
Supercritical Water Gasification of Biomass: A Literature and Technology Overview


Abstract
:1. Introduction
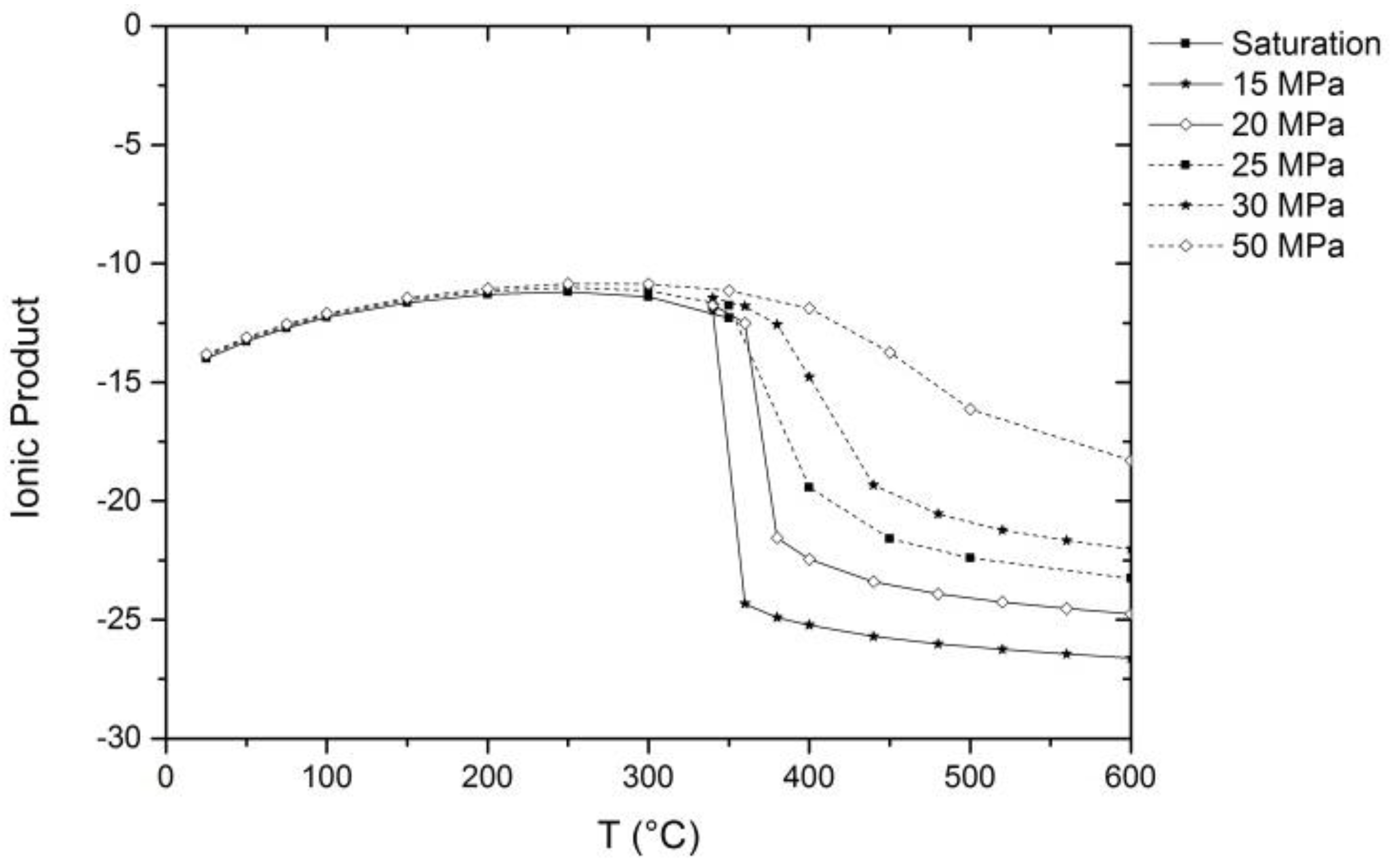
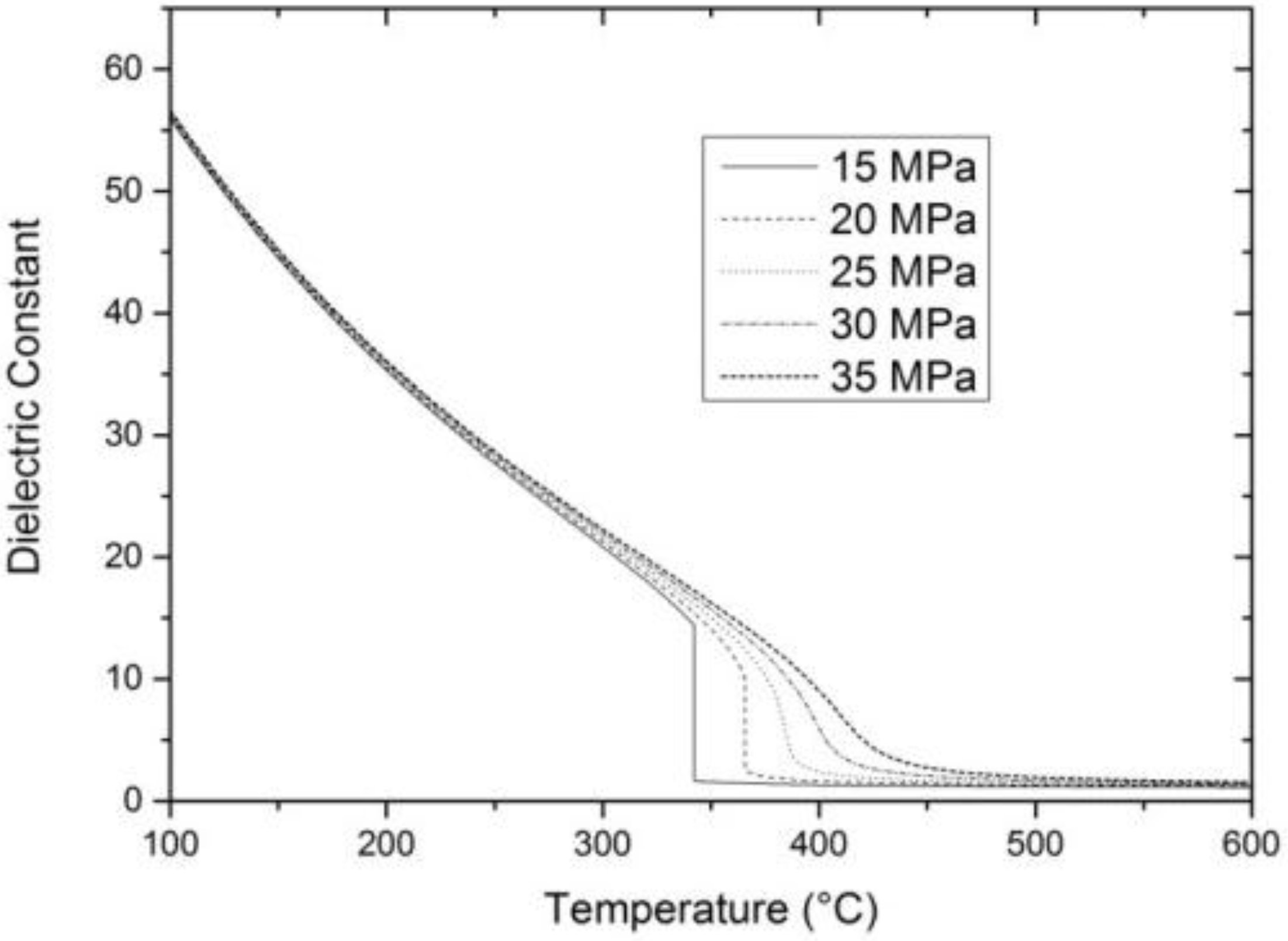
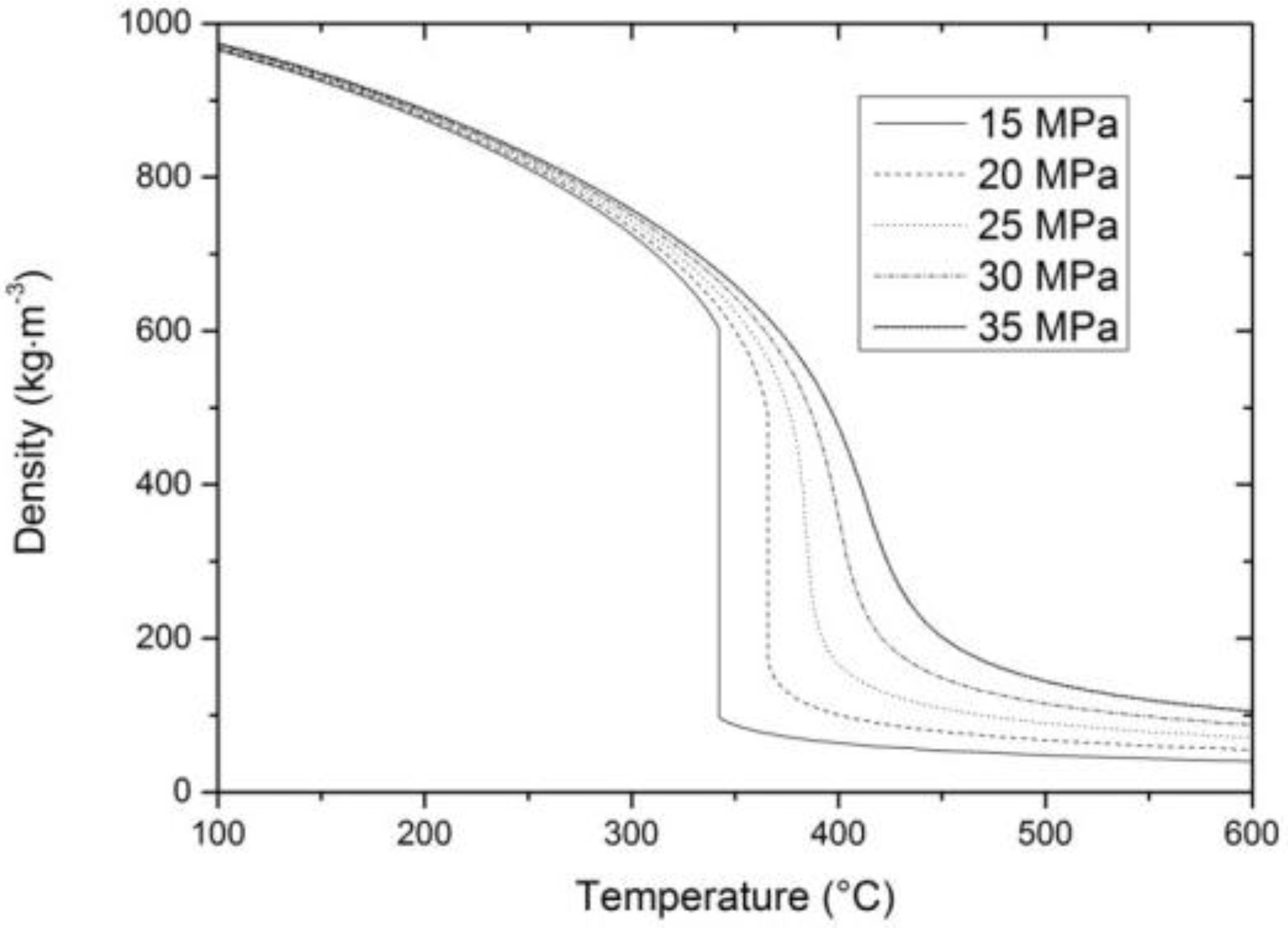
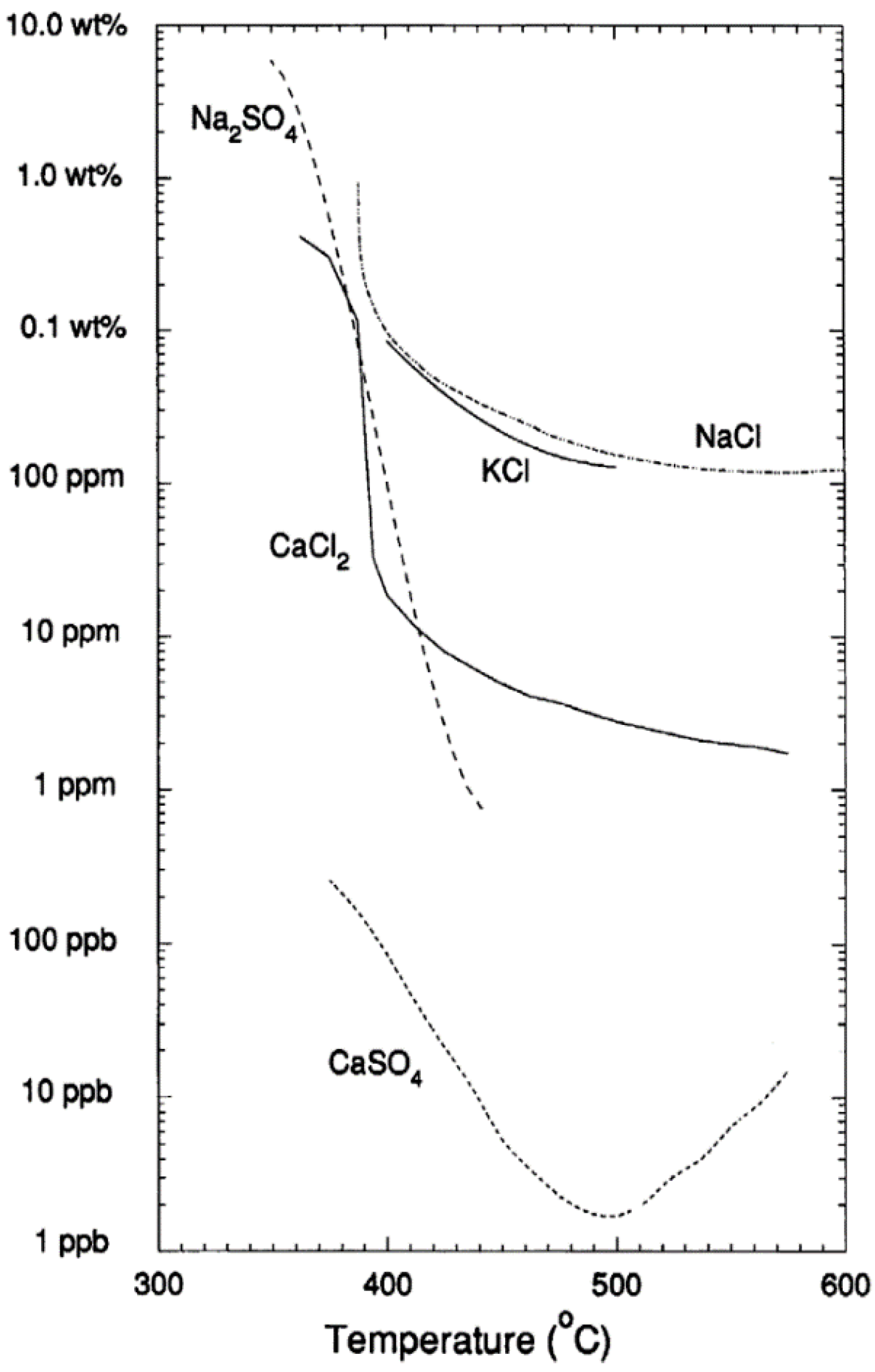
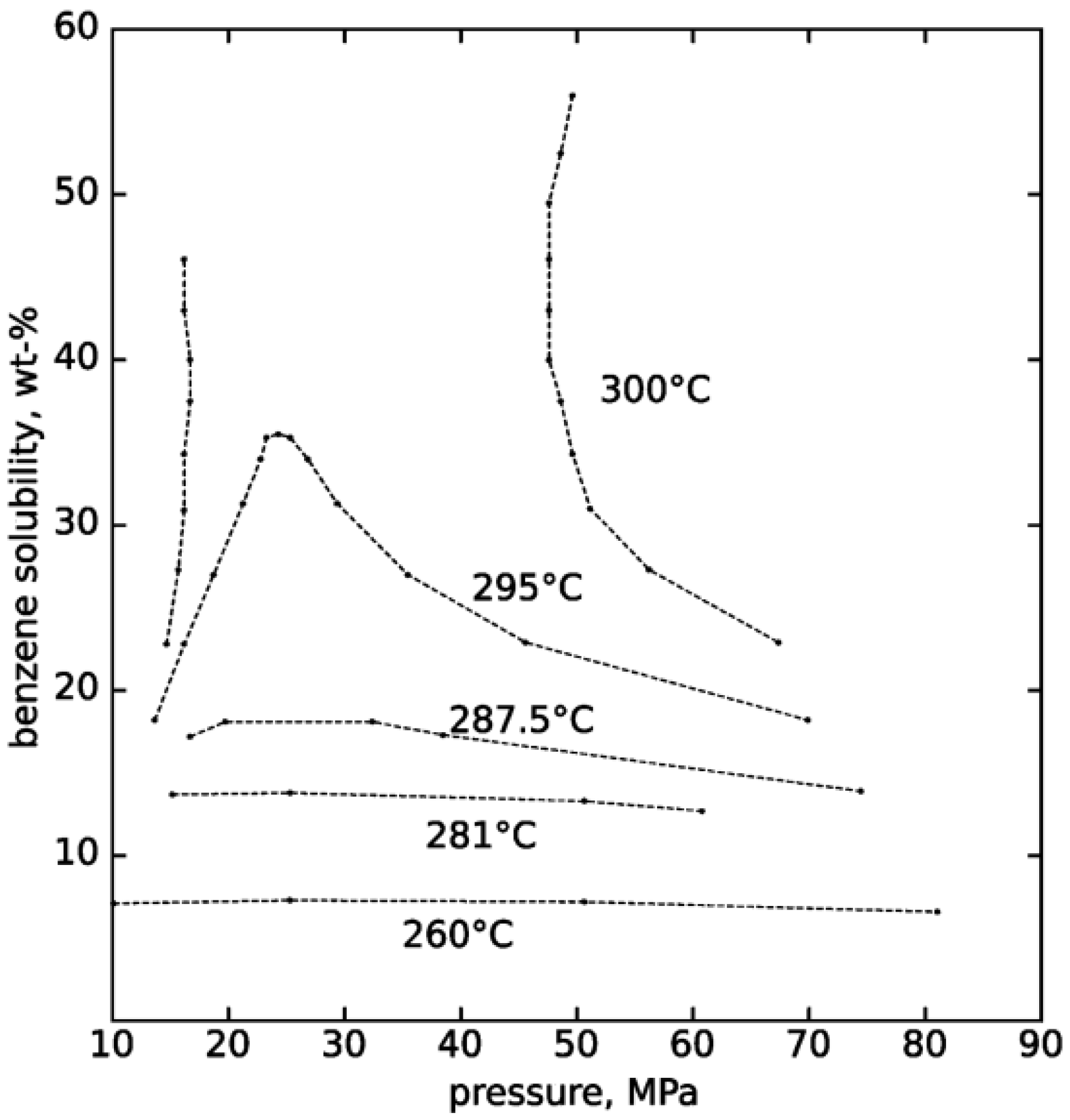
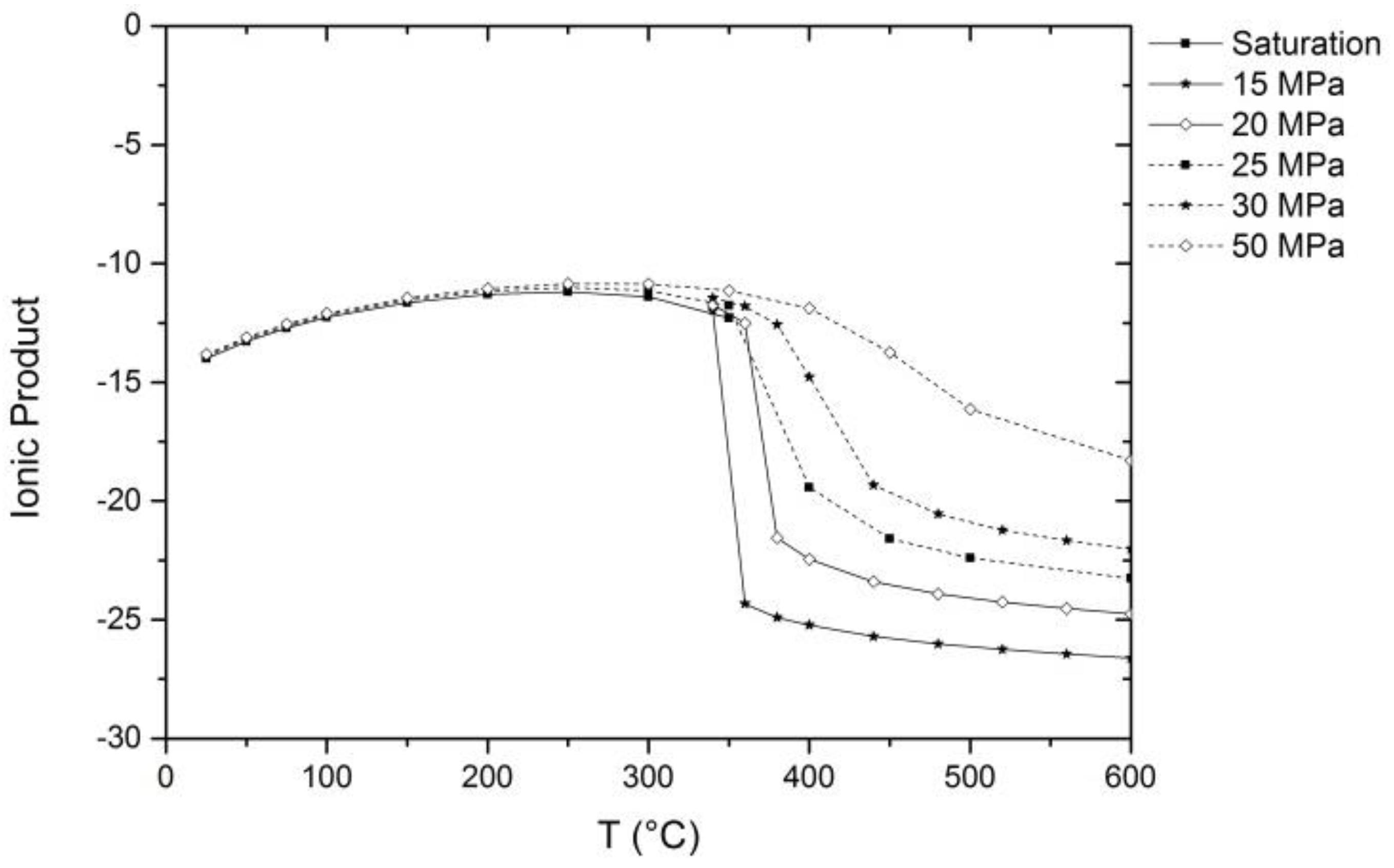
2. Properties of Near-Critical and Supercritical Water
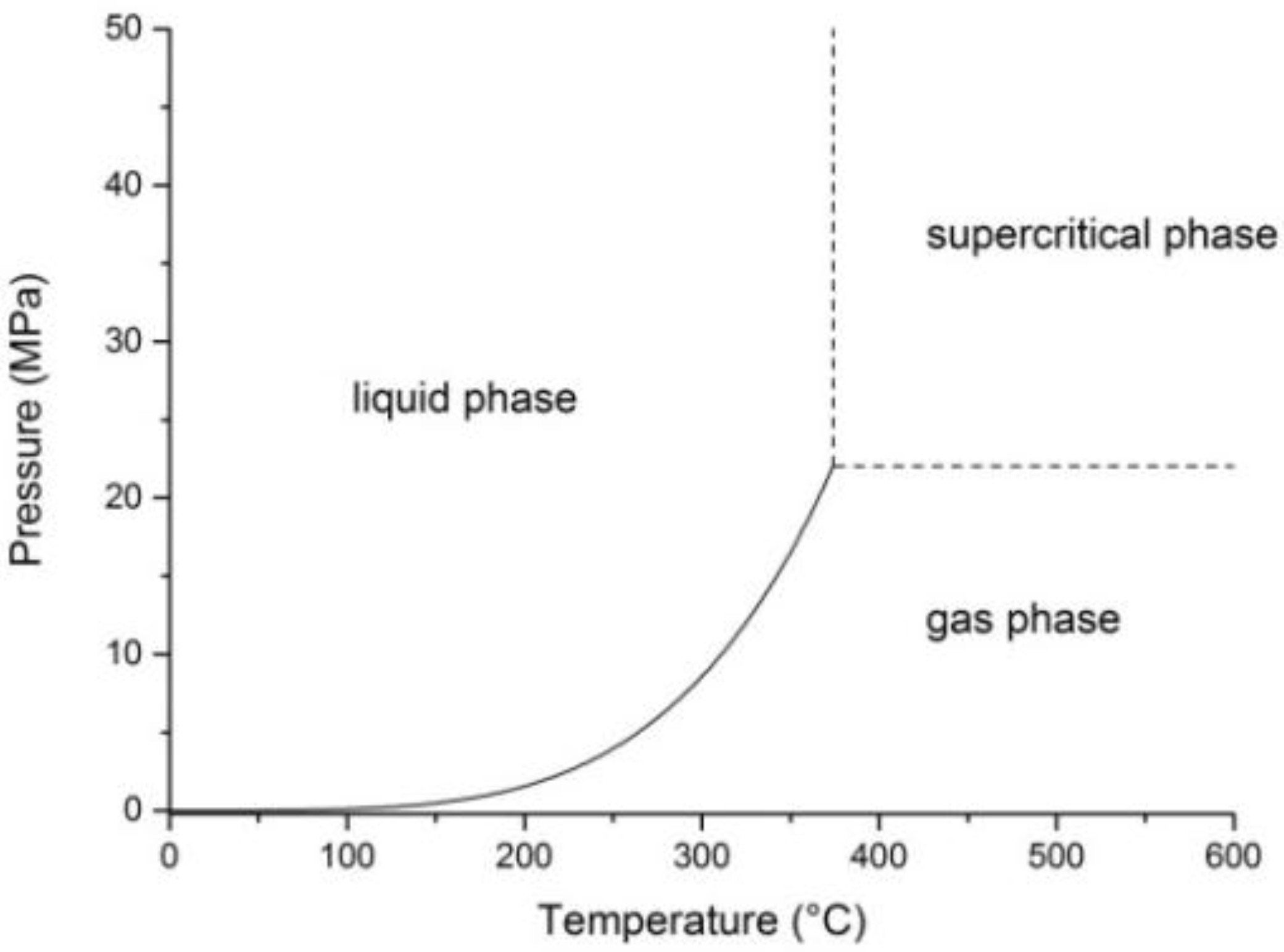
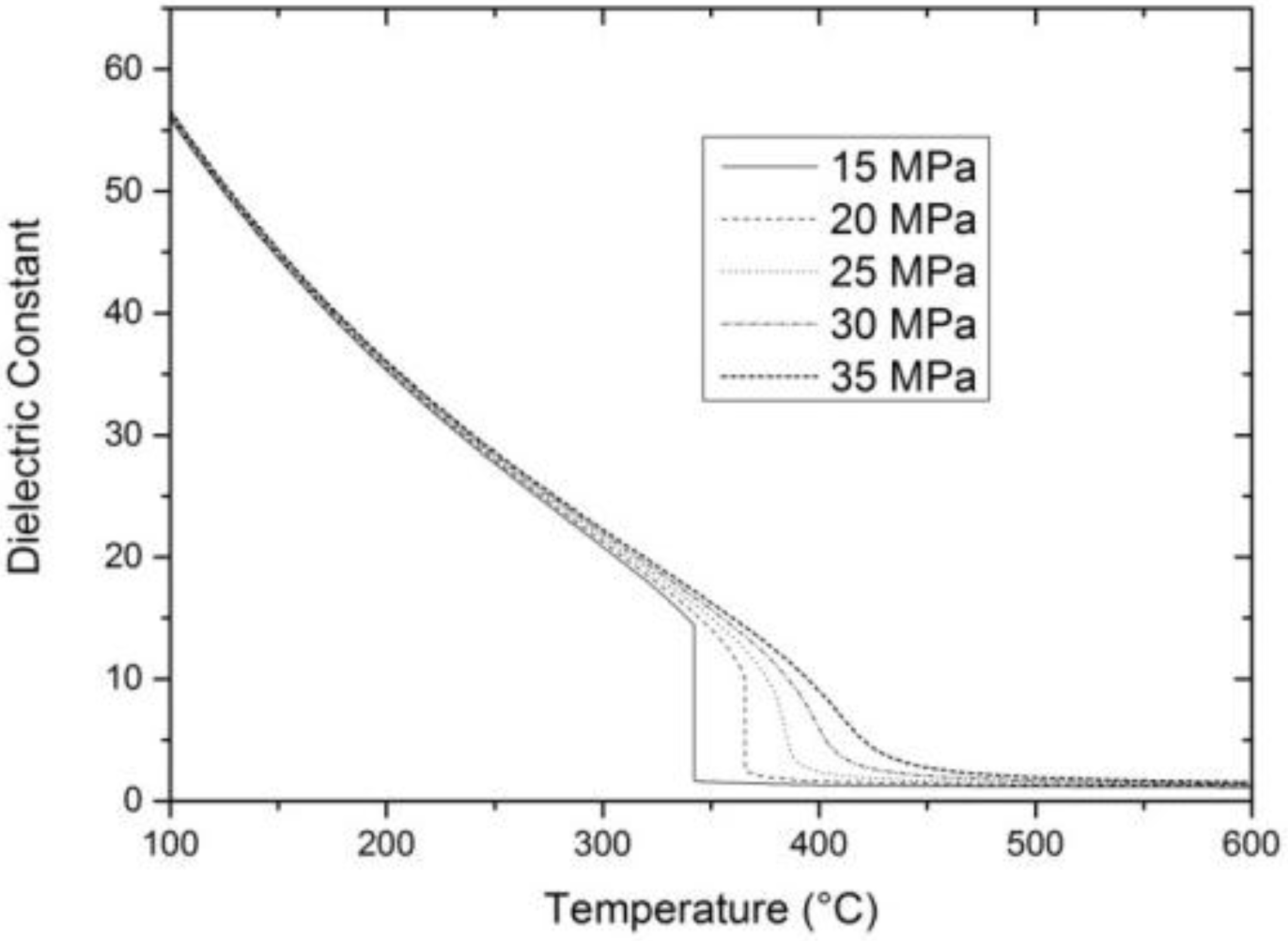

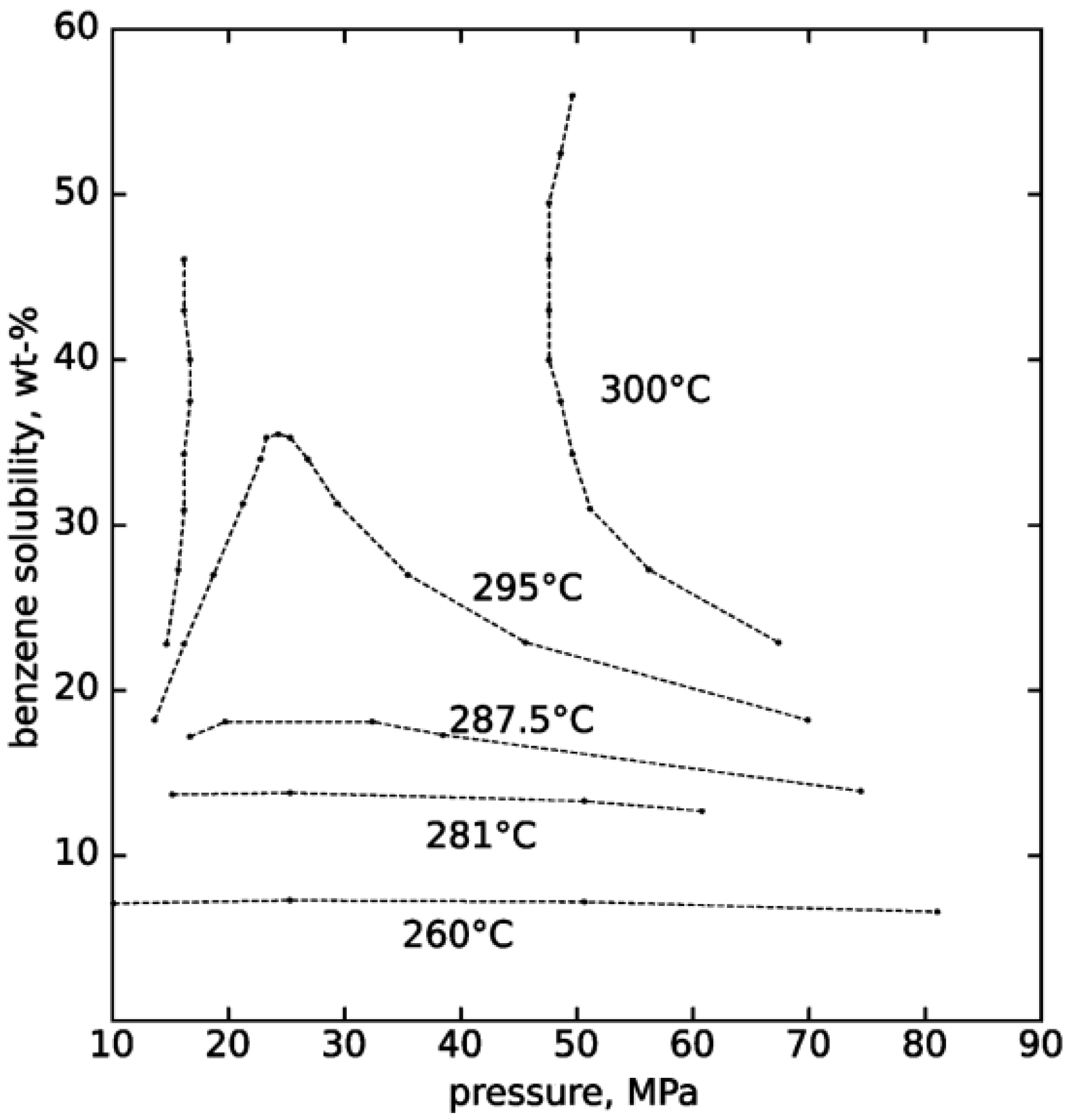

3. Hydrothermal Conversion of Organic Feedstocks
3.1. Carbonization
3.2. Oxidation
3.3. Liquefaction
3.4. Gasification
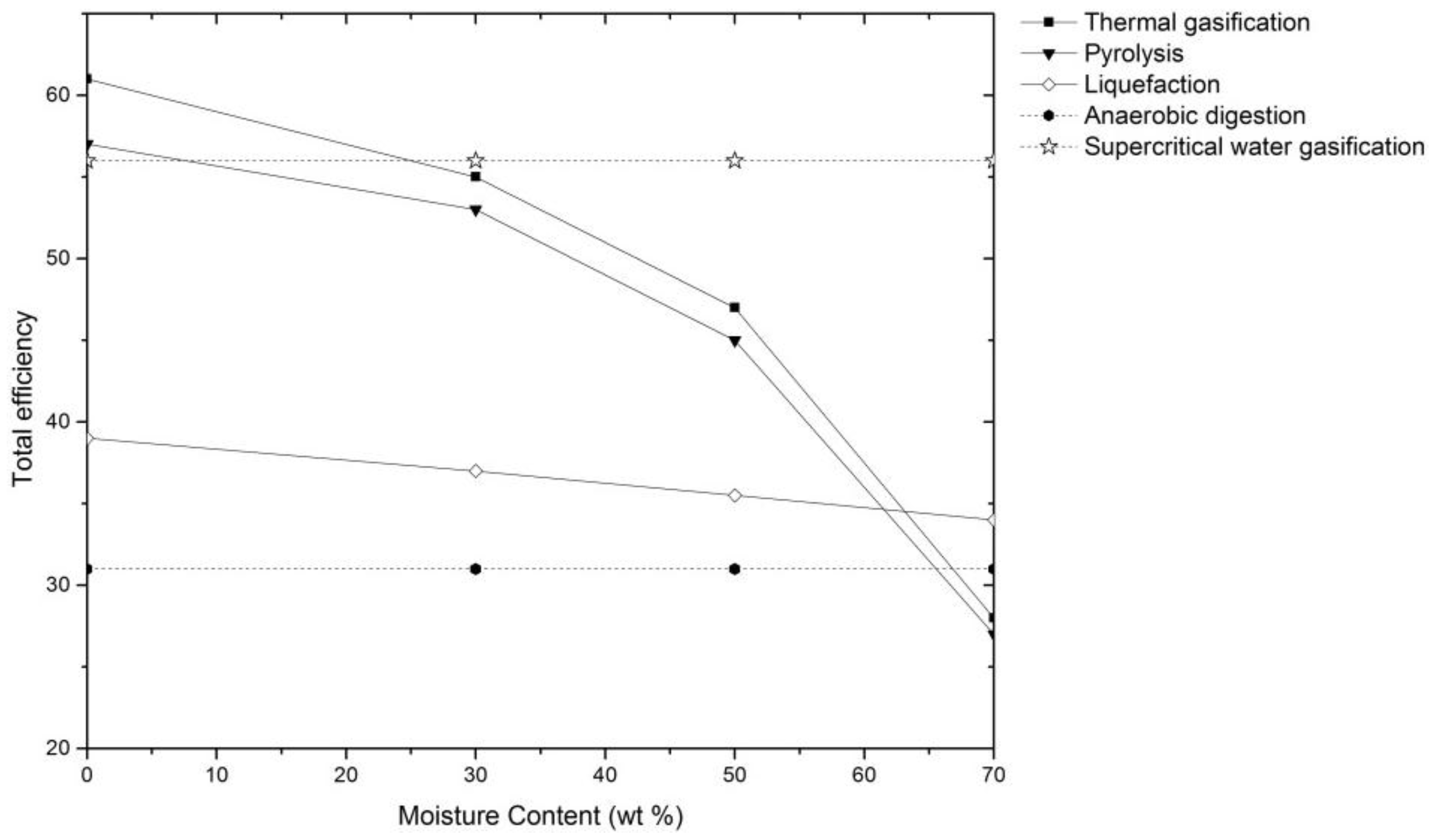
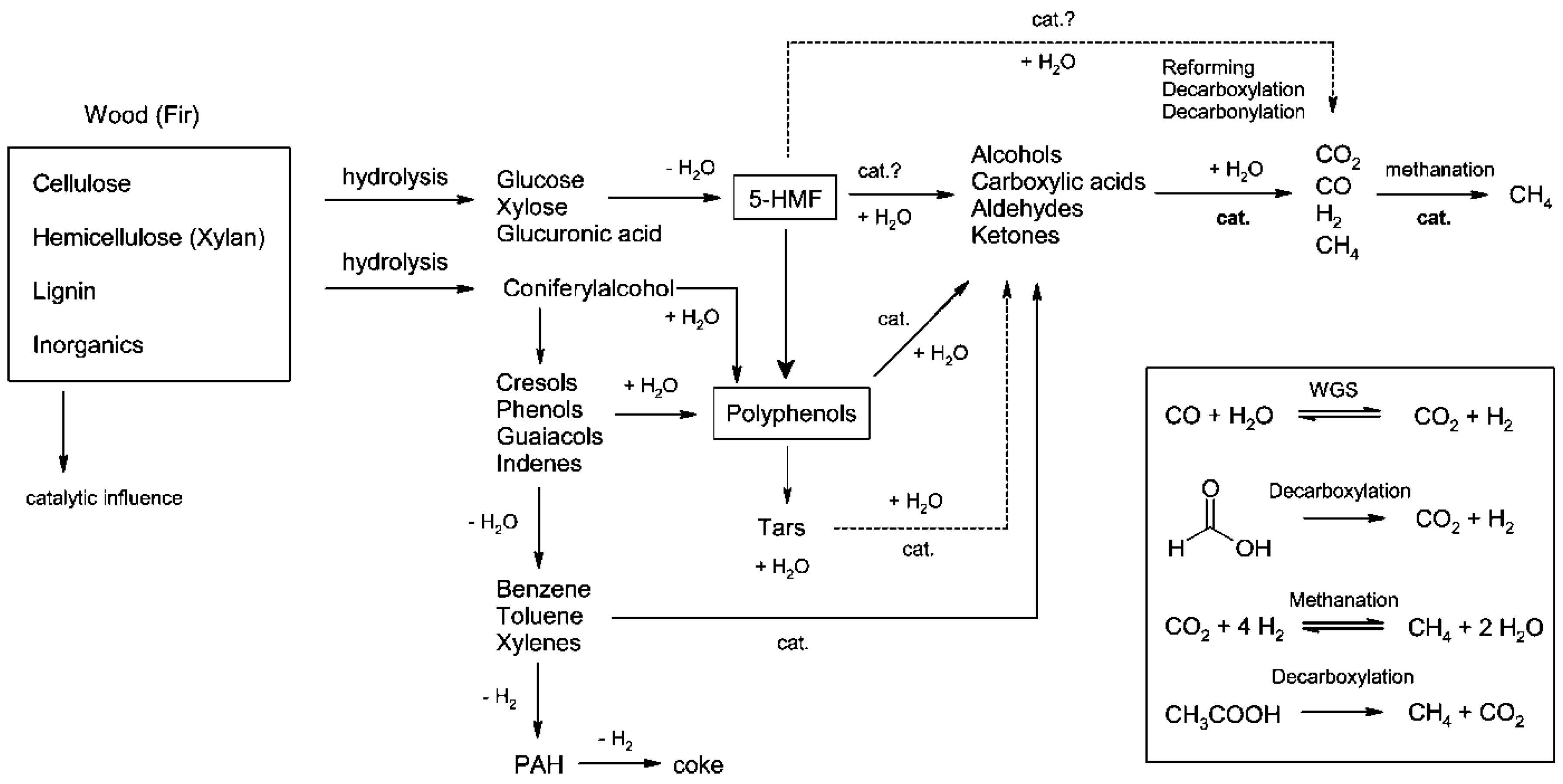
4. Supercritical Water Gasification of Biomass: Experimental Approaches
4.1. Understanding the Chemistry
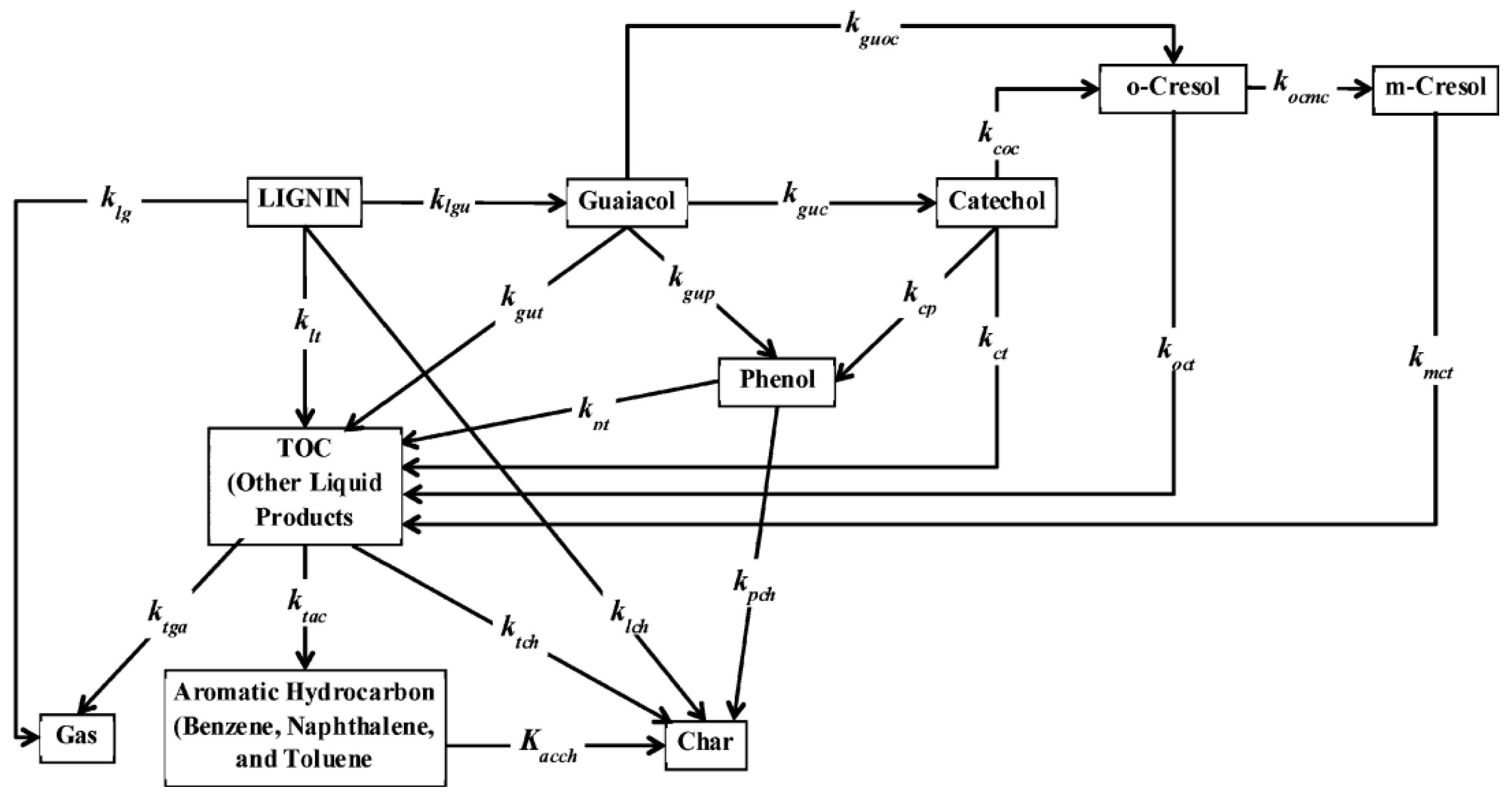
4.1.1. Biopolymers
4.1.2. Monomers
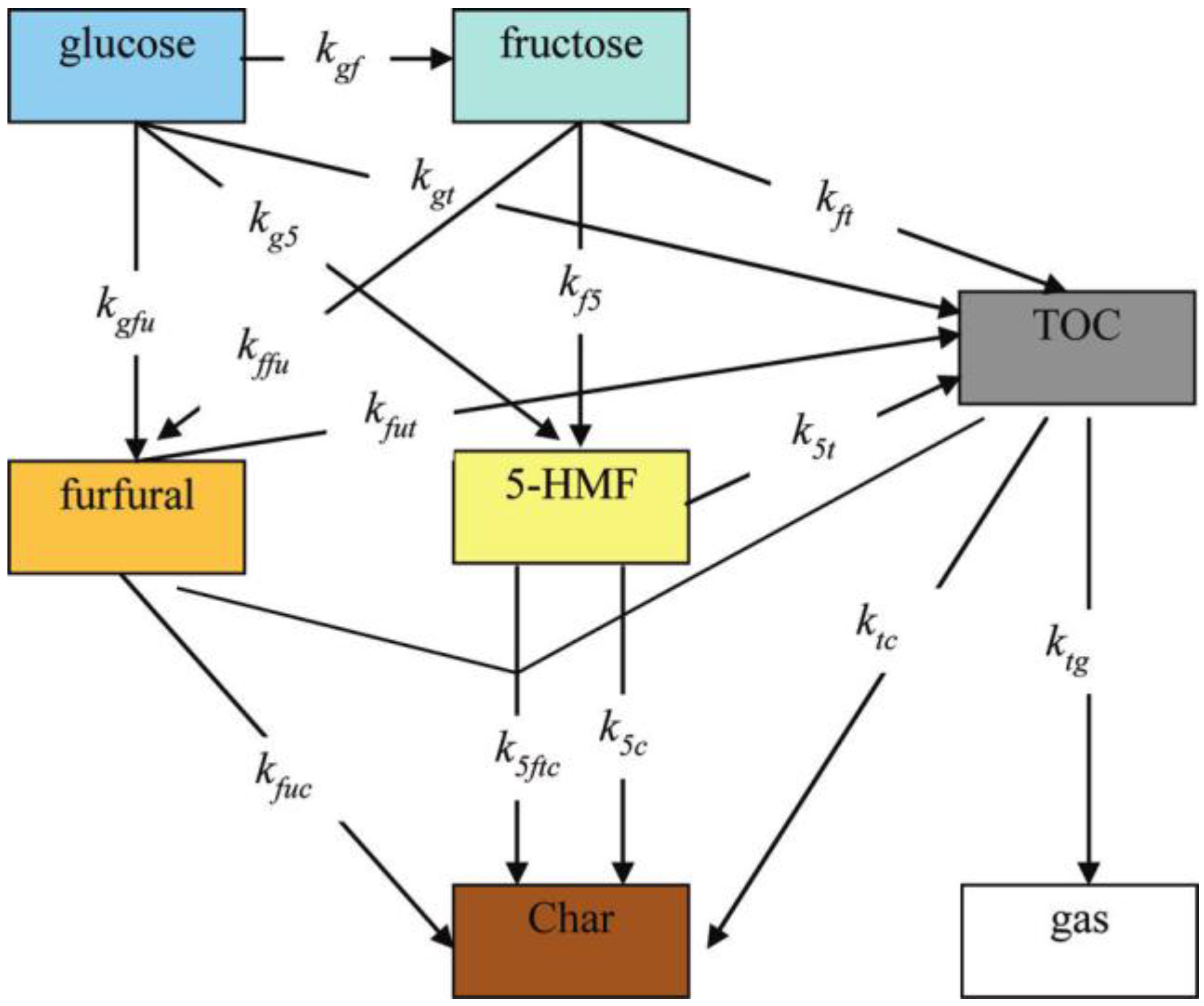
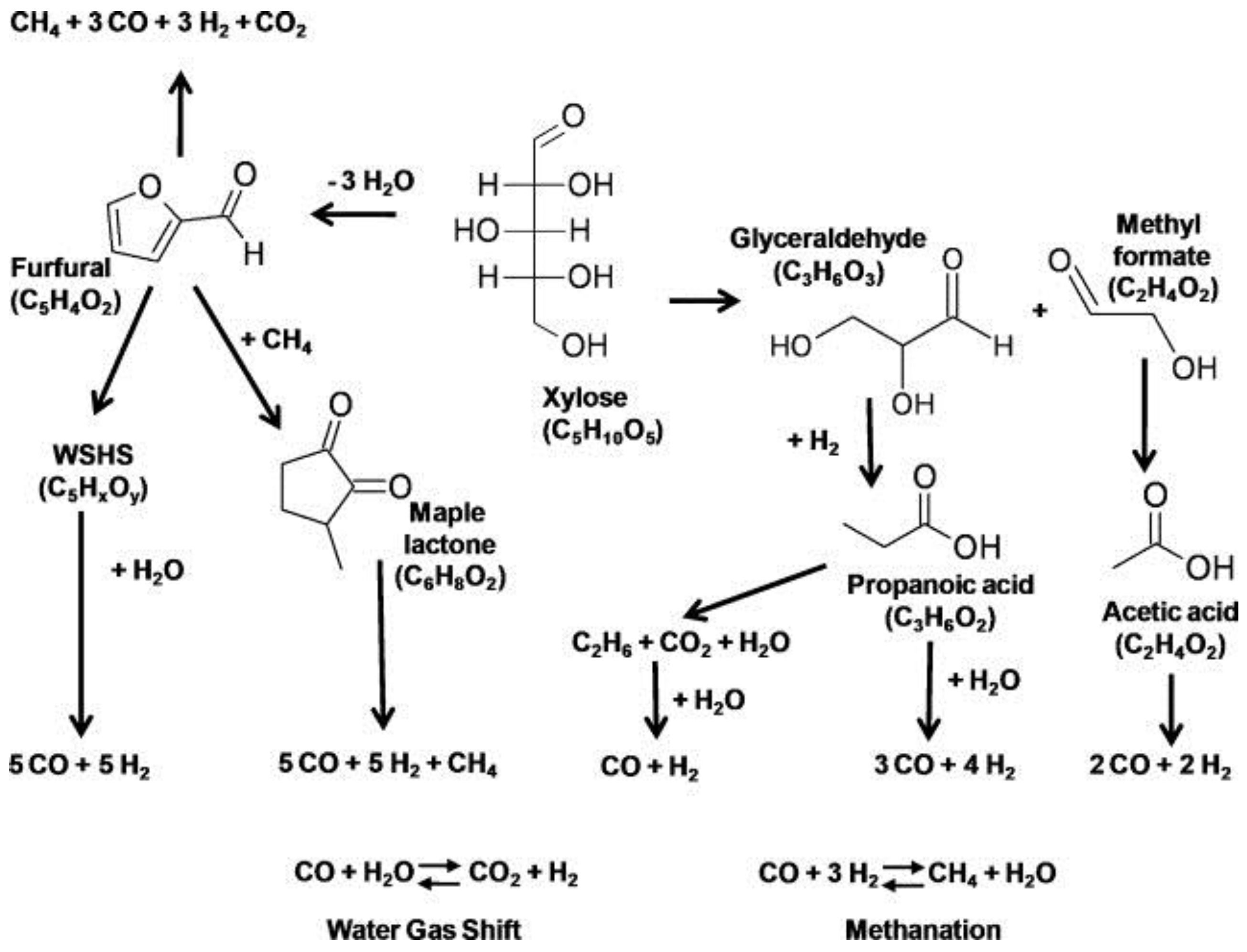
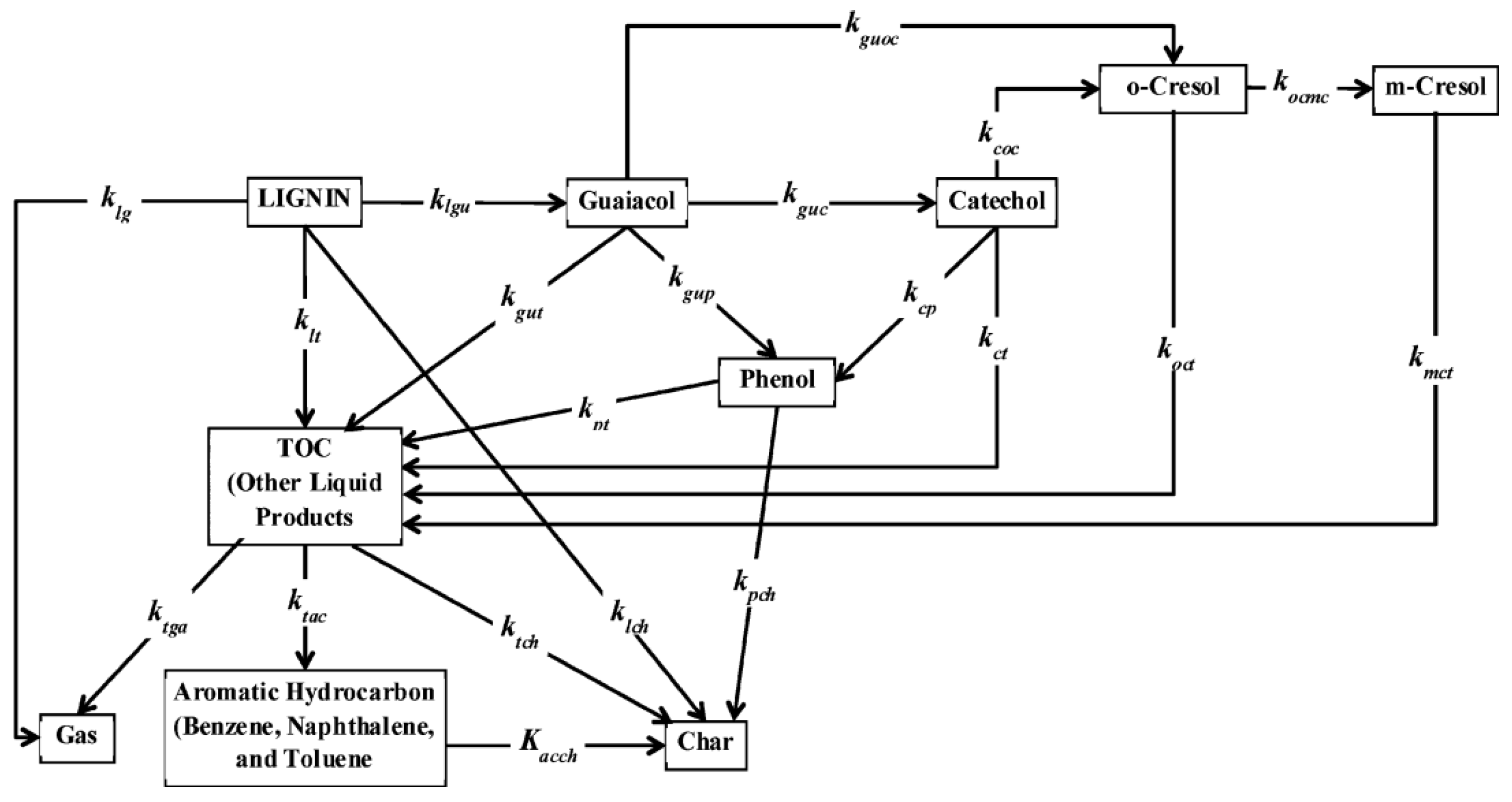
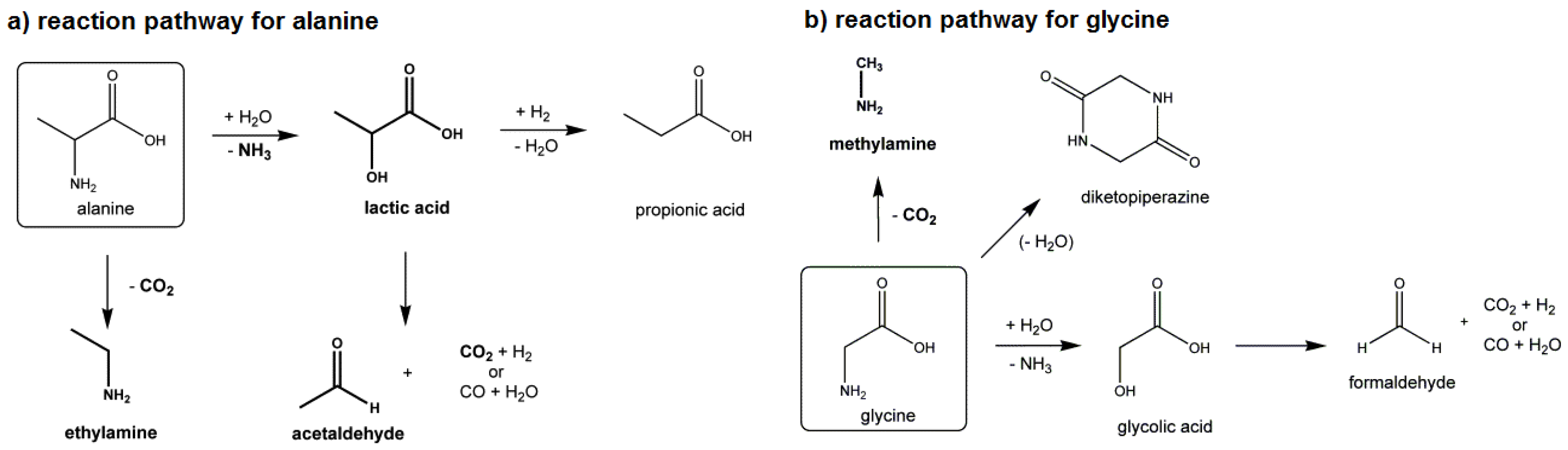
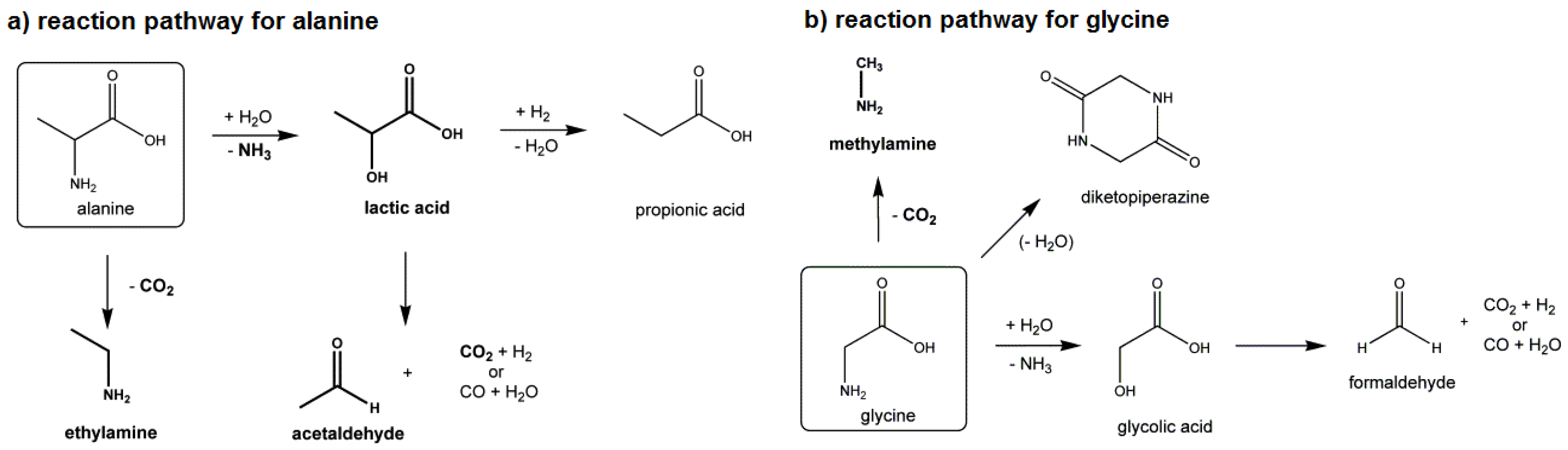
4.1.3. Amino Acids
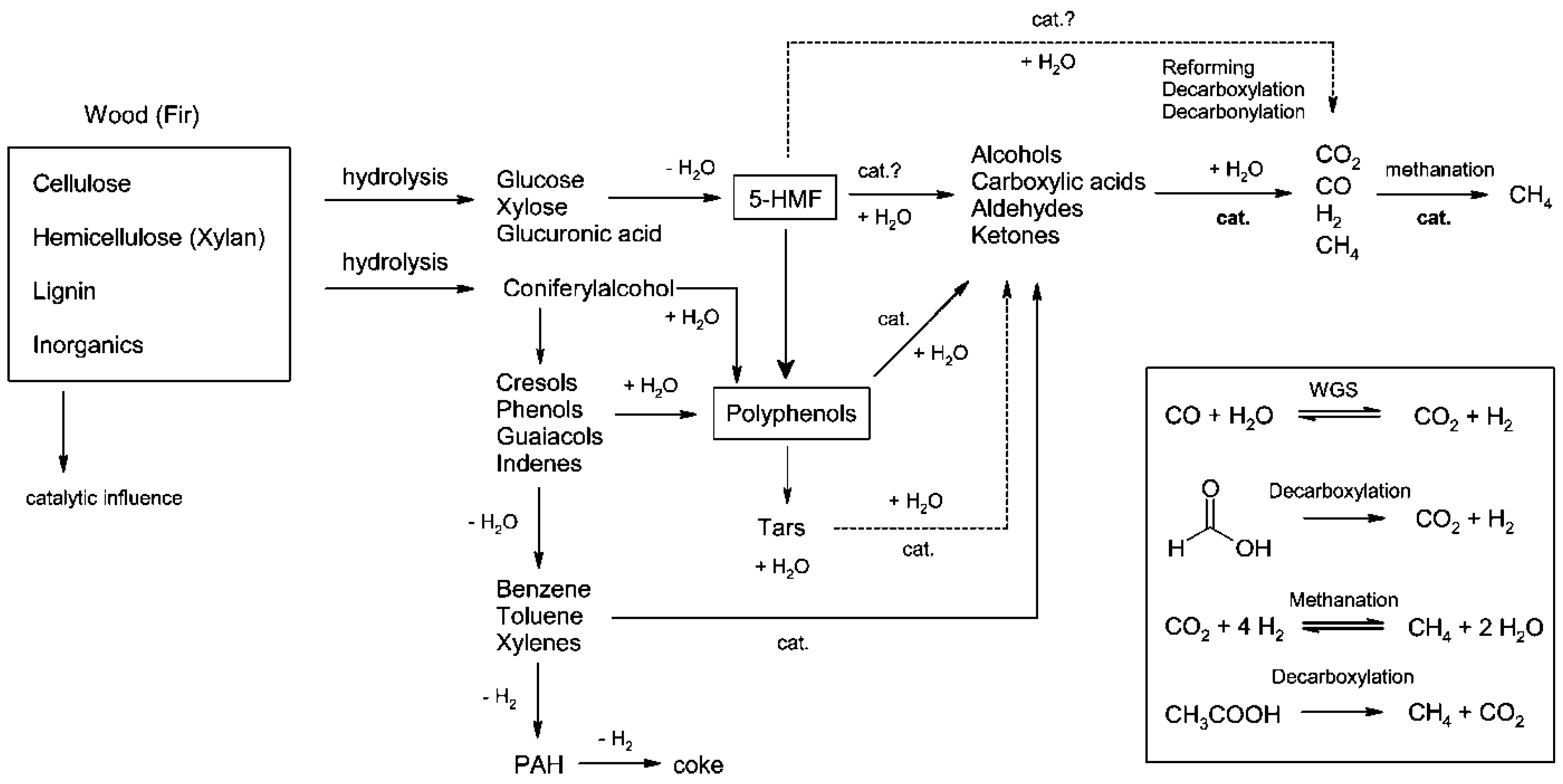
4.1.4. Intermediates
4.1.5. Mixtures
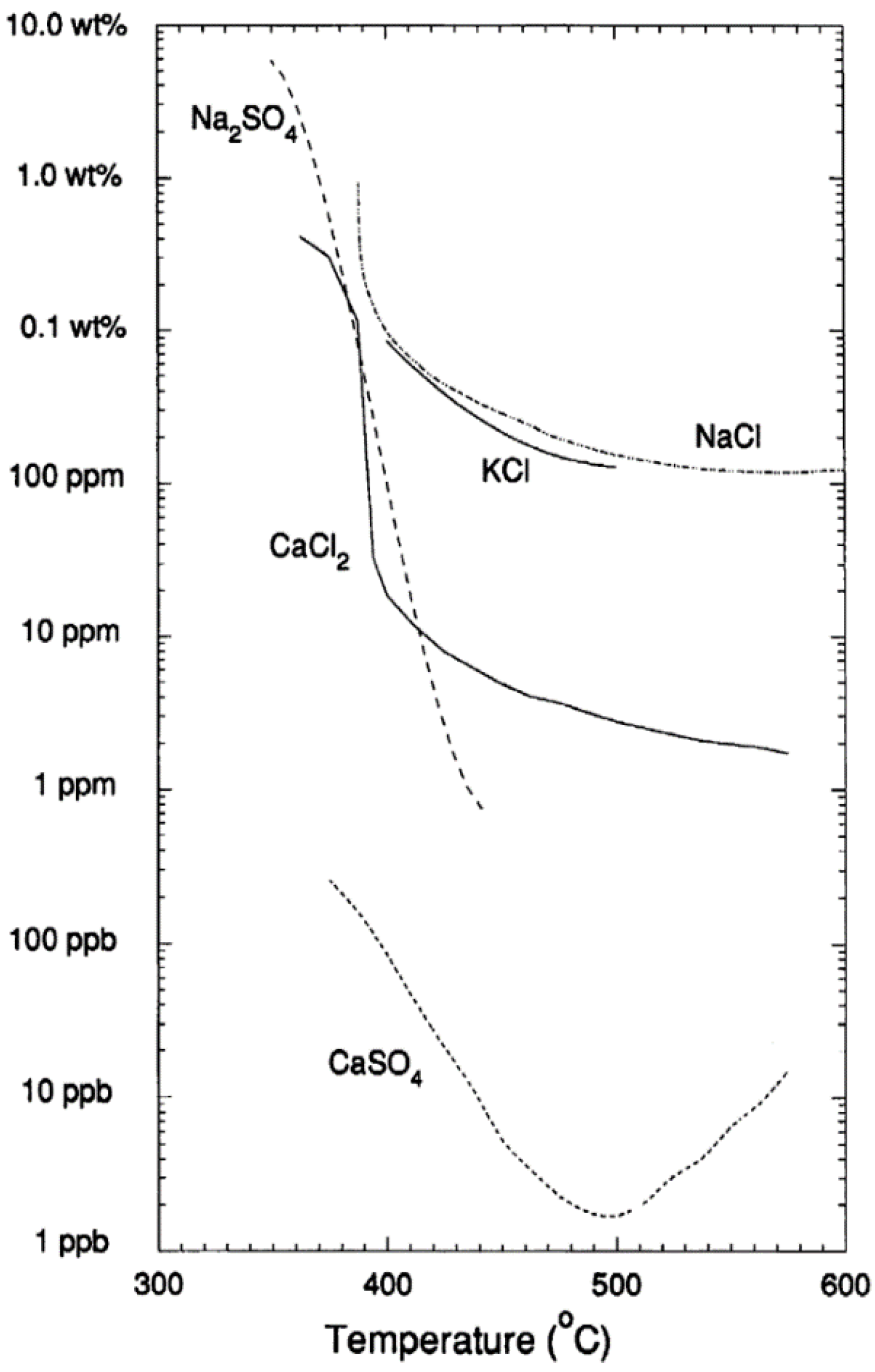
4.1.6. Effect of Salts and the Role as Homogeneous Catalysts
4.1.7. Effect of Heterogeneous Catalysts
4.1.8. Behavior of Heteroatoms
4.1.9. Gas Phase Reactions
