DNA Damage and Repair in Human Cancer: Molecular Mechanisms and Contribution to Therapy-Related Leukemias

Abstract
:1. Introduction
2. Results and Discussion
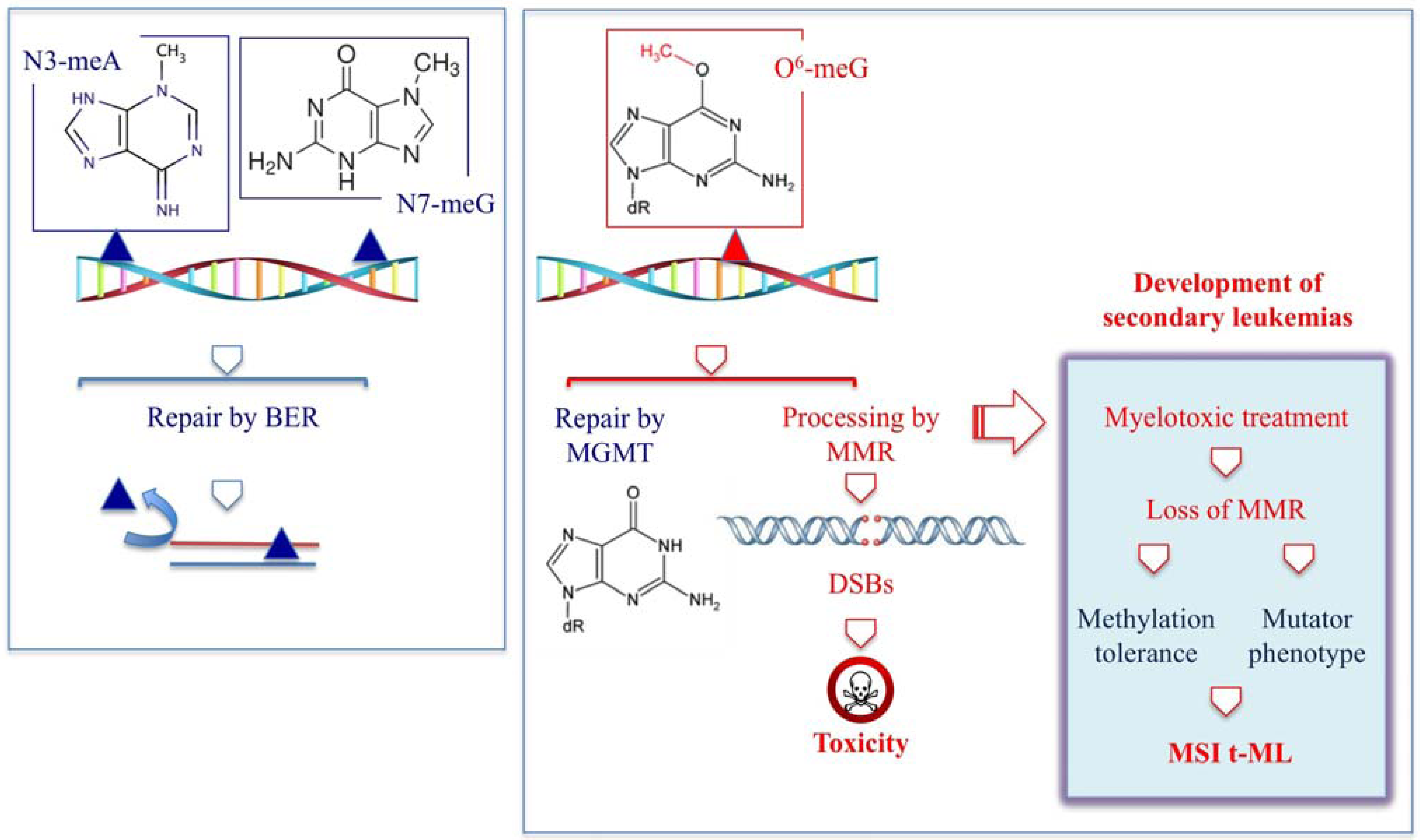
2.1. t-MN Following Therapy with Alkylating Agents

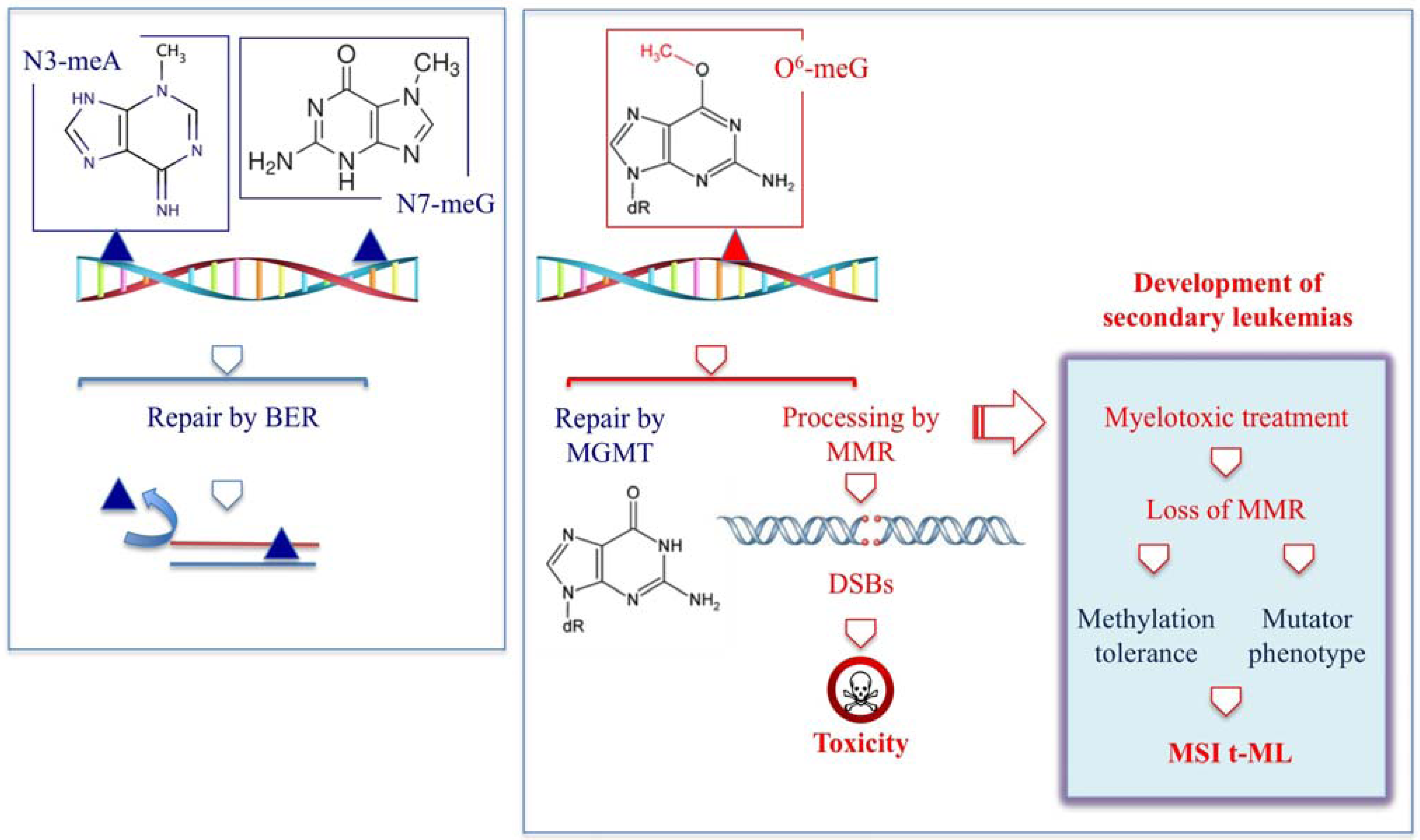{kind=link}
| Monofunctional agents | DNA adduct | DNA repair | ||
|---|---|---|---|---|
| Dacarbazine | Procarbazine | Temozolomide | ||
 |  |  | O6–meG → | MGMT, MMR |
| DSBs → | FANC pathway (HR) | |||
| 3–meA; 7-meG → | BER | |||
| Drug | DNA adduct | DNA repair | References |
|---|---|---|---|
| BCNU, CCNU, ACNU | |||
 |
|
| [19,20,21] |
| Cyclophosphamide | |||
 |
|
| [22,23,24,25,26,27,28] |
| Chlorambucil | |||
 |
|
| [29,30,31,32] |
| Melphalan | |||
 |
|
| [30,31,32,33] |
| Busulfan | |||
 |
|
| [20,21,34,35,36] |
| Platinum compounds | |||
 |
|
| [37,38,39,40,41] |
2.2. t-MN Following Therapy with Topoisomerase Inhibitors
| Drugs | Chemical Structure | DNA adduct | DHA repair | References |
|---|---|---|---|---|
| Topoisomerase II | ||||
| Etoposide |  | DSBs | NHEJ, HR | [50,51] |
| Doxorubicin |  |
| HR, NER, NHEJ, BER ? | [52,53,54] |
| Epirubicin |  |
| NHEJ, HR, NER, BER ? | |
| Mitoxantrone |  |
| NHEJ, HR | [55] |
| Topoisomerase I | ||||
| Camptothecin |  | SSBs, DSBs | Tdp1, HR, NHEJ | [56] |
| No patients | Primary cancer /disease | Drugs | References |
|---|---|---|---|
| 2 t-APL | seminoma, breast cancer | Etoposide, cisplatin, bleomycin; 4-epi-doxorubicin, cyclophosphamide, methotrexate, 5-fluorouracil, RT | [73] |
| 106 t-APL | 60 breast carcinoma; 15 non-Hodgkin’s lymphoma; 4 other hematologic; malignancies; 25 various solid tumors; 1 multiple sclerosis; 1chronic poly-radiculoneuritis | RT, RT+CT; Alkylating agents (cyclophosphamide, ifosfamide, chlorambucil, dacarbazine, melphalan, CCNU); topoII inhibitors (Doxorubicin, epirubicin, daunorubicin, mitoxantrone, VP16, VM26); antimetabolites (5-FU, methotrexate, cytarabine), vincristine, bleomycin, cisplatin, | [67] |
