
Molecular Level Factors Affecting the Efficiency of Organic Chromophores for p-Type Dye Sensitized Solar Cells

 , , ,
, , ,
Abstract
:1. Introduction
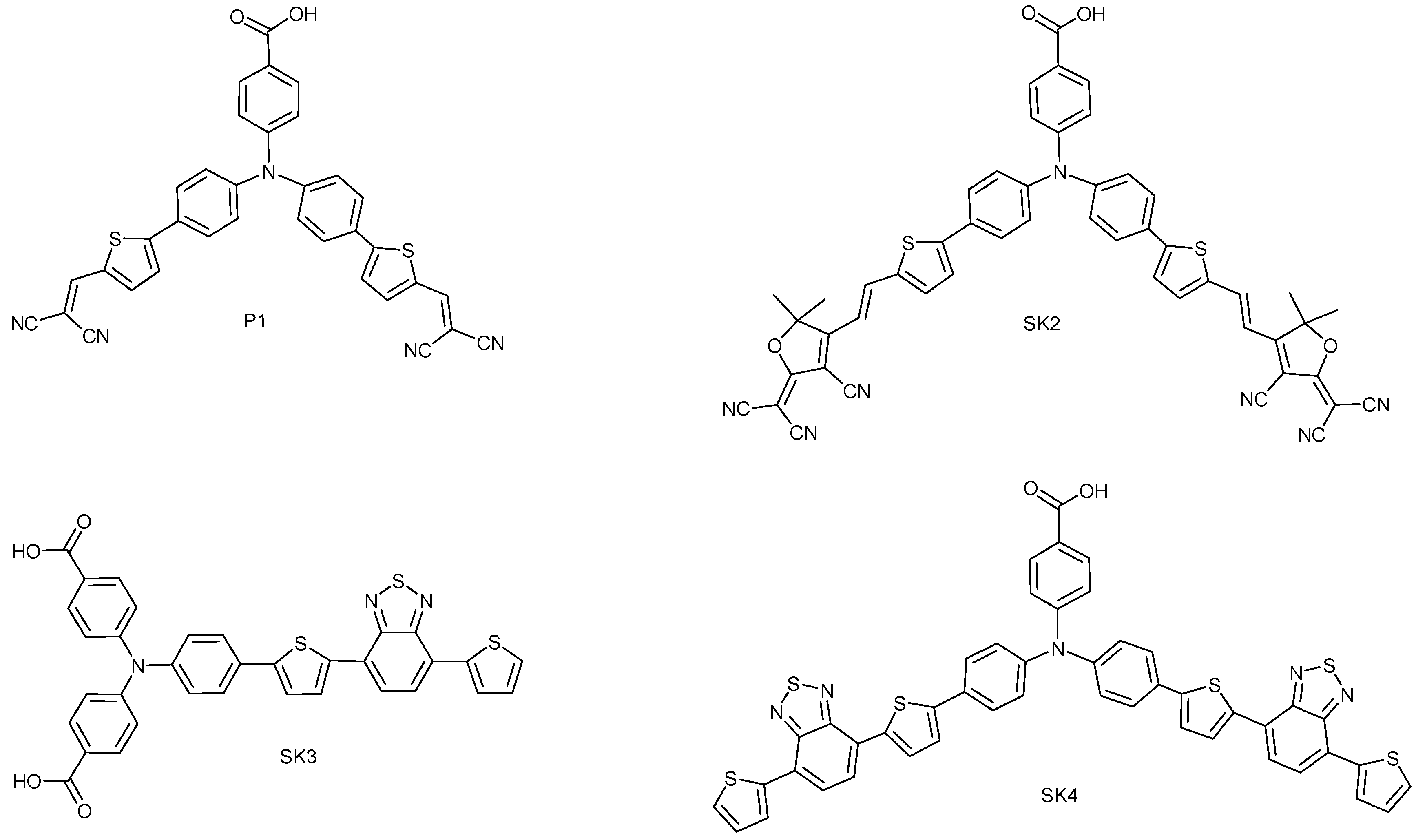
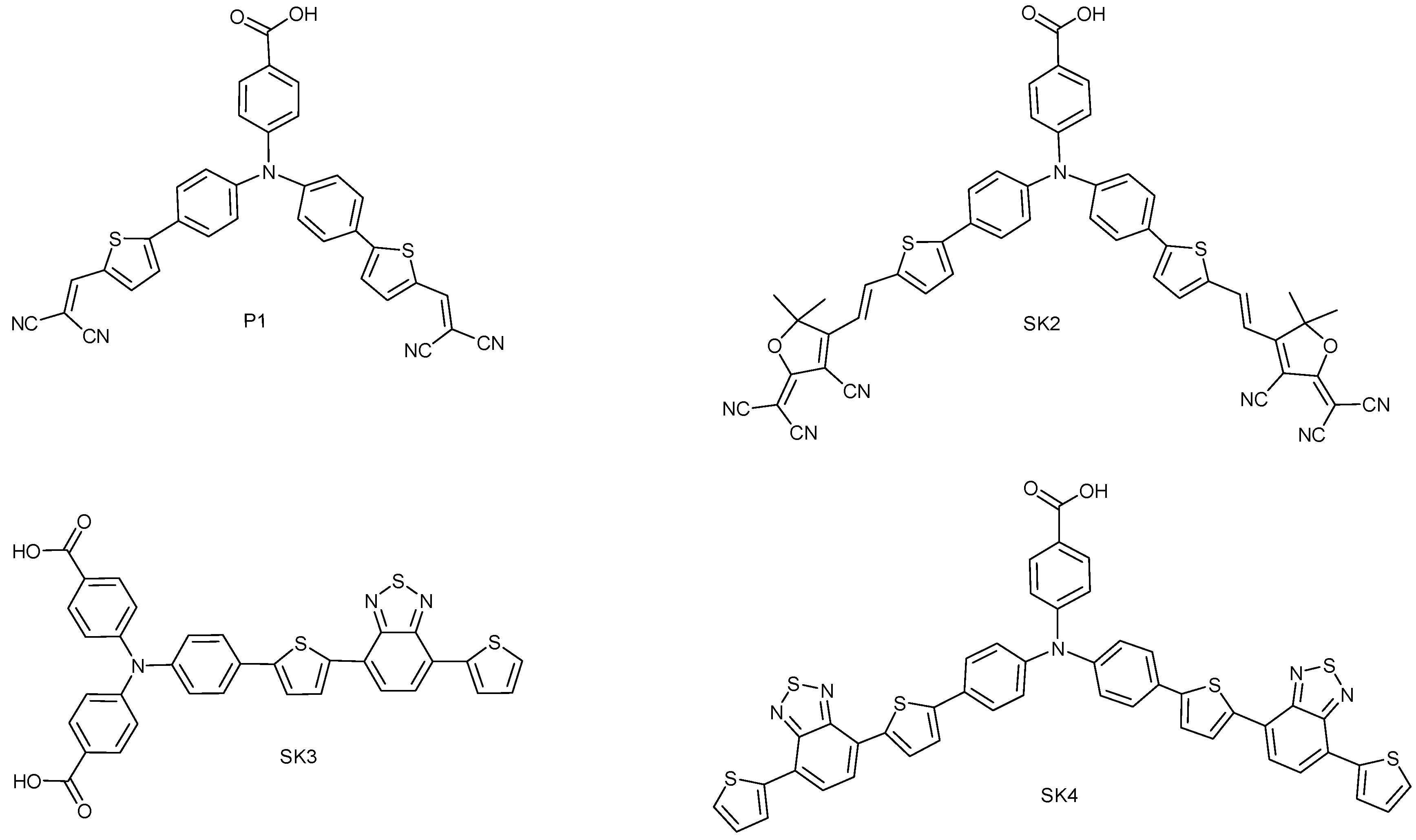
2. Results and Discussion
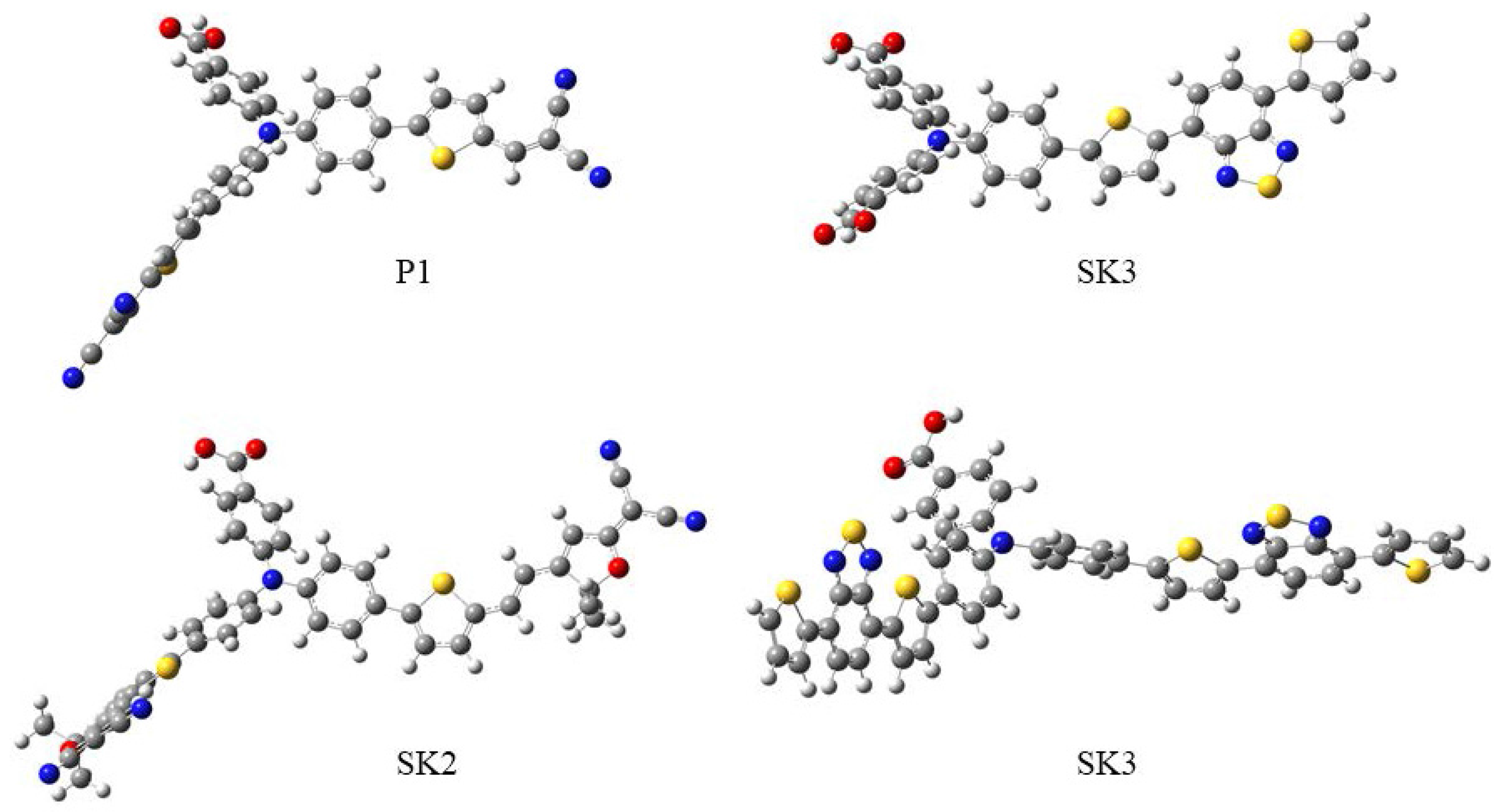
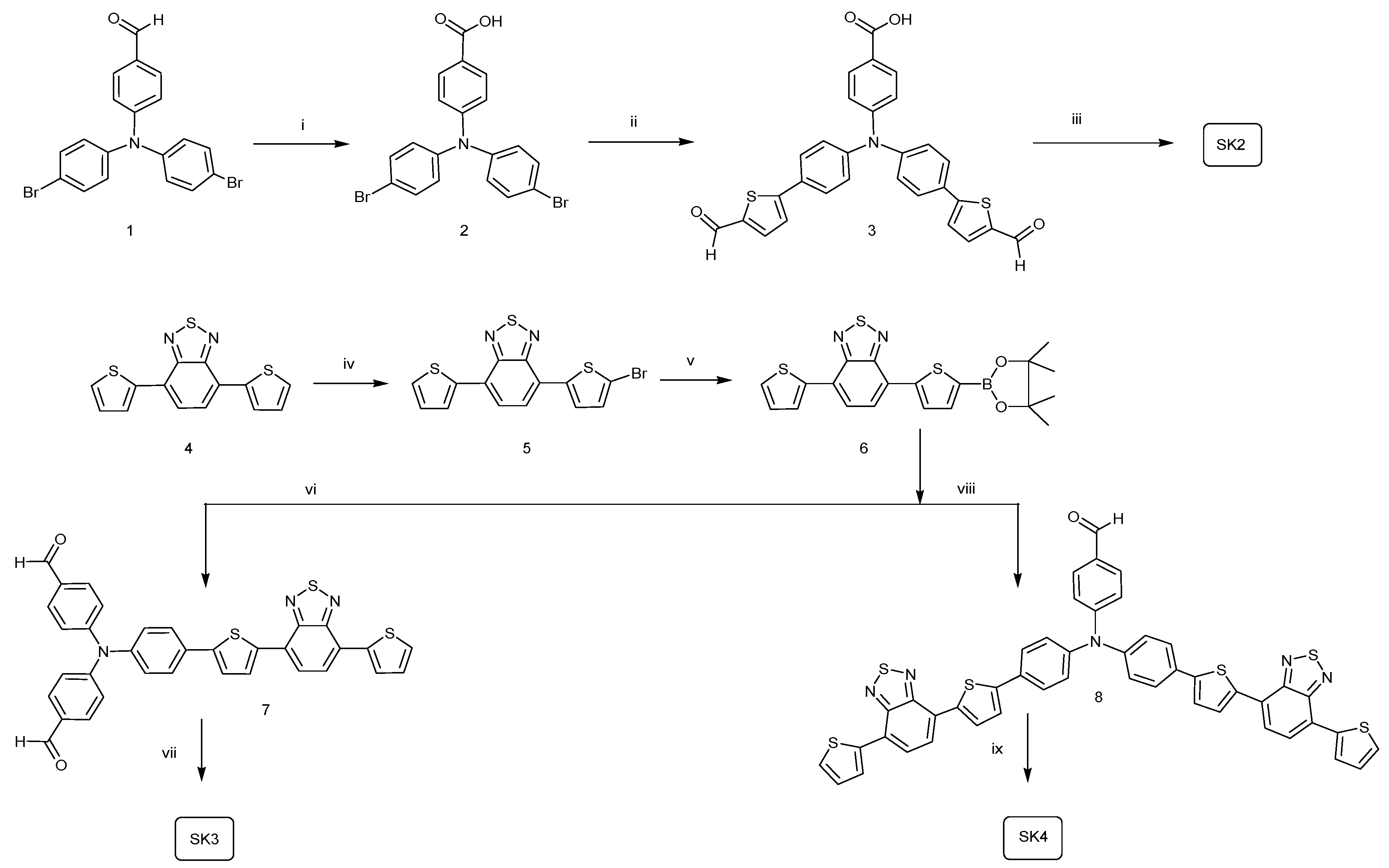
2.1. Synthesis

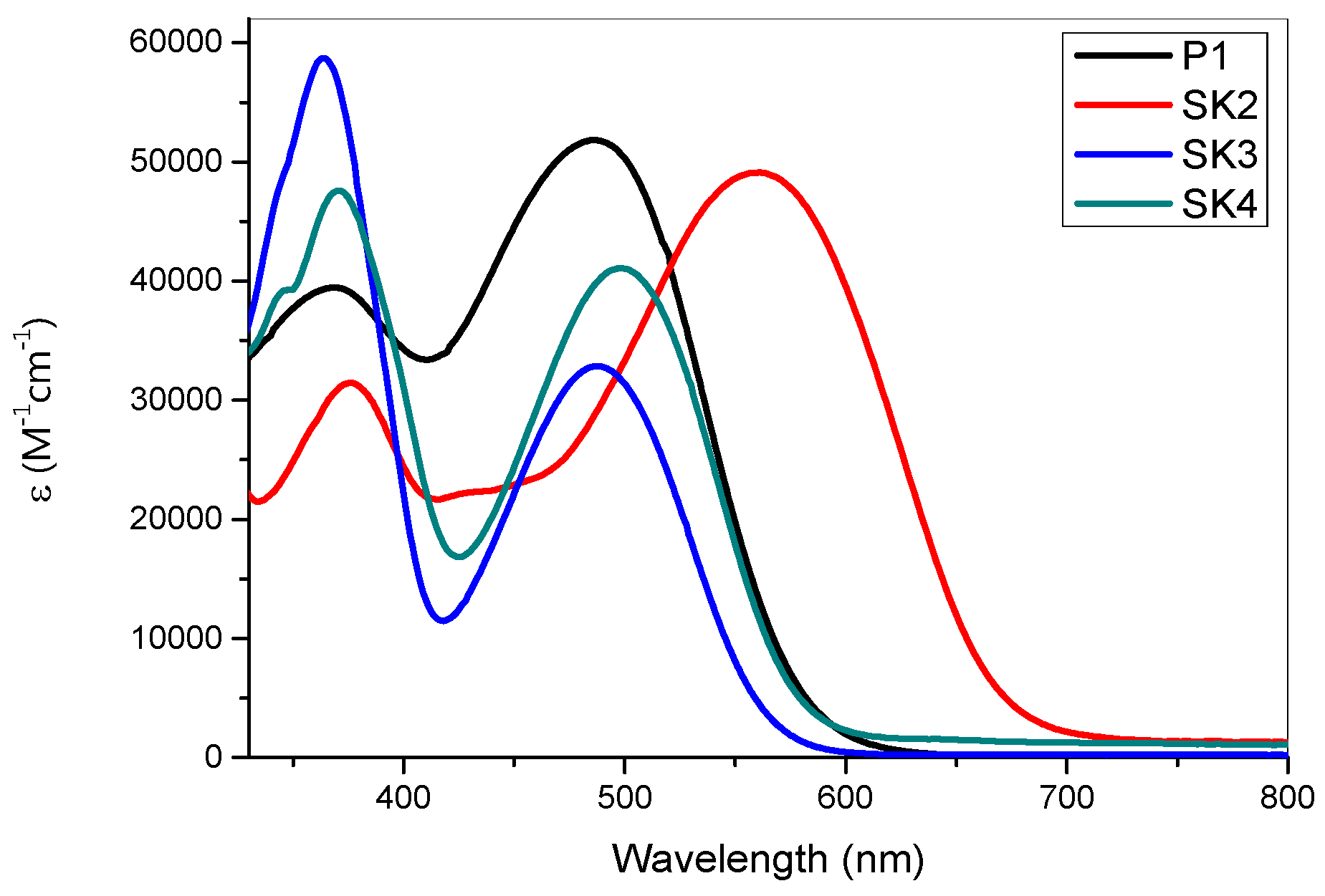
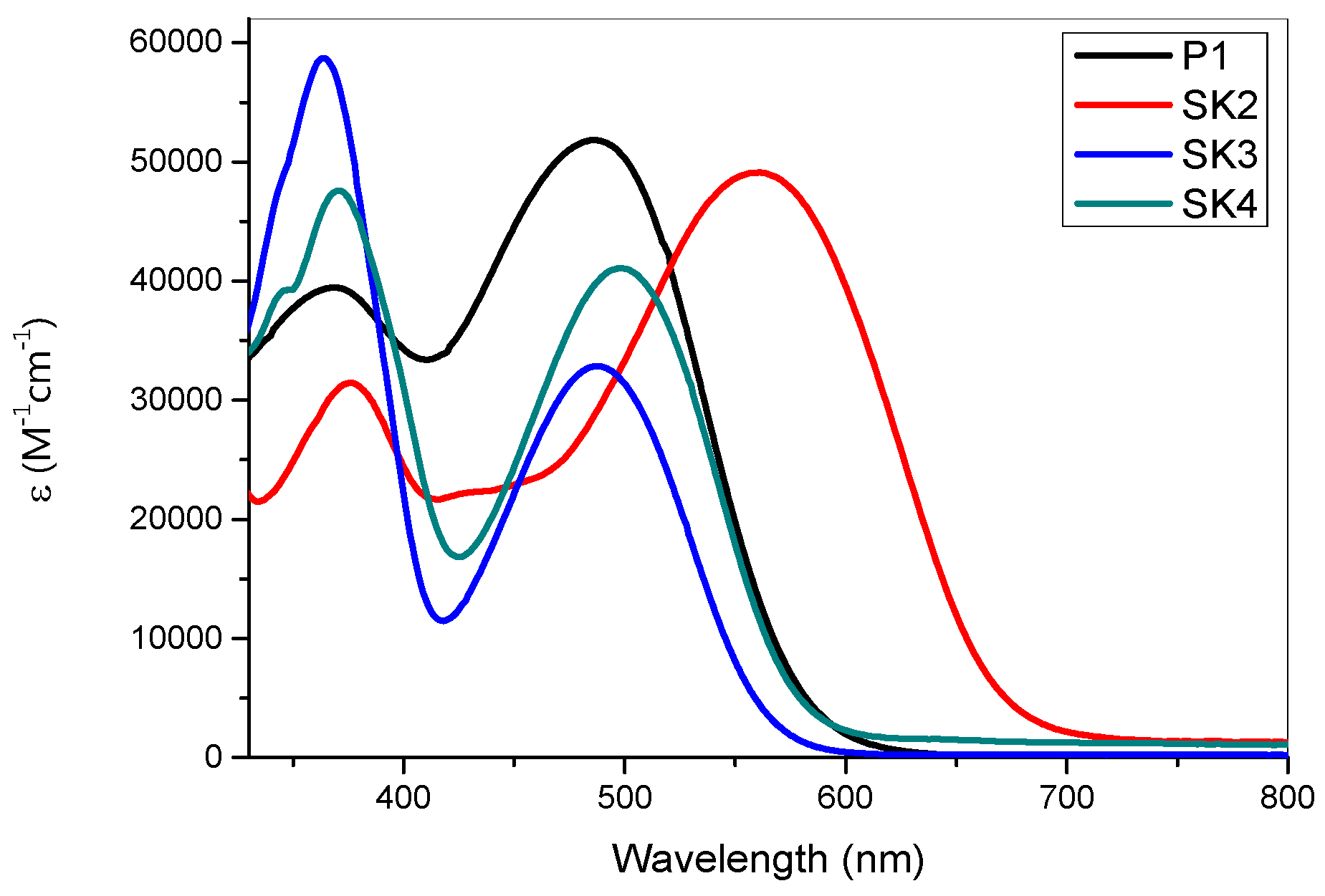
2.2. Spectroscopic and Electrochemical Properties

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dye | λabs(nm) (ε)(104 M−1⋅cm−1) | λem (nm) | E°° (eV) b | E(HOMO) vs. NHE(V) (vs. Vacuum) (eV) c | E(LUMO) vs. NHE (V) (vs. Vacuum) (eV) c |
|---|---|---|---|---|---|
| P1 | 372 (3.97); 489 (5.17) | 611 | 2.14 | 1.34(−6.0) | −0.62(−4.0) |
| SK2 | 373 (3.12); 559 (4.90) | 674 | 1.91 | 1.26(−5.9) | −0.22(−4.4) |
| SK3 | 357 (5.87); 488 (3.28) | 609 | 2.21 | 1.17(−5.8) | −0.85(−3.8) |
| SK4 | 365 (4.76); 498 (4.11) | 621 | 2.17 | 1.16(−5.8) | −0.86(−3.7) |
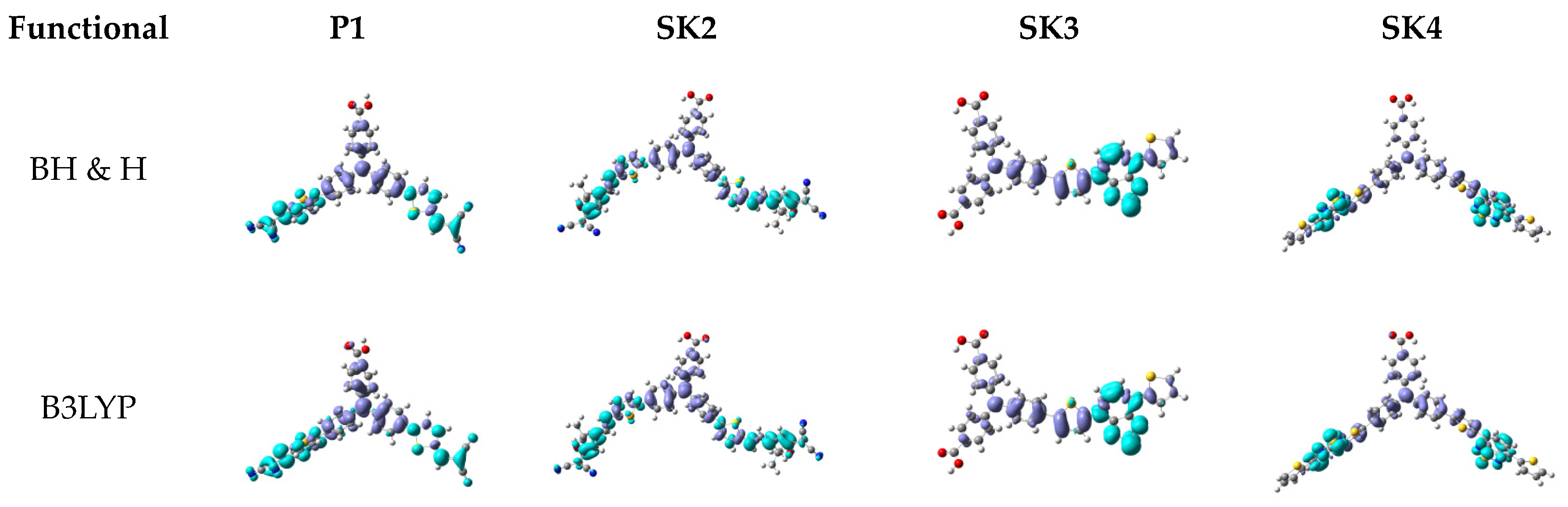
2.3. Computational Investigation

| Dye | Eexp(eV) [λmax(nm)] | E°pBH & H(eV) [λmax(nm)] | E°pB3LYP (eV) [λmax(nm)] | |ΔEBH & H|(eV) | |ΔEB3LYP|(eV) |
|---|---|---|---|---|---|
| P1 | 2.53 [489] | 2.79 [443] | 2.30 [540] | 0.26 | 0.23 |
| SK2 | 2.22 [559] | 2.86 [433] | 2.08 [595] | 0.64 | 0.14 |
| SK3 | 2.54 [488] | 2.62 [473] | 2.14 [580] | 0.08 | 0.40 |
| SK4 | 2.49 [498] | 2.48 [501] | 1.92 [614] | 0.01 | 0.57 |
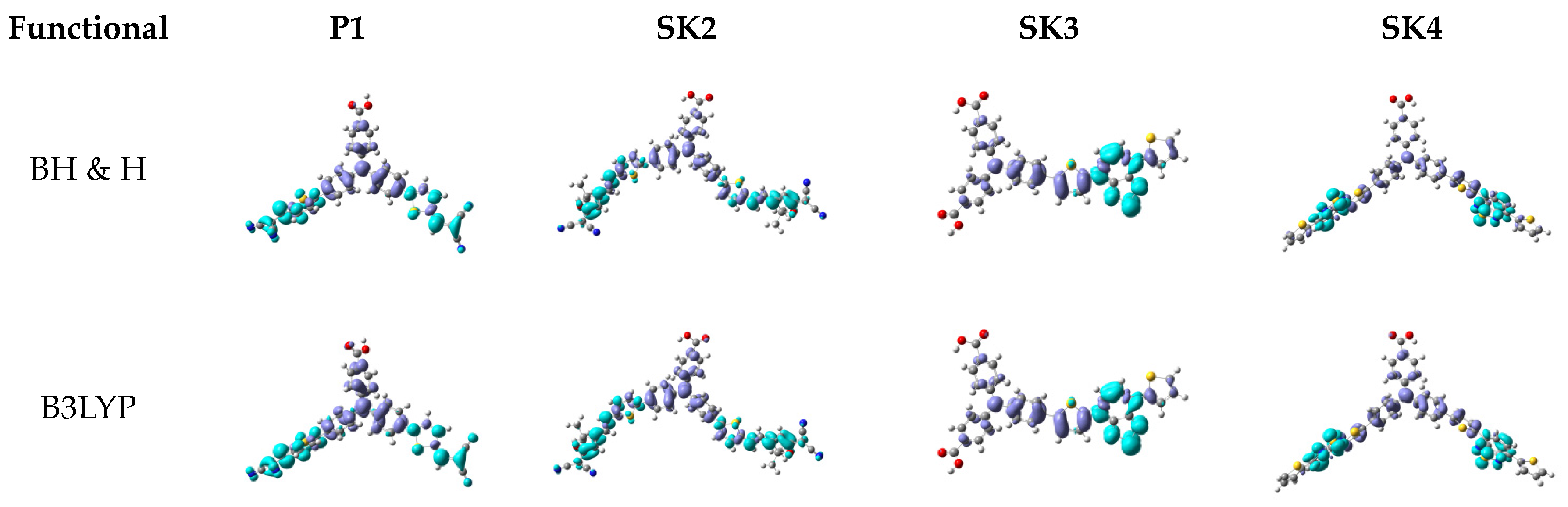
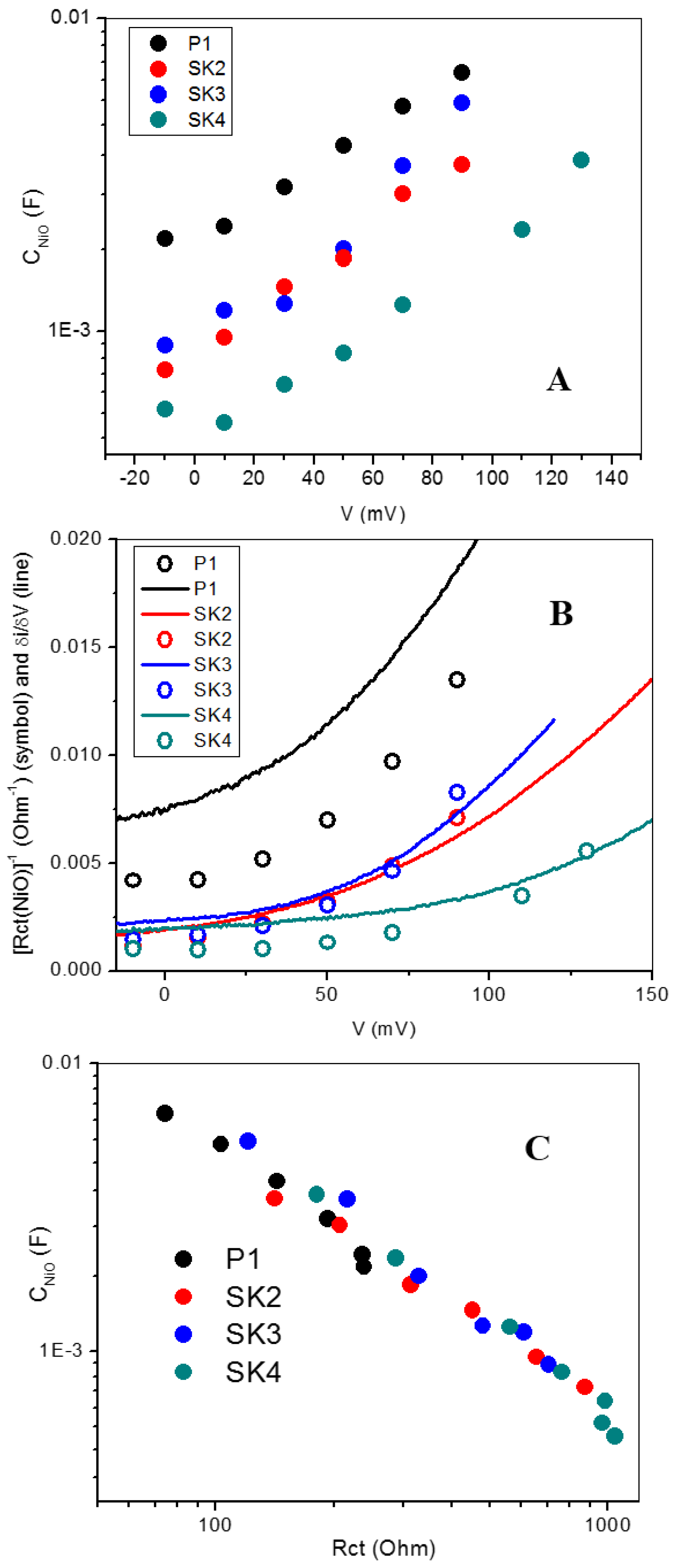
2.4. Photoelectrochemical Investigation in DSSCs
| Dye | Jsc (mA/cm2) | Voc (mV) | FF | PCE (%) |
|---|---|---|---|---|
| P1 | 1.14 | 95 | 0.32 | 0.035 |
| SK2 | 0.51 | 81 | 0.33 | 0.014 |
| SK3 | 0.54 | 82 | 0.33 | 0.015 |
| SK4 | 0.43 | 134 | 0.32 | 0.018 |

