The Essential Toxin: Impact of Zinc on Human Health

Abstract
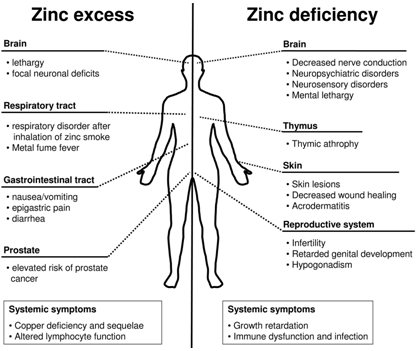
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
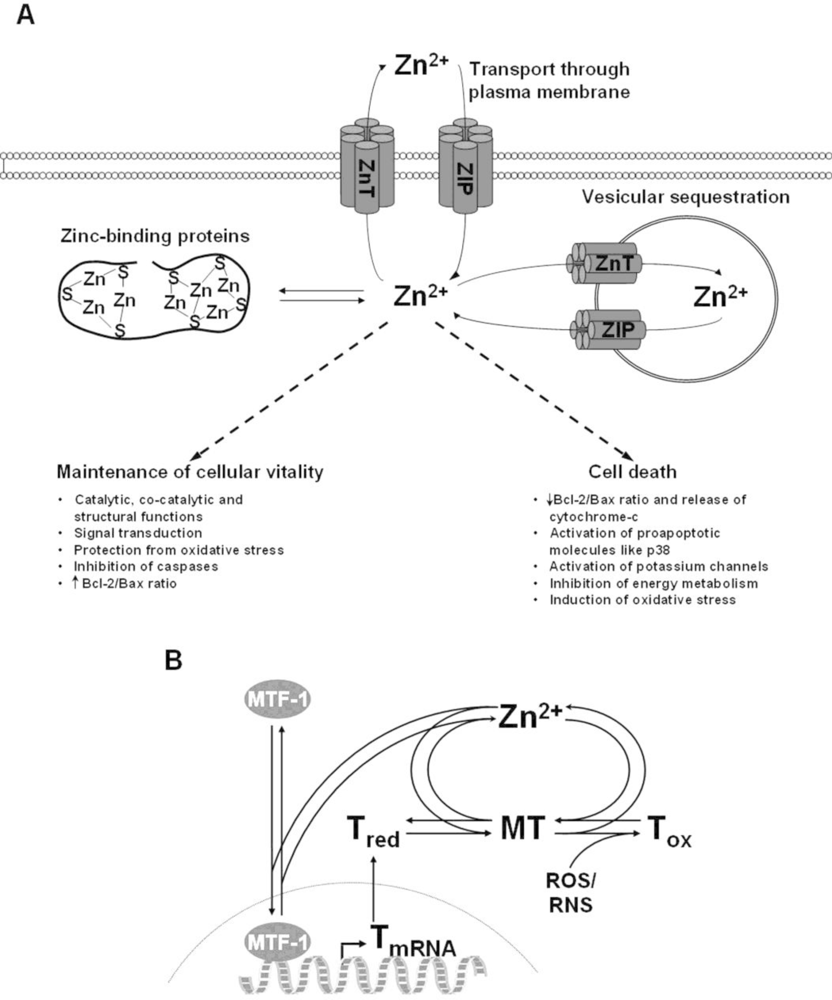
2. Zinc Homeostasis
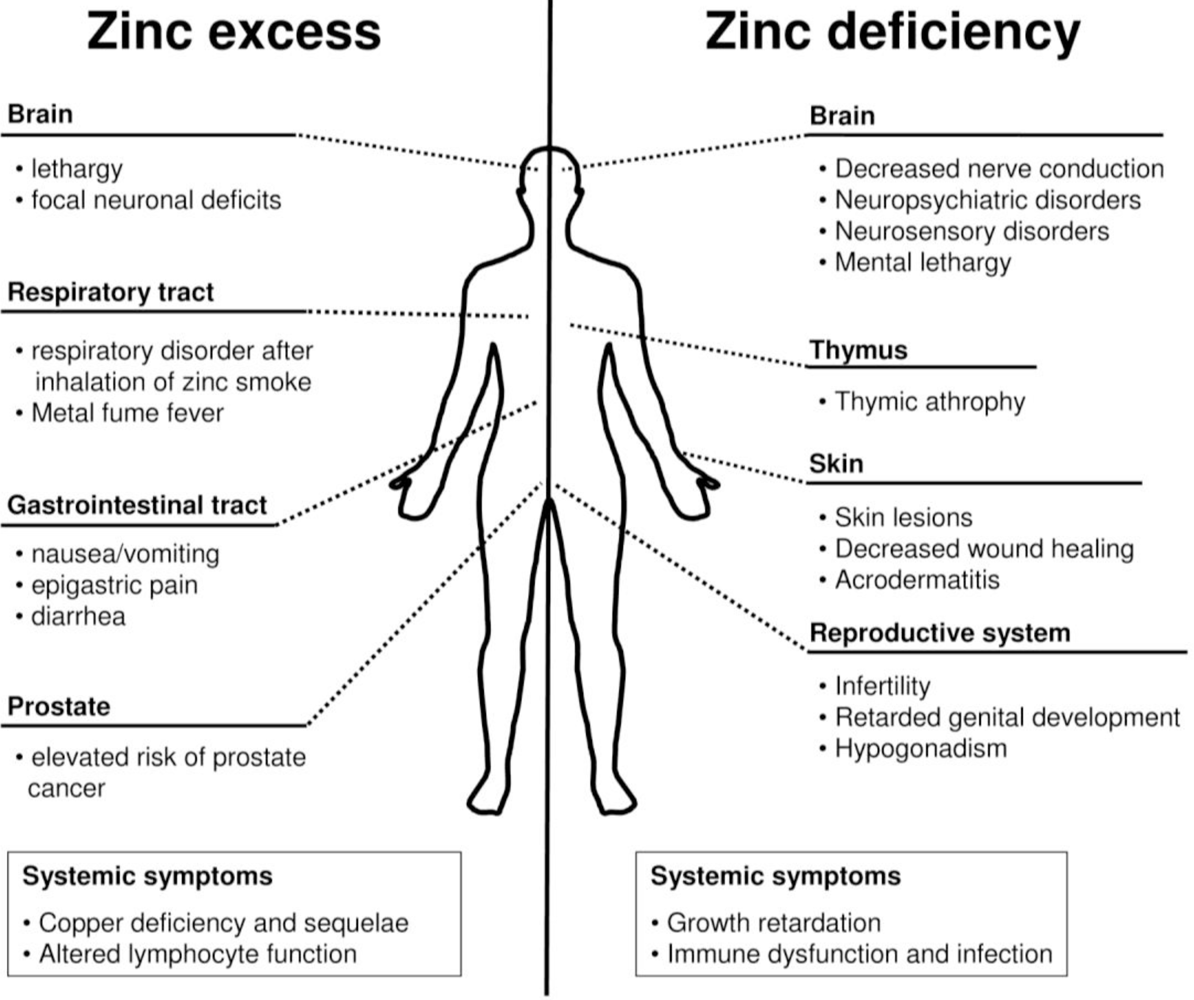
3. Exposure to Zinc