| 17 t-APL | Langerhans cell histiocytosis | Etoposide | [74] |
| 6 t-APL vs. 35 de novo APL | Breast cancer, multiple sclerosis | Mitoxantrone | [69] |
| 11 t-MN vs. 10 relapse | 108 APL | ATRA+ consolidation therapy (VP16, mitoxantrone, etoposide, daunorubicin, idarubicin, methotrexate, prednisolone) | [75] |
| 17 t-MN | 918 APL | ATRA+ consolidation therapy; (idarubicin, mitoxantrone) | [76] |
2.3. Antimetabolites
| Chemical structure | DHA adduct | DNA repair | |
|---|---|---|---|
| 5-Fluorouracil |  |
|
|
| Fludarabine |  |
|
|
| Azathioprine |  |
|
|
2.4. Occupational Exposure to Chemotherapeutic Drugs
3. Conclusions
Acknowledgments
References
- Godley, L.A.; Larson, R.A. Therapy-related myeloid leukemia. Semin. Oncol. 2008, 35, 418–429. [Google Scholar] [CrossRef]
- Pedersen-Bjergaard, J.; Christiansen, D.H.; Desta, F.; Andersen, M.K. Alternative genetic pathways and cooperating genetic abnormalities in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia 2006, 20, 1943–1949. [Google Scholar] [CrossRef]
- Leone, G.; Fianchi, L.; Pagano, L.; Voso, M.T. Incidence and susceptibility to therapy-related myeloid neoplasms. Chem. Biol. Interact. 2010, 184, 39–45. [Google Scholar] [CrossRef]
- Kayser, S.; Dohner, K.; Krauter, J.; Kohne, C-H.; Horst, H.A.; Held, G.; Lilienfeld-Toal, M.; Wilhelm, S.; Kündgen, A.; Götze, K.; et al. The impact of therapy-related acute myeloid leukemia (AML) on outcome in 2,853 adult patients with newly diagnosed AML. Blood 2011, 117, 2137–2145. [Google Scholar]
- Sill, H.; Olipitz, W.; Zebisch, A.; Schulz, E.; Wölfler, A. Therapy-related myeloid neoplasms: Pathobiology and clinical characteristics. Br. J. Pharmacol. 2011, 162, 792–805. [Google Scholar] [CrossRef]
- WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. (Eds.) IARC Press: Lyon, France, 2008.