4.2. Real Biomass Experiments
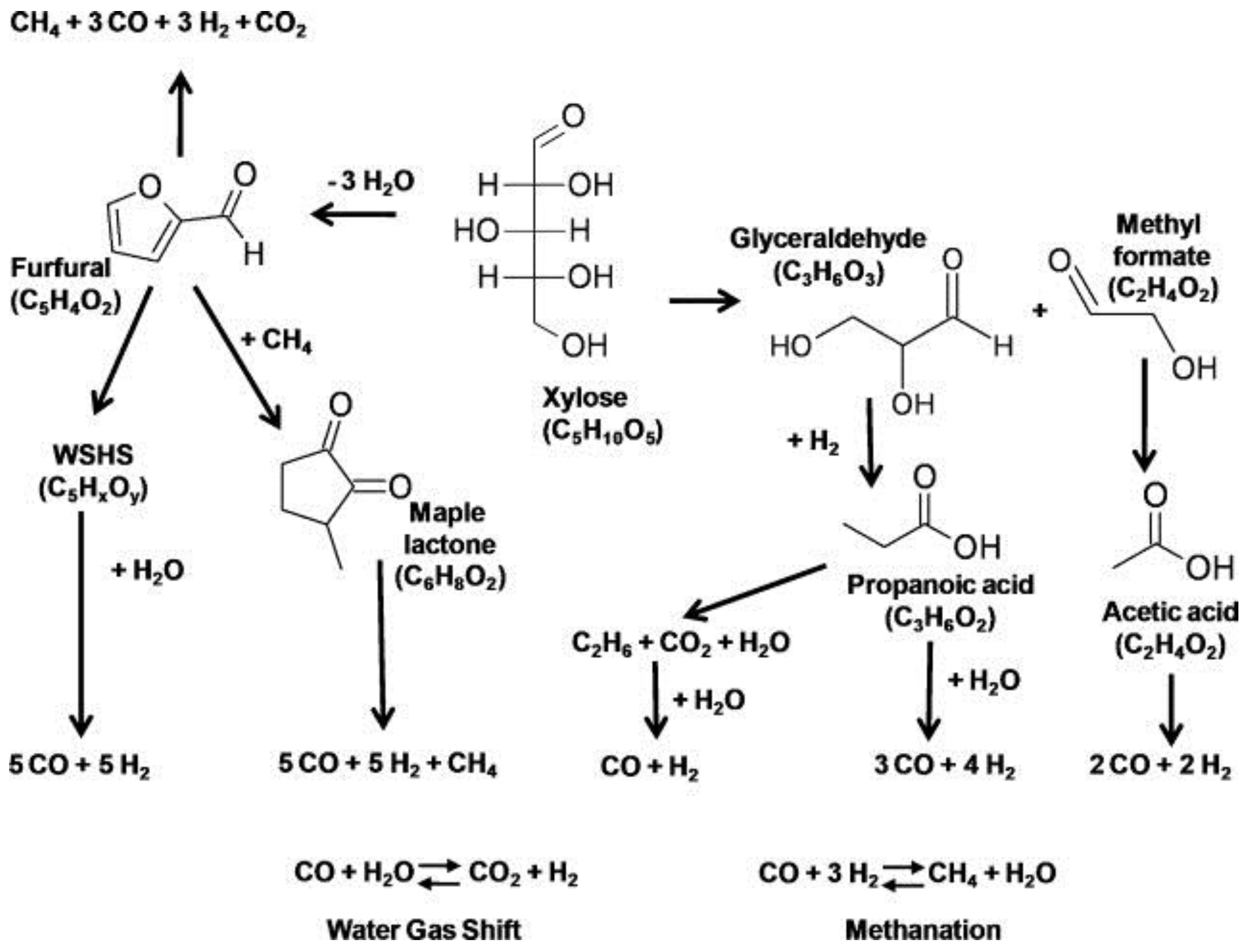
4.2.1. Carbohydrates
4.2.2. Algae
4.2.3. Sludge
4.2.4. Manure

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feedstock | wt% | Temperature (°C) | Pressure (MPa) | Reactor type | Residence time | CGE (%) | Mole fraction of gas products (%) | Reference | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CH4 | CO2 | H2 | CO | ||||||||
| Corn Starch | 10.4 | ~700 | 28 | Tubular with a carbon catalyst | 2.18 h | 91 | 22 | 38 | 37 | 2 | [75] |
| Microalgae (Chlorella Vulgaris) | 7.3 | 600 | 24 | Quartz Capillary | 2 min | 53 | 25 | 26 | 7 | 22 | [94] |
| Sewage Sludge | 10 | 540 | 25 | Fluidized Bed | N/A | 32 | 13 | 43 | 26 | 13.5 | [98] |
| Swine Manure | 13.2 | 405 | 30.1 | Batch reactor with nickel catalyst | 36 min | 75.8 | 46 | 43 | 10.7 | 0.1 | [13] |
| Chicken Manure | 10 | 600 | 25 | Flow reactor with carbon catalyst | 1.7 min | 90 | 21.2 | 45.4 | 28.7 | 0 | [77] |
5. Supercritical Water Gasification of Biomass: Modeling Approaches
5.1. Kinetic Modeling
- in most of these works, all of the reactions are assumed to be first order reactions;
- the temperature effect on the rate constants has not been sufficiently investigated;
- most of these works only incorporate the decomposition rates and the formation of intermediates, but do not show the whole gasification routes;