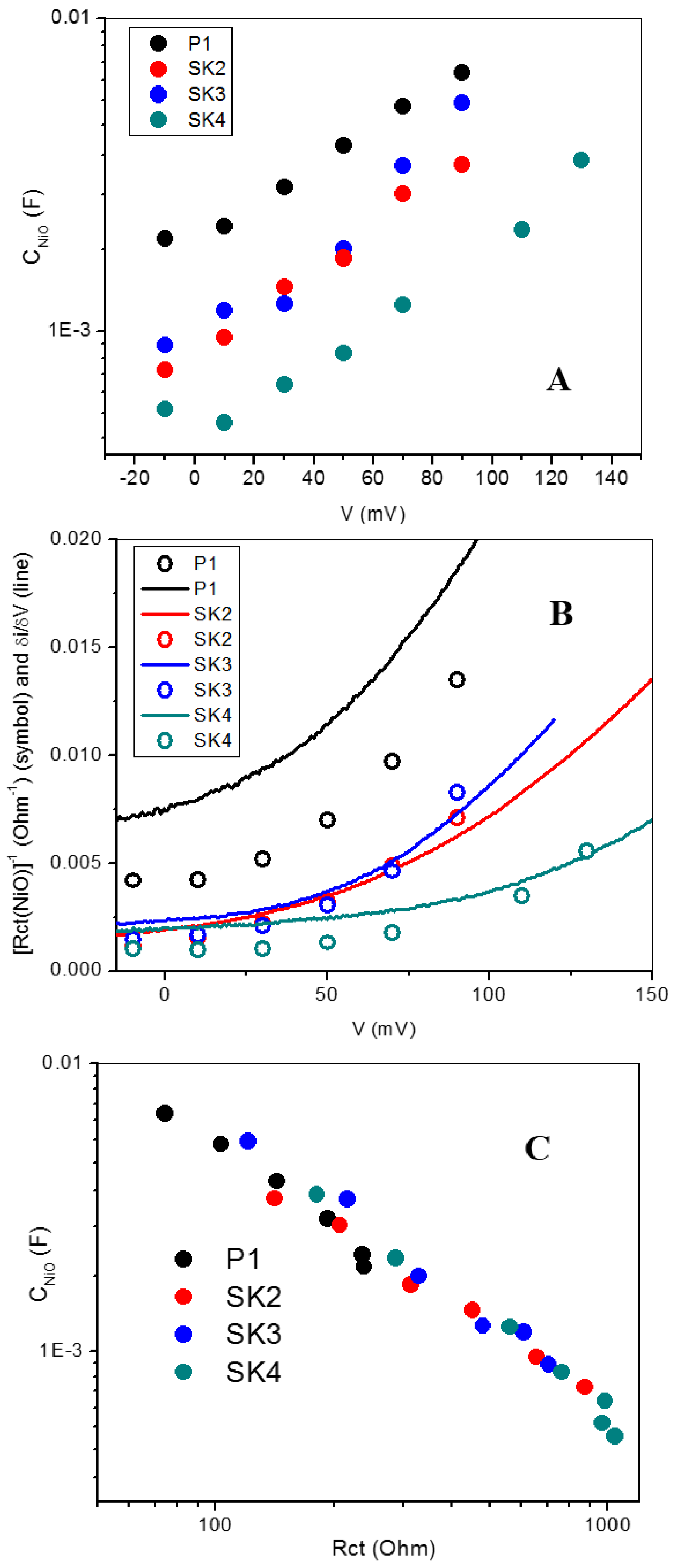
3. Experimental Section
3.1. General Remarks
3.2. Synthesis of the p-Type Sensitizers
3.2.1. 4-[Bis-(4-bromo-phenyl)-amino]-benzoic Acid (2)
3.2.2. 4-{Bis-[4-(5-formyl-thiophen-2-yl)-phenyl]-amino}-benzoic Acid (3)
3.2.3. 4-(Bis(4-(5-((E)-2-(4-cyano-5-(dicyanomethylene)-2,2-dimethyl-2,5-dihydrofuran-3-yl)vinyl)thiophen-2-yl)phenyl)amino)benzoic Acid (SK2)
3.2.4. 4-(5-Bromothiophen-2-yl)-7-(thiophen-2-yl)benzo[1,2,5]thiadiazole (5)
3.2.5. 4-(5-(4,4,5,5-Tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-2-yl)-7-(thiophen-2-yl)benzo[c][1,2,5]thiadiazole (6)
3.2.6. 4,4′-(4-(5-(7-(Thiophen-2-yl)benzo[c][1,2,5]thiadiazol-4-yl)thiophen-2-yl)phenylazanediyl)dibenzaldehyde (7)
3.2.7. 4-(Bis(4-(5-(7-(thiophen-2-yl)benzo[c][1,2,5]thiadiazol-4-yl)thiophen-2-yl)phenyl)amino)benzaldehyde (8)
3.2.8. 4,4′-(4-(5-(7-(Thiophen-2-yl)benzo[c][1,2,5]thiadiazol-4-yl)thiophen-2-yl)phenylazanediyl)dibenzoic Acid (SK3)
3.2.9. 4-(Bis(4-(5-(7-(thiophen-2-yl)benzo[c][1,2,5]thiadiazol-4-yl)thiophen-2-yl)phenyl)amino)benzoic Acid (SK4)
3.3. Solar Cell Assembly
3.4. Spectroscopic, Electrochemical, and Photoelectrochemical Measurements
3.5. Computational Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- O′Regan, B.; Gratzel, M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films. Nature 1991, 353, 737–740. [Google Scholar] [CrossRef]
- International Energy Agency (IEA). Potential for Building Integrated Photovoltaics; Report IEA PVPS Task 7; NET Ltd.: St. Ursen, Switzerland, 2002. [Google Scholar]
- Giordano, F.; Guidobaldi, A.; Petrolati, E.; Vesce, L.; Riccitelli, R.; Reale, A.; Brown, T.M.; di Carlo, A. Realization of high performance large area Z-series-interconnected opaque dye solar cell modules. Prog. Photovolt. Res. Appl. 2013, 21, 1653–1658. [Google Scholar] [CrossRef]
