Largest Magnetic Moments in the Half-Heusler Alloys XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te): A First-Principles Study


1
School of Physical Science and Technology, Southwest University, Chongqing 400715, China
2
College of Physics and Information Technology, Chongqing Normal University, Chongqing 401331, China
3
Institute for Superconducting & Electronic Materials (ISEM), University of Wollongong, Wollongong 2500, Australia
*
Author to whom correspondence should be addressed.
Materials 2017, 10(9), 1078; https://doi.org/10.3390/ma10091078
Submission received: 7 August 2017
/
Revised: 11 September 2017
/
Accepted: 12 September 2017
/
Published: 14 September 2017
(This article belongs to the Special Issue Intermetallic Alloys: Fabrication, Properties and Applications 2017)
Abstract
:A recent theoretical work indicates that intermetallic materials LiMnZ (Z = N, P) with a half-Heusler structure exhibit half-metallic (HM) behaviors at their strained lattice constants, and the magnetic moments of these alloys are expected to reach as high as 5 μB per formula unit. (Damewood et al. Phys. Rev. B 2015, 91, 064409). This work inspired us to find new Heusler-based half-metals with the largest magnetic moment. With the help of the first-principles calculation, we reveal that XCrZ (X = K, Rb, Cs; Z = S, Se, Te) alloys show a robust, half-metallic nature with a large magnetic moment of 5 μB at their equilibrium and strained lattice constants in their most stable phases, while the excellent HM nature of LiCrZ (Z = S, Se, Te) alloys can be observed in one of their metastable phases. Moreover, the effects of uniform strain in LiCrZ (Z = S, Se, Te) alloys in type II arrangement have also been discussed.
1. Introduction
In recent years, the fast development of spintronics [1] has caused more and more concern for researchers. Extensive applications (e.g., single spin electron sources [2] and spin injections [3]) have been envisaged [4]. One ideal choice for spintronics is the source of the spin-polarized charge carriers (SPCC). In this quest for materials, half-Heusler half-metallic alloys [5] are a noticeable family of intermetallic materials with 1:1:1 composition and have fully SPCC at the Fermi level.
The half-Heusler family has become one of the research hot-spots in intermetallic materials systems because the concept of half-metallic (HM) behaviors arose from the theoretical calculations by de Groot et al. for the well-known NiMnSb [6] in the 1980s. Then, quite a lot of half-Heusler alloys [7,8,9,10,11,12,13,14,15] had been predicted to be HM materials (HMMs). Recently, the electronic, magnetic, and stability properties were systematically investigated by first principles calculation in half-Heusler alloys of LiMnZ (Z = N, P, Si) [16]. Damewood et al. found that these LiMnZ (Z = N, P, Si) alloys show HM behaviors with large semiconducting-type band-gaps and magnetic moments (>3 μB per formula unit) at their strained lattice constants. To our best knowledge, based on the the Slater-Pauling (S-P) and generalized electron-filling rules [17,18,19,20], the largest magnetic moment of the half-Heusler-type alloy should be 5 μB per formula unit. Due to the large semiconducting-type band-gaps and the large magnetic moments, LiMnZ (Z = N, P, Si) alloys may be good candidates for spintronic materials for devices operating at or above room temperature.
Based on the above information, it is necessary for us to further explore new HM half-Heuselr alloys, XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te), with the largest magnetic moment (5 μB per formula unit) and large semiconducting-type band-gaps. In this work, first-principles calculations have been used to exhibit a theoretical study of the structural, electronic, magnetic, and HM properties of the XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te) half-Heusler alloys in three possible arrangements.
2. Computational Details
The electronic-structure and magnetism calculations were performed via CASTEP code on the basis of the pseudo-potential method with a plane-wave basis set [21,22]. The ultrasoft pseudo-potential [23] has been used to describe the interactions between the atomic core and the valence electrons. A most common generalized gradient approximation (GGA) [24] has been selected to describe the electron exchange-correlation. A plane-wave basis set cut-off of 450 eV and a mesh of 12 × 12 × 12 k-points was used for Brillouin zone integrations. The convergence tolerance for the calculations was selected as a difference in total energy within 1 × 10−6 eV·atom−1.
To determine the real-space bonding analysis, the electron localization function (ELF) was calculated using the CASTEP code. The ELF is a real-space indicator of the extent to which electrons are localized and display a strong Pauli repulsion, and therefore it can locate bonding and non-bonding electron pairs in the real-space of the crystal-structure [25,26].
3. Results and Discussion
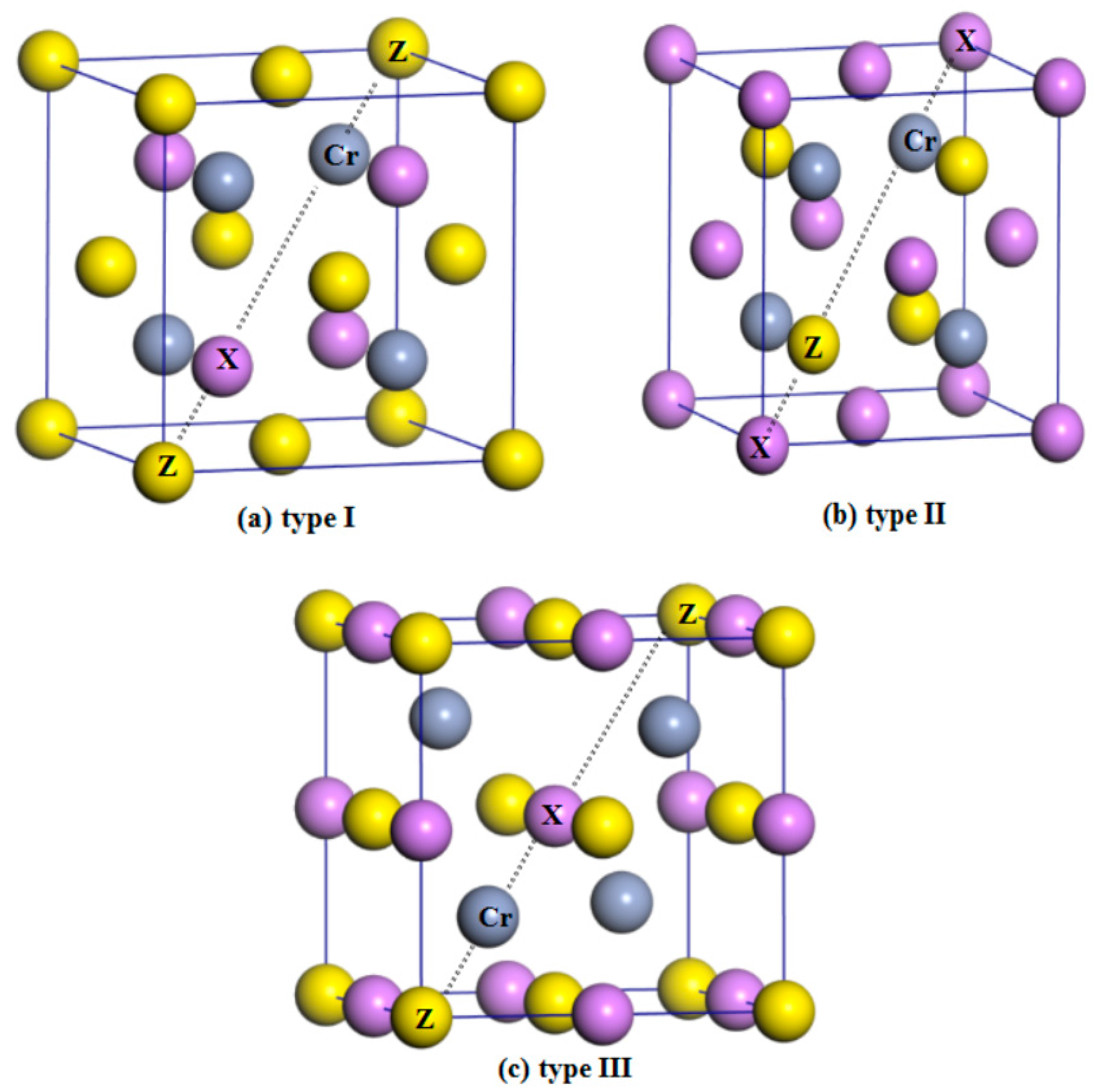
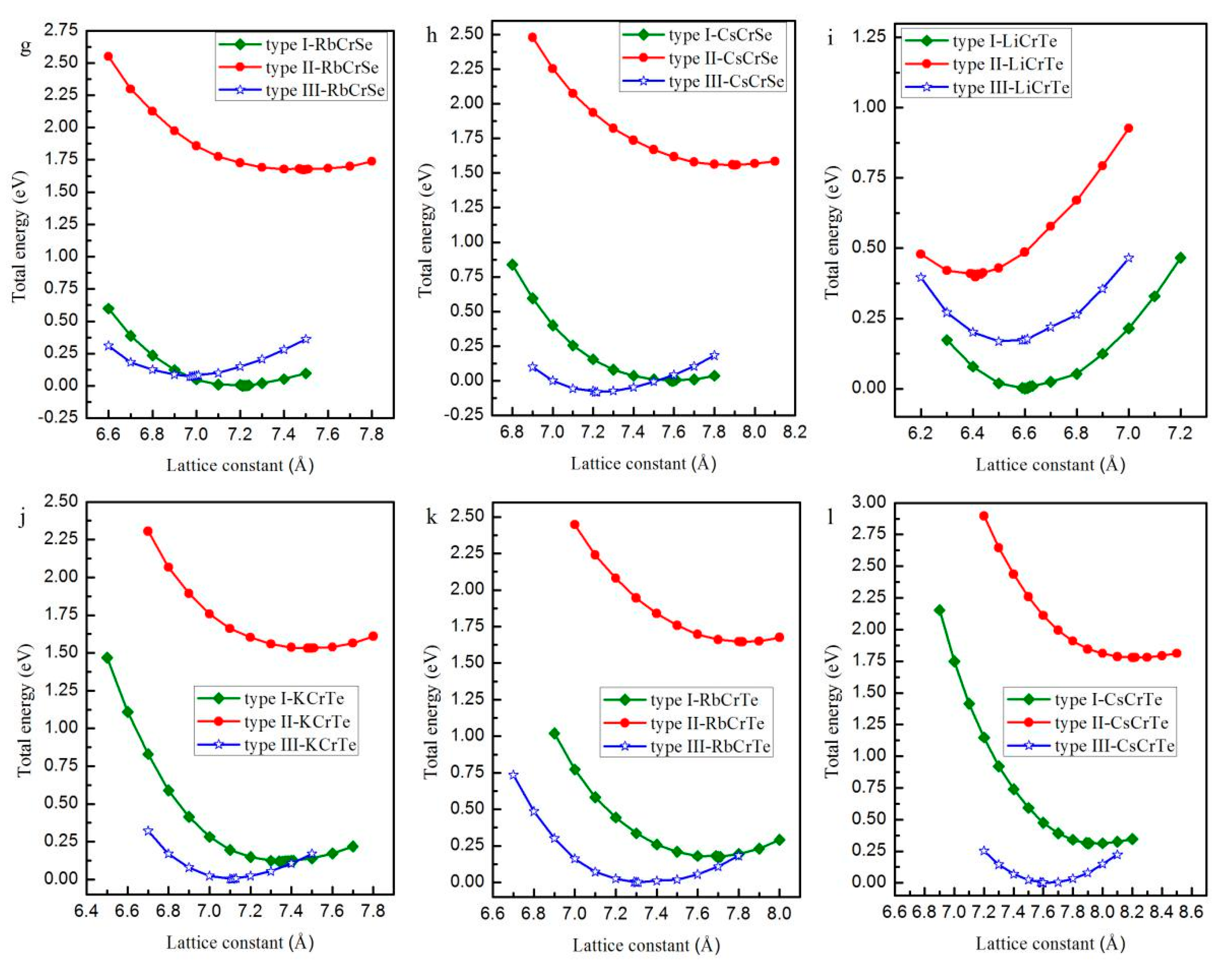
Normally, half-Heusler alloys have a formula of XYZ. In this work, X is the Li, K, Rb, and Cs atoms, Y is the Cr atom, and Z is the main-group-element atoms S, Se, and Te. As shown in Figure 1, in the half-Heusler alloys XCrZ with the C1b structure, three possible arrangements have been taken into consideration: type I = [4c (0.25, 0.25, 0.25), 4d (0.75, 0.75, 0.75), 4a (0, 0, 0)], type II = [4a (0, 0, 0), 4d (0.75, 0.75, 0.75), 4c (0.25, 0.25, 0.25)] and type III = [4b (0.5, 0.5, 0.5), 4d (0.75, 0.75, 0.75), 4a (0, 0, 0)]. To obtain the equilibrium lattice structures of the XCrZ, the geometry optimization is firstly performed in their three possible arrangements. The achieved total energy-lattice constant curves for XCrZ are shown in Figure 2. Obviously, KCrS, RbCrS, CsCrS, KcrSe, and RbCrSe alloys have their lowest energies in type I instead of type II and III. For the CsCrSe, KCrTe, RbCrTe, and CsCrTe (LiCrS, LiCrSe, and LiCrTe) alloys, the structure of type III (type II), with the lowest energy, is the most stable among the three arrangements.
Table 1 shows for all XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te) the calculated total magnetic moments and the sizes of the semiconducting-type band-gaps. For the semiconducting-type band-gaps, the Fermi level (EF) just falls within the gap in the spin-down band, indicating semiconductor properties. As can be observed in Table 1, the values of the total magnetic moment per formula unit (Mt) are 5 μB for XCrZ (X = K, Rb, Cs; Z = S, Se, Te) in their most stable phases (arrangements). However, the calculated magnetic moments per formula unit for LiCrZ (Z = S, Se, Te) are not the integral Bohr magneton for the most stable phase. As is known, for the Heusler-type HMMs, their calculated Mt is usually an integer value [27,28,29,30]. The non-inter values of Mt for LiCrZ (Z = S, Se, Te) in type II indicate that they are not HMMs in the most stable phase. Also, as shown in Table 1, all XCrZ alloys have very large semiconducting-type band-gaps (>2 eV) in their most stable phase, except for LiCrZ (Z = S, Se, Te). It means that they may maintain their magnetic and HM behaviors at room temperature.