3.1. Exposure by Inhalation
3.2. Dermal Exposure
3.3. Oral Exposure
Gastrointestinal Effects
Zinc-Induced Copper Deficiency
Zinc Supplementation and Cancer
Immunological Effects
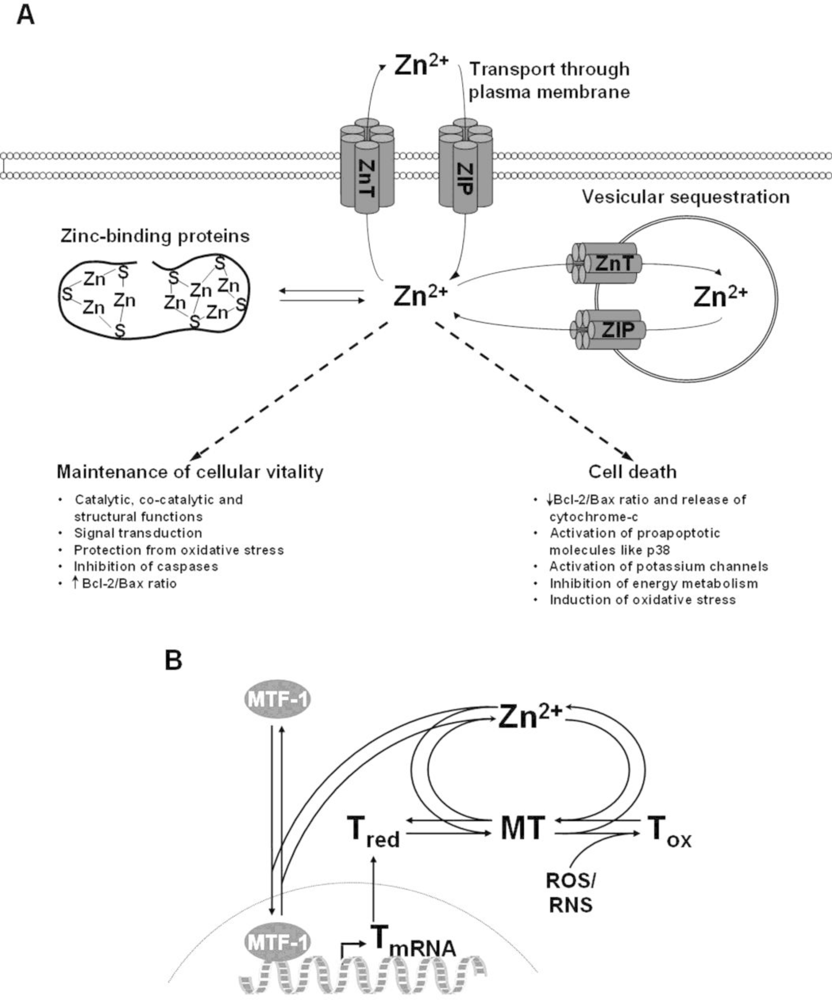
4. The Role of Zinc in Cell Death
4.1. Impact of Zinc on Apoptosis