- Mathew, S.; Yella, A.; Gao, P.; Humphry-Baker, R.; CurchodBasile, F.E.; Ashari-Astani, N.; Tavernelli, I.; Rothlisberger, U.; NazeeruddinMd, K.; Grätzel, M. Dye-sensitized solar cells with 13% efficiency achieved through the molecular engineering of porphyrin sensitizers. Nat. Chem. 2014, 6, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Green, M.A. Third Generation Photovoltaics: Advanced Solar Energy Conversion; Springer-Verlag: Heidelberg, Germany, 2003. [Google Scholar]
- Hagfeldt, A.; Boschloo, G.; Sun, L.; Kloo, L.; Pettersson, H. Dye-Sensitized Solar Cells. Chem. Rev. 2010, 110, 6595–6663. [Google Scholar] [CrossRef] [PubMed]
- Lide, D.R. Handbook of Chemistry and Physics, 90th ed.; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Odobel, F.; Pellegrin, Y.; Gibson, E.A.; Hagfeldt, A.; Smeigh, A.L.; Hammarström, L. Recent advances and future directions to optimize the performances of p-type dye-sensitized solar cells. Coord. Chem. Rev. 2012, 256, 2414–2423. [Google Scholar] [CrossRef]
- Morandeira, A.; Boschloo, G.; Hagfeldt, A.; Hammarström, L. Coumarin 343-NiO Films as Nanostructured Photocathodes in Dye-Sensitized Solar Cells: Ultrafast Electron Transfer, Effect of the I3−/I− Redox Couple and Mechanism of Photocurrent Generation. J. Phys. Chem. C 2008, 112, 9530–9537. [Google Scholar] [CrossRef]
- Zhu, H.; Hagfeldt, A.; Boschloo, G. Photoelectrochemistry of Mesoporous NiO Electrodes in Iodide/Triiodide Electrolytes. J. Phys. Chem. C 2007, 111, 17455–17458. [Google Scholar] [CrossRef]
- Borgström, M.; Blart, E.; Boschloo, G.; Mukhtar, E.; Hagfeldt, A.; Hammarström, L.; Odobel, F. Sensitized Hole Injection of Phosphorus Porphyrin into NiO: Toward New Photovoltaic Devices. J. Phys. Chem. B 2005, 109, 22928–22934. [Google Scholar] [CrossRef] [PubMed]