- the models which include entire gasification routes incorporate the lumped kinetic model, which neglects the composition of intermediates.
| Compound | Type of Reaction | Activation Energy (kJ·mol−1) | Pre-Exp. Factor (s−1) | Temperature Range (°C) | Pressure (Mpa) | Reference |
|---|---|---|---|---|---|---|
| Cellulose | Conversion | 145.9 | 1 × 1011.9 | 320–370 | 25 | [31] |
| Cellulose | Conversion | 547.9 | 1 × 1044.6 | 370–400 | 25 | [31] |
| Cellobiose | Conversion | 96.4 | 1.28 × 108 | 300–350 | 25 | [110] |
| Cellobiose | Conversion | 96.4 | 1.48 × 108 | 350–400 | 30 | [110] |
| Cellobiose | Hydrolysis | 108.6 | 1.10 × 109 | 300–350 | 25 | [110] |
| Cellobiose | Hydrolysis | 108.6 | 1.15 × 109 | 350–400 | 30 | [110] |
| Cellobiose | Pyrolysis to glucosyl-eryhtrose and glycoaldehyde | 30.4 | 17.68 | 300–350 | 25 | [110] |
| Cellobiose | Pyrolysis to glucosyl-eryhtrose and glycoaldehyde | 30.4 | 57.13 | 350–400 | 30 | [110] |
| Cellobiose | Pyrolysis to glucosyl-glycoaldehyde and eryhtrose | 69.3 | 82,730.74 | 300–350 | 25 | [110] |
| Cellobiose | Pyrolysis to glucosyl-glycoaldehyde and eryhtrose | 69.3 | 83,456.10 | 350–400 | 30 | [110] |
| Glucose | Decomposition | 121 | 1.33 × 1010 | 175–400 | 25 | [43] |
| Glucose | Decomposition | 96 | 1.23 × 108 | 300–350 | 25 | [111] |
| Glucose | Isomerization to fructose | 112.75 | 2.99 × 109 | 300–400 | 25–30 | [42] |
| Fructose | Decomposition to acids | 130.94 | 7.48 × 1010 | 300–400 | 25–30 | [42] |