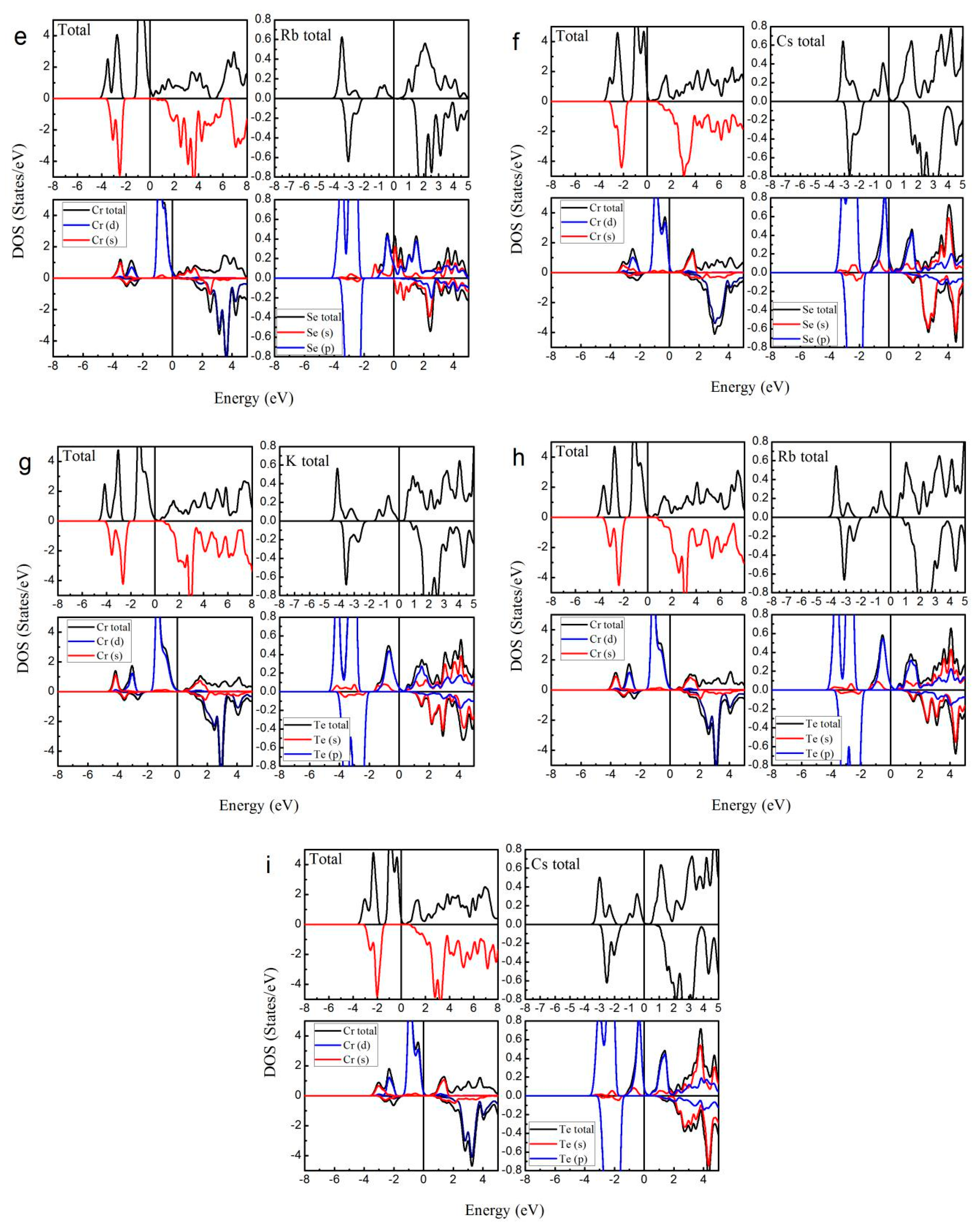
To further confirm the possible half-metallicity of XCrZ (X = K, Rb, Cs; Z = S, Se, Te), we show in Figure 3 the calculated total and atom-projected DOS for KCrS, RbCrS, CsCrS, KCrSe, and RbCrSe in the type I arrangement and CsCrSe, KCrTe, RbCrTe, and CsCrTe in the type III arrangement, respectively. Obviously, it can be found that all these mentioned alloys show half-metallic behaviors: in the majority spin (spin-up) channel, the energy bands exhibit a metallic overlap with the EF, whereas in the minority spin (spin-down) direction, an energy gap is opened and the EF locates within the gap.
It is common sense that the DOS can be widely used to discuss the bonding/anti-bonding states and the gap formation, and we should also point out that a similar analytical approach used in this work can be observed in [31,32].
Figure 3 shows the total density of states (TDOS) and the partial density of states (PDOS) of XCrZ (X = K, Rb, Cs; Z = S, Se, Te) in the most stable phase. Obviously, in both spin channels, the main contributions to the TDOS around the Fermi level arose from the 3d states of the Cr atom. The X and Z atoms have a rather lower PDOS than the Cr atom.
As shown in Figure 3, in the spin-up channel the main peaks of the Cr and Z atoms occurred in the range from −2 eV to 0 eV, and −4 eV to −2 eV, respectively. Meanwhile, between the −4 eV and −2 eV states, similar-shaped, hybridized peaks can also be found in the Cr atoms. In the spin-down channel, in the same energy range (−4 eV~−2 eV), for the Cr and Z atoms, such hybridized peaks appeared at the same time. Therefore, the hybridization between the Cr and Z atoms that formed strong bonding states range from −4 eV to −2 eV. Above the EF, in the spin-down channel, the anti-bonding can be found at around 3 eV, and in the spin-up channel, no opposite energy states are observed. Moreover, similar to the LiMnZ alloys [16], the bonding-antibonding states led to the formation of a semiconducting-type band-gap in the spin-down channel.
For the type I and type III arrangements, the semiconducting-type band-gaps of the XCrZ (X = K, Rb, Cs; Z = S, Se, Te) alloys are very large. However, compared to the type I and type III arrangements, the type II arrangement does not form a large semiconducting-type band-gap (see Table 1) because the Cr and Z are second neighbors in a cubic environment.
In addition, as mentioned above, the calculated Mt, 5 μB for XCrZ (X = K, Rb, Cs; Z = S, Se, Te) follows the modified S-P rule recently presented by Damewood et al. [16],
here Zt is the number of total of valence electrons in XCrZ (X = K, Rb, Cs; Z = S, Se, Te), respectively.
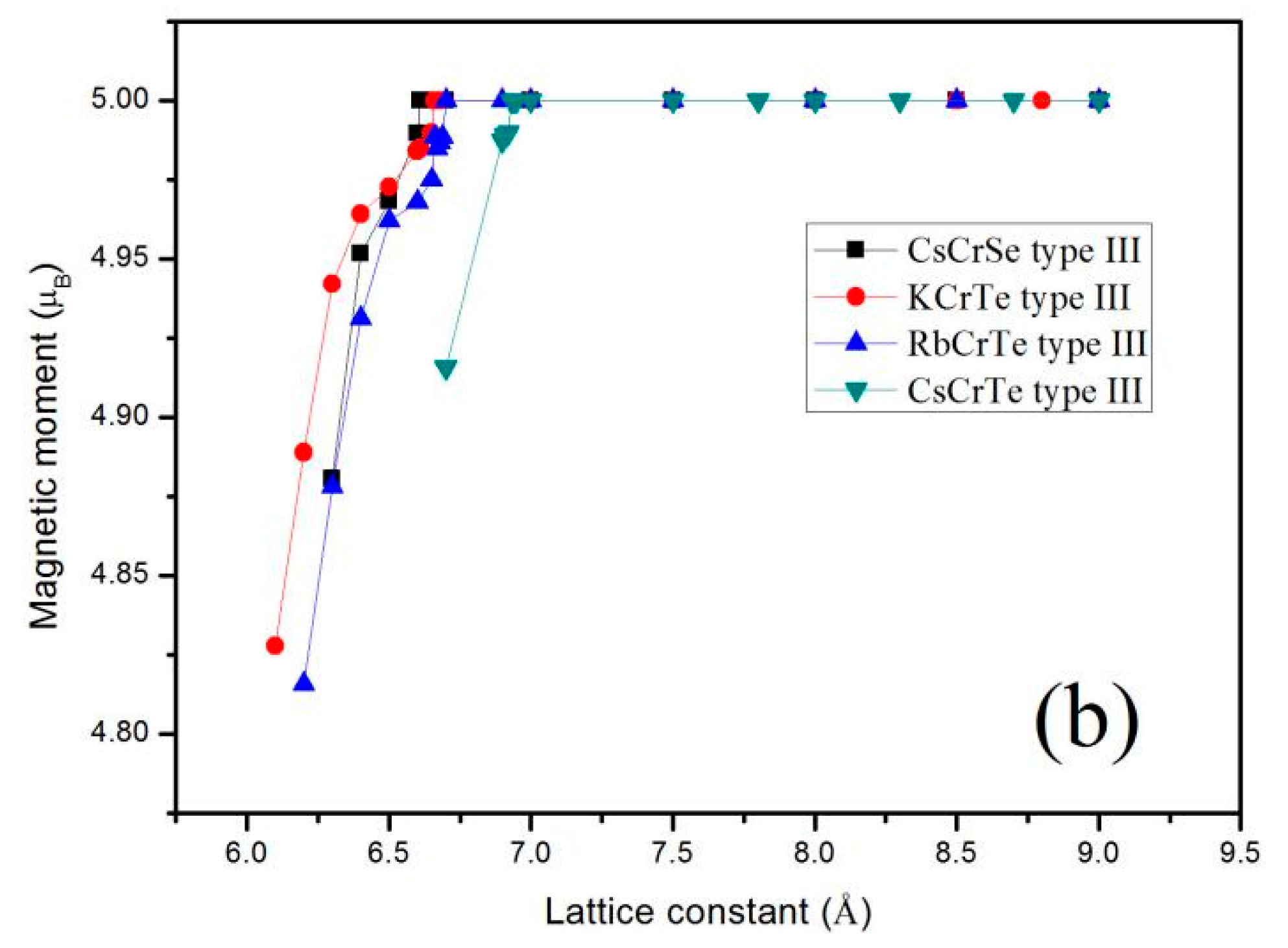
Mt = (Zt − 8)·μB,
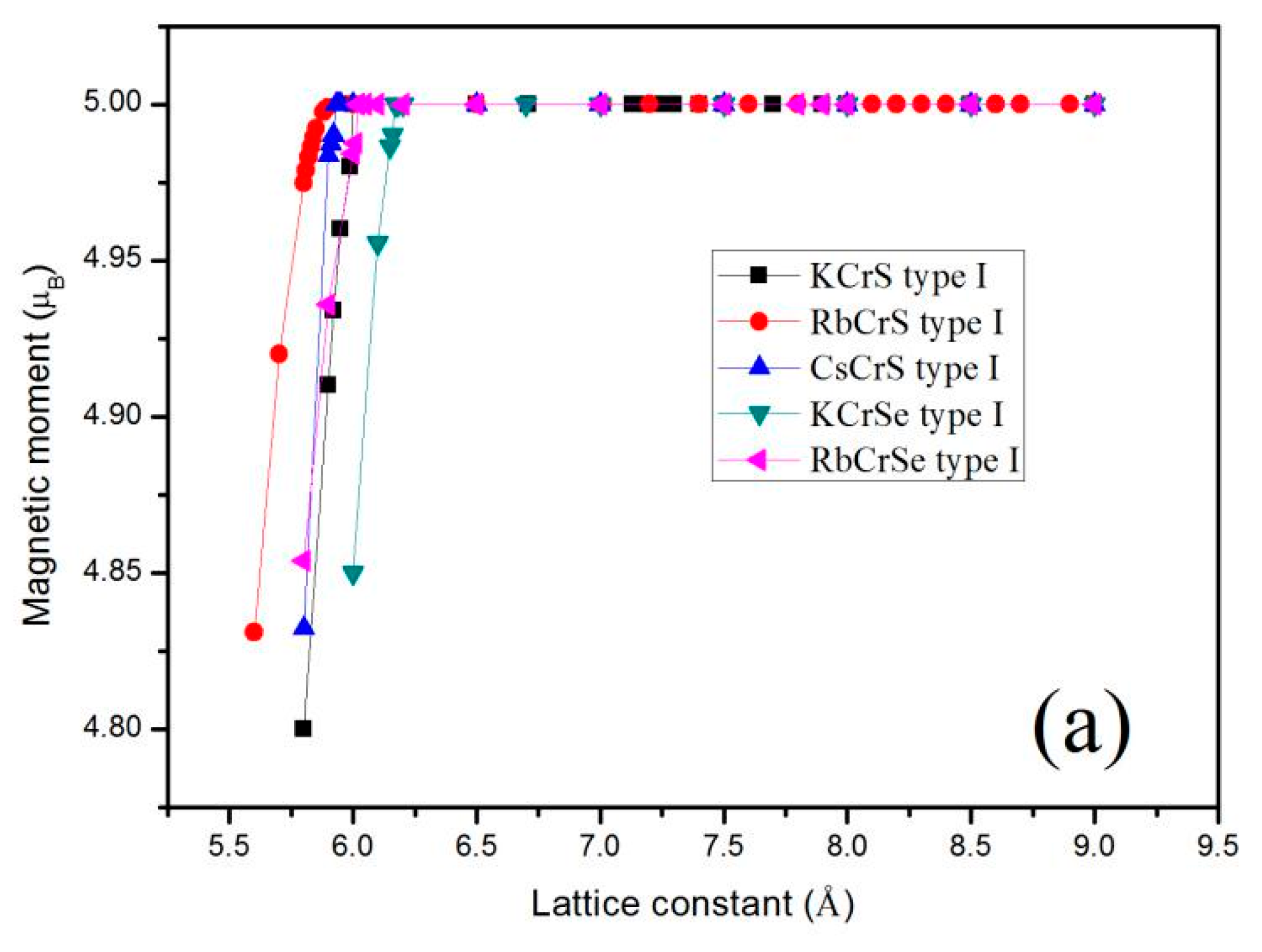
Furthermore, the total Mt for the XCrZ (X = K, Rb, Cs; Z = S, Se, Te) alloys at their strained lattice constants with the most stable phase has been calculated, and the results are shown in Figure 4. Obviously, for all these mentioned alloys, the total Mt of 5.00 μB/f.u remained constant within an expansion and contraction of less than 0.01 µB over a large range of lattice constant values. That is to say, the half-metallic behavior of these alloys is quite robust. When the lattice constants are compressed to the critical value 5.99 Å for KCrS, 5.84 Å for RbCrS, 5.92 Å for CsCrS, 6.16 Å for KCrSe, 6.01 Å for RbCrSe, 6.60 Å for CsCrSe, 6.65 Å for KCrTe, 6.69 Å for RbCrTe, and 6.92 Å for CsCrTe, respectively, the semiconducting-type band-gap in the spin-down channel closed and thus the integer value of the total magnetic moments disappeared.