- Pedersen-Bjergaard, J.; Rowley, J. The balanced and the unbalanced chromosome aberrations of acute myeloid leukemia may develop in different ways and may contribute differently to malignant transformation. Blood 1994, 83, 2780–2786. [Google Scholar]
- Pedersen-Bjergaard, J.; Andersen, M.K.; Andersen, M.T.; Christiansen, D.H. Genetics of therapy-related myelodysplasia and acute myeloid leukemia. Leukemia 2008, 22, 240–248. [Google Scholar] [CrossRef]
- Karran, P.; Bignami, M. DNA damage tolerance, mismatch repair and genome instability. Bioessays 1994, 16, 833–839. [Google Scholar] [CrossRef]
- Karran, P.; Offman, J.; Bignami, M. Human mismatch repair, drug-induced DNA damage, and secondary cancer. Biochimie 2003, 85, 1149–1160. [Google Scholar] [CrossRef]
- Bignami, M.; Casorelli, I.; Karran, P. Mismatch repair and response to DNA-damaging antitumour therapies. Eur. J. Cancer 2003, 39, 2142–2149. [Google Scholar] [CrossRef]
- Ben-Yehuda, D.; Krichevsky, S.; Caspi, O.; Rund, D.; Polliack, A.; Abeliovich, D.; Zelig, O.; Yahalom, V.; Paltiel, O.; Or, R.; et al. Microsatellite instability and p53 mutations in therapy-related leukemia suggest mutator phenotype. Blood 1996, 88, 4296–4303. [Google Scholar]
- Das-Gupta, E.P.; Seedhouse, C.H.; Russell, N.H. Microsatellite instability occurs in defined subsets of patients with acute myeloblastic leukaemia. Br. J. Haematol. 2001, 114, 307–312. [Google Scholar] [CrossRef]
- Casorelli, I.; Offman, J.; Mele, L.; Pagano, L.; Sica, S.; D’Errico, M.; Giannini, G.; Leone, G.; Bignami, M.; Karran, P. Drug treatment in the development of mismatch repair defective acute leukemia and myelodysplastic syndrome. DNA Repair 2003, 2, 547–559. [Google Scholar] [CrossRef]
- Offman, J.; Gascoigne, K.; Bristow, F.; Macpherson, P.; Bignami, M.; Casorelli, I.; Leone, G.; Sica, S.; Halil, O.; Cummins, D.; et al. Repeated sequences in CASPASE-5 and FANCD2 but not NF1 are targets for mutation in microsatellite-unstable acute leukemia/myelodysplastic syndrome. Mol. Cancer Res. 2005, 3, 251–260. [Google Scholar] [CrossRef]
- Wimmer, K.; Kratz, C.P. Constitutional mismatch repair-deficiency syndrome. Haematologica 2010, 95, 699–701. [Google Scholar] [CrossRef]
- Sheikhha, M.; Tobal, K.; Liu, Y.J. High level of microsatellite instability but not hypermethylation of mismatch repair genes in therapy-related and secondary acute myeloid leukaemia and myelodysplastic syndrome. Br. J. Haematol. 2002, 117, 359–365. [Google Scholar] [CrossRef]
- Worrillow, L.J.; Smith, G.; Scott, K.; Andersson, M.; Ashcroft, J.; Dores, G.M.; Glimelius, B.; Holowaty, E.; Jackson, G.H.; Jones, G.L.; et al. Polymorphic MLH1 and risk of cancer after methylating chemotherapy for Hodgkin lymphoma. J. Med. Genet. 2008, 45, 142–146. [Google Scholar]
- Drabløs, F.; Feyzi, E.; Aas, P.A.; Vaagbø, C.B.; Kavli, B.; Bratlie, M.S.; Peña-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA repair mechanisms and medical significance. DNA Repair 2004, 3, 1389–1407. [Google Scholar] [CrossRef]
- Friedman, H.S.; Johnson, S.P.; Dong, Q.; Schold, S.C.; Rasheed, B.K.; Bigner, S.H.; Ali-Osman, F.; Dolan, E.; Colvin, O.M.; Houghton, P.; et al. Methylator resistance mediated by mismatch repair deficiency in a glioblastoma multiforme xenograft. Cancer Res. 1997, 57, 2933–2936. [Google Scholar]
- McNeill, D.R.; Lam, W.; DeWeese, T.L.; Cheng, Y.-C.; Wilson, D.M. Impairment of APE1 function enhances cellular sensitivity to clinically relevant alkylators and antimetabolites. Mol. Cancer Res. 2009, 7, 897–906. [Google Scholar] [CrossRef]
- Cai, Y.; Wu, M.H.; Ludeman, S.M.; Grdina, D.J.; Dolan, M.E. Role of O6-alkylguanine-DNA alkyltransferase in protecting against cyclophosphamide-induced toxicity and mutagenicity. Cancer Res. 1999, 59, 3059–3063. [Google Scholar]
- Hemminki, K. DNA-binding products of nornitrogen mustard, a metabolite of cyclophosphamide. Chem. Biol. Interact. 1987, 61, 75–88. [Google Scholar] [CrossRef]