- Gibson, E.A.; Smeigh, A.L.; le Pleux, L.; Fortage, J.; Boschloo, G.; Blart, E.; Pellegrin, Y.; Odobel, F.; Hagfeldt, A.; Hammarström, L. A p-Type NiO-Based Dye-Sensitized Solar Cell with an Open-Circuit Voltage of 0.35 V. Angew. Chem. Int. Ed. 2009, 48, 4402–4405. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Zhu, H.; Edvinsson, T.; Boschloo, G.; Hagfeldt, A.; Sun, L. Design of an Organic Chromophore for P-Type Dye-Sensitized Solar Cells. J. Am. Chem. Soc. 2008, 130, 8570–8571. [Google Scholar] [CrossRef] [PubMed]
- Abbotto, A.; Manfredi, N.; Marinzi, C.; de Angelis, F.; Mosconi, E.; Yum, J.H.; Zhang, X.X.; Nazeeruddin, M.K.; Gratzel, M. Di-branched di-anchoring organic dyes for dye-sensitized solar cells. Energy Environ. Sci. 2009, 2, 1094–1101. [Google Scholar] [CrossRef]
- Abbotto, A.; Leandri, V.; Manfredi, N.; de Angelis, F.; Pastore, M.; Yum, J.H.; Nazeeruddin, M.K.; Gratzel, M. Bis-Donor-Bis-Acceptor Tribranched Organic Sensitizers for Dye-Sensitized Solar Cells. Eur. J. Org. Chem. 2011, 2011, 6195–6205. [Google Scholar] [CrossRef]
- Manfredi, N.; Cecconi, B.; Abbotto, A. Multi-Branched Multi-Anchoring Metal-Free Dyes for Dye-Sensitized Solar Cells. Eur. J. Org. Chem. 2014, 2014, 7069–7086. [Google Scholar] [CrossRef]
- Nattestad, A.; Mozer, A.J.; Fischer, M.K.R.; Cheng, Y.B.; Mishra, A.; Bauerle, P.; Bach, U. Highly efficient photocathodes for dye-sensitized tandem solar cells. Nat. Mater. 2010, 9, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.L.; Zhang, Z.; Chen, D.; Bauerle, P.; Bach, U.; Cheng, Y.-B. Sensitization of nickel oxide: Improved carrier lifetime and charge collection by tuning nanoscale crystallinity. Chem. Commun. 2012, 48, 9885–9887. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Gibson, E.A.; Qin, P.; Boschloo, G.; Gorlov, M.; Hagfeldt, A.; Sun, L. Double-Layered NiO Photocathodes for p-Type DSSCs with Record IPCE. Adv. Mater. 2010, 22, 1759–1762. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Linder, M.; Brinck, T.; Boschloo, G.; Hagfeldt, A.; Sun, L. High Incident Photon-to-Current Conversion Efficiency of p-Type Dye-Sensitized Solar Cells Based on NiO and Organic Chromophores. Adv. Mater. 2009, 21, 2993–2996. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, H.B.; Zhong, C.; Li, C.M. Rational design of triphenylamine dyes for highly efficient p-type dye sensitized solar cells. Dyes Pigment. 2014, 105, 97–104. [Google Scholar] [CrossRef]
- Yen, Y.-S.; Chen, W.-T.; Hsu, C.-Y.; Chou, H.-H.; Lin, J.T.; Yeh, M.-C.P. Arylamine-Based Dyes for p-Type Dye-Sensitized Solar Cells. Org. Lett. 2011, 13, 4930–4933. [Google Scholar] [CrossRef] [PubMed]
- Melikian, G.; Rouessac, F.P.; Alexandre, C. Synthesis of Substituted Dicyanomethylendihydrofurans. Synth. Commun. 1995, 25, 3045–3051. [Google Scholar] [CrossRef]
- Yassar, A.; Videlot, C.; Jaafari, A. Synthesis and photovoltaic properties of mono-substituted quaterthiophenes bearing strong electron-withdrawing group. Sol. Energy Mater. Sol. Cells 2006, 90, 916–922. [Google Scholar] [CrossRef]
- Barrette, W.C.; Johnson, H.W.; Sawyer, D.T. Voltammetric evaluation of the effective acidities (pKa′) for Broensted acids in aprotic solvents. Anal. Chem. 1984, 56, 1890–1898. [Google Scholar] [CrossRef] [PubMed]
- Bockris, J.O.M.; Khan, S.U.M. Surface Electrochemistry—A Molecular Level Approach; Kluwer Academic/Plenum Publishers: New York, NY, USA, 1993. [Google Scholar]
- Scrascia, A.; Pastore, M.; Yin, L.; Anna Picca, R.; Manca, M.; Guo, Y.-C.; de Angelis, F.; Della Sala, F.; Cingolani, R.; Gigli, G.; et al. Organic Dyes Containing A Triple Bond Spacer for Dye Sensitized Solar Cells: A Combined Experimental and Theoretical Investigation. Curr. Org. Chem. 2011, 15, 3535–3543. [Google Scholar] [CrossRef]