Previous studies have shown that ternary alloys, including the Li atom, are good candidates in optoelectronic and spintronic applications [33,34]. Although the LiCrZ alloys presented in the current work are not HMMs (see Table 1) in the most stable arrangement (type I), the other arrangements (type II and III) should also be reported here to check the electronic, magnetic, and half-metallic properties. We hope to search for a metastable (type II or III) of the LiCrZ (Z = S, Se, Te) alloys exhibiting HM behavior with the largest magnetic moment (5 μB) and a large semiconducting-type band-gap (>2 eV).
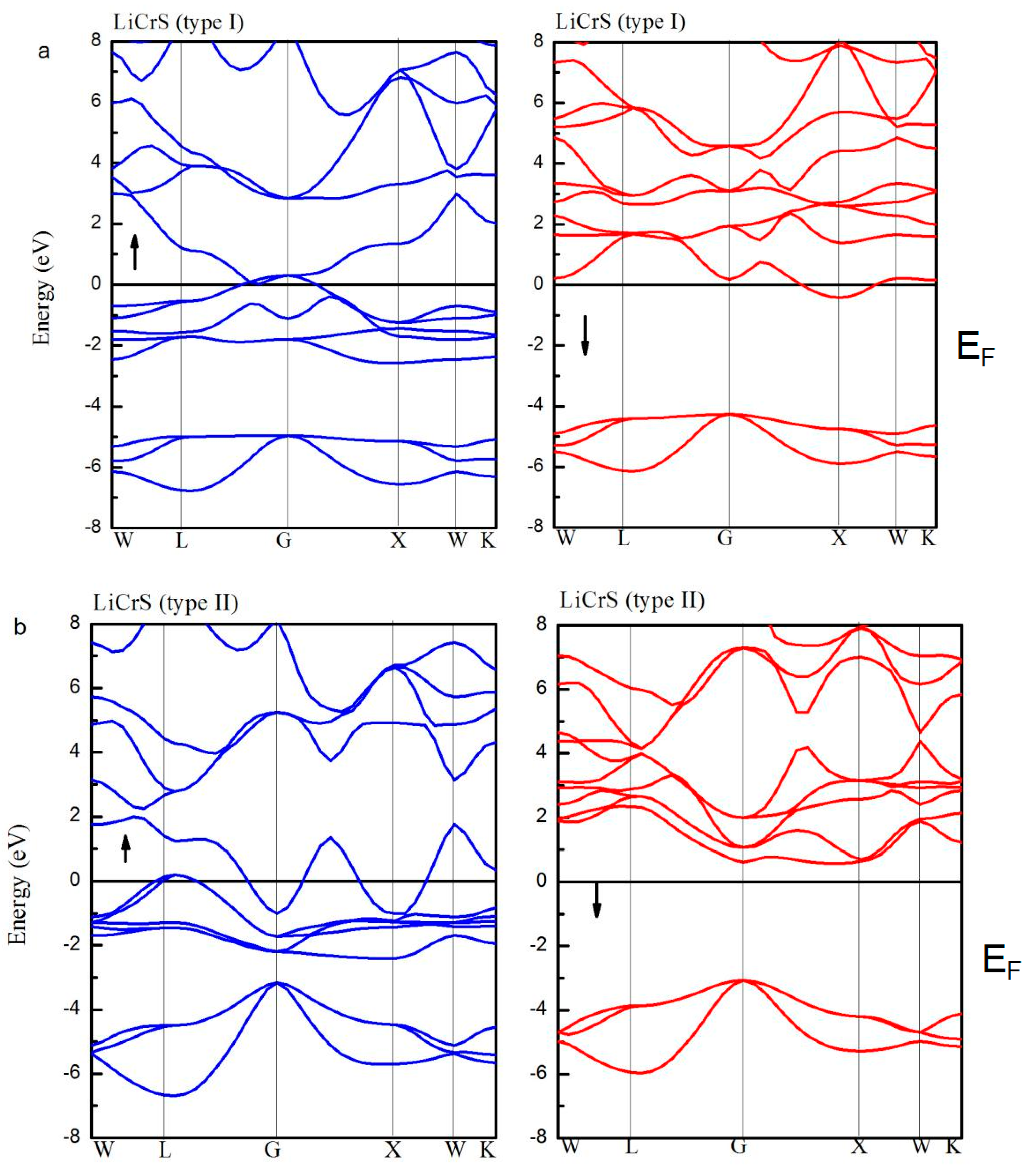
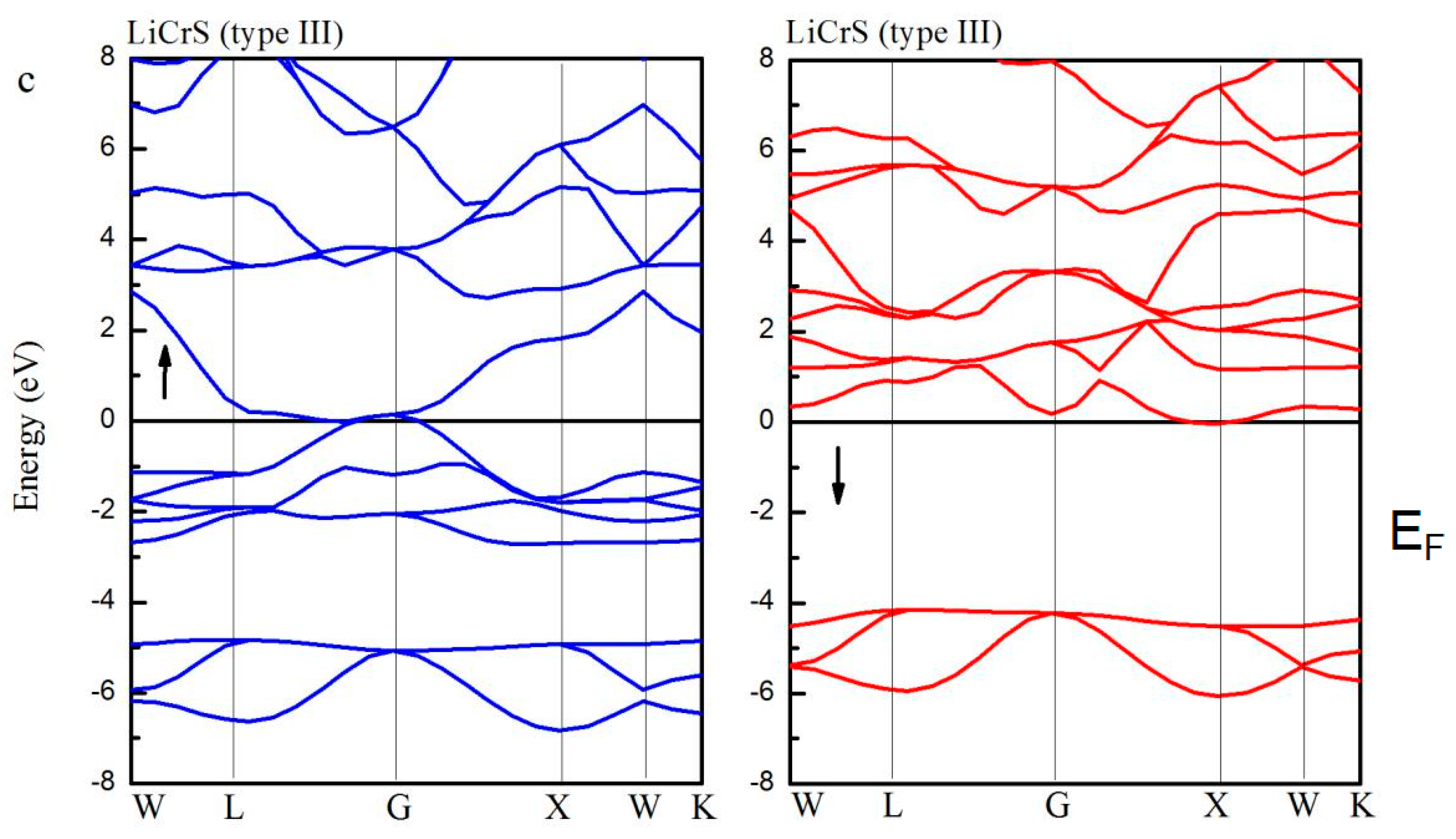
Figure 5 shows the spin-up (blue lines) and spin-down (red lines) band structures for the LiCrS alloy in the three atomic (type I, type II, and type III) arrangements. Definitely, in the type I and III arrangements, in both the spin channels, the EF overlaps with the energy bands. However, the LiCrS in the type II arrangement exhibits a half-metallic nature, namely, the majority spin electrons show metallic behaviors and the minority spin electrons exhibit semiconducting properties. Similar behaviors were also discovered in the LiCrSe and LiCrTe alloys. The indirect semiconducting-type band-gaps of the LiCrZ (Z = S, Se, Te) alloys in the type II arrangement are 3.62 eV, 3.15 eV, and 2.10 eV, respectively, and are also listed in Table 1.
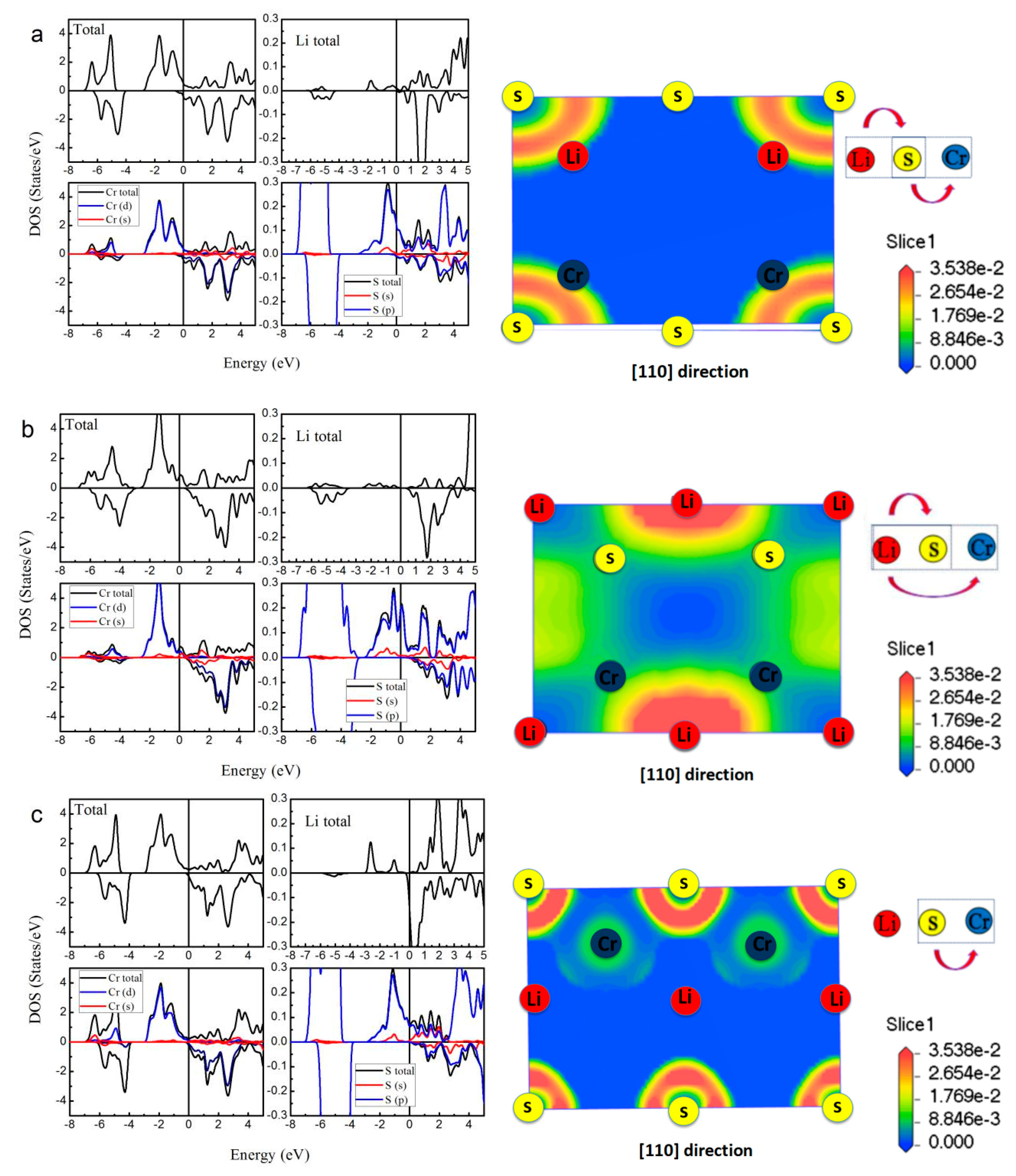
As a representative of all the LiCrZ alloys, in Figure 6, we display the calculated total and atom-projected DOS, and the ELF graphs of the LiCrS alloy in the three atomic arrangements (type I, type II, and type III). For the type II arrangement, in the minority spin channel, the antibonding peak is shifted high above EF due to the exchange splitting [18], whereas for the case of the type I and III arrangements, the minority DOS, together with the energy gap, moves to low energy. Moreover, we also perform the ELF maps project on the (1 1 0) plane of the LiCrS in types I, II, and III, respectively. The high- and low- ELF values in the graphs of ELF correspond to areas of localized electrons and the area around the maxima, respectively [35]. As shown in Figure 6a,c, for the type I and type III arrangements, the regions of the highest ELF value are all around the main-group S atom along the S-Cr bound axes, indicating their sharing behavior and the occurrence of the covalent bond. We should note that, for the type I and III arrangements, the S and Cr atoms are nearest neighbors and show strong S-Cr covalent-hybridization. However, for the type II arrangement, the S and Cr atoms sit by the second neighbor sites and show nearly no S-Cr covalent-hybridization.
In the half-Heusler alloys LiCrZ (Z = S, Se, Te), through the DOS and the ELF maps, we can summarize the electronic structure into the following two features: (i) under the strong S-Cr covalent-hybridization, LiCrZ (Z = S, Se, Te) alloys with both type I and type III arrangements show typically metallic band structure; (ii) under less S-Cr covalent-hybridization, LiCrZ with type II arrangement takes on excellent half-metallic band behavior with largest magnetic moment 5 μB at the equilibrium lattice constant.