- D’Incalci, M.; Bonfanti, M.; Pifferi, A.; Mascellani, E.; Tagliabue, G.; Berger, D.; Fiebig, H.H. The antitumour activity of alkylating agents is not correlated with the levels of glutathione, glutathione transferase and O6-Alkylguanine-DNA-alkyltransferase of human tumour xenografts. Eur. J. Cancer 1998, 34, 1749–1755. [Google Scholar]
- Shiraishi, A.; Sakumi, K.; Sekiguchi, M. Increased susceptibility to chemotherapeutic alkylating agents of mice deficient in DNA repair methyltransferase. Carcinogenesis 2000, 21, 1879–1883. [Google Scholar] [CrossRef]
- Hansen, R.J.; Nagasubramanian, R.; Delaney, S.M.; Samson, L.D.; Dolan, M.E. Role of O6-methylguanine-DNA methyltransferase in protecting from alkylating agent-induced toxicity and mutations in mice. Carcinogenesis 2007, 28, 1111–1116. [Google Scholar]
- Nagasubramanian, R.; Hansen, R.J.; Delaney, S.M.; Cherian, M.M.; Samson, L.D.; Kogan, S.C.; Dolan, M.E. Survival and tumorigenesis in O6-methylguanine DNA methyltransferase-deficient mice following cyclophosphamide exposure. Mutagenesis 2008, 23, 341–346. [Google Scholar] [CrossRef]
- Loeber, R.; Michaelson, E.; Fang, Q.; Campbell, C.; Pegg, A.E.; Tretyakova, N. Cross-linking of the DNA repair protein O6-alkylguanine DNA alkyltransferase to DNA in the presence of antitumor nitrogen mustards. Chem. Res. Toxicol. 2008, 21, 787–795. [Google Scholar] [CrossRef]
- Rajski, S.R.; Williams, R.M. DNA Cross-linking agents as antitumor drugs. Chem. Rev. 1998, 98, 2723–2796. [Google Scholar]
- Osborne, M.; Wilman, D.; Lawley, P. Alkylation of DNA by the nitrogen mustard bis(2-chloro ethyl)methylamine. Chem. Res. Toxicol. 1995, 8, 316–320. [Google Scholar] [CrossRef]
- Florea-Wang, D.; Pawlowicz, A.J.; Sinkkonen, J.; Kronberg, L.; Vilpo, J.; Hovinen, J. Reactions of 4-(Bis(2-chloroethyl) amino) benzenebutanoic acid (chlorambucil) with DNA. Chem. Biodiver. 2009, 6, 1002–1013. [Google Scholar] [CrossRef]
- Balcome, S.; Park, S.; Quirk Dorr, D.R.; Hafner, L.; Phillips, L.; Tretyakova, N. Adenine-containing DNA-DNA cross-links of antitumor nitrogen mustards. Chem. Res. Toxicol. 2004, 17, 950–962. [Google Scholar] [CrossRef]
- Grant, D.F.; Bessho, T.; Reardon, J.T. Nucleotide excision repair of melphalan monoadducts. Cancer Res. 1998, 58, 5196–5200. [Google Scholar]
- Ponti, M.; Souhami, R.L.; Fox, B.W.; Hartley, J. DNA interstrand crosslinking and sequence selectivity of dimethanesulphonates. Br. J. Cancer 1991, 63, 743–747. [Google Scholar] [CrossRef]
- Iwamoto, T.; Hiraku, Y.; Oikawa, S.; Mizutani, H.; Kojima, M.; Kawanishi, S. DNA intrastrand cross-link at the 5'-GA-3' sequence formed by busulfan and its role in the cytotoxic effect. Cancer Sci. 2004, 95, 454–458. [Google Scholar] [CrossRef]
- McManus, F.; Fang, Q.; Booth, J.; Noronha, A.; Pegg, A.; Wilds, C. Synthesis and characterization of an O6-2'-deoxyguanosine-alkyl-O6-2'-deoxyguanosine interstrand cross-link in a 5'-GNC motif and repair by human O6-alkylguanine-DNA alkyltransferase. Org. Biomol. Chem. 2010, 8, 4414–4426. [Google Scholar] [CrossRef]
- Jung, Y.; Lippard, S.J. Direct cellular responses to platinum-induced DNA damage. Chem. Rev. 2007, 107, 1387–1407. [Google Scholar]
- Branch, P.; Masson, M.; Aquilina, G.; Bignami, M.; Karran, P. Spontaneous development of drug resistance: Mismatch repair and p53 defects in resistance to cisplatin in human tumor cells. Oncogene. 2000, 19, 3138–3145. [Google Scholar] [CrossRef]
- Fink, D.; Nebel, S.; Aebi, S.; Zheng, H.; Cenni, B.; Nehmé, A.; Christen, R.D.; Howell, S.B. The role of DNA mismatch repair in platinum drug resistance. Cancer Res. 1996, 56, 4881–4886. [Google Scholar]
- Köberle, B.; Masters, J.R.; Hartley, J.A.; Wood, R.D. Defective repair of cisplatin-induced DNA damage caused by reduced XPA protein in testicular germ cell tumours. Curr. Biol. 1999, 9, 273–276. [Google Scholar] [CrossRef]
- Ferry, K.V.; Hamilton, T.C.; Johnson, S.W. Increased nucleotide excision repair in cisplatin-resistant ovarian cancer cells: Role of ercc1-xpf. Biochem. Pharmacol. 2000, 60, 1305–1313. [Google Scholar]