- Mba, M.; D′Acunzo, M.; Salice, P.; Carofiglio, T.; Maggini, M.; Caramori, S.; Campana, A.; Aliprandi, A.; Argazzi, R.; Carli, S.; et al. Sensitization of Nanocrystalline TiO2 with Multibranched Organic Dyes and Co(III)/(II) Mediators: Strategies to Improve Charge Collection Efficiency. J. Phys. Chem. C 2013, 117, 19885–19896. [Google Scholar] [CrossRef]
- Ji, Z.Q.; Natu, G.; Huang, Z.J.; Wu, Y.Y. Linker effect in organic donor-acceptor dyes for p-type NiO dye sensitized solar cells. Energy Environ. Sci. 2011, 4, 2818–2821. [Google Scholar] [CrossRef]
- Barolo, C.; Nazeeruddin, M.K.; Fantacci, S.; Di Censo, D.; Comte, P.; Liska, P.; Viscardi, G.; Quagliotto, P.; de Angelis, F.; Ito, S.; et al. Synthesis, Characterization, and DFT-TDDFT Computational Study of a Ruthenium Complex Containing a Functionalized Tetradentate Ligand. Inorg. Chem. 2006, 45, 4642–4653. [Google Scholar] [CrossRef] [PubMed]
- Qin, P.; Wiberg, J.; Gibson, E.A.; Linder, M.; Li, L.; Brinck, T.; Hagfeldt, A.; Albinsson, B.; Sun, L.C. Synthesis and Mechanistic Studies of Organic Chromophores with Different Energy Levels for p-Type Dye-Sensitized Solar Cells. J. Phys. Chem. C 2010, 114, 4738–4748. [Google Scholar] [CrossRef]
- Petersson, G.A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H.P.; Izmaylov, A.F.; Bloino, J.; Zheng, G.; Sonnenberg, J.L.; Hada, M.; et al. Gaussian 09, revision A.02.; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Autschbach, J. Charge-Transfer Excitations and Time-Dependent Density Functional Theory: Problems and Some Proposed Solutions. Chemphyschem 2009, 10, 1757–1760. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, T.; Seth, M.; Krykunov, M.; Autschbach, J.; Wang, F. Is charge transfer transitions really too difficult for standard density functionals or are they just a problem for time-dependent density functional theory based on a linear response approach. J. Mol. Struct. THEOCHEM 2009, 914, 106–109. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Koch, W.; Holthausen, M. A Chemist’s Guide to Density Functional Theory, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2001. [Google Scholar]
- Gregg, B.A. Excitonic Solar Cells. J. Phys. Chem. B 2003, 107, 4688–4698. [Google Scholar] [CrossRef]
- Zhen, C.-G.; Becker, U.; Kieffer, J. Tuning Electronic Properties of Functionalized Polyhedral Oligomeric Silsesquioxanes: A DFT and TDDFT Study. J. Phys. Chem. A 2009, 113, 9707–9714. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.-G.; Zhen, C.-G.; Jeong, E.J.; Kieffer, J.; Kim, J. Organic Dye Design Tools for Efficient Photocurrent Generation in Dye-Sensitized Solar Cells: Exciton Binding Energy and Electron Acceptors. Adv. Funct. Mater. 2012, 22, 1606–1612. [Google Scholar] [CrossRef] [Green Version]
- Choi, H.; Baik, C.; Kang, S.O.; Ko, J.; Kang, M.-S.; Nazeeruddin, M.K.; Grätzel, M. Highly Efficient and Thermally Stable Organic Sensitizers for Solvent-Free Dye-Sensitized Solar Cells. Angew. Chem. Int. Ed. 2008, 47, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Bisquert, J.; Fabregat Santiago, F. Impedance spectroscopy: A general introduction and application to dye-sensitized solar cells. In Dye Sensitized Solar Cells; Kalyanasundaram, K., Ed.; CRC Press: Boca Raton, FL, USA, 2010. [Google Scholar]
- Bisquert, J. Chemical capacitance of nanostructured semiconductors: Its origin and significance for nanocomposite solar cells. Phys. Chem. Chem. Phys. 2003, 5, 5360–5364. [Google Scholar] [CrossRef]