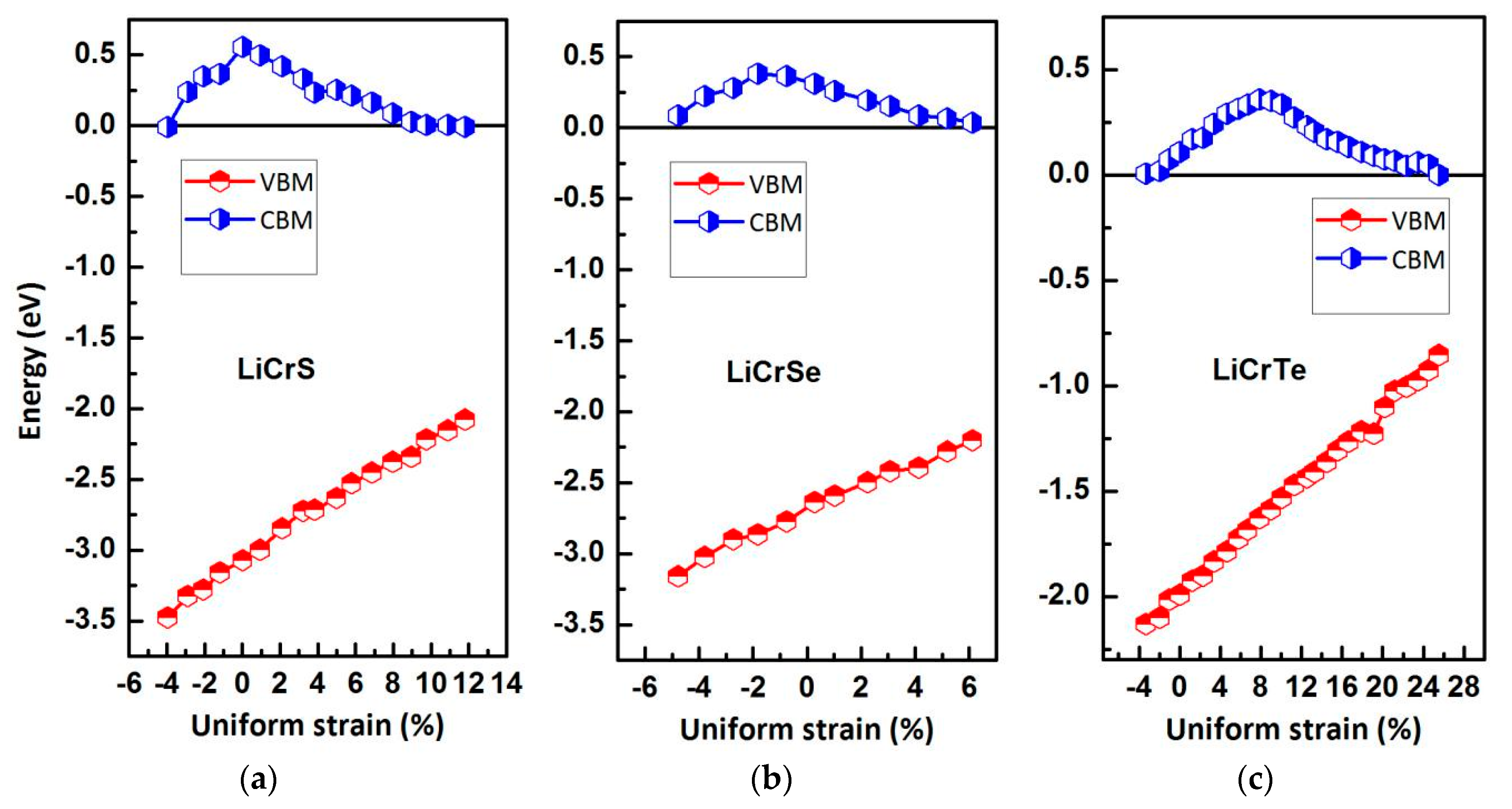
In the spintronic devices, the Heusler type HM multilayers or thin films are often touched by researchers; however, the actual and ideal lattice constants are usually inconsistent. The change of the lattice constants will lead to significant changes of the electronic, magnetic, and HM properties of the equilibrium state. Hence, we need to examine the HM stability for the LiCrZ (Z = S, Se, Te) in the type II arrangement at the strained lattice constants. The band-structure calculations at the strained lattice constants were performed for the LiCrZ (Z = S, Se, Te) alloys in the type II arrangement. In this discussion, the values of CBM and VBM for the LiCrZ (Z = S, Se, Te) alloys in the minority spin channel have been recorded to show the HM behavior for clarity, as plotted in Figure 7. Obviously, the HM states are kept for the lattice constants of 5.57–6.50 Å for LiCrS, 5.71–6.36 Å for LiCrSe, and 6.15–7.94 Å for LiCrTe, respectively. That is, these three alloys can maintain their half-metallicity when their lattice constants are changed by −4.2% to 11.68%, −4.83% to 6%, and −3.45% to 24.64% relative to the equilibrium lattice constants.
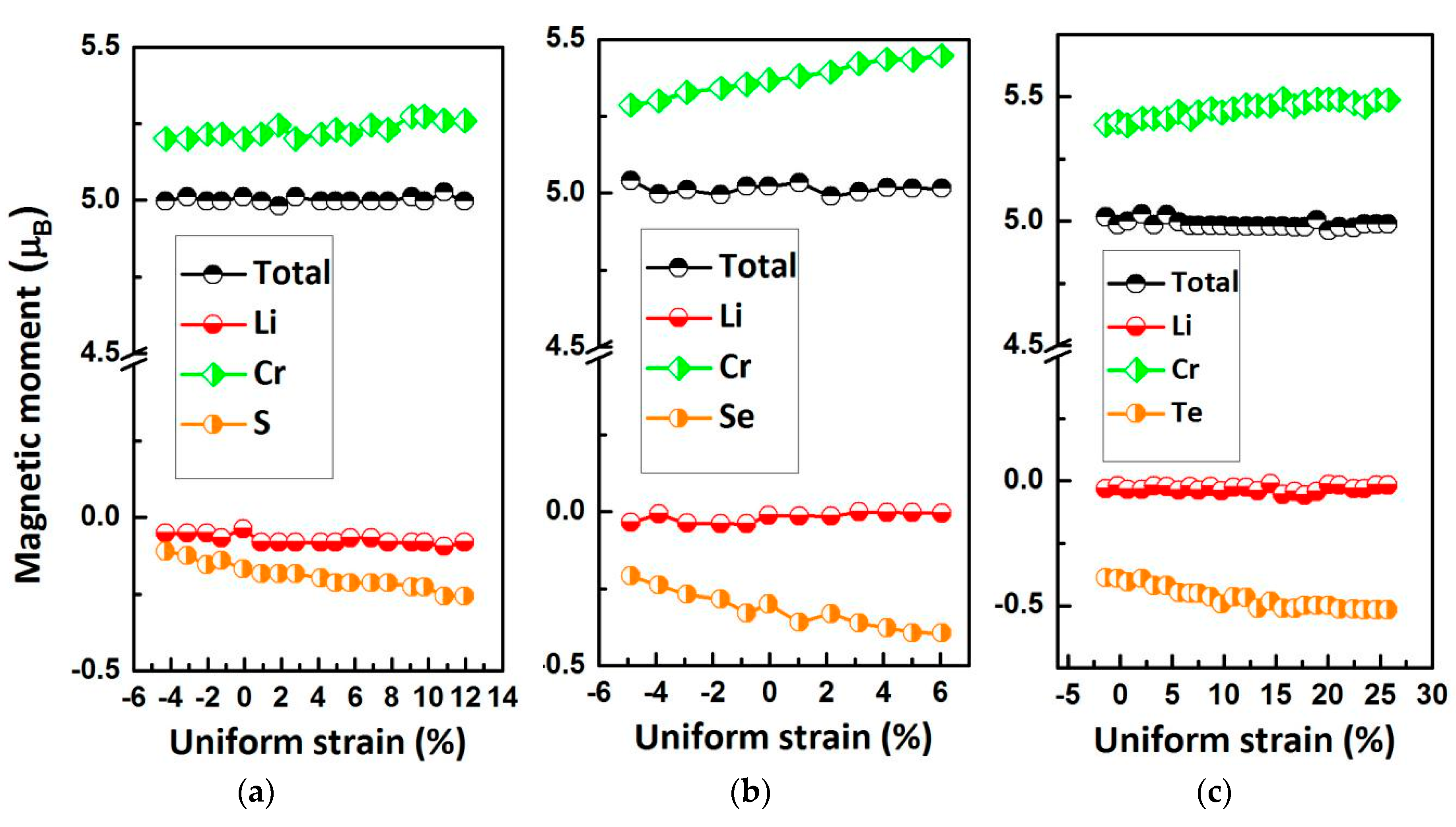
In Figure 8, we show the relationship between the Mt and the lattice constant of type II LiCrZ (Z = S, Se, Te). Obviously, the total Mt is always a fixed integer value 5 μB in the whole variational range. The atomic Mt of Cr and Z are sensitive to lattice distortion. The Mt of the Cr atom increases with increasing lattice constants, whereas for the Z (Z = S, Se, Te) atom they continuously decrease.
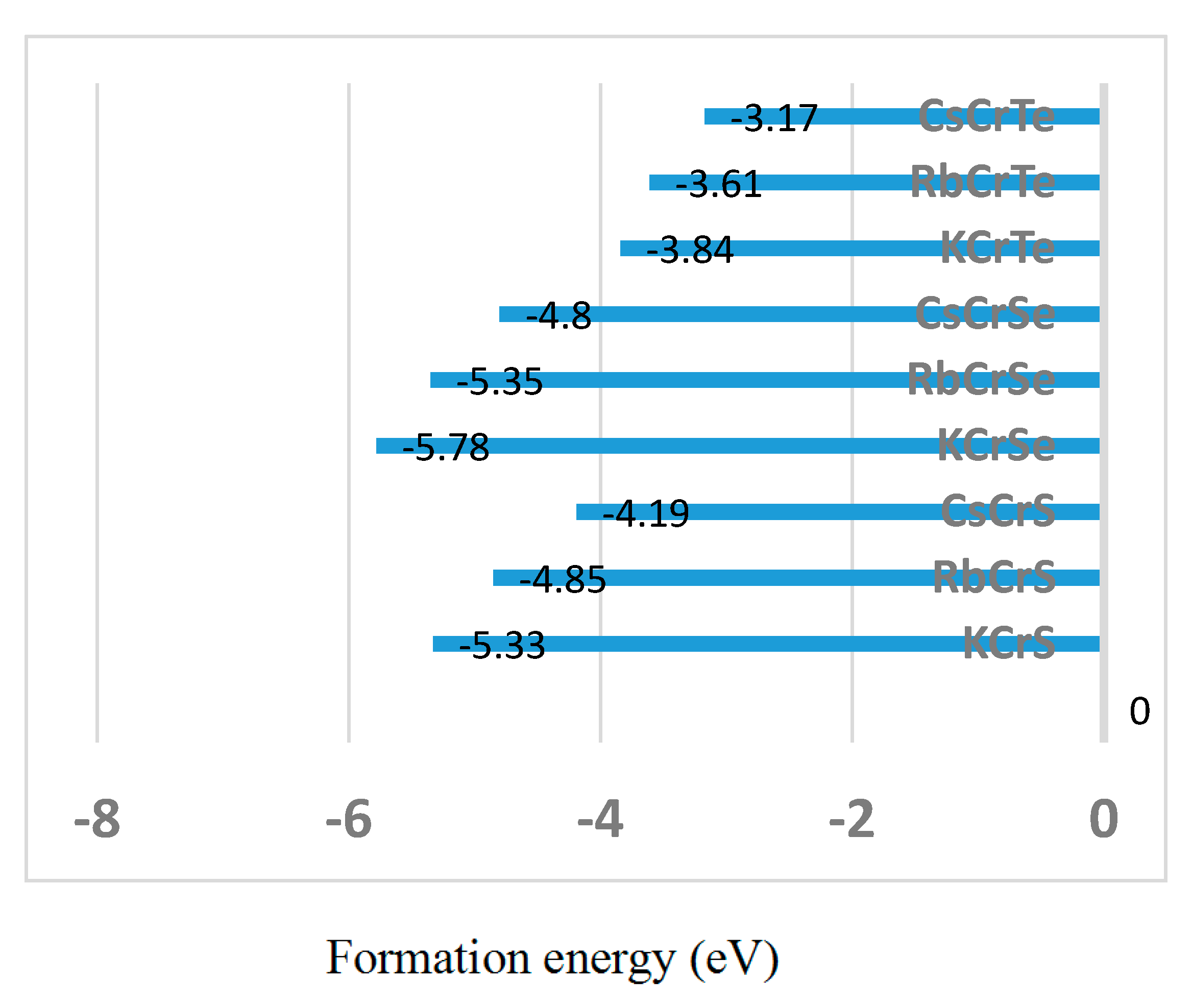
Our work suggests that the XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te) half-Heusler alloys are useful in spintonic applications. For the half-Heusler type XCrZ (X = K, Rb, Cs; Z = S, Se, Te) alloys, their formation energies have been calculated based on the following formula:
where is the total energy of XCrZ per formula unit, and , , and are the total energies per atom of each element in the bulk for the X, Cr, and Zr, respectively. The results have been shown in Figure 9; the negative formation energies indicate that these alloys are expected to be stable. Therefore, they have the large change to be synthesized by normal equilibrium methods (e.g., arc-melting). However, for LiCrZ (Z = S, Se, Te), some non-equilibrium methods (e.g., rapid quenching) can be selected to prepare these meta-stable compounds [36].
4. Conclusions
A first principles calculation was used to predict a series of new half-Heusler-based, half-metallic materials XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te) with the largest magnetic moment (5 μB) and large semiconducting-type band-gaps (>2 eV). In detail, for XCrZ (X = K, Rb, Cs; Z = S, Se, Te), the HM nature of these alloys appeared at their equilibrium and strained lattice constants and in their most stable phases. However, for the LiCrZ alloys, the HM behaviors of these alloys did not appear in the most stable phase but in one of the metastable phases. The half-metallicity of the XCrZ alloys is robust against uniform strain, which makes these alloys very stable with respect to the spin polarization properties.