| Glucose | Conversion to 5-HMF | 114.40 | 1.49 × 108 | 300–400 | 25 | [44] |
| Glucose | Conversion to non-furfural organics | 137.40 | 2.05 × 1011 | 300–400 | 25 | [44] |
| TOC | Conversion to gas products | 27.92 | 0.78 | 300–400 | 25 | [44] |
| TOC | Conversion to char | 17.29 | 0.04 | 300–400 | 25 | [44] |
| Xylose | Conversion to furfural | 120.1 | 1.20 × 1012 | 450–650 | 25 | [47] |
| Furfural | Conversion to WSHS | 55.6 | 5.70 × 103 | 450–650 | 25 | [47] |
| WSHS | Conversion to gas | 138.9 | 1.60 × 108 | 450–650 | 25 | [47] |
| Guaiacol | Conversion | 40.0 | 1.87 × 103 | 380–400 | 30 | [48] |
5.2. Computational Fluid Dynamics Modeling
5.3. Thermodynamic Equilibrium Modeling
| Investigated compounds | Type of approach | Type of EoS or software used for the calculations | Considered phases | Significant contribution | Reference |
|---|---|---|---|---|---|
| Methanol, glucose, cellulose, starch, sawdust | Gibbs free energy minimization (GFEM) | Peng-Robinson EoS | Only gas phase | The first paper with a thermodynamic model | [118] |
| Glucose | GFEM | Duan EoS | Only gas phase | The introduction of additional constraints | [124] |
| Wood sawdust | GFEM | Duan EoS | Gas phase and solid carbon | The introduction of solid carbon | [122] |
| Poultry manure | GFEM | HSC chemistry 6.12 | Multi-Phase | The first paper which took the inorganic elements into account | [79] |