4.2. Role of Zinc in Neuronal Death
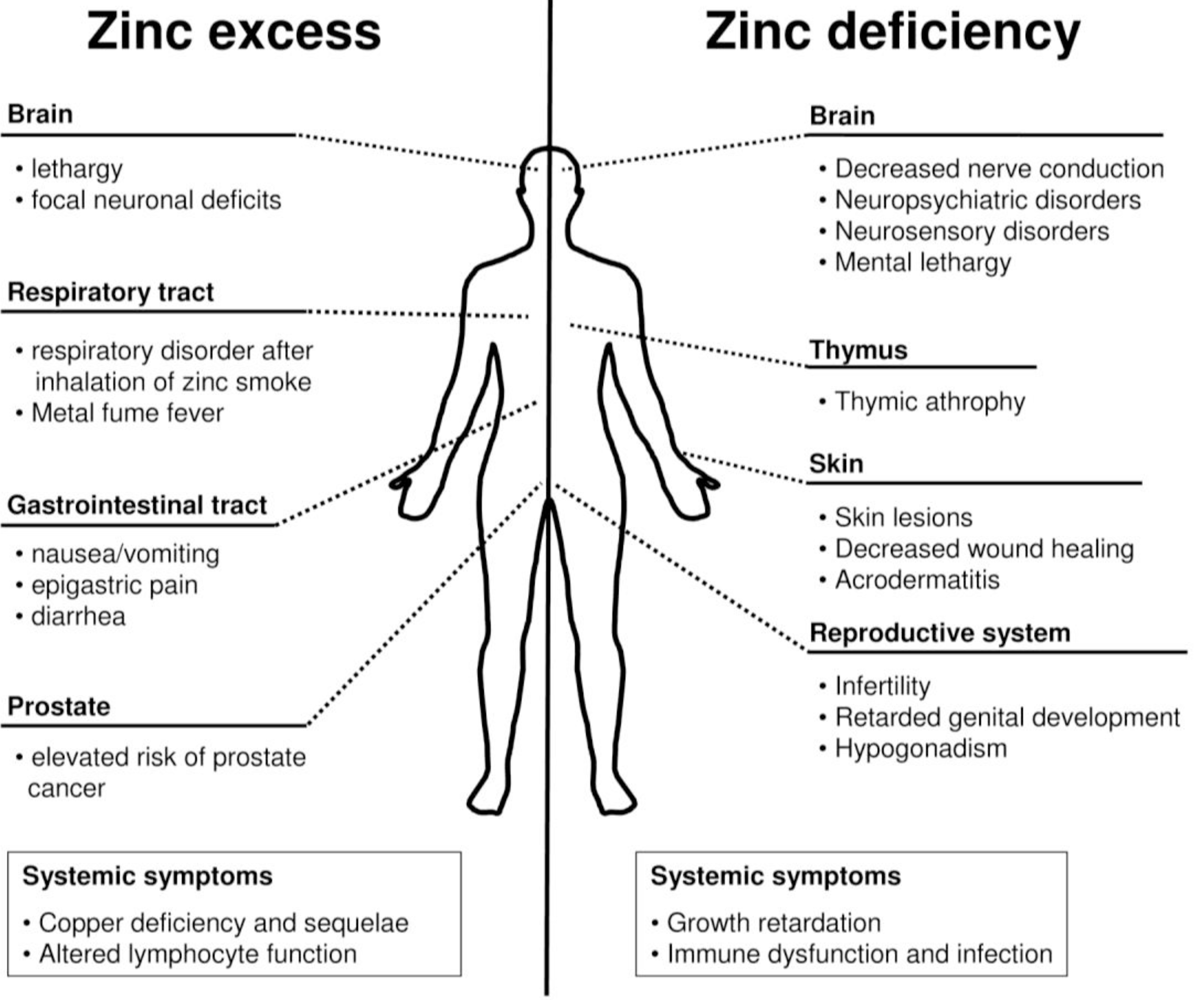
5. Zinc Deficiency
6. Conclusions
References
- Fosmire, GJ. Zinc toxicity. Am. J. Clin. Nutr 1990, 51, 225–227. [Google Scholar]
- U. S. National Library of Medicine, Toxnet Database.