- Deans, A.J.; West, S.C. DNA interstrand crosslink repair and cancer. Nat. Rev. Cancer 2011, 11, 467–480. [Google Scholar] [CrossRef]
- Povirk, L.; Shuker, D. DNA damage and mutagenesis induced by nitrogen mustards. Mutat. Res. 1994, 318, 205–226. [Google Scholar] [CrossRef]
- Scardocci, A.; Guidi, F.; D’Alo, F.; Gumiero, D.; Fabiani, E.; DiRuscio, A.; Martini, M.; Larocca, L.M.; Zollino, M.; Hohaus, S.; et al. Reduced BRCA1 expression due to promoter hypermethylation in therapy-related acute myeloid leukaemia. Br. J. Cancer 2006, 95, 1108–1113. [Google Scholar] [CrossRef]
- Casorelli, I.; Tenedini, E.; Tagliafico, E.; Blasi, M.F.; Giuliani, A.; Crescenzi, M.; Pelosi, E.; Testa, U.; Peschle, C.; Mele, L.; et al. Identification of a molecular signature for leukemic promyelocytes and their normal counterparts: Focus on DNA repair genes. Leukemia 2006, 20, 1978–1988. [Google Scholar]
- Li, L.; Li, M.; Sun, C.; Francisco, L.; Chakraborty, S.; Sabado, M.; McDonald, T.; Gyorffy, J.; Chang, K.; Wang, S.; et al. Altered hematopoietic cell gene expression precedes development of therapy-related myelodysplasia/acute myeloid leukemia and identifies patients at risk. Cancer Cell 2011, 20, 591–605. [Google Scholar]
- Efthimiou, M.; Stephanou, G.; Demopoulos, N.; Nikolaropoulos, S.S. Aneugenic potential of the anticancer drugs melphalan and chlorambucil. The involvement of apoptosis and chromosome segregation regulating proteins. J. Appl. Toxicol. 2011. [Google Scholar] [CrossRef]
- Cobo, M.; Isla, D.; Massuti, B.; Montes, A.; Sanchez, J.M.; Provencio, M.; Viñolas, N.; Paz-Ares, L.; Lopez-Vivanco, G.; Muñoz, M.A.; et al. Customizing cisplatin based on quantitative excision repair cross-complementing 1 mRNA expression: A phase III trial in non-small-cell lung cancer. J. Clin. Oncol. 2007, 25, 2747–2754. [Google Scholar]
- Travis, L.B.; Andersson, M.; Gospodarowicz, M.; van Leewen, F.E.; Bergfeld, K.; Lynch, C.F.; Curtis, R.E.; Kohler, B.A.; Wiklund, T.; Storm, H.; et al. Treatment-associated leukemia following testicular cancer. J. Natl. Cancer Inst. 2000, 92, 1165–1171. [Google Scholar]
- McClendon, K.; Osheroff, N. DNA topoisomerase II, genotoxicity, and cancer. Mutat. Res. 2007, 623, 83–97. [Google Scholar] [CrossRef]
- Fortune, J.; Osheroff, N. Topoisomerase II as a target for anticancer drugs: When enzymes stop being nice. Prog. Nucleic Acid Res. Mol. Biol. 2000, 64, 221–253. [Google Scholar] [CrossRef]
- Spencer, D.M.S.; Bilardi, R.; Koch, T.H.; Post, G.C.; Nafie, J.W.; Kimura, K.-I.; Cutts, S.M.; Phillips, D.R. DNA repair in response to anthracycline-DNA adducts: A role for both homologous recombination and nucleotide excision repair. Mutat. Res. 2011, 638, 110–121. [Google Scholar]
- Zeman, S.; Phillips, D.; Rothers, D. Characterization of covalent adriamycin-DNA adducts. Proc. Natl. Acad. Sci. USA 1998, 95, 11561–11565. [Google Scholar] [CrossRef]
- Bilardi, R.; Kimura, K.-I.; Phillips, D.R.; Cutts, S.M. Processing of anthracycline-DNA adducts via DNA replication and interstrand crosslink repair pathways. Biochem. Pharmacol. 2012, 83, 1241–1250. [Google Scholar] [CrossRef]
- Mansour, O.C.; Evison, B.J.; Sleebs, B.E.; Watson, K.G.; Nudelman, A.; Rephaeli, A.; Buck, D.P.; Collins, J.G.; Bilardi, R.A.; Phillips, D.R.; et al. New anthracenedione derivatives with improved biological activity by virtue of stable drug-DNA adduct formation. J. Med. Chem. 2010, 53, 6851–6866. [Google Scholar]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA Topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef]
- Parker, B.S.; Cutts, S.M.; Cullinane, C.; Phillips, D.R. Formaldehyde activation of mitoxantrone yields CpG and CpA specific DNA adducts. Nucleic Acids Res. 2000, 28, 982–990. [Google Scholar] [CrossRef]
- Taatjes, D.J.; Gaudiano, G.; Resing, K.; Koch, T.H. Redox pathway leading to the alkylation of DNA by the anthracycline, antitumor drugs adriamycin and daunomycin. J. Med. Chem. 1997, 40, 1276–1286. [Google Scholar] [CrossRef]
- Cutts, S.M.; Nudelman, A.; Rephaeli, A.; Phillips, D.R. The power and potential of doxorubicin-DNA adducts. IUBMB Life 2005, 57, 73–81. [Google Scholar] [CrossRef]