| Methanol, ethanol, glycerol, glucose and cellulose | GFEM | Ideal gas | Gas phase and solid carbon | No need for initial guesses for the mole amount of product species | [123] |
| Methanol, glucose, sewage sludge | Reaction equilibria | Peng-Robinson EoS | Gas phase and solid carbon | The first paper which used the reaction equilibria method | [126] |
| Glycerol and microalgae | GFEM | Peng-Robinson EoS | Gas phase and solid carbon | Char formation conditions have been examined | [125] |
| Glucose and cellulose | GFEM | Virial EoS and ideal gas | Only gas phase | Introduction of the entropy maximization | [120] |
| Pig-Cow manure mix | GFEM | FactSage 5.4.1 and SimuSage 1.12 | Multi-Phase | The first paper which investigated the partitioning behavior of elements | [12] |
| Microalgae | GFEM | Peng-Robinson EoS for gases and revised HKF EoS for the aqueous compounds | Multi-Phase | The first paper which proposed a multi-phase mathematical model for both subcritical and supercritical regions | [128] |
5.4. Process Modeling
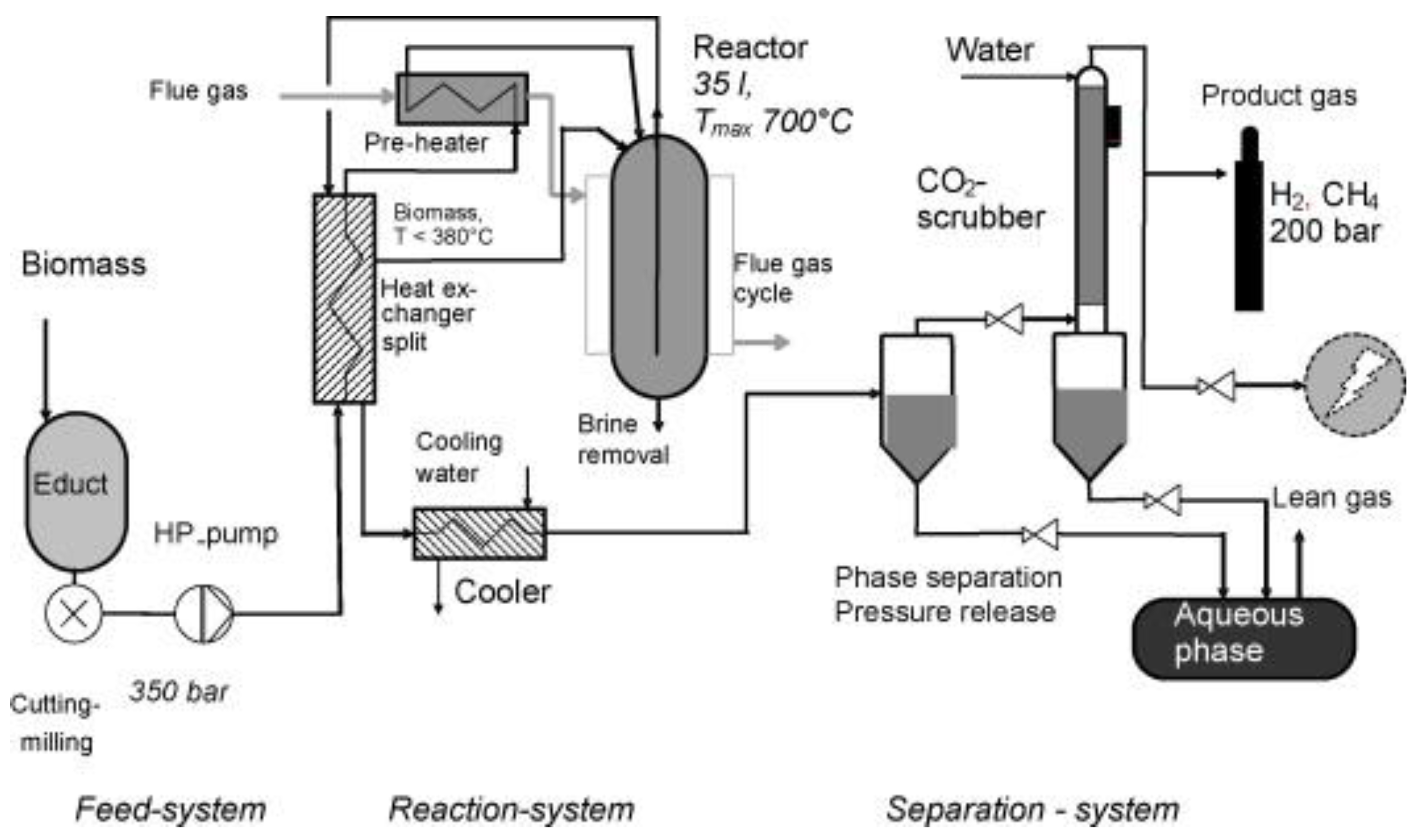
6. Process Challenges and Reactor Technology Aspects for Industrial Applications
7. Conclusions
Acknowledgments
Authors Contributions
Conflicts of Interest
References
- U.S. Energy Information Administration International Energy Outlook 2013; Report Number: DOE/EIA-0484(2013); U.S. Department of Energy: Washington, DC, USA, 2013.