Acknowledgments
Z.X. Cheng thanks the Australian Research Council for support. Funding for this research was provided by: Scientific Research Project for High Level Talent in Colleges and Universities of Hebei Province (No. GCC 2014042), Natural Science Foundation of Hebei Province (No. E2016202383), Chongqing City Funds for Distinguished Young Scientists (No. cstc2014jcyjjq50003), and the Program for Leading Talents in Science and Technology Innovation of Chongqing City (No. CSTCKJCXLJRC19).
Author Contributions
Zhenxiang Cheng and Guodong Liu designed the project, Xiaotian Wang performed the calculations and prepared the manuscript, and all authors analyzed the data and discussed the results.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Žutić, I.; Fabian, J.; Sarma, S.D. Spintronics: Fundamentals and applications. Rev. Mod. Phys. 2004, 76, 323–410. [Google Scholar] [CrossRef]
- Park, J.H.; Vescovo, E.; Kim, H.; Kwon, C. Direct evidence for a half-metallic ferromagnet. Nature 1998, 392, 794–796. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Wu, M.W. Schottky-barrier-induced spin relaxation in spin injection. Phys. Rev. B 2005, 72, 153301. [Google Scholar] [CrossRef]
- Hashemifar, S.J.; Kratzer, P.; Scheffler, M. Preserving the half-metallicity at the Heusler alloy Co2MnSi (001) surface: A density functional theory study. Phys. Rev. Lett. 2005, 94, 096402. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wray, L.A.; Xia, Y.; Xu, S.; Jia, S.; Cava, R.J.; Hasan, M.Z. Half-Heusler ternary compounds as new multifunctional experimental platforms for topological quantum phenomena. Nat. Mater. 2010, 9, 546–549. [Google Scholar] [CrossRef] [PubMed]
- De Groot, R.A.; Mueller, F.M.; Van Engen, P.G.; Buschow, K.H.J. New class of materials: Half-metallic ferromagnets. Phys. Rev. Lett. 1983, 50, 2024. [Google Scholar] [CrossRef]
- Khalaf Alzyadi, J.M.; Jolan, M.H.; Yao, K.L. Surface half-metallicity of half-Heusler compound FeCrSe and interface half-metallicity of FeCrSe/Gap. J. Magn. Magn. Mater. 2016, 403, 8–13. [Google Scholar] [CrossRef]
- Lakdja, A.; Rozale, H.; Chahed, A.; Benhelal, O. Ferromagnetism in the half-heusler XCsBa compounds from first-principles calculations (X = C, Si, and Ge). J. Alloys Compd. 2013, 564, 8–12. [Google Scholar] [CrossRef]
- Lakdja, A.; Rozale, H.; Sayede, A.; Chahed, A. Origin of ferromagnetism in the half-Heusler XRbCs compounds (X = N, P and As). J. Magn. Magn. Mater. 2014, 354, 235–238. [Google Scholar] [CrossRef]
- Sedeek, K.; Hantour, H.; Makram, N.; Said, S.A. Observation of strong ferromagnetism in the half-heusler compound cotisb system. J. Magn. Magn. Mater. 2016, 407, 218–223. [Google Scholar] [CrossRef]
- Rozale, H.; Amar, A.; Lakdja, A.; Moukadem, A.; Chahed, A. Half-metallicity in the half-Heusler RbSrC, RbSrSi and RbSrGe compounds. J. Magn. Magn. Mater. 2013, 336, 83–87. [Google Scholar] [CrossRef]
- Białek, B.; Lee, J.I.; Kim, M. The surface electronic properties of newly designed half-metallic ferromagnets: GeKCa and SnKCa. Comput. Mater. Sci. 2014, 81, 510–516. [Google Scholar] [CrossRef]
- Umamaheswari, R.; Vijayalakshmi, D.; Kalpana, G. First-principles calculation of structural, electronic and magnetic properties of half-Heusler LiCaC and NaCaC compounds. Phys. B Condens. Matter 2014, 448, 256–259. [Google Scholar] [CrossRef]
- Chen, J.; Gao, G.Y.; Yao, K.L.; Song, M.H. Half-metallic ferromagnetism in the half-Heusler compounds GeKCa and SnKCa from first-principles calculations. J. Alloys Compd. 2011, 509, 10172–10178. [Google Scholar] [CrossRef]
- Ahmad, M.; Murtaza, G.; Khenata, R.; Omran, S.B.; Bouhemadou, A. Structural, elastic, electronic, magnetic and optical properties of RbSrX (X = C, Si, Ge) half-Heusler compounds. J. Magn. Magn. Mater. 2015, 377, 204–210. [Google Scholar] [CrossRef]
- Damewood, L.; Busemeyer, B.; Shaughnessy, M.; Fong, C.Y.; Yang, L.H.; Felser, C. Stabilizing and increasing the magnetic moment of half-metals: The role of Li in half-Heusler LiMnZ (Z = N, P, Si). Phys. Rev. B 2015, 91, 064409. [Google Scholar] [CrossRef]
- Zhang, X.M.; Xu, G.Z.; Du, Y.; Liu, E.K.; Liu, Z.Y.; Liu, G.D.; Wu, G.H. Phase stability, magnetism and generalized electron-filling rule of vanadium-based inverse Heusler compounds. Europhys. Lett. 2013, 104, 27012. [Google Scholar] [CrossRef]
- Fecher, G.H.; Kandpal, H.C.; Wurmehl, S.; Felser, C.; Schönhense, G. Slater-pauling rule and curie temperature of Co2-based Heusler compounds. J. Appl. Phys. 2006, 99, 08J106. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, X.; Cheng, Z. Electronic, magnetic, mechanical, half-metallic and highly dispersive zero-gap half-metallic properties of rare-earth-element-based quaternary Heusler compounds. J. Alloys Compd. 2017, 718, 63–74. [Google Scholar] [CrossRef]
- Gao, G.Y.; Hu, L.; Yao, K.L.; Luo, B.; Liu, N. Large half-metallic gaps in the quaternary Heusler alloys CoFeCrZ (Z = Al, Si, Ga, Ge): A first-principles study. J. Alloys Compd. 2013, 551, 539–543. [Google Scholar] [CrossRef]
- Payne, M.C.; Teter, M.P.; Allan, D.C.; Arias, T.A.; Joannopoulos, J.D. Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients. Rev. Mod. Phys. 1992, 64, 1045. [Google Scholar] [CrossRef]
- Segall, M.D.; Lindan, P.J.; Probert, M.A.; Pickard, C.J.; Hasnip, P.J.; Clark, S.J.; Payne, M.C. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892–7895. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Edgecombe, K.E.A. Simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, Z.; Wang, J.; Wang, L.; Yu, Z.; Fang, C.; Liu, G. Origin of the half-metallic band-gap in newly designed quaternary Heusler compounds ZrVTiZ (Z = Al, Ga). RSC Adv. 2016, 6, 57041–57047. [Google Scholar] [CrossRef]
- Birsan, A. Small interfacial distortions lead to significant changes of the half-metallic and magnetic properties in Heusler alloys: The case of the new CoFeZrSi compound. J. Alloys Compd. 2017, 710, 393–398. [Google Scholar] [CrossRef]
- Wei, X.P.; Zhang, Y.L.; Wang, T.; Sun, X.W.; Song, T.; Guo, P.; Deng, J.B. Stability, electronic and magnetic properties investigations on Zr2YZ (Y = Co, Cr, V and Z = Al, Ga, In, Pb, Sn, Tl) compounds. Mater. Res. Bull. 2017, 86, 139–145. [Google Scholar] [CrossRef]
- Wang, X.T.; Cui, Y.T.; Liu, X.F.; Liu, G.D. Electronic structures and magnetism in the Li2AgSb-type Heusler alloys, Zr2CoZ (Z = Al, Ga, In, Si, Ge, Sn, Pb, Sb): A first-principles study. J. Magn. Magn. Mater. 2015, 394, 50–59. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Liu, Z.H.; Liu, G.D.; Ma, X.Q. Half-metallic fully compensated ferrimagnetism in C1b-type half heusler compounds Mn2Si1−xGex. J. Magn. Magn. Mater. 2015, 387, 67–71. [Google Scholar] [CrossRef]
- Zhang, Y.J.; Liu, Z.H.; Liu, E.K.; Liu, G.D.; Ma, Q.X.; Wu, G.H. Towards fully compensated ferrimagnetic spin gapless semiconductors for spintronic applications. Europhys. Lett. 2015, 111, 37009. [Google Scholar] [CrossRef]
- Kieven, D.; Klenk, R.; Naghavi, S.; Felser, C.; Gruhn, T. I-II-V half-Heusler compounds for optoelectronics: Ab initio calculations. Phys. Rev. B 2010, 81, 075208. [Google Scholar] [CrossRef]
- Jungwirth, T.; Novák, V.; Martí, X.; Cukr, M.; Máca, F.; Shick, A.B.; Zemek, J. Demonstration of molecular beam epitaxy and a semiconducting band structure for I-Mn-V compounds. Phys. Rev. B 2011, 83, 035321. [Google Scholar] [CrossRef]
- Zhang, X.M.; Xu, G.Z.; Liu, E.K.; Liu, Z.Y.; Wang, W.H.; Wu, G.H. On the influence of tetrahedral covalent-hybridization on electronic band structure of topological insulators from first principles. J. Appl. Phys. 2015, 117, 045706. [Google Scholar] [CrossRef]
- Luo, H.; Xin, Y.; Liu, B.; Meng, F.; Liu, H.; Liu, E.; Wu, G. Competition of L2 1 and XA structural ordering in Heusler alloys X2CuAl (X = Sc, Ti, V, Cr, Mn, Fe, Co, Ni). J. Alloys Compd. 2016, 665, 180–185. [Google Scholar] [CrossRef]
Figure 1.
Crystal structures of the three arrangements within the half-Heusler alloys XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te) noted as type I (a); type II (b) and type III (c).
Figure 1.
Crystal structures of the three arrangements within the half-Heusler alloys XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te) noted as type I (a); type II (b) and type III (c).
Figure 2.
The total energy as a function of the lattice constant in the three atomic arrangements type I, type II, and type III for half-Heusler alloys LiCrS (a); KCrS (b); RbCrS (c); CsCrS (d); LiCrSe (e); KCrSe (f); RbCrSe (g); CsCrSe (h); LiCrTe (i); KCrTe (j); RbCrTe (k) and CsCrTe (l), respectively.
Figure 2.
The total energy as a function of the lattice constant in the three atomic arrangements type I, type II, and type III for half-Heusler alloys LiCrS (a); KCrS (b); RbCrS (c); CsCrS (d); LiCrSe (e); KCrSe (f); RbCrSe (g); CsCrSe (h); LiCrTe (i); KCrTe (j); RbCrTe (k) and CsCrTe (l), respectively.
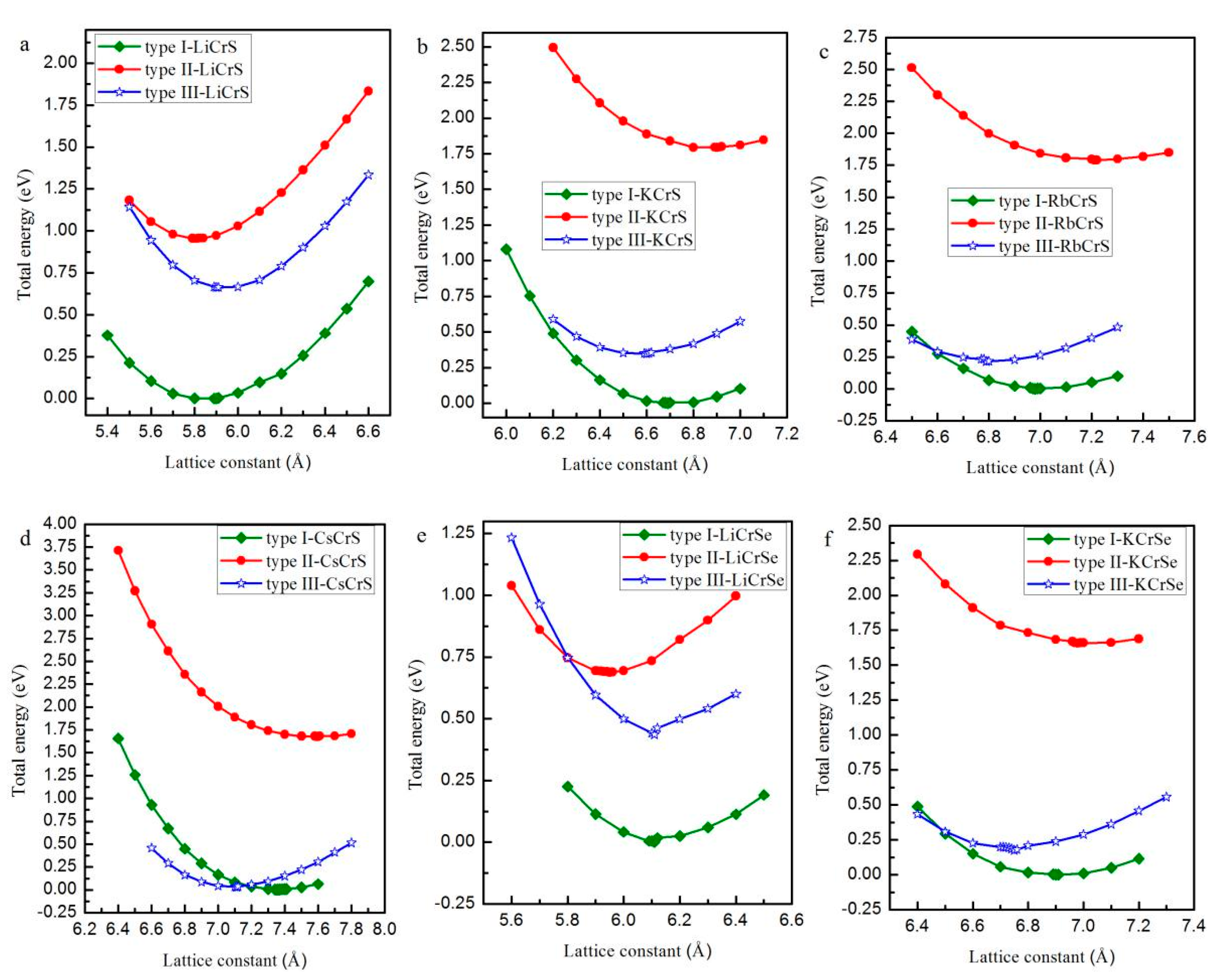
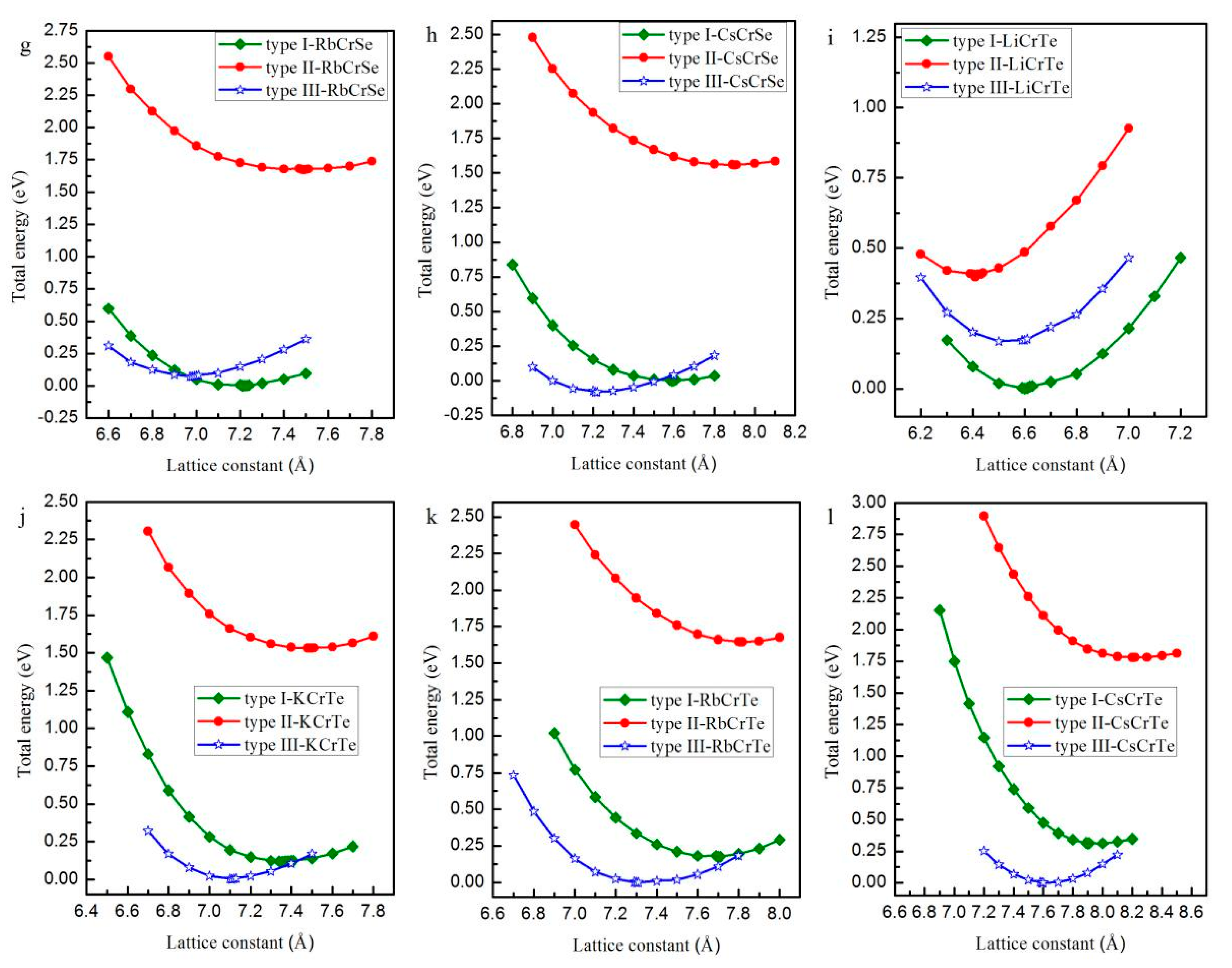
Figure 3.