- Parker, B.S.; Buley, T.; Evison, B.J.; Cutts, S.M.; Neumann, G.M.; Iskander, M.N.; Phillips, D.R. A molecular understanding of mitoxantrone-DNA adduct formation: Effect of cytosine methylation and flanking sequences. J. Biol. Chem. 2004, 279, 18814–18823. [Google Scholar]
- Doroshow, J.H. Role of hydrogen peroxide and hydroxyl radical formation in the killing of Ehrlich tumor cells by anticancer quinones. Proc Nat. Acad. Sci. USA 1986, 83, 4514–4518. [Google Scholar] [CrossRef]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef]
- Novak, R.F.; Kharasch, E.D. Mitoxantrone: Propensity for free radical formation and lipid peroxidation—Implications for cardiotoxicity. Invest. New Drugs 1985, 3, 95–99. [Google Scholar]
- Stoddart, A.; McNerney, M.E.; Bartom, E.; Bergerson, R.; Young, D.J.; Qian, Z.; Wang, J.; Fernald, A.A.; Davis, E.M.; Larson, R.A.; et al. Genetic pathways leading to therapy-related myeloid neoplasms. Mediterr. J. Hematol. Infect. Dis. 2011, 3. [Google Scholar] [CrossRef]
- Andersen, M.; Johansson, B.; Larsen, S.; Pederson-Bjergaard, J. Chromosomal abnormalities in secondary MDS and AML. Relationship to drugs and radiation with specific emphasis on the balanced rearrangements. Haematologica 1998, 83, 483–488. [Google Scholar]
- Pulsoni, A.; Pagano, L.; lo Coco, F.; Avvisati, G.; Mele, L.; di Bona, E.; Invernizzi, R.; Leoni, F.; Marmont, F.; Mele, A.; et al. Clinicobiological features and outcome of acute promyelocytic leukemia occurring as a second tumor: The GIMEMA experience. Blood 2002, 100, 1972–1976. [Google Scholar]
- Beaumont, M.; Sanz, M.; Carli, P.M.; Maloisel, F.; Thomas, X.; Detourmignies, L.; Guerci, A.; Gratecos, N.; Rayon, C.; San Miguel, J.; et al. Therapy-related acute promyelocytic leukemia. J. Clin. Oncol. 2003, 21, 2123–2137. [Google Scholar]
- Hasan, S.K.; Mays, A.N.; Ottone, T.; Ledda, A.; La Nasa, G.; Cattaneo, C.; Borlenghi, E.; Melillo, L.; Montefusco, E.; Cervera, J.; et al. Molecular analysis of t(15;17) genomic breakpoints in secondary acute promyelocytic leukemia arising after treatment of multiple sclerosis. Blood 2008, 112, 3383–3390. [Google Scholar] [CrossRef]
- Mistry, A.R.; Felix, C.; Whitmarsh, R.J.; Mason, A.; Reiter, A.; Cassinat, B.; Parry, A.; Walz, C.; Wiemels, J.L.; Segal, M.R.; et al. DNA topoisomerase II in therapy-related acute promyelocytic leukemia. N. Engl. J. Med. 2005, 352, 1529–1538. [Google Scholar]
- Hasan, S.K.; Ottone, T.; Schlenk, R.F.; Xiao, Y.; Wiemels, J.L.; Mitra, M.E.; Bernasconi, P.; Di Raimondo, F.; Stanghellini, M.T.; Marco, P.; et al. Analysis of t (15;17) chromosomal breakpoint sequences in therapy-related versus de novo acute promyelocytic leukemia: Association of DNA breaks with specific DNA motifs at PML and RARA loci. Genes Chromosom. Cancer 2010, 732, 726–732. [Google Scholar]
- Mays, A.N.; Osheroff, N.; Xiao, Y.; Wiemels, J.L.; Felix, C.; Byl, J.A.W.; Saravanamuttu, K.; Peniket, A.; Corser, R.; Chang, C.; et al. Evidence for direct involvement of epirubicin in the formation of chromosomal translocations in t(15;17) therapy-related acute promyelocytic leukemia. Blood 2010, 115, 326–330. [Google Scholar]
- Joannides, M.; Mays, N.; Mistry, R.; Hasan, S.K.; Reiter, A.; Wiemels, J.L.; Felix, C.; Coco, F.L.; Osheroff, N.; Solomon, E.; et al. Molecular pathogenesis of secondary acute promyelocytic leukemia. Mediterr. J. Hematol. Infect. Dis. 2011, 3. [Google Scholar] [CrossRef]
- Hoffmann, L.; Möller, P.; Pedersen-Bjergaard, J.; Waage, A.; Pedersen, M.; Hirsch, F. Therapy-related acute promyelocytic leukemia with t(15;17) (q22;q12) following chemotherapy with drugs targeting DNA topoisomerase II. A report of two cases and a review of the literature. Ann. Oncol. 1995, 6, 781–788. [Google Scholar]
- Kudo, K.; Yoshida, H.; Kiyoi, H.; Numata, S.; Horibe, K.; Naoe, T. Etoposide-related acute promyelocytic leukemia. Leukemia 1998, 12, 1171–1175. [Google Scholar]