- Jones, MM; Schoenheit, JE; Weaver, AD. Pretreatment and heavy metal LD50 values. Toxicol. Appl. Pharmacol 1979, 49, 41–44. [Google Scholar]
- Vallee, BL; Falchuk, KH. The biochemical basis of zinc physiology. Physiol. Rev 1993, 73, 79–118. [Google Scholar]
- Wastney, ME; Aamodt, RL; Rumble, WF; Henkin, RI. Kinetic analysis of zinc metabolism and its regulation in normal humans. Am. J. Physiol 1986, 251, R398–R408. [Google Scholar]
- Bentley, PJ; Grubb, BR. Experimental dietary hyperzincemia tissue disposition of excess zinc in rabbits. Trace. Elem. Med 1991, 8, 202–207. [Google Scholar]
- He, LS; Yan, XS; Wu, DC. Age-dependent variation of zinc-65 metabolism in LACA mice. Int J. Radiat. Biol 1991, 60, 907–916. [Google Scholar]
- Llobet, JM; Domingo, JL; Colomina, MT; Mayayo, E; Corbella, J. Subchronic oral toxicity of zinc in rats. Bull. Environ. Contam. Toxicol 1988, 41, 36–43. [Google Scholar]
- Scott, BJ; Bradwell, AR. Identification of the serum binding proteins for iron, zinc, cadmium, nickel, and calcium. Clin. Chem 1983, 29, 629–633. [Google Scholar]
- Lichten, LA; Cousins, RJ. Mammalian zinc transporters: nutritional and physiologic regulation. Annu. Rev. Nutr 2009, 29, 153–176. [Google Scholar]
- Haase, H; Maret, W. Intracellular zinc fluctuations modulate protein tyrosine phosphatase activity in insulin/insulin-like growth factor-1 signaling. Exp. Cell Res 2003, 291, 289–298. [Google Scholar]
- Taylor, KM; Vichova, P; Jordan, N; Hiscox, S; Hendley, R; Nicholson, RI. ZIP7-mediated intracellular zinc transport contributes to aberrant growth factor signaling in antihormone-resistant breast cancer Cells. Endocrinology 2008, 149, 4912–4920. [Google Scholar]
- Chimienti, F; Aouffen, M; Favier, A; Seve, M. Zinc homeostasis-regulating proteins: new drug targets for triggering cell fate. Curr. Drug Targets 2003, 4, 323–338. [Google Scholar]
- Tapiero, H; Tew, KD. Trace elements in human physiology and pathology: zinc and metallothioneins. Biomed. Pharmacother 2003, 57, 399–411. [Google Scholar]
- Krezel, A; Maret, W. Dual nanomolar and picomolar Zn(II) binding properties of metallothionein. J. Am. Chem. Soc 2007, 129, 10911–10921. [Google Scholar]
- Laity, JH; Andrews, GK. Understanding the mechanisms of zinc-sensing by metal-response element binding transcription factor-1 (MTF-1). Arch. Biochem. Biophys 2007, 463, 201–210. [Google Scholar]
- Maret, W. Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid. Redox. Signal 2006, 8, 1419–1441. [Google Scholar]
- Toxicological Profile for Zinc; Agency for Toxic Substances and Disease Registry Division of Toxicology and Environmental Medicine: Atlanta, GA, USA, 2005.
- Homma, S; Jones, R; Qvist, J; Zapol, WM; Reid, L. Pulmonary vascular lesions in the adult respiratory distress syndrome caused by inhalation of zinc chloride smoke: a morphometric study. Hum. Pathol 1992, 23, 45–50. [Google Scholar]
- Freitag, A; Caduff, B. ARDS caused by military zinc fumes exposure. Schweiz Med. Wochenschr 1996, 126, 1006–1010. [Google Scholar]
- Johnson, FA; Stonehill, RB. Chemical pneumonitis from inhalation of zinc chloride. Dis. Chest 1961, 40, 619–624. [Google Scholar]
- Zerahn, B; Kofoed-Enevoldsen, A; Jensen, BV; Molvig, J; Ebbehoj, N; Johansen, JS; Kanstrup, IL. Pulmonary damage after modest exposure to zinc chloride smoke. Respir. Med 1999, 93, 885–890. [Google Scholar]
- Vogelmeier, C; Konig, G; Bencze, K; Fruhmann, G. Pulmonary involvement in zinc fume fever. Chest 1987, 92, 946–948. [Google Scholar]
- Rohrs, LC. Metal-fume fever from inhaling zinc oxide. AMA Arch. Ind. Health 1957, 16, 42–47. [Google Scholar]
- Brown, JJ. Zinc fume fever. Br. J. Radiol 1988, 61, 327–329. [Google Scholar]
- Martin, CJ; Le, XC; Guidotti, TL; Yalcin, S; Chum, E; Audette, RJ; Liang, C; Yuan, B; Zhang, X; Wu, J. Zinc exposure in Chinese foundry workers. Am. J. Ind. Med 1999, 35, 574–580. [Google Scholar]
- Sturgis, CC; Drinker, P; Thompson, RM. Metal fume fever: I. Clinical observations on the effect of the experimental inhalation of zinc oxide by two apparently normal persons. J. Ind. Hyg 1927, 9, 88–97. [Google Scholar]
- Hammond, JW. Metal fume fever in crushed stone industry. J. Ind. Hyg 1944, 26, 117–119. [Google Scholar]
- Blanc, P; Wong, H; Bernstein, MS; Boushey, HA. An experimental human model of metal fume fever. Ann. Intern. Med 1991, 114, 930–936. [Google Scholar]
- Drinker, P; Thompson, RM; Finn, JL. Metal fume fever: IV. Threshold doses of zinc oxide, preventive measures, and the chronic effects of repeated exposures. J. Ind. Hyg 1927, 9, 331–345. [Google Scholar]
- Marquart, H; Smid, T; Heederik, D; Visschers, M. Lung function of welders of zinc-coated mild steel: cross-sectional analysis and changes over five consecutive work shifts. Am. J. Ind. Med 1989, 16, 289–296. [Google Scholar]
- OSHA, Occupational Safety and Health Standards; Occupational Safety and Health Administration: Washington, DC, USA, 2003; Volume 29, CFR 1910.1000, pp. Table Z-1.