- Process: The potential for biomass in the energy mix. Filtr. Sep. 2006, 43, 28–30.
- Basu, P.; Mettanant, V. Biomass gasification in supercritical water—a review. Int. J. Chem. React. Eng. 2009, 7, 1–61. [Google Scholar]
- European Commission Biomass—Green Energy for Europe; Report Number: EUR21350; European Commission: Brussels, Belgium, 2005.
- Matsumura, Y.; Minowa, T. Fundamental design of a continuous biomass gasification process using a supercritical water fluidized bed. Int. J. Hydrog. Energy 2004, 29, 701–707. [Google Scholar] [CrossRef]
- Amin, S.; Reid, R.; Modell, M. Reforming and decomposition of glucose in an aqueous phase. In Proceedings of the Intersociety Conference on Environmental Systems, San Francisco, CA, USA, 21–24 July 1975.
- Barner, H.E.; Huang, C.Y.; Johnson, T.; Jacobs, G.; Martch, M.A.; Killilea, W.R. Supercritical water oxidation: An emerging technology. J. Hazard. Mater. 1992, 31, 1–17. [Google Scholar] [CrossRef]
- Brunner, G. Near critical and supercritical water. Part I. Hydrolytic and hydrothermal processes. J. Supercrit. Fluids 2009, 47, 373–381. [Google Scholar] [CrossRef]
- Peterson, A.A.; Vogel, F.; Lachance, R.P.; Fröling, M.; Antal, M.J.J.; Tester, J.W. Thermochemical biofuel production in hydrothermal media: A review of sub- and supercritical water technologies. Energy Environ. Sci. 2008, 1, 32–65. [Google Scholar] [CrossRef]
- Kruse, A.; Dinjus, E. Hot compressed water as reaction medium and reactant: Properties and synthesis reactions. J. Supercrit. Fluids 2007, 39, 362–380. [Google Scholar] [CrossRef]
- Wagner, W.; Pruß, A. The IAPWS formulation 1995 for the thermodynamic properties of ordinary water substance for general and scientific use. J. Phys. Chem. Ref. Data 2002, 31, 387–535. [Google Scholar] [CrossRef]
- Yakaboylu, O.; Harinck, J.; Gerton Smit, K.G.; de Jong, W. Supercritical water gasification of manure: A thermodynamic equilibrium modeling approach. Biomass Bioenergy 2013, 59, 253–263. [Google Scholar] [CrossRef]
- Waldner, M.H. Catalytic Hydrothermal Gasification of Biomass for the Production of Synthetic Natural Gas. Ph.D. Thesis, Eidgenössische Technische Hochschule/Paul Scherrer Institute, Zürich, Switzerland, 2007. [Google Scholar]
- Masaru Watanabe, T.S. Chemical reactions of C(1) compounds in near-critical and supercritical water. Chem. Rev. 2004, 104, 5803–5821. [Google Scholar] [CrossRef] [PubMed]
- Armellini, F.J. Phase Equilibria and Precipitation Phenomena of Sodium Chloride and Sodium Sulfate in Sub- and Supercritical Water. Ph.D. Thesis, Massachusetts Institute of Technology, Cambridge, MA, USA, 1993. [Google Scholar]
- Connolly, J.F. Solubility of hydrocarbons in water near the critical solution temperatures. J. Chem. Eng. Data 1966, 11, 13–16. [Google Scholar] [CrossRef]
- Marshall, W.L.; Franck, E.U. Ion product of water substance, 0–1000 °C, 1–10,000 bars New International Formulation and its background. J. Phys. Chem. Ref. Data 1981, 10. [Google Scholar] [CrossRef]
- Webley, P.A.; Tester, J.W. Fundamental kinetics of methane oxidation in supercritical water. Energy Fuels 1991, 5, 411–419. [Google Scholar] [CrossRef]
- Thermophysical Properties of Fluid Systems. Available online: http://webbook.nist.gov/chemistry/fluid/ (accessed on 6 June 2014).
- Castello, D. Supercritical Water Gasification of Biomass. Ph.D. Thesis, University of Trento, Trento, Italy, 2013. [Google Scholar]
- Coronella, C.J.; Lynam, J.G.; Reza, M.T.; Uddin, M.H. Hydrothermal carbonization of lignocellulosic biomass. In Application of Hydrothermal Reactions to Biomass Conversion; Jin, F., Ed.; Springer: Berlin, Germany, 2014; pp. 275–311. [Google Scholar]
- Onwudili, J.A. Hydrothermal gasification of biomass for hydrogen production. In Application of Hydrothermal Reactions to Biomass Conversion; Jin, F., Ed.; Springer: Berlin, Germany, 2014; pp. 219–246. [Google Scholar]
- Toor, S.S.; Rosendahl, L.A.; Hoffmann, J.; Pedersen, T.H.; Nielsen, R.P.; Søgaard, E.G. Hydrothermal liquefaction of biomass. In Application of Hydrothermal Reactions to Biomass Conversion; Jin, F., Ed.; Springer: Berlin, Germany, 2014; pp. 189–217. [Google Scholar]
- Xu, C.C.; Shao, Y.; Yuan, Z.; Cheng, S.; Feng, S.; Nazari, L.; Tymchyshyn, M. Hydrothermal Liquefaction of Biomass in Hot-Compressed Water, Alcohols, and Alcohol-Water Co-solvents for Biocrude Production. In Application of Hydrothermal Reactions to Biomass Conversion; Jin, F., Ed.; Springer: Berlin, Germany, 2014; pp. 171–187. [Google Scholar]
- Bermejo, M.D.; Cocero, M.J. Supercritical water oxidation: A technical review. AIChE J. 2006, 52, 3933–3951. [Google Scholar] [CrossRef]
- Hodes, M.; Marrone, P.A.; Hong, G.T.; Smith, K.A.; Tester, J.W. Salt precipitation and scale control in supercritical water oxidation—Part A: Fundamentals and research. J. Supercrit. Fluids 2004, 29, 265–288. [Google Scholar] [CrossRef]
- Cortright, R.D.; Davda, R.R.; Dumesic, J.A. Hydrogen from catalytic reforming of biomass-derived hydrocarbons in liquid water. Nature 2002, 418, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Gassner, M.; Vogel, F.; Heyen, G.; Maréchal, F. Optimal process design for the polygeneration of SNG, power and heat by hydrothermal gasification of waste biomass: Process optimisation for selected substrates. Energy Environ. Sci. 2011, 4, 1742–1758. [Google Scholar] [CrossRef]
- Van der Meijden, C.M.; Veringa, H.J.; Rabou, L.P.L.M. The production of synthetic natural gas (SNG): A comparison of three wood gasification systems for energy balance and overall efficiency. Biomass Bioenergy 2010, 34, 302–311. [Google Scholar] [CrossRef]
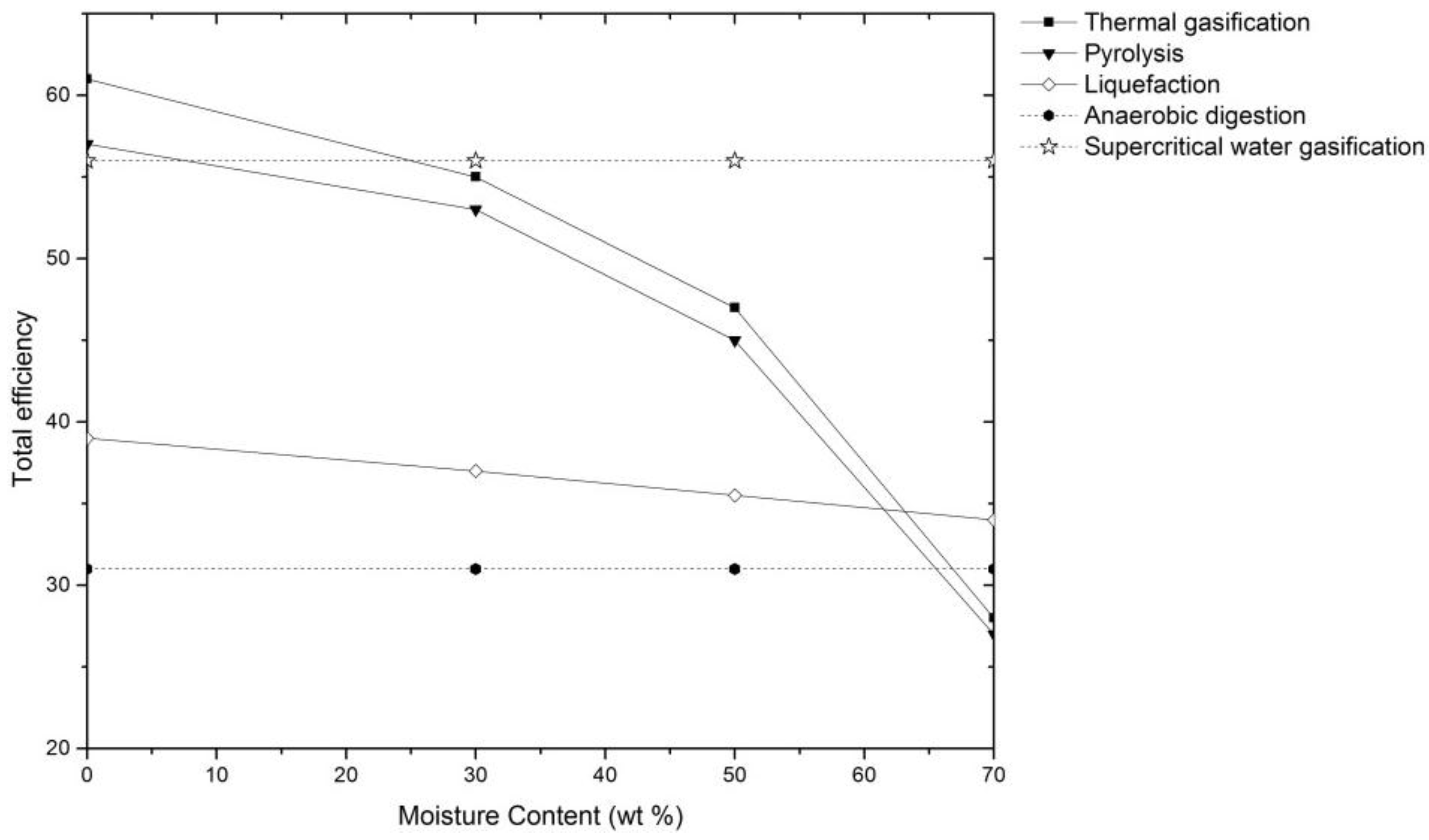
- Yoshida, Y.; Dowaki, K.; Matsumura, Y.; Matsuhashi, R.; Li, D.; Ishitani, H.; Komiyama, H. Comprehensive comparison of efficiency and CO2 emissions between biomass energy conversion technologies—Position of supercritical water gasification in biomass technologies. Biomass Bioenergy 2003, 25, 257–272. [Google Scholar] [CrossRef]
- Sasaki, M.; Adschiri, T.; Arai, K. Kinetics of cellulose conversion at 25 MPa in sub- and supercritical water. AIChE J. 2004, 50, 192–202. [Google Scholar] [CrossRef]
- Sasaki, M.; Kabyemela, B.; Malaluan, R.; Hirose, S.; Takeda, N.; Adschiri, T.; Arai, K. Cellulose hydrolysis in subcritical and supercritical water. J. Supercrit. Fluids 1998, 13, 261–268. [Google Scholar] [CrossRef]
- Resende, F.L.P.; Neff, M.E.; Savage, P.E. Noncatalytic gasification of cellulose in supercritical water. Energy Fuels 2007, 21, 3637–3643. [Google Scholar] [CrossRef]
- Yong, T.L.-K.; Matsumura, Y. Kinetic analysis of lignin hydrothermal conversion in sub- and supercritical Water. Ind. Eng. Chem. Res. 2013, 52, 5626–5639. [Google Scholar] [CrossRef]
- Yong, T.L.-K.; Matsumura, Y. Reaction kinetics of the lignin conversion in supercritical water. Ind. Eng. Chem. Res. 2012, 51, 11975–11988. [Google Scholar] [CrossRef]
- Resende, F.L.P.; Fraley, S.A.; Berger, M.J.; Savage, P.E. Noncatalytic gasification of lignin in supercritical water. Energy Fuels 2008, 22, 1328–1334. [Google Scholar] [CrossRef]
- Lee, I.-G.; Kim, M.-S.; Ihm, S.-K. Gasification of glucose in supercritical water. Ind. Eng. Chem. Res. 2002, 41, 1182–1188. [Google Scholar] [CrossRef]
- Hao, X.H.; Guo, L.J.; Mao, X.; Zhang, X.M.; Chen, X.J. Hydrogen production from glucose used as a model compound of biomass gasified in supercritical water. Int. J. Hydrog. Energy 2003, 28, 55–64. [Google Scholar] [CrossRef]
- Williams, P.T.; Onwudili, J. Composition of products from the supercritical water gasification of glucose: A model biomass compound. Ind. Eng. Chem. Res. 2005, 44, 8739–8749. [Google Scholar] [CrossRef]
- Goodwin, A.K.; Rorrer, G.L. Conversion of glucose to hydrogen-rich gas by supercritical water in a microchannel reactor. Ind. Eng. Chem. Res. 2008, 47, 4106–4114. [Google Scholar] [CrossRef]
- Güngören Madenoğlu, T.; Sağlam, M.; Yüksel, M.; Ballice, L. Simultaneous effect of temperature and pressure on catalytic hydrothermal gasification of glucose. J. Supercrit. Fluids 2013, 73, 151–160. [Google Scholar] [CrossRef]
- Kabyemela, B.M.; Adschiri, T.; Malaluan, R.M.; Arai, K. Glucose and fructose decomposition in subcritical and supercritical water: Detailed reaction pathway, mechanisms, and kinetics. Ind. Eng. Chem. Res. 1999, 38, 2888–2895. [Google Scholar] [CrossRef]
- Matsumura, Y.; Yanachi, S.; Yoshida, T. Glucose decomposition kinetics in water at 25 MPa in the temperature range of 448–673 K. Ind. Eng. Chem. Res. 2006, 45, 1875–1879. [Google Scholar] [CrossRef]
- Chuntanapum, A.; Matsumura, Y. Char formation mechanism in supercritical water gasification process: A study of model compounds. Ind. Eng. Chem. Res. 2010, 49, 4055–4062. [Google Scholar] [CrossRef]
- Kruse, A.; Gawlik, A. Biomass conversion in water at 330–410 °C and 30–50 MPa. Identification of key compounds for indicating different chemical reaction pathways. Ind. Eng. Chem. Res. 2003, 42, 267–279. [Google Scholar] [CrossRef]
- Aida, T.M.; Shiraishi, N.; Kubo, M.; Watanabe, M.; Smith, R.L. Reaction kinetics of D-xylose in sub- and supercritical water. J. Supercrit. Fluids 2010, 55, 208–216. [Google Scholar] [CrossRef]
- Goodwin, A.K.; Rorrer, G.L. Reaction rates for supercritical water gasification of xylose in a micro-tubular reactor. Chem. Eng. J. 2010, 163, 10–21. [Google Scholar] [CrossRef]
- Wahyudiono, W; Kanetake, T.; Sasaki, M.; Goto, M. Decomposition of a lignin model compound under hydrothermal conditions. Chem. Eng. Technol. 2007, 30, 1113–1122. [Google Scholar] [CrossRef]
- Wahyudiono, W; Sasaki, M.; Goto, M. Thermal decomposition of guaiacol in sub- and supercritical water and its kinetic analysis. J. Mater. Cycles Waste Manag. 2011, 13, 68–79. [Google Scholar] [CrossRef]
- Dileo, G.J.; Neff, M.E.; Savage, P.E. Gasification of guaiacol and phenol in supercritical water. Energy Fuels 2007, 21, 2340–2345. [Google Scholar] [CrossRef]
- Klingler, D.; Berg, J.; Vogel, H. Hydrothermal reactions of alanine and glycine in sub- and supercritical water. J. Supercrit. Fluids 2007, 43, 112–119. [Google Scholar] [CrossRef]
- Dileo, G.J.; Neff, M.E.; Kim, S.; Savage, P.E. Supercritical water gasification of phenol and glycine as models for plant and protein biomass. Energy Fuels 2008, 22, 871–877. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, J.; Zhao, L.; Sheng, C. Decomposition of formic acid in supercritical water. Energy Fuels 2010, 24, 95–99. [Google Scholar] [CrossRef]
- Shin, K.S.; Cho, H.Y.; Nam, Y.W.; Lee, D.S. Hydrothermal decomposition of formic acid in sub- and supercritical water. Environ. Eng. Res. 1998, 3, 61–66. [Google Scholar]
- Aida, T.M.; Ikarashi, A.; Saito, Y.; Watanabe, M.; Smith, R.L.; Arai, K. Dehydration of lactic acid to acrylic acid in high temperature water at high pressures. J. Supercrit. Fluids 2009, 50, 257–264. [Google Scholar] [CrossRef]
- Mok, W.S.; Antal, M.J.; Jones, M. Formation of acrylic acid from lactic acid in supercritical water. J. Org. Chem. 1989, 54, 4596–4602. [Google Scholar] [CrossRef]
- Chuntanapum, A.; Matsumura, Y. Formation of tarry material from 5-HMF in subcritical and supercritical water. Ind. Eng. Chem. Res. 2009, 48, 9837–9846. [Google Scholar] [CrossRef]
- Chuntanapum, A.; Yong, T.L.-K.; Miyake, S.; Matsumura, Y. Behavior of 5-HMF in subcritical and supercritical water. Ind. Eng. Chem. Res. 2008, 47, 2956–2962. [Google Scholar] [CrossRef]