Calculated total and atom-projected density of states (DOS) for (a) type I KCrS; (b) type I RbCrS; (c) type I CsCrS; (d) type I KCrSe; (e) type I RbCrSe; (f) type III CsCrSe; (g) type III KCrTe; (h) type III RbCrTe; and (i) type III CsCrTe. (The Fermi level EF was set as X = 0).
Figure 3.
Calculated total and atom-projected density of states (DOS) for (a) type I KCrS; (b) type I RbCrS; (c) type I CsCrS; (d) type I KCrSe; (e) type I RbCrSe; (f) type III CsCrSe; (g) type III KCrTe; (h) type III RbCrTe; and (i) type III CsCrTe. (The Fermi level EF was set as X = 0).
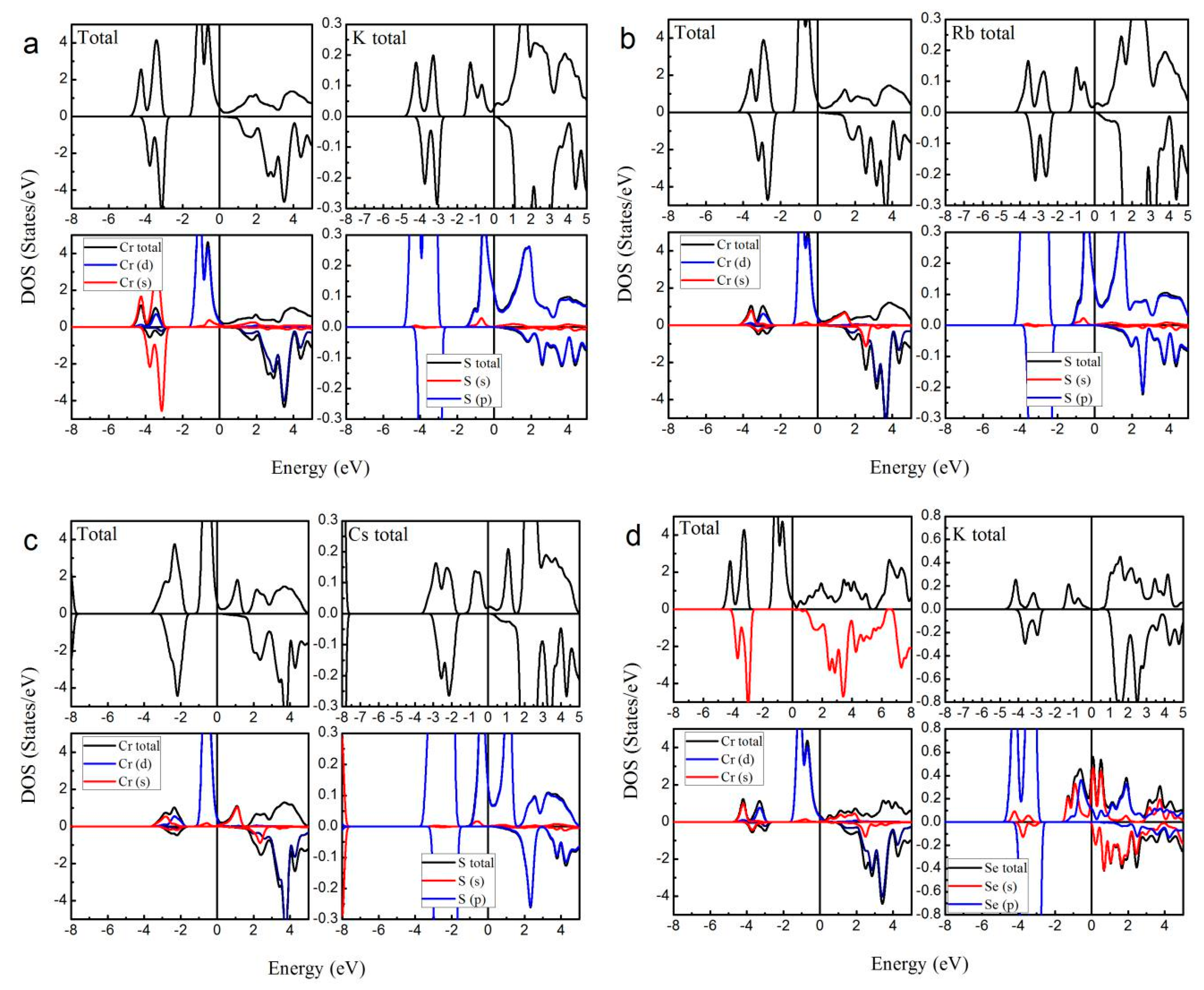
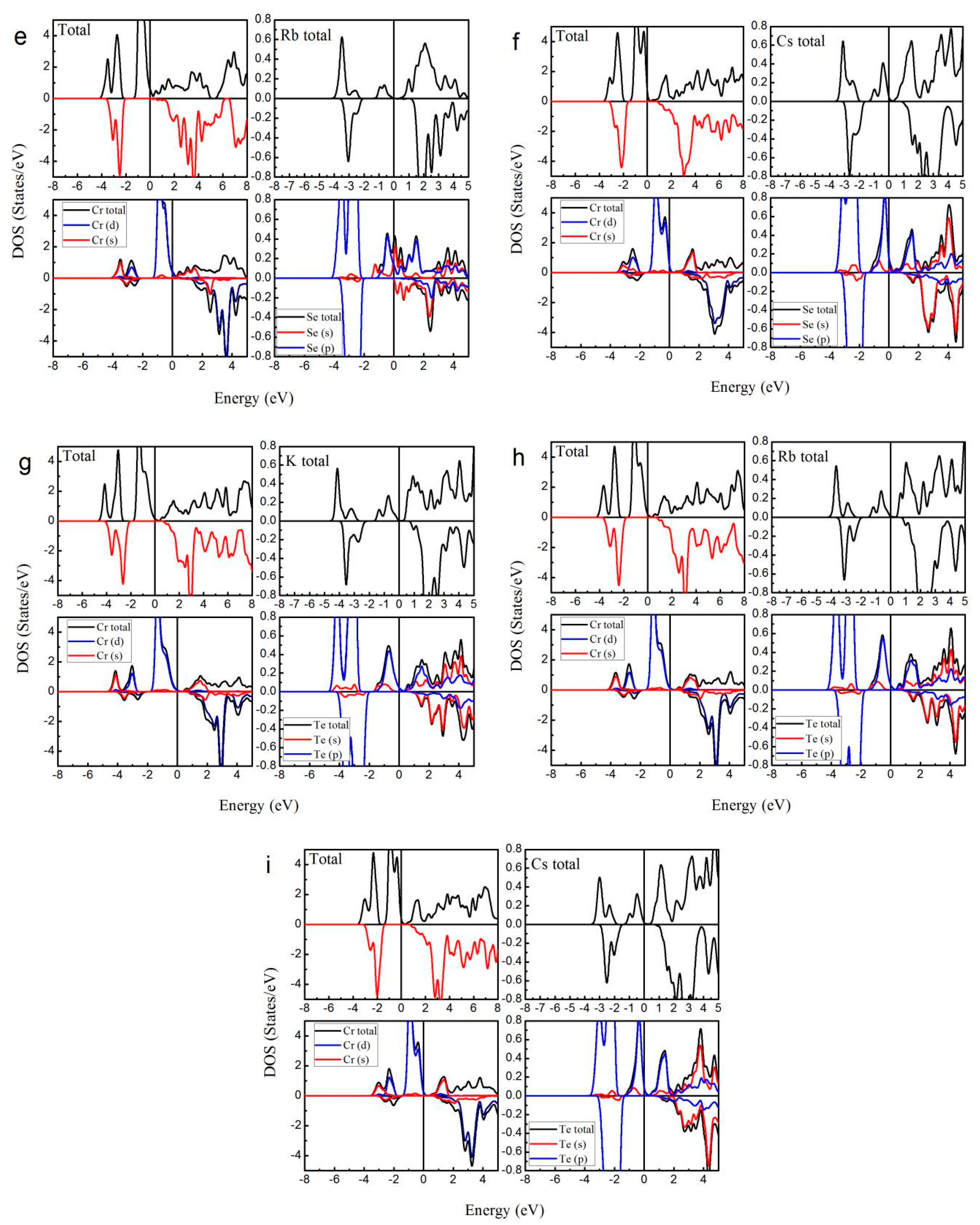
Figure 4.
Calculated total magnetic moments as functions of the lattice constant for KCrS, RbCrS, CsCrS, KCrSe, RbCrSe (a) and CsCrSe, KCrTe, RbCrTe, CsCrTe (b), respectively, with their most stable phase.
Figure 4.
Calculated total magnetic moments as functions of the lattice constant for KCrS, RbCrS, CsCrS, KCrSe, RbCrSe (a) and CsCrSe, KCrTe, RbCrTe, CsCrTe (b), respectively, with their most stable phase.