- Agren, MS. Percutaneous absorption of zinc from zinc oxide applied topically to intact skin in man. Dermatologica 1990, 180, 36–39. [Google Scholar]
- Agren, MS; Krusell, M; Franzen, L. Release and absorption of zinc from zinc oxide and zinc sulfate in open wounds. Acta Dermato.-Venereol 1991, 71, 330–333. [Google Scholar]
- Lansdown, AB. Interspecies variations in response to topical application of selected zinc compounds. Food. Chem. Toxicol 1991, 29, 57–64. [Google Scholar]
- Agren, MS; Franzen, L; Chvapil, M. Effects on wound healing of zinc oxide in a hydrocolloid dressing. J. Am. Acad. Dermatol 1993, 29, 221–227. [Google Scholar]
- Lansdown, AB. Influence of zinc oxide in the closure of open skin wounds. Int. J. Cosmet. Sci 1993, 15, 83–85. [Google Scholar]
- Stromberg, HE; Agren, MS. Topical zinc oxide treatment improves arterial and venous leg ulcers. Br. J. Dermatol 1984, 111, 461–468. [Google Scholar]
- Trumbo, P; Yates, AA; Schlicker, S; Poos, M. Dietary reference intakes: vitamin A, vitamin K, arsenic, boron, chromium, copper, iodine, iron, manganese, molybdenum, nickel, silicon, vanadium, and zinc. J. Am. Diet. Assoc 2001, 101, 294–301. [Google Scholar]
- Brown, MA; Thom, JV; Orth, GL; Cova, P; Juarez, J. Food poisoning involving zinc contamination. Arch. Environ. Health 1964, 8, 657–660. [Google Scholar]
- Fox, MRS. Zinc excess. In Zinc in Human Biology; Mills, CF, Ed.; Springer Verlag: New York, NY, USA, 1989; pp. 366–368. [Google Scholar]
- Porea, TJ; Belmont, JW; Mahoney, DH, Jr. Zinc-induced anemia and neutropenia in an adolescent. J. Pediatr 2000, 136, 688–690. [Google Scholar]
- Samman, S; Roberts, DC. The effect of zinc supplements on plasma zinc and copper levels and the reported symptoms in healthy volunteers. Med. J. Aust 1987, 146, 246–249. [Google Scholar]
- Haase, H; Overbeck, S; Rink, L. Zinc supplementation for the treatment or prevention of disease: current status and future perspectives. Exp. Gerontol 2008, 43, 394–408. [Google Scholar]
- Callender, GR; Gentzkow, CJ. Acute poisoning by the zinc and antimony content of limeade prepared in a galvanized iron can. Military Surgeon 1937, 80, 67–71. [Google Scholar]
- Lewis, MR; Kokan, L. Zinc gluconate: acute ingestion. J. Toxicol. Clin. Toxicol 1998, 36, 99–101. [Google Scholar]
- Liu, CH; Lee, CT; Tsai, FC; Hsu, SJ; Yang, PM. Gastroduodenal corrosive injury after oral zinc oxide. Ann. Emerg. Med 2006, 47, 296. [Google Scholar]
- Magee, AC; Matrone, G. Studies on growth, copper metabolism of rats fed high levels of zinc. J. Nutr 1960, 72, 233–242. [Google Scholar]
- Ogiso, T; Moriyama, K; Sasaki, S; Ishimura, Y; Minato, A. Inhibitory effect of high dietary zinc on copper absorption in rats. Chem. Pharm. Bull. (Tokyo) 1974, 22, 55–60. [Google Scholar]
- Van Campen, DR. Copper interference with the intestinal absorption of zinc-65 by rats. J. Nutr 1969, 97, 104–108. [Google Scholar]
- Igic, PG; Lee, E; Harper, W; Roach, KW. Toxic effects associated with consumption of zinc. Mayo. Clin. Proc 2002, 77, 713–716. [Google Scholar]
- Ogiso, T; Ogawa, N; Miura, T. Inhibitory effect of high dietary zinc on copper absorption in rats. II. Binding of copper and zinc to cytosol proteins in the intestinal mucosa. Chem. Pharm. Bull 1979, 27, 515–521. [Google Scholar]
- Oestreicher, P; Cousins, RJ. Copper and zinc absorption in the rat: mechanism of mutual antagonism. J. Nutr 1985, 115, 159–166. [Google Scholar]
- Fischer, PW; Giroux, A; L’Abbe, MR. The effect of dietary zinc on intestinal copper absorption. Am. J. Clin. Nutr 1981, 34, 1670–1675. [Google Scholar]