- Imagawa, J.; Harada, Y.; Shimomura, T.; Tanaka, H.; Okikawa, Y.; Hyodo, H.; Kimura, A.; Harada, H. Clinical and genetic features of therapy-related myeloid neoplasms after chemotherapy for acute promyelocytic leukemia. Blood 2010, 116, 6018–6022. [Google Scholar] [CrossRef]
- Montesinos, P.; Gonzalez, J.D.; Gonzalez, J.; Rayon, C.; de Lisa, E.; Amigo, M.L.; Ossenkoppele, G.J.; Peñarrubia, M.J. Therapy-related myeloid neoplasms in patients with acute promyelocytic leukemia treated with all-trans-retinoic acid and anthracycline-based chemotherapy. J. Clin. Oncol. 2010, 28, 3872–3879. [Google Scholar]
- Strick, R.; Strissel, P.L.; Borgers, S.; Smith, S.L.; Rowley, J.D. Dietary bioflavonoids induce cleavage in the gene and may contribute to infant leukemia. Proc. Natl. Acad. Sci. USA 2000, 97, 4790–4795. [Google Scholar]
- Kunz, C.; Focke, F.; Saito, Y.; Schuermann, D.; Lettieri, T.; Selfridge, J.; Schär, P. Base excision by thymine DNA glycosylase mediates DNA-directed cytotoxicity of 5-fluorouracil. PLoS Biol. 2009, 7. [Google Scholar] [CrossRef]
- Cortellino, S.; Turner, D.; Masciullo, V.; Schepis, F.; Albino, D.; Daniel, R.; Skalka, A.M.; Meropol, N.J.; Alberti, C.; Larue, L.; et al. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc. Natl. Acad. Sci. USA 2003, 100, 15071–15076. [Google Scholar]
- An, Q.; Robins, P.; Lindahl, T.; Barnes, D.E. 5-fluorouracil incorporated into DNA is excised by the Smug1 DNA glycosylase to reduce drug cytotoxicity. Cancer Res. 2007, 67, 940–945. [Google Scholar]
- Pettersen, H.S.; Visnes, T.; Vågbø, C.B.; Svaasand, E.K.; Doseth, B.; Slupphaug, G.; Kavli, B.; Krokan, H.E. UNG-initiated base excision repair is the major repair route for 5-fluorouracil in DNA, but 5-fluorouracil cytotoxicity depends mainly on RNA incorporation. Nucleic Acids Res. 2011, 39, 8430–8444. [Google Scholar]
- Fischer, F.; Baerenfaller, K.; Jiricny, J. 5-fluorouracil is efficiently removed from DNA by the base excision and mismatch repair systems. Gastroenterology 2007, 133, 1858–1868. [Google Scholar] [CrossRef]
- Li, L.S.; Morales, J.C.; Veigl, M.; Sedwick, D.; Greer, S.; Meyers, M.; Wagner, M.; Fishel, R.; Boothman, D.A. DNA mismatch repair (MMR)-dependent 5-fluorouracil cytotoxicity and the potential for new therapeutic targets. Br. J. Pharmacol. 2009, 158, 679–692. [Google Scholar] [CrossRef]
- Smith, M.R.; Neuberg, D.; Flinn, I.W.; Grever, M.R.; Lazarus, H.M.; Rowe, J.M.; Dewald, G.; Bennett, J.M.; Paietta, E.M.; Byrd, J.C.; et al. Incidence of therapy-related myeloid neoplasia after initial therapy for chronic lymphocytic leukemia with fludarabine-cyclophosphamide versus fludarabine: Long-term follow-up of US Intergroup Study E2997. Blood 2011, 118, 3525–3527. [Google Scholar]
- Huang, P.; Chubb, S.; Plunkett, S. Termination of DNA synthesis by 9-beta-D arabinofuranosyl-2-fluoroadenine. J. Biol. Chem. 1990, 265, 16617–16625. [Google Scholar]
- De Campos-Nebel, M.; Larripa, I.; González-Cid, M. Non-homologous end joining is the responsible pathway for the repair of fludarabine-induced DNA double strand breaks in mammalian cells. Mutat. Res. 2008, 646, 8–16. [Google Scholar] [CrossRef]
- Bulgar, D.; Snell, M.; Donze, J.R.; Kirkland, E.B.; Li, L.; Yang, S.; Xu, Y.; Gerson, S.L.; Liu, L. Targeting base excision repair suggests a new therapeutic strategy of fludarabine for the treatment of chronic lymphocytic leukemia. Leukemia 2010, 24, 1795–1799. [Google Scholar] [CrossRef]
- Li, L.; Keating, M.J.; Plunkett, W.; Yang, L.Y. Fludarabine-mediated repair inhibition of cisplatin-induced DNA lesions in human chronic myelogenous leukemia-blast crisis K562 cells: Induction of synergistic cytotoxicity independent of reversal of apoptosis resistance. Mol. Pharmacol. 1997, 52, 798–806. [Google Scholar]
- Pepper, C.; Lowe, H.; Fegan, C.; Thurieau, C.; Thurston, D.E.; Hartley, J.; Delavault, P. Fludarabine-mediated suppression of the excision repair enzyme ERCC1 contributes to the cytotoxic synergy with the DNA minor groove crosslinking agent SJG-136 (NSC 694501) in chronic lymphocytic leukaemia cells. Br. J. Cancer 2007, 97, 253–259. [Google Scholar] [CrossRef]