- Yoshida, T.; Matsumura, Y. Gasification of cellulose, xylan, and lignin mixtures in supercritical water. Ind. Eng. Chem. Res. 2001, 40, 5469–5474. [Google Scholar] [CrossRef]
- Yoshida, T.; Oshima, Y.; Matsumura, Y. Gasification of biomass model compounds and real biomass in supercritical water. Biomass Bioenergy 2004, 26, 71–78. [Google Scholar] [CrossRef]
- Weiss-Hortala, E.; Kruse, A.; Ceccarelli, C.; Barna, R. Influence of phenol on glucose degradation during supercritical water gasification. J. Supercrit. Fluids 2010, 53, 42–47. [Google Scholar] [CrossRef]
- Goodwin, A.K.; Rorrer, G.L. Conversion of xylose and xylose-phenol mixtures to hydrogen-rich gas by supercritical water in an isothermal microtube flow reactor. Energy Fuels 2009, 23, 3818–3825. [Google Scholar] [CrossRef]
- Castello, D.; Kruse, A.; Fiori, L. Supercritical water gasification of glucose/phenol mixtures as model compounds for ligno-cellulosic biomass. Chem. Eng. Trans. 2014, 37, 193–198. [Google Scholar]
- Sınaǧ, A.; Kruse, A.; Schwarzkopf, V. Key compounds of the hydropyrolysis of glucose in supercritical water in the presence of K2CO3. Ind. Eng. Chem. Res. 2003, 42, 3516–3521. [Google Scholar] [CrossRef]
- Kruse, A.; Faquir, M. Hydrothermal biomass gasification—effects of salts, backmixing and their interaction. Chem. Eng. Technol. 2007, 30, 749–754. [Google Scholar] [CrossRef]
- Kruse, A.; Meier, D.; Rimbrecht, P.; Schacht, M. Gasification of pyrocatechol in supercritical water in the presence of potassium hydroxide. Ind. Eng. Chem. Res. 2000, 39, 4842–4848. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, S.; Wang, Y.; Zhang, J.; Xu, D.; Gong, Y. Gasification of two and three-components mixture in supercritical water: Influence of NaOH and initial reactants of acetic acid and phenol. Int. J. Hydrog. Energy 2012, 37, 2278–2286. [Google Scholar] [CrossRef]
- Minowa, T.; Zhen, F.; Ogi, T. Cellulose decomposition in hot-compressed water with alkali or nickel catalyst. J. Supercrit. Fluids 1998, 13, 253–259. [Google Scholar] [CrossRef]
- Muangrat, R.; Onwudili, J.A.; Williams, P.T. Influence of alkali catalysts on the production of hydrogen-rich gas from the hydrothermal gasification of food processing waste. Appl. Catal. B Environ. 2010, 100, 440–449. [Google Scholar] [CrossRef]
- Muangrat, R.; Onwudili, J.A.; Williams, P.T. Alkali-promoted hydrothermal gasification of biomass food processing waste: A parametric study. Int. J. Hydrog. Energy 2010, 35, 7405–7415. [Google Scholar] [CrossRef]
- Onwudili, J.A.; Lea-Langton, A.R.; Ross, A.B.; Williams, P.T. Catalytic hydrothermal gasification of algae for hydrogen production: Composition of reaction products and potential for nutrient recycling. Bioresour. Technol. 2013, 127, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Yildiz Bircan, S.; Kamoshita, H.; Kanamori, R.; Ishida, Y.; Matsumoto, K.; Hasegawa, Y.; Kitagawa, K. Behavior of heteroatom compounds in hydrothermal gasification of biowaste for hydrogen production. Appl. Energy 2011, 88, 4874–4878. [Google Scholar] [CrossRef]
- Matsumura, Y.; Minowa, T.; Potic, B.; Kersten, S.; Prins, W.; van Swaaij, W.; Vandebeld, B.; Elliott, D.; Neuenschwander, G.; Kruse, A.; et al. Biomass gasification in near- and super-critical water: Status and prospects. Biomass Bioenergy 2005, 29, 269–292. [Google Scholar] [CrossRef]
- Zöhrer, H. Hydrothermal gasification of fermentation residues for SNG-production. Ph.D. Thesis, Swiss Federal Institute of Technology in Zurich, Zürich, Switzerland, 2013. [Google Scholar]
- Antal, M.J.; Allen, S.G.; Schulman, D.; Xu, X.; Divilio, R.J. Biomass gasification in supercritical water. Ind. Eng. Chem. Res. 2000, 39, 4040–4053. [Google Scholar] [CrossRef]
- Byrd, A.J.; Pant, K.K.; Gupta, R.B. Hydrogen production from glycerol by reforming in supercritical water over Ru/Al2O3 catalyst. Fuel 2008, 87, 2956–2960. [Google Scholar] [CrossRef]
- Nakamura, A.; Kiyonaga, E.; Yamamura, Y.; Shimizu, Y.; Minowa, T.; Noda, Y.; Matsumura, Y. Gasification of catalyst-suspended chicken manure in supercritical water. J. Chem. Eng. Jpn. 2008, 41, 433–440. [Google Scholar] [CrossRef]
- Waldner, M.H.; Vogel, F. Renewable production of methane from woody biomass by catalytic hydrothermal gasification. Ind. Eng. Chem. Res. 2005, 44, 4543–4551. [Google Scholar] [CrossRef]
- Yanagida, T.; Minowa, T.; Nakamura, A.; Matsumura, Y.; Noda, Y. Behavior of inorganic elements in poultry manure during supercritical water gasification. J. Jpn. Inst. Energy 2008, 87, 731–736. [Google Scholar] [CrossRef]
- Yanik, J.; Ebale, S.; Kruse, A.; Saglam, M.; Yüksel, M. Biomass gasification in supercritical water: II. Effect of catalyst. Int. J. Hydrog. Energy 2008, 33, 4520–4526. [Google Scholar] [CrossRef]
- Boukis, N.; Diem, V.; Habicht, W.; Dinjus, E. Methanol reforming in supercritical water. Ind. Eng. Chem. Res. 2003, 42, 728–735. [Google Scholar] [CrossRef]
- Gadhe, J.B.; Gupta, R.B. Hydrogen production by methanol reforming in supercritical water: Suppression of methane formation. Ind. Eng. Chem. Res. 2005, 44, 4577–4585. [Google Scholar] [CrossRef]
- Yu, D.; Aihara, M.; Antal, M.J. Hydrogen production by steam reforming glucose in supercritical water. Energy Fuels 1993, 7, 574–577. [Google Scholar] [CrossRef]
- Castello, D.; Kruse, A.; Fiori, L. Biomass gasification in supercritical and subcritical water: The effect of the reactor material. Chem. Eng. J. 2013, 228, 535–544. [Google Scholar] [CrossRef]
- Guan, Q.; Wei, C.; Savage, P.E. Kinetic model for supercritical water gasification of algae. Phys. Chem. Chem. Phys. 2012, 14, 3140–3147. [Google Scholar] [CrossRef] [PubMed]
- Resende, F.L.P.; Savage, P.E. Kinetic model for noncatalytic supercritical water gasification of cellulose and lignin. AIChE J. 2010, 56, 2412–2420. [Google Scholar]
- Araki, K.; Fujiwara, H.; Sugimoto, K.; Oshima, Y.; Koda, S. Kinetics of water-gas shift reaction in supercritical water. J. Chem. Eng. Jpn. 2004, 37, 443–448. [Google Scholar] [CrossRef]
- Rice, S.F.; Steeper, R.R.; Aiken, J.D. Water density effects on homogeneous water-gas shift reaction kinetics. J. Phys. Chem. A 1998, 102, 2673–2678. [Google Scholar] [CrossRef]
- Sato, T.; Kurosawa, S.; Smith, R.L., Jr.; Adschiri, T.; Arai, K. Water gas shift reaction kinetics under noncatalytic conditions in supercritical water. J. Supercrit. Fluids 2004, 29, 113–119. [Google Scholar] [CrossRef]
- Kruse, A.; Dinjus, E. Hydrogen from methane and supercritical water. Angew. Chem. Int. Ed. 2003, 42, 909–911. [Google Scholar] [CrossRef]
- Modell, M. Gasification and liquefaction of forest products in supercritical water. In Fundamentals of Thermochemical Biomass Conversion; Overend, R., Milne, T., Mudge, L., Eds.; Elsevier: Amsterdam, The Netherlands, 1985; pp. 95–119. [Google Scholar]
- D’Jesús, P.; Boukis, N.; Kraushaar-Czarnetzki, B.; Dinjus, E. Gasification of corn and clover grass in supercritical water. Fuel 2006, 85, 1032–1038. [Google Scholar] [CrossRef]
- Stucki, S.; Vogel, F.; Ludwig, C.; Haiduc, A.G.; Brandenberger, M. Catalytic gasification of algae in supercritical water for biofuel production and carbon capture. Energy Environ. Sci. 2009, 2, 535–541. [Google Scholar] [CrossRef]
- Chakinala, A.G.; Brilman, D.W.F.; van Swaaij, W.P.M.; Kersten, S.R.A. Catalytic and non-catalytic supercritical water gasification of microalgae and glycerol. Ind. Eng. Chem. Res. 2010, 49, 1113–1122. [Google Scholar] [CrossRef]
- Guan, Q.; Savage, P.E.; Wei, C. Gasification of alga Nannochloropsis sp. in supercritical water. J. Supercrit. Fluids 2012, 61, 139–145. [Google Scholar] [CrossRef]
- Bagnoud-Velásquez, M.; Brandenberger, M.; Vogel, F.; Ludwig, C. Continuous catalytic hydrothermal gasification of algal biomass and case study on toxicity of aluminum as a step toward effluents recycling. Catal. Today 2014, 223, 35–43. [Google Scholar] [CrossRef]
- Xu, X.; Antal, M.J. Gasification of sewage sludge and other biomass for hydrogen production in supercritical water. Environ. Prog. 1998, 17, 215–220. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, L.; Cao, W.; Jin, H.; Guo, S.; Zhang, X. Hydrogen production by sewage sludge gasification in supercritical water with a fluidized bed reactor. Int. J. Hydrog. Energy 2013, 38, 12991–12999. [Google Scholar] [CrossRef]
- Zhai, Y.; Wang, C.; Chen, H.; Li, C.; Zeng, G.; Pang, D.; Lu, P. Digested sewage sludge gasification in supercritical water. Waste Manag. Res. 2013, 31, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Yong, T.L.-K.; Matsumura, Y. Catalytic gasification of poultry manure and eucalyptus wood mixture in supercritical water. Ind. Eng. Chem. Res. 2012, 51, 5685–5690. [Google Scholar] [CrossRef]
- Sasaki, M.; Furukawa, M.; Minami, K.; Adschiri, T.; Arai, K. Kinetics and mechanism of cellobiose hydrolysis and retro-aldol condensation in subcritical and supercritical water. Ind. Eng. Chem. Res. 2002, 41, 6642–6649. [Google Scholar] [CrossRef]
- Brock, E.E.; Savage, P.E. Detailed chemical kinetics model for supercritical water oxidation of C1 compounds and H2. AIChE J. 1995, 41, 1874–1888. [Google Scholar] [CrossRef]
- Brock, E.E.; Savage, P.E.; Barker, J.R. A reduced mechanism for methanol oxidation in supercritical water. Chem. Eng. Sci. 1998, 53, 857–867. [Google Scholar] [CrossRef]
- Brock, E.E.; Oshima, Y.; Savage, P.E.; Barker, J.R. Kinetics and mechanism of methanol oxidation in supercritical water. J. Phys. Chem. 1996, 100, 15834–15842. [Google Scholar] [CrossRef]
- Helling, R.K.; Tester, J.W. Oxidation kinetics of carbon monoxide in supercritical water. Energy Fuels 1987, 1, 417–423. [Google Scholar] [CrossRef]
- Tester, J.W.; Webley, P.A.; Holgate, H.R. Revised global kinetic measurements of methanol oxidation in supercritical water. Ind. Eng. Chem. Res. 1993, 32, 236–239. [Google Scholar] [CrossRef]
- Ederer, H.J.; Kruse, A.; Mas, C.; Ebert, K.H. Modelling of the pyrolysis of tert-butylbenzene in supercritical water. J. Supercrit. Fluids 1999, 15, 191–204. [Google Scholar] [CrossRef]
- Bühler, W.; Dinjus, E.; Ederer, H.J.; Kruse, A.; Mas, C. Ionic reactions and pyrolysis of glycerol as competing reaction pathways in near- and supercritical water. J. Supercrit. Fluids 2002, 22, 37–53. [Google Scholar] [CrossRef]
- Castello, D.; Fiori, L. Kinetics modeling and main reaction schemes for the supercritical water gasification of methanol. J. Supercrit. Fluids 2012, 69, 64–74. [Google Scholar] [CrossRef]
- Kabyemela, B.M.; Takigawa, M.; Adschiri, T.; Malaluan, R.M.; Arai, K. Mechanism and Kinetics of Cellobiose Decomposition in Sub- and Supercritical Water. Ind. Eng. Chem. Res. 1998, 37, 357–361. [Google Scholar] [CrossRef]
- Kabyemela, B.M.; Adschiri, T.; Malaluan, R.M.; Arai, K. Kinetics of glucose epimerization and decomposition in subcritical and supercritical water. Ind. Eng. Chem. Res. 1997, 36, 1552–1558. [Google Scholar] [CrossRef]
- Yoshida, T.; Matsumura, Y. Reactor development for supercritical water gasification of 4.9 wt% glucose solution at 673 K by using computational fluid dynamics. Ind. Eng. Chem. Res. 2009, 48, 8381–8386. [Google Scholar] [CrossRef]
- Goodwin, A.K.; Rorrer, G.L. Modeling of supercritical water gasification of xylose to hydrogen-rich gas in a hastelloy microchannel reactor. Ind. Eng. Chem. Res. 2011, 50, 7172–7182. [Google Scholar] [CrossRef]
- Wei, L.; Lu, Y.; Wei, J. Hydrogen production by supercritical water gasification of biomass: Particle and residence time distribution in fluidized bed reactor. Int. J. Hydrog. Energy 2013, 38, 13117–13124. [Google Scholar] [CrossRef]