- Fiske, DN; McCoy, HE, III; Kitchens, CS. Zinc-induced sideroblastic anemia: report of a case, review of the literature, and description of the hematologic syndrome. Am. J. Hematol 1994, 46, 147–150. [Google Scholar]
- Prohaska, JR. Biochemical changes in copper deficiency. J. Nutr. Biochem 1990, 1, 452–461. [Google Scholar]
- Sandstead, HH. Requirements and toxicity of essential trace elements, illustrated by zinc and copper. Am. J. Clin. Nutr 1995, 61, 621S–624S. [Google Scholar]
- Irving, JA; Mattman, A; Lockitch, G; Farrell, K; Wadsworth, LD. Element of caution: a case of reversible cytopenias associated with excessive zinc supplementation. CMAJ 2003, 169, 129–131. [Google Scholar]
- Prasad, AS; Brewer, GJ; Schoomaker, EB; Rabbani, P. Hypocupremia induced by zinc therapy in adults. JAMA 1978, 240, 2166–2168. [Google Scholar]
- Broun, ER; Greist, A; Tricot, G; Hoffman, R. Excessive zinc ingestion: a reversible cause of sideroblastic anemia and bone marrow depression. JAMA 1990, 265, 1441–1443. [Google Scholar]
- Frieden, E. The copper connection. Semin. Hematol 1983, 20, 114–117. [Google Scholar]
- Willis, MS; Monaghan, SA; Miller, ML; McKenna, RW; Perkins, WD; Levinson, BS; Bhushan, V; Kroft, SH. Zinc-induced copper deficiency: a report of three cases initially recognized on bone marrow examination. Am. J. Clin. Pathol 2005, 123, 125–131. [Google Scholar]
- Williams, DM. Copper deficiency in humans. Semin. Hematol 1983, 20, 118–128. [Google Scholar]
- Williams, DM; Lynch, RE; Lee, GR; Cartwright, GE. Superoxide dismutase activity in copper-deficient swine. Proc. Soc. Exp. Biol. Med 1975, 149, 534–536. [Google Scholar]
- Foster, M; Petocz, P; Samman, S. Effects of zinc on plasma lipoprotein cholesterol concentrations in humans: A meta-analysis of randomised controlled trials. Atherosclerosis 2009. [Google Scholar]
- Black, MR; Medeiros, DM; Brunett, E; Welke, R. Zinc supplements and serum lipids in young adult white males. Am. J. Clin. Nutr 1988, 47, 970–975. [Google Scholar]
- Hooper, PL; Visconti, L; Garry, PJ; Johnson, GE. Zinc lowers high-density lipoprotein-cholesterol levels. JAMA 1980, 244, 1960–1961. [Google Scholar]
- Freeland-Graves, JH; Friedman, BJ; Han, WH; Shorey, RL; Young, R. Effect of zinc supplementation on plasma high-density lipoprotein cholesterol and zinc. Am. J. Clin. Nutr 1982, 35, 988–992. [Google Scholar]
- Reiser, S; Powell, AS; Yang, C; Canary, J. Effect of copper intake on blood cholesterol and its lipoprotein distribution in men. Nutr. Rep. Int 1987, 36, 641–649. [Google Scholar]
- Klevay, LM; Inman, L; Johnson, LK; Lawler, M; Mahalko, JR; Milne, DB; Lukaski, HC; Bolonchuk, W; Sandstead, HH. Increased cholesterol in plasma in a young man during experimental copper depletion. Metabolism 1984, 33, 1112–1118. [Google Scholar]
- Beyersmann, D; Hartwig, A. Carcinogenic metal compounds: recent insight into molecular and cellular mechanisms. Arch. Toxicol 2008, 82, 493–512. [Google Scholar]
- Costello, LC; Franklin, RB. Novel role of zinc in the regulation of prostate citrate metabolism and its implications in prostate cancer. Prostate 1998, 35, 285–296. [Google Scholar]
- Habib, FK. Zinc and the steroid endocrinology of the human prostate. J. Steroid. Biochem 1978, 9, 403–407. [Google Scholar]
- Zaichick, V; Sviridova, TV; Zaichick, SV. Zinc in the human prostate gland: normal, hyperplastic and cancerous. Int. Urol. Nephrol 1997, 29, 565–574. [Google Scholar]
- Costello, LC; Liu, Y; Zou, J; Franklin, RB. Evidence for a zinc uptake transporter in human prostate cancer cells which is regulated by prolactin and testosterone. J. Biol. Chem 1999, 274, 17499–17504. [Google Scholar]
- Franklin, RB; Feng, P; Milon, B; Desouki, MM; Singh, KK; Kajdacsy-Balla, A; Bagasra, O; Costello, LC. hZIP1 zinc uptake transporter down regulation and zinc depletion in prostate cancer. Mol. Cancer 2005, 4, 32. [Google Scholar]
- Jarrard, DF. Does zinc supplementation increase the risk of prostate cancer? Arch. Ophthalmol 2005, 123, 102–103. [Google Scholar]
- Leitzmann, MF; Stampfer, MJ; Wu, K; Colditz, GA; Willett, WC; Giovannucci, EL. Zinc supplement use and risk of prostate cancer. J. Natl. Cancer Inst 2003, 95, 1004–1007. [Google Scholar]
- Honscheid, A; Rink, L; Haase, H. T-lymphocytes: a target for stimulatory and inhibitory effects of zinc ions. Endocr. Metab. Immune Disord. Drug Targets 2009, 9, 132–144. [Google Scholar]
- Rink, L; Gabriel, P. Zinc and the immune system. Proc. Nutr. Soc 2000, 59, 541–552. [Google Scholar]
- Delafuente, JC. Nutrients and immune responses. Rheum. Dis. Clin. North Am 1991, 17, 203–212. [Google Scholar]
- Fraker, PJ; DePasquale-Jardieu, P; Zwickl, CM; Luecke, RW. Regeneration of T-cell helper function in zinc-deficient adult mice. Proc. Nat. Acad. Sci. USA 1978, 75, 5660–5664. [Google Scholar]
- Haase, H; Rink, L. Functional significance of zinc-related signaling pathways in immune cells. Annu. Rev. Nutr 2009, 29, 133–152. [Google Scholar]
- Wellinghausen, N; Driessen, C; Rink, L. Stimulation of human peripheral blood mononuclear cells by zinc and related cations. Cytokine 1996, 8, 767–771. [Google Scholar]
- Bulgarini, D; Habetswallner, D; Boccoli, G; Montesoro, E; Camagna, A; Mastroberardino, G; Rosania, C; Testa, U; Peschle, C. Zinc modulates the mitogenic activation of human peripheral blood lymphocytes. Ann. Ist. Super. Sanita 1989, 25, 463–470. [Google Scholar]
- Wellinghausen, N; Martin, M; Rink, L. Zinc inhibits interleukin-1-dependent T cell stimulation. Eur. J. Immunol 1997, 27, 2529–2535. [Google Scholar]
- Campo, CA; Wellinghausen, N; Faber, C; Fischer, A; Rink, L. Zinc inhibits the mixed lymphocyte culture. Biol. Tr. Elem. Res 2001, 79, 15–22. [Google Scholar]
- Faber, C; Gabriel, P; Ibs, KH; Rink, L. Zinc in pharmacological doses suppresses allogeneic reaction without affecting the antigenic response. Bone Marrow Transplant 2004, 33, 1241–1246. [Google Scholar]
- Duchateau, J; Delepesse, G; Vrijens, R; Collet, H. Beneficial effects of oral zinc supplementation on the immune response of old people. Am. J. Med 1981, 70, 1001–1004. [Google Scholar]
- Cummings, JE; Kovacic, JP. The ubiquitous role of zinc in health and disease. J. Vet. Emerg. Crit. Care 2009, 19, 215–240. [Google Scholar]
- Formigari, A; Irato, P; Santon, A. Zinc, antioxidant systems and metallothionein in metal mediated-apoptosis: biochemical and cytochemical aspects. Comp. Biochem. Physiol. Pt. C 2007, 146, 443–459. [Google Scholar]
- Haase, H; Watjen, W; Beyersmann, D. Zinc induces apoptosis that can be suppressed by lanthanum in C6 rat glioma cells. Biol. Chem 2001, 382, 1227–1234. [Google Scholar]
- Truong-Tran, AQ; Carter, J; Ruffin, RE; Zalewski, PD. The role of zinc in caspase activation and apoptotic cell death. Biometals 2001, 14, 315–330. [Google Scholar]
- Fraker, PJ; Telford, WG. A reappraisal of the role of zinc in life and death decisions of cells. Proc. Soc. Exp. Biol. Med 1997, 215, 229–236. [Google Scholar]
- Watjen, W; Haase, H; Biagioli, M; Beyersmann, D. Induction of apoptosis in mammalian cells by cadmium and zinc. Environ. Health Perspect 2002, 110, 865–867. [Google Scholar]
- Kim, YH; Kim, EY; Gwag, BJ; Sohn, S; Koh, JY. Zinc-induced cortical neuronal death with features of apoptosis and necrosis: mediation by free radicals. Neuroscience 1999, 89, 175–182. [Google Scholar]
- McLaughlin, B; Pal, S; Tran, MP; Parsons, AA; Barone, FC; Erhardt, JA; Aizenman, E. p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J. Neurosci 2001, 21, 3303–3311. [Google Scholar]
- Wiseman, DA; Wells, SM; Wilham, J; Hubbard, M; Welker, JE; Black, SM. Endothelial response to stress from exogenous Zn2+ resembles that of NO-mediated nitrosative stress, and is protected by MT-1 overexpression. Am. J. Physiol. Cell Physiol 2006, 291, C555–568. [Google Scholar]
- Brown, AM; Kristal, BS; Effron, MS; Shestopalov, AI; Ullucci, PA; Sheu, KF; Blass, JP; Cooper, AJ. Zn2+ inhibits alpha-ketoglutarate-stimulated mitochondrial respiration and the isolated alpha-ketoglutarate dehydrogenase complex. J. Biol. Chem 2000, 275, 13441–13447. [Google Scholar]
- Sheline, CT; Behrens, MM; Choi, DW. Zinc-induced cortical neuronal death: contribution of energy failure attributable to loss of NAD(+) and inhibition of glycolysis. J. Neurosci 2000, 20, 3139–3146. [Google Scholar]
- Feng, P; Li, T; Guan, Z; Franklin, RB; Costello, LC. The involvement of Bax in zinc-induced mitochondrial apoptogenesis in malignant prostate cells. Mol. Cancer 2008, 7, 25. [Google Scholar]
- Dineley, KE; Votyakova, TV; Reynolds, IJ. Zinc inhibition of cellular energy production: implications for mitochondria and neurodegeneration. J. Neurochem 2003, 85, 563–570. [Google Scholar]
- Feng, P; Li, TL; Guan, ZX; Franklin, RB; Costello, LC. Direct effect of zinc on mitochondrial apoptogenesis in prostate cells. Prostate 2002, 52, 311–318. [Google Scholar]
- Mills, DA; Schmidt, B; Hiser, C; Westley, E; Ferguson-Miller, S. Membrane potential-controlled inhibition of cytochrome c oxidase by zinc. J. Biol. Chem 2002, 277, 14894–14901. [Google Scholar]
- Bitanihirwe, BK; Cunningham, MG. Zinc: the brain’s dark horse. Synapse 2009, 63, 1029–1049. [Google Scholar]
- Cui, L; Takagi, Y; Sando, K; Wasa, M; Okada, A. Nitric oxide synthase inhibitor attenuates inflammatory lesions in the skin of zinc-deficient rats. Nutrition 2000, 16, 34–41. [Google Scholar]
- Cui, L; Takagi, Y; Wasa, M; Sando, K; Khan, J; Okada, A. Nitric oxide synthase inhibitor attenuates intestinal damage induced by zinc deficiency in rats. J. Nutr 1999, 129, 792–798. [Google Scholar]
- Oteiza, PI; Clegg, MS; Zago, MP; Keen, CL. Zinc deficiency induces oxidative stress and AP-1 activation in 3T3 cells. Free Radical Biol. Med 2000, 28, 1091–1099. [Google Scholar]
- Williams, RJP. The biochemistry of zinc. Polyhedron 1987, 6, 61–69. [Google Scholar]
- Perry, DK; Smyth, MJ; Stennicke, HR; Salvesen, GS; Duriez, P; Poirier, GG; Hannun, YA. Zinc is a potent inhibitor of the apoptotic protease, caspase-3. A novel target for zinc in the inhibition of apoptosis. J. Biol. Chem 1997, 272, 18530–18533. [Google Scholar]
- Maret, W; Jacob, C; Vallee, BL; Fischer, EH. Inhibitory sites in enzymes: zinc removal and reactivation by thionein. Proc. Nat. Acad. Sci. USA 1999, 96, 1936–1940. [Google Scholar]
- Stennicke, HR; Salvesen, GS. Biochemical characteristics of caspases-3, -6, -7, and -8. J. Biol. Chem 1997, 272, 25719–25723. [Google Scholar]
- Clegg, MS; Hanna, LA; Niles, BJ; Momma, TY; Keen, CL. Zinc deficiency-induced cell death. IUBMB Life 2005, 57, 661–669. [Google Scholar]
- Fukamachi, Y; Karasaki, Y; Sugiura, T; Itoh, H; Abe, T; Yamamura, K; Higashi, K. Zinc suppresses apoptosis of U937 cells induced by hydrogen peroxide through an increase of the Bcl-2/Bax ratio. Biochem. Biophys. Res. Commun 1998, 246, 364–369. [Google Scholar]