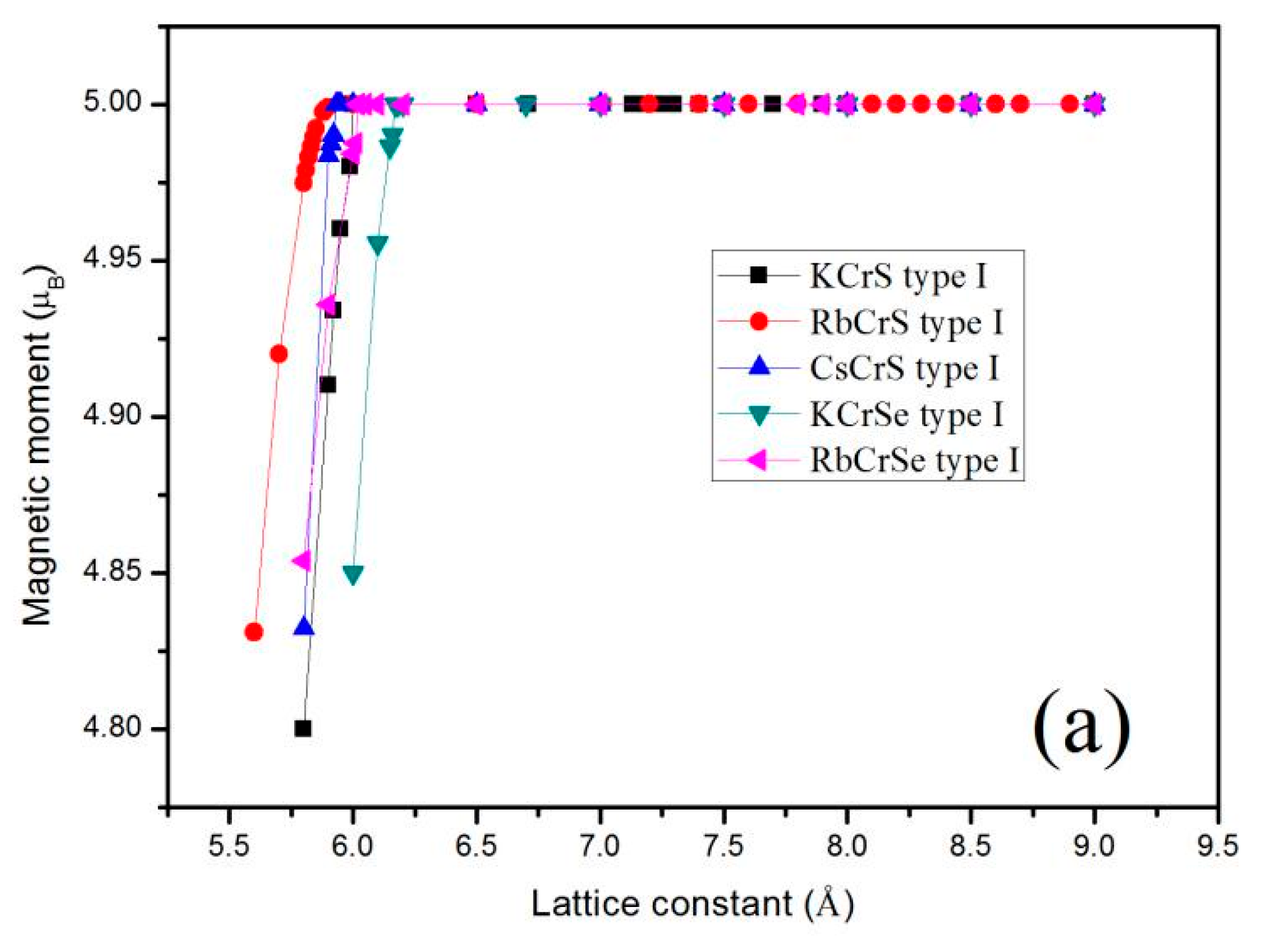
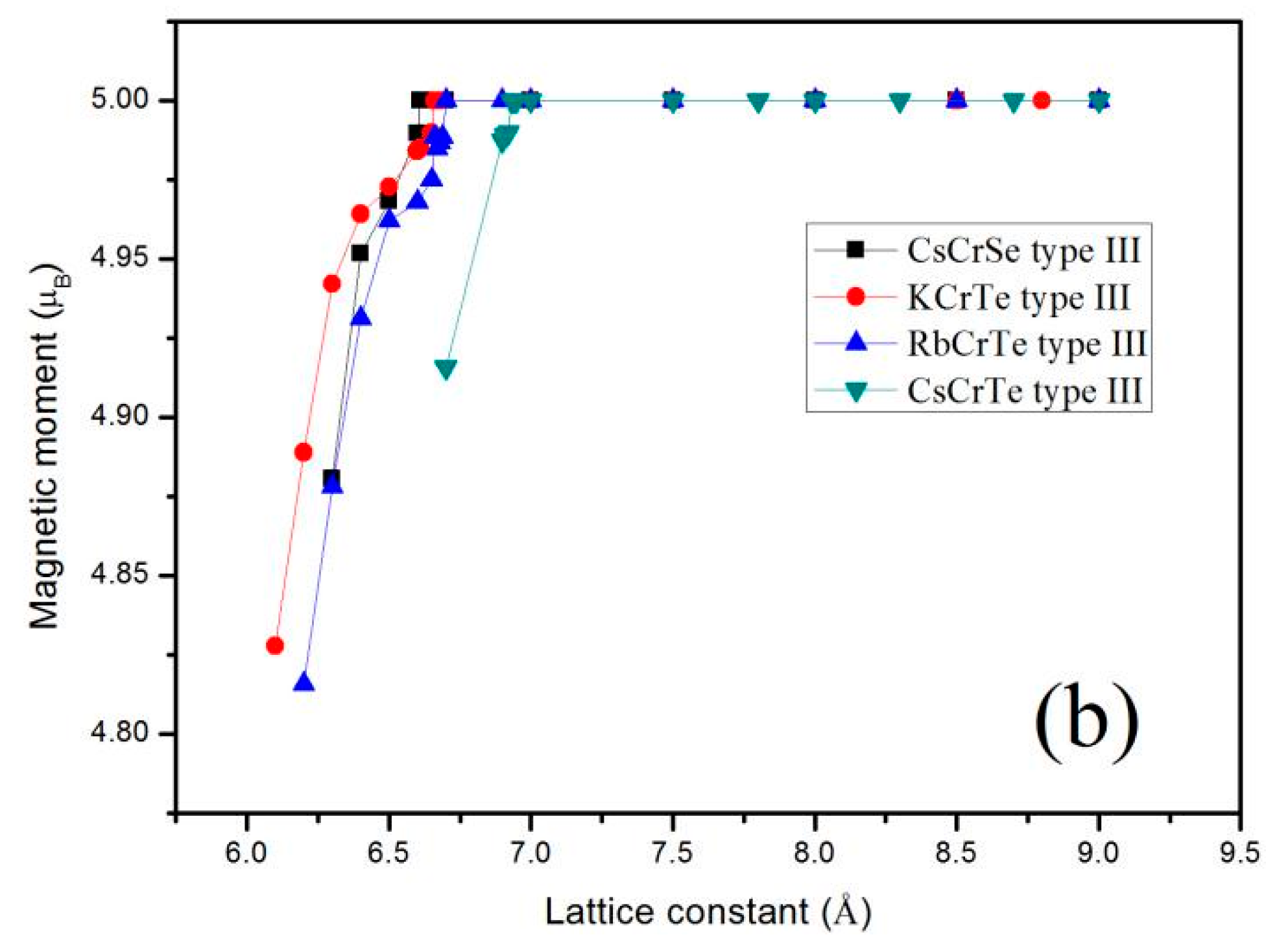
Figure 5.
The spin-up (blue lines) and spin-down (red lines) band structures for LiCrS alloy in the three atomic (type I (a); type II (b); and type III (c)) arrangements. (The Fermi level EF was set as Y = 0).
Figure 5.
The spin-up (blue lines) and spin-down (red lines) band structures for LiCrS alloy in the three atomic (type I (a); type II (b); and type III (c)) arrangements. (The Fermi level EF was set as Y = 0).
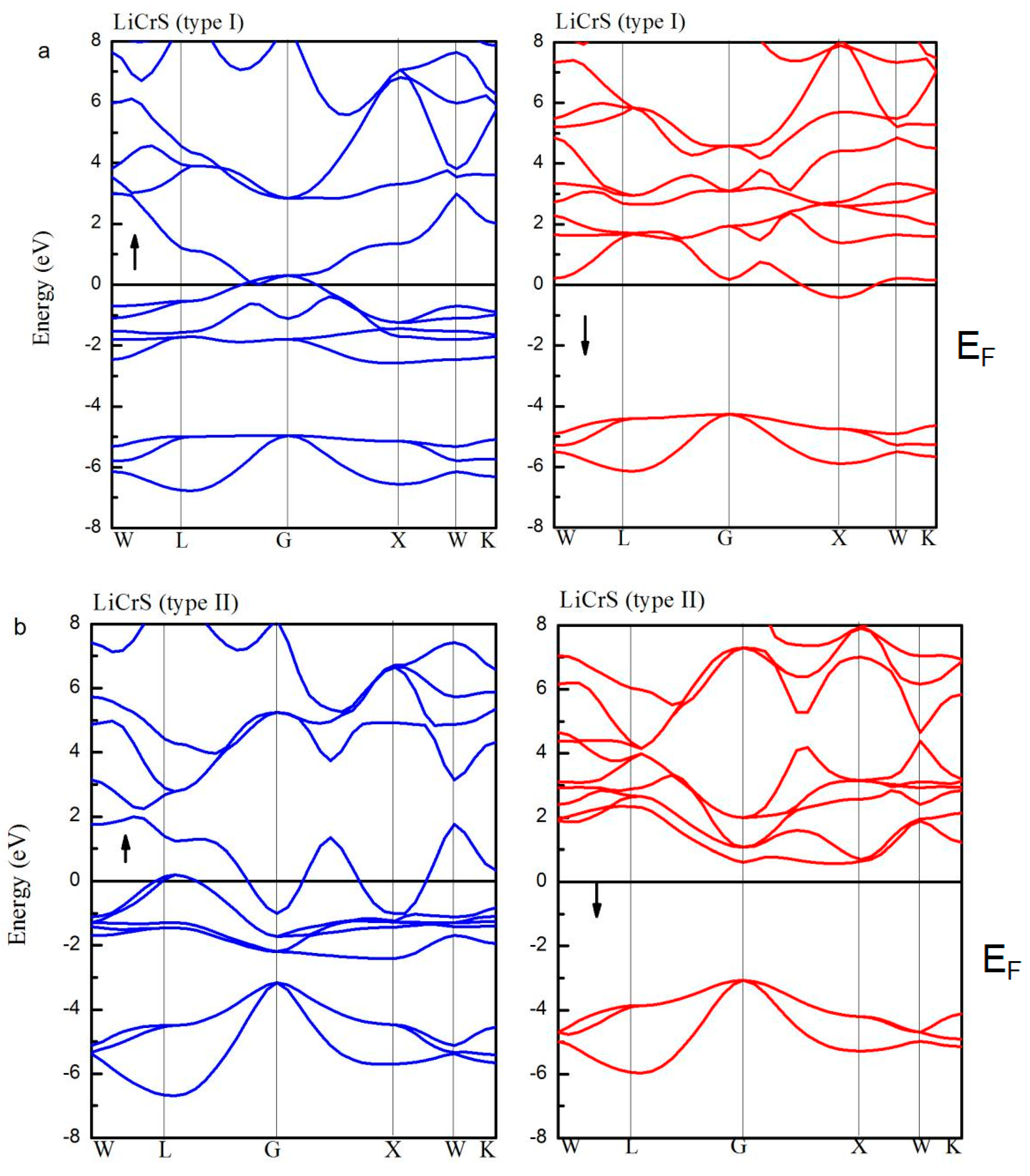

Figure 6.
Calculated total and atom-projected DOS, and the electron localization function (ELF) graphs of the LiCrS alloy in the three atomic arrangements: type I (a); type II (b); and type III (c). (The Fermi level EF was set as X = 0).
Figure 6.
Calculated total and atom-projected DOS, and the electron localization function (ELF) graphs of the LiCrS alloy in the three atomic arrangements: type I (a); type II (b); and type III (c). (The Fermi level EF was set as X = 0).
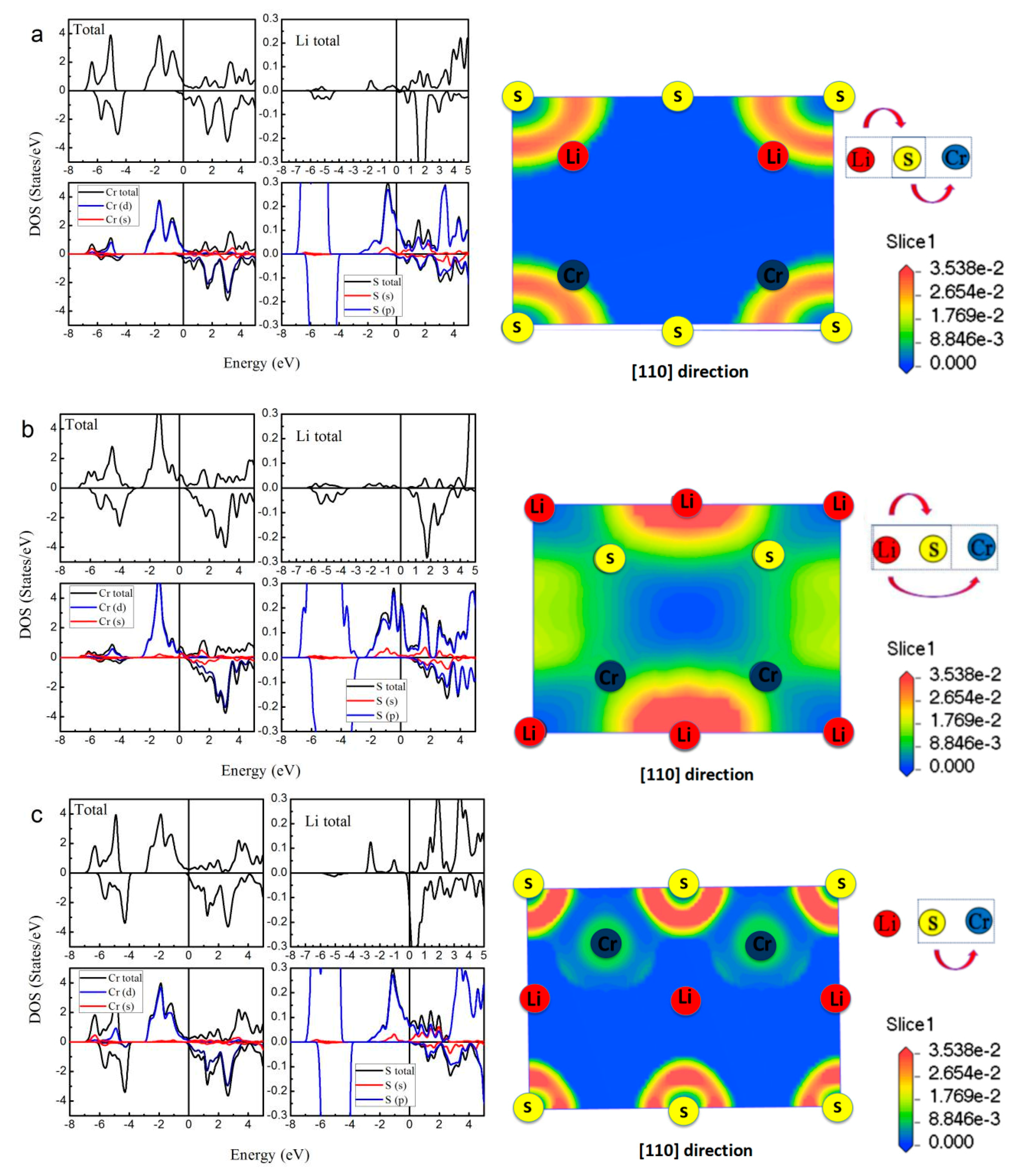
Figure 7.
Calculated valence band maximum (VBM) and conduction band minimum (CBM) as a function of the lattice constants for the LiCrS (a), LiCrSe (b) and LiCrTe (c) alloys in the type II arrangement. The black circle and the red circle represent the VBM and CBM, respectively. (The Fermi level EF was set as Y = 0).