- Lefebvre, J.F.; Sun, X.Z.; Calladine, J.A.; George, M.W.; Gibson, E.A. Promoting charge-separation in p-type dye-sensitized solar cells using bodipy. Chem. Commun. 2014, 50, 5258–5260. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhao, N.; Sun, Y.; Miao, F.; Liu, Y.; Liu, X.; Zhang, Y.; Ai, W.; Song, G.; Shen, X.; et al. Highly selective red- and green-emitting two-photon fluorescent probes for cysteine detection and their bio-imaging in living cells. Chem. Commun. 2012, 48, 3442–3444. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Lee, M.J.; Song, H.M.; Song, B.J.; Seo, K.D.; Pastore, M.; Anselmi, C.; Fantacci, S.; de Angelis, F.; Nazeeruddin, M.K.; et al. Organic dyes incorporating low-band-gap chromophores based on π-extended benzothiadiazole for dye-sensitized solar cells. Dyes Pigments 2011, 91, 192–198. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karamshuk, S.; Caramori, S.; Manfredi, N.; Salamone, M.; Ruffo, R.; Carli, S.; Bignozzi, C.A.; Abbotto, A. Molecular Level Factors Affecting the Efficiency of Organic Chromophores for p-Type Dye Sensitized Solar Cells. Energies 2016, 9, 33. https://doi.org/10.3390/en9010033
Karamshuk S, Caramori S, Manfredi N, Salamone M, Ruffo R, Carli S, Bignozzi CA, Abbotto A. Molecular Level Factors Affecting the Efficiency of Organic Chromophores for p-Type Dye Sensitized Solar Cells. Energies. 2016; 9(1):33. https://doi.org/10.3390/en9010033
Chicago/Turabian StyleKaramshuk, Svitlana, Stefano Caramori, Norberto Manfredi, Matteo Salamone, Riccardo Ruffo, Stefano Carli, Carlo A. Bignozzi, and Alessandro Abbotto. 2016. "Molecular Level Factors Affecting the Efficiency of Organic Chromophores for p-Type Dye Sensitized Solar Cells" Energies 9, no. 1: 33. https://doi.org/10.3390/en9010033