- Withag, J.A.M.; Sallevelt, J.L.H.P.; Brilman, D.W.F.; Bramer, E.A.; Brem, G. Heat transfer characteristics of supercritical water in a tube: Application for 2D and an experimental validation. J. Supercrit. Fluids 2012, 70, 156–170. [Google Scholar] [CrossRef]
- Sallevelt, J.L.H.P.; Withag, J.A.M.; Bramer, E.A.; Brilman, D.W.F.; Brem, G. One-dimensional model for heat transfer to a supercritical water flow in a tube. J. Supercrit. Fluids 2012, 68, 1–12. [Google Scholar] [CrossRef]
- Withag, J.A.M. On the Gasification of Wet Biomass in Supercritical Water. Ph.D. Thesis, University of Twente, Enschede, The Netherlands, 2013. [Google Scholar]
- Tang, H.; Kitagawa, K. Supercritical water gasification of biomass: Thermodynamic analysis with direct Gibbs free energy minimization. Chem. Eng. J. 2005, 106, 261–267. [Google Scholar] [CrossRef]
- Freitas, A.C.; Guirardello, R. Thermodynamic analysis of supercritical water gasification of microalgae biomass for hydrogen and syngas production. Chem. Eng. Trans. 2013, 32, 553–558. [Google Scholar]
- Freitas, A.C.D.; Guirardello, R. Supercritical water gasification of glucose and cellulose for hydrogen and syngas production. Chem. Eng. Trans. 2012, 27, 361–366. [Google Scholar]
- Freitas, A.C.D.; Guirardello, R. Comparison of several glycerol reforming methods for hydrogen and syngas production using Gibbs energy minimization. Int. J. Hydrog. Energy 2014, 39, 17969–17984. [Google Scholar] [CrossRef]
- Lu, Y.; Guo, L.; Zhang, X.; Yan, Q. Thermodynamic modeling and analysis of biomass gasification for hydrogen production in supercritical water. Chem. Eng. J. 2007, 131, 233–244. [Google Scholar] [CrossRef]
- Voll, F.A.P.; Rossi, C.C.R.S.; Silva, C.; Guirardello, R.; Souza, R.O.M.A.; Cabral, V.F.; Cardozo-Filho, L. Thermodynamic analysis of supercritical water gasification of methanol, ethanol, glycerol, glucose and cellulose. Int. J. Hydrog. Energy 2009, 34, 9737–9744. [Google Scholar] [CrossRef]
- Yan, Q.; Guo, L.; Lu, Y. Thermodynamic analysis of hydrogen production from biomass gasification in supercritical water. Energy Convers. Manag. 2006, 47, 1515–1528. [Google Scholar] [CrossRef]
- Castello, D.; Fiori, L. Supercritical water gasification of biomass: Thermodynamic constraints. Bioresour. Technol. 2011, 102, 7574–7582. [Google Scholar] [CrossRef] [PubMed]
- Letellier, S.; Marias, F.; Cezac, P.; Serin, J.P. Gasification of aqueous biomass in supercritical water: A thermodynamic equilibrium analysis. J. Supercrit. Fluids 2010, 51, 353–361. [Google Scholar] [CrossRef]
- Marias, F.; Letellier, S.; Cezac, P.; Serin, J.P. Energetic analysis of gasification of aqueous biomass in supercritical water. Biomass Bioenergy 2011, 35, 59–73. [Google Scholar] [CrossRef]
- Yakaboylu, O.; Harinck, J.; Smit, K.G.; de Jong, W. Supercritical water gasification of biomass: A thermodynamic model for the prediction of product compounds at equilibrium state. Energy Fuels 2014, 28, 2506–2522. [Google Scholar] [CrossRef]
- Kozeschnik, E. A numerical model for evaluation of unconstrained and compositionally constrained thermodynamic equilibria. Calphad 2000, 24, 245–252. [Google Scholar] [CrossRef]
- Feng, W.; van der Kooi, H.J.; de Swaan Arons, J. Biomass conversions in subcritical and supercritical water: Driving force, phase equilibria, and thermodynamic analysis. Chem. Eng. Process. Process Intensif. 2004, 43, 1459–1467. [Google Scholar] [CrossRef]
- Luterbacher, J.S.; Fröling, M.; Vogel, F.; Maréchal, F.; Tester, J.W. Hydrothermal gasification of waste biomass: Process design and life cycle asessment. Environ. Sci. Technol. 2009, 43, 1578–1583. [Google Scholar] [CrossRef] [PubMed]
- Gassner, M.; Vogel, F.; Heyen, G.; Maréchal, F. Optimal process design for the polygeneration of SNG, power and heat by hydrothermal gasification of waste biomass: Thermo-economic process modelling and integration. Energy Environ. Sci. 2011, 4, 1726–1741. [Google Scholar] [CrossRef]
- Gutiérrez Ortiz, F.J.; Ollero, P.; Serrera, A.; Sanz, A. Thermodynamic study of the supercritical water reforming of glycerol. Int. J. Hydrog. Energy 2011, 36, 8994–9013. [Google Scholar] [CrossRef]
- Fiori, L.; Valbusa, M.; Castello, D. Supercritical water gasification of biomass for H2 production: Process design. Bioresour. Technol. 2012, 121, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Withag, J.A.M.; Smeets, J.R.; Bramer, E.A.; Brem, G. System model for gasification of biomass model compounds in supercritical water—A thermodynamic analysis. J. Supercrit. Fluids 2012, 61, 157–166. [Google Scholar] [CrossRef]
- Kruse, A. Hydrothermal biomass gasification. J. Supercrit. Fluids 2009, 47, 391–399. [Google Scholar] [CrossRef]
- Tolman, R. Process for Converting Sewage Sludge and Municipal Solid Wastes to Clean Fuels; Report Number: P600-01-12; California Energy Commission: Sacramento, CA, USA, 2001. [Google Scholar]
- Marrone, P.A.; Hong, G.T. Corrosion control methods in supercritical water oxidation and gasification processes. J. Supercrit. Fluids 2009, 51, 83–103. [Google Scholar] [CrossRef]
- Elliott, D.C. Catalytic hydrothermal gasification of biomass. Biofuels Bioprod. Biorefin. 2008, 2, 254–265. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, S.Z.; Xu, D.H.; Gong, Y.M.; Ma, H.H.; Tang, X.Y. Review of catalytic supercritical water gasification for hydrogen production from biomass. Renew. Sustain. Energy Rev. 2010, 14, 334–343. [Google Scholar] [CrossRef]
- Zöhrer, H.; de Boni, E.; Vogel, F. Hydrothermal processing of fermentation residues in a continuous multistage rig—Operational challenges for liquefaction, salt separation, and catalytic gasification. Biomass Bioenergy 2014, 65, 51–63. [Google Scholar] [CrossRef]
- Zöhrer, H.; Vogel, F. Hydrothermal catalytic gasification of fermentation residues from a biogas plant. Biomass Bioenergy 2013, 53, 138–148. [Google Scholar] [CrossRef]
- Karayıldırım, T.; Sınağ, A.; Kruse, A. Char and Coke Formation as Unwanted Side Reaction of the Hydrothermal Biomass Gasification. Chem. Eng. Technol. 2008, 31, 1561–1568. [Google Scholar] [CrossRef]
- Zöhrer, H.; Mayr, F.; Vogel, F. Stability and performance of ruthenium catalysts based on refractory oxide supports in supercritical water conditions. Energy Fuels 2013, 27, 4739–4747. [Google Scholar] [CrossRef]
- Matsumura, Y.; Harada, M.; Nagata, K.; Kikuchi, Y. Effect of heating rate of biomass feedstock on carbon gasification efficiency in supercritical water gasification. Chem. Eng. Commun. 2006, 193, 649–659. [Google Scholar] [CrossRef]
- Sınaǧ, A.; Kruse, A.; Rathert, J. Influence of the heating rate and the type of catalyst on the formation of key intermediates and on the generation of gases during hydropyrolysis of glucose in supercritical water in a batch reactor. Ind. Eng. Chem. Res. 2004, 43, 502–508. [Google Scholar] [CrossRef]
- Boukis, N.; Galla, U.; Müller, H.; Dinjus, E. Biomass gasification in supercritical water. Experimental progress achieved with the VERENA pilot plant. In Proceedings of the 15th European Conference & Exhibition, 7–11 May 2007; Volume 7, pp. 1013–1016.
- Boukis, N.; Galla, U.; D’Jesus, P.; Müller, H.; Dinjus, E. Gasification of wet biomass in supercritical water. Results of pilot plant experiments. In Proceedings of the 14th European Biomass Conference, Paris, France, 17–21 October 2005; pp. 964–967.
- Chen, J.; Lu, Y.; Guo, L.; Zhang, X.; Xiao, P. Hydrogen production by biomass gasification in supercritical water using concentrated solar energy: System development and proof of concept. Int. J. Hydrog. Energy 2010, 35, 7134–7141. [Google Scholar] [CrossRef]
- Liao, B.; Guo, L.; Lu, Y.; Zhang, X. Solar receiver/reactor for hydrogen production with biomass gasification in supercritical water. Int. J. Hydrog. Energy 2013, 38, 13038–13044. [Google Scholar] [CrossRef]
- Lu, Y.; Zhao, L.; Guo, L. Technical and economic evaluation of solar hydrogen production by supercritical water gasification of biomass in China. Int. J. Hydrog. Energy 2011, 36, 14349–14359. [Google Scholar] [CrossRef]
- Xiao, P.; Guo, L.; Zhang, X.; Zhu, C.; Ma, S. Continuous hydrogen production by biomass gasification in supercritical water heated by molten salt flow: System development and reactor assessment. Int. J. Hydrog. Energy 2013, 38, 12927–12937. [Google Scholar] [CrossRef]
- Harinck, J.; Smit, K.G. A Process and a Reaction Apparatus for the Gasification of Wet Biomass. WO Application WO/2013/030026, 7 March 2013. [Google Scholar]
- Hong, G.T.; Killilea, W.R.; Thomason, T.B. Method for Solids Separation in a Wet Oxidation Type Process. U.S. Patent 4,822,497, 18 April 1989. [Google Scholar]
- Daman, E.L. Process and Apparatus for Supercritical Water Oxidation. U.S. Patent 5,723,045, 3 March 1998. [Google Scholar]
- Daman, E.L. Process and Apparatus for Supercritical Water Oxidation. U.S. Patent 5,571,423, 5 November 1996. [Google Scholar]
- Marrone, P.A.; Hodes, M.; Smith, K.A.; Tester, J.W. Salt precipitation and scale control in supercritical water oxidation—Part B: Commercial/full-scale applications. J. Supercrit. Fluids 2004, 29, 289–312. [Google Scholar] [CrossRef]
- Xu, D.H.; Wang, S.Z.; Gong, Y.M.; Guo, Y.; Tang, X.Y.; Ma, H.H. A novel concept reactor design for preventing salt deposition in supercritical water. Chem. Eng. Res. Des. 2010, 88, 1515–1522. [Google Scholar] [CrossRef]
- Schubert, M.; Regler, J.W.; Vogel, F. Continuous salt precipitation and separation from supercritical water. Part 1: Type 1 salts. J. Supercrit. Fluids 2010, 52, 99–112. [Google Scholar] [CrossRef]
- Karlsruhe, F. Device for treating Flowable Materials in Super-Critical Water, e.g., for Treating Effluent, Comprises Cylindrical Reactor with Pressure Lines for Introducing Educt and Removing Product. German Patent DE20220307, 30 April 2003. [Google Scholar]
- Potic, B.; Kersten, S.R.A.; Ye, M.; van der Hoef, M.A.; Kuipers, J.A.M.; van Swaaij, W.P.M. Fluidization with hot compressed water in micro-reactors. Chem. Eng. Sci. 2005, 60, 5982–5990. [Google Scholar] [CrossRef]
- Lu, Y.; Jin, H.; Guo, L.; Zhang, X.; Cao, C.; Guo, X. Hydrogen production by biomass gasification in supercritical water with a fluidized bed reactor. Int. J. Hydrog. Energy 2008, 33, 6066–6075. [Google Scholar] [CrossRef]
- Cao, C.; Guo, L.; Jin, H.; Guo, S.; Lu, Y.; Zhang, X. The influence of alkali precipitation on supercritical water gasification of glucose and the alkali recovery in fluidized-bed reactor. Int. J. Hydrog. Energy 2013, 38, 13293–13299. [Google Scholar] [CrossRef]
- Harinck, J.; Smit, K.G. A Reaction Apparatus and a Process for the Gasification of Wet Biomass. WO Application WO/2013/030027, 7 March 2013. [Google Scholar]
- Harinck, J.; Smit, K.G. A Process for the Gasification of Wet Biomass. WO Application WO/2013/030028, 7 March 2013. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yakaboylu, O.; Harinck, J.; Smit, K.G.; De Jong, W. Supercritical Water Gasification of Biomass: A Literature and Technology Overview. Energies 2015, 8, 859-894. https://doi.org/10.3390/en8020859
Yakaboylu O, Harinck J, Smit KG, De Jong W. Supercritical Water Gasification of Biomass: A Literature and Technology Overview. Energies. 2015; 8(2):859-894. https://doi.org/10.3390/en8020859
Chicago/Turabian StyleYakaboylu, Onursal, John Harinck, K. G. Smit, and Wiebren De Jong. 2015. "Supercritical Water Gasification of Biomass: A Literature and Technology Overview" Energies 8, no. 2: 859-894. https://doi.org/10.3390/en8020859