Figure 7.
Calculated valence band maximum (VBM) and conduction band minimum (CBM) as a function of the lattice constants for the LiCrS (a), LiCrSe (b) and LiCrTe (c) alloys in the type II arrangement. The black circle and the red circle represent the VBM and CBM, respectively. (The Fermi level EF was set as Y = 0).
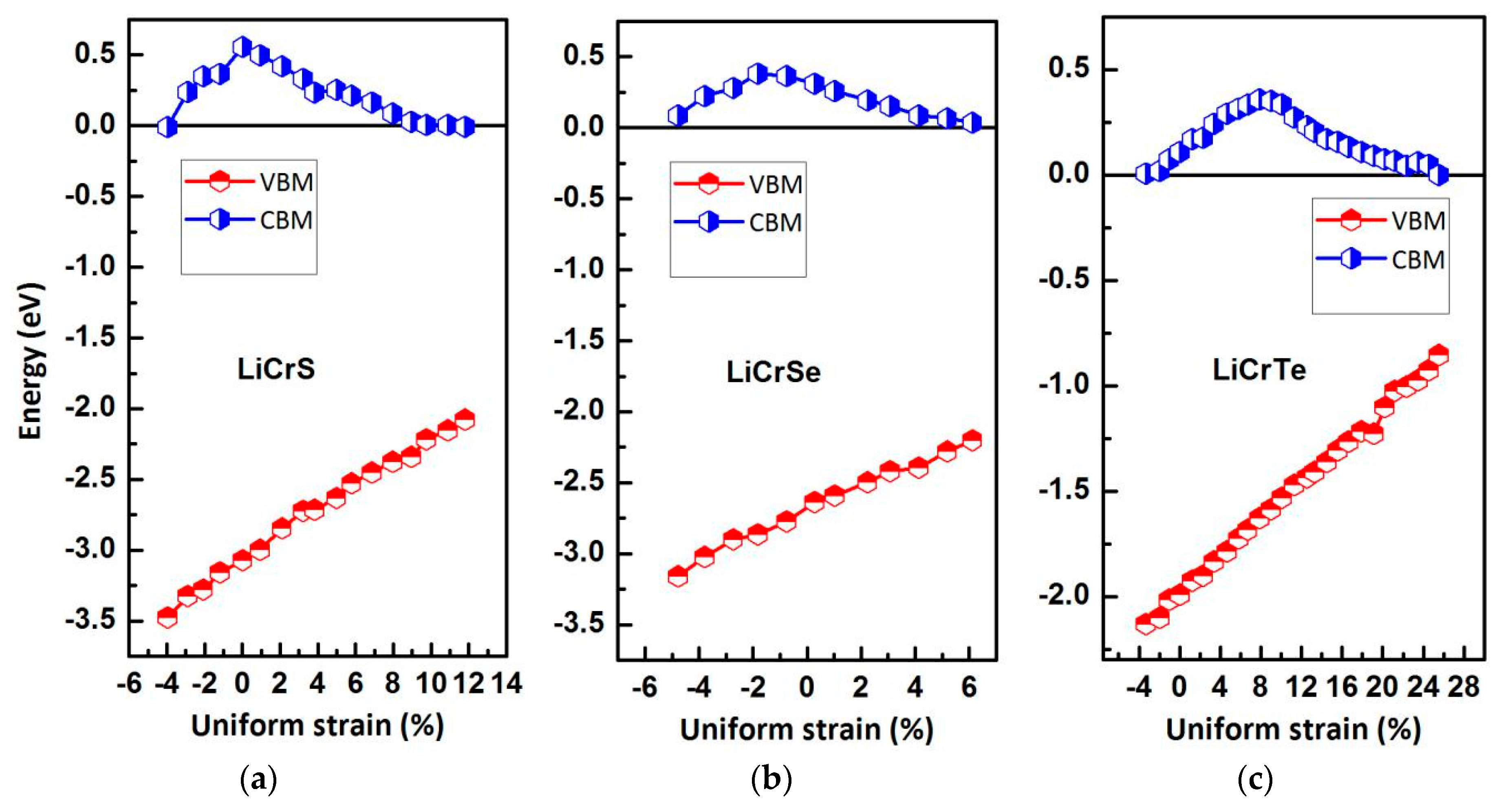
Figure 8.
Total and atomic magnetic moments as functions of the lattice constants for the LiCrS (a), LiCrSe (b) and LiCrTe (c) alloys in type II arrangement.
Figure 8.
Total and atomic magnetic moments as functions of the lattice constants for the LiCrS (a), LiCrSe (b) and LiCrTe (c) alloys in type II arrangement.
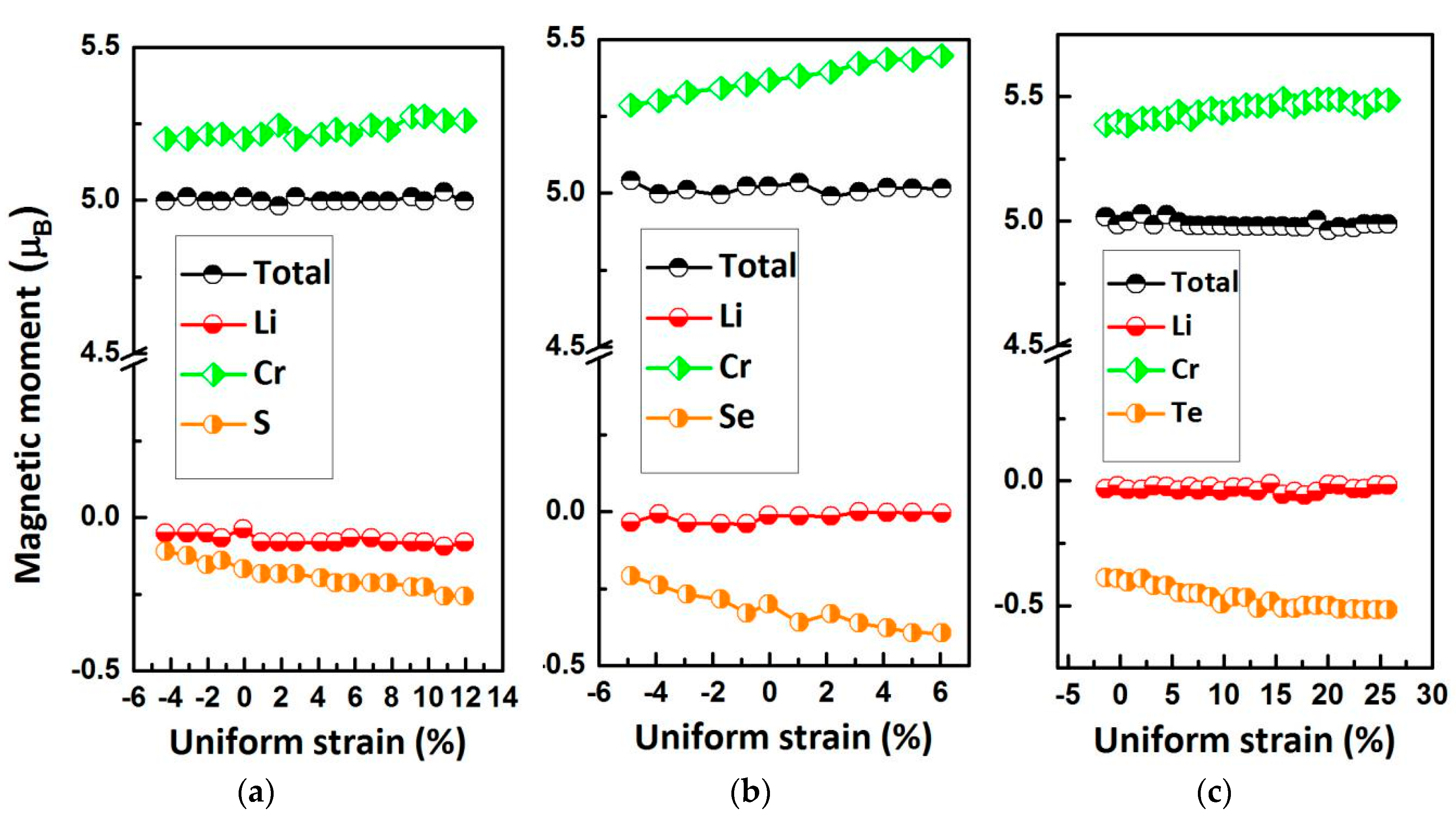
Figure 9.
Calculated formation energies (eV) for the XCrZ (X = K, Rb, Cs; Z = S, Se, Te) alloys with the most stable phase.
Figure 9.
Calculated formation energies (eV) for the XCrZ (X = K, Rb, Cs; Z = S, Se, Te) alloys with the most stable phase.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimized lattice constants (a), calculated total and the atomic magnetic moments (Mt) per formula unit, and sizes of the semiconducting-type band-gap (Gap) at different crystal structures for XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te). Yes (or no) indicates that this alloy is (or is not) a half metal.
Table 1.
Optimized lattice constants (a), calculated total and the atomic magnetic moments (Mt) per formula unit, and sizes of the semiconducting-type band-gap (Gap) at different crystal structures for XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te). Yes (or no) indicates that this alloy is (or is not) a half metal.
| Alloy | Type | a (Å) | Mtot (μB) | MX | MCr | MZ | Gap (eV) | HM Behavior |
|---|---|---|---|---|---|---|---|---|
| LiCrS | Type I | 5.89 | 4.82 | −0.08 | 5.04 | −0.14 | - | No |
| Type II | 5.82 | 5.00 | −0.08 | 5.22 | −0.12 | 3.62 | Yes | |
| Type III | 5.90 | 4.97 | −0.02 | 5.18 | −0.20 | - | No | |
| LiCrSe | Type I | 6.19 | 4.92 | −0.08 | 5.16 | −0.16 | - | No |
| Type II | 6.00 | 5.00 | −0.04 | 5.36 | −0.32 | 3.15 | Yes | |
| Type III | 6.11 | 4.97 | 0.04 | 5.30 | −0.36 | - | No | |
| LiCrTe | Type I | 6.60 | 4.95 | −0.02 | 5.30 | −0.32 | - | No |
| Type II | 6.37 | 5.00 | −0.02 | 5.42 | −0.40 | 2.10 | Yes | |
| Type III | 6.60 | 4.94 | 0.02 | 5.40 | −0.48 | - | No | |
| KCrS | Type I | 6.71 | 5.00 | −0.12 | 5.34 | −0.22 | 3.15 | Yes |
| Type II | 6.90 | 5.00 | −0.32 | 5.54 | −0.24 | 2.14 | Yes | |
| Type III | 6.60 | 5.00 | −0.24 | 5.40 | −0.18 | 3.41 | Yes | |
| KCrSe | Type I | 6.91 | 5.00 | −0.26 | 5.38 | −0.12 | 2.85 | Yes |
| Type II | 6.98 | 5.00 | −0.12 | 5.58 | −0.46 | 2.09 | Yes | |
| Type III | 6.75 | 5.00 | −0.14 | 5.46 | −0.34 | 3.07 | Yes | |
| KCrTe | Type I | 7.35 | 5.00 | −0.06 | 5.46 | −0.42 | 2.87 | Yes |
| Type II | 7.41 | 5.00 | −0.08 | 5.66 | −0.58 | 1.87 | Yes | |
| Type III | 7.11 | 5.00 | −0.10 | 5.54 | −0.46 | 2.92 | Yes | |
| RbCrS | Type I | 7.01 | 5.00 | −0.14 | 5.38 | −0.24 | 2.65 | Yes |
| Type II | 7.21 | 5.00 | −0.30 | 5.56 | −0.26 | 1.69 | Yes | |
| Type III | 6.79 | 5.00 | −0.24 | 5.40 | −0.18 | 3.19 | Yes | |
| RbCrSe | Type I | 7.21 | 5.00 | −0.12 | 5.42 | −0.30 | 2.33 | Yes |
| Type II | 7.49 | 5.00 | −0.08 | 5.64 | −0.56 | 1.49 | Yes | |
| Type III | 6.98 | 5.00 | −0.16 | 5.46 | −0.30 | 2.72 | Yes | |
| RbCrTe | Type I | 7.63 | 5.00 | −0.04 | 5.50 | −0.46 | 2.40 | Yes |
| Type II | 7.81 | 5.00 | −0.06 | 5.68 | −0.62 | 1.54 | Yes | |
| Type III | 7.30 | 5.00 | −0.12 | 5.54 | −0.42 | 2.71 | Yes | |
| CsCrS | Type I | 7.35 | 5.00 | −0.14 | 5.42 | −0.28 | 2.09 | Yes |
| Type II | 7.59 | 5.00 | −0.26 | 5.58 | −0.30 | 1.41 | Yes | |
| Type III | 7.11 | 5.00 | −0.22 | 5.40 | −0.18 | 2.83 | Yes | |
| CsCrSe | Type I | 7.54 | 5.00 | −0.02 | 5.48 | −0.46 | 2.01 | Yes |
| Type II | 7.90 | 5.00 | −0.04 | 5.68 | −0.64 | 1.31 | Yes | |
| Type III | 7.21 | 5.00 | −0.14 | 5.42 | −0.28 | 2.61 | Yes | |
| CsCrTe | Type I | 7.98 | 5.00 | −0.02 | 5.52 | −0.50 | 2.09 | Yes |
| Type II | 8.21 | 5.00 | −0.02 | 5.72 | −0.70 | 1.31 | Yes | |
| Type III | 7.59 | 5.00 | −0.12 | 5.54 | −0.42 | 2.29 | Yes |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wang, X.; Cheng, Z.; Liu, G. Largest Magnetic Moments in the Half-Heusler Alloys XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te): A First-Principles Study. Materials 2017, 10, 1078. https://doi.org/10.3390/ma10091078
AMA Style
Wang X, Cheng Z, Liu G. Largest Magnetic Moments in the Half-Heusler Alloys XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te): A First-Principles Study. Materials. 2017; 10(9):1078. https://doi.org/10.3390/ma10091078
Chicago/Turabian StyleWang, Xiaotian, Zhenxiang Cheng, and Guodong Liu. 2017. "Largest Magnetic Moments in the Half-Heusler Alloys XCrZ (X = Li, K, Rb, Cs; Z = S, Se, Te): A First-Principles Study" Materials 10, no. 9: 1078. https://doi.org/10.3390/ma10091078
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.