwiński. 2018. "Comparative Physicochemical and Electrochemical Characterization of the Structure and Composition of Thin Pd Binary and Ternary Codeposits with Pt, Ru, and Rh" Materials 11, no. 5: 798. https://doi.org/10.3390/ma11050798