- Swann, P.; Waters, T.; Moulton, D.; Xu, Y.; Zheng, Q.; Edwards, M.; Mace, R. Role of postreplicative DNA mismatch repair in the cytotoxic action of thioguanine. Science 1996, 273, 1109–1111. [Google Scholar]
- Donovan, P.O.; Perrett, C.M.; Zhang, X.; Montaner, B.; Harwood, C.A.; Mcgregor, J.M.; Walker, S.L.; Hanaoka, F.; Karran, P. Azathioprine and UVA light generate mutagenic oxidative DNA damage. Science 2008, 309, 1871–1874. [Google Scholar]
- Ren, X.; Li, F.; Jeffs, G.; Zhang, X.; Xu, Y.Z.; Karran, P. Guanine sulphinate is a major stable product of photochemical oxidation of DNA 6-thioguanine by UVA irradiation. Nucleic Acids Res. 2010, 38, 1832–1840. [Google Scholar] [CrossRef]
- Gueranger, Q.; Kia, A.; Frith, D.; Karran, P. Crosslinking of DNA repair and replication proteins to DNA in cells treated with 6-thioguanine and UVA. Nucleic Acids Res. 2011, 39, 5057–5066. [Google Scholar] [CrossRef]
- Karran, P.; Attard, N. Thiopurines in current medical practice: Molecular mechanisms and contributions to therapy-related cancer. Nat. Rev. Cancer 2008, 8, 24–36. [Google Scholar] [CrossRef]
- Euvrard, S.; Kanitakis, J.; Claudy, A. Skin cancers after organ transplantation. N. Engl. J. Med. 2003, 348, 1681–1691. [Google Scholar] [CrossRef]
- Falck, K.; Gröhn, P.; Sorsa, M.; Vainio, H.; Heinonen, E.; Holsti, L. Mutagenicity in urine of nurses handling cytostatic drugs. Lancet 1979, 1, 1250–1251. [Google Scholar]
- Norppa, H.; Sorsa, M.; Vainio, H.; Gröhn, P.; Heinonen, E.; Holsti, L.; Nordman, E. Increased sister chromatid exchange frequencies in lymphocytes of nurses handling cytostatic drugs. Scand. J. Work Environ. Health 1980, 6, 299–301. [Google Scholar] [CrossRef]
- El-Ebiary, A.A.; Abuelfadl, A.A.; Sarhan, N.I. Evaluation of genotoxicity induced by exposure to antineoplastic drugs in lymphocytes of oncology nurses and pharmacists. J. Appl. Toxicol. 2011. [Google Scholar] [CrossRef]
- Mader, R.M.; Kokalj, A.; Kratochvil, E.; Pilger, A.; Rüdiger, H.W. Longitudinal biomonitoring of nurses handling antineoplastic drugs. J. Clin. Nurs. 2008, 18, 263–269. [Google Scholar]
- Jakab, M.; Major, J.; Tompa, A. Follow up genotoxicological monitoring of nurses handling antineoplastic drugs. J. Toxicol. Environ. Health 2011, 62, 37–41. [Google Scholar]
- Skov, T.; Maarup, B.; Olsen, J.; Rørth, M.; Winthereik, H.; Lynge, E. Leukaemia and reproductive outcome among nurses handling antineoplastic drugs. Br. J. Ind. Med. 1992, 49, 855–861. [Google Scholar]
- Gunnarsdottir, H.; Aspelund, T.; Karlsson, T.; Rafnsson, V. Occupational risk factors for breast cancer among nurses. Int. J. Occup. Environ. Health 1997, 3, 254–258. [Google Scholar]
- Ellis, N.A.; Huo, D.; Yildiz, O.; Worrillow, L.J.; Banerjee, M.; Le Beau, M.M.; Larson, R.A.; Allan, J.M.; Onel, K. MDM2 SNP309 and TP53 Arg72Pro interact to alter therapy-related acute myeloid leukemia susceptibility. Blood 2008, 112, 741–749. [Google Scholar]
- Cano, K.E.; Li, L.; Bhatia, S.; Bhatia, R.; Forman, S.J.; Chen, Y. NMR-based metabolomic analysis of the molecular pathogenesis of therapy-related myelodysplasia/acute myeloid leukemia. J. Proteome Res. 2011, 10, 2873–2881. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Casorelli, I.; Bossa, C.; Bignami, M. DNA Damage and Repair in Human Cancer: Molecular Mechanisms and Contribution to Therapy-Related Leukemias. Int. J. Environ. Res. Public Health 2012, 9, 2636-2657. https://doi.org/10.3390/ijerph9082636
Casorelli I, Bossa C, Bignami M. DNA Damage and Repair in Human Cancer: Molecular Mechanisms and Contribution to Therapy-Related Leukemias. International Journal of Environmental Research and Public Health. 2012; 9(8):2636-2657. https://doi.org/10.3390/ijerph9082636
Chicago/Turabian StyleCasorelli, Ida, Cecilia Bossa, and Margherita Bignami. 2012. "DNA Damage and Repair in Human Cancer: Molecular Mechanisms and Contribution to Therapy-Related Leukemias" International Journal of Environmental Research and Public Health 9, no. 8: 2636-2657. https://doi.org/10.3390/ijerph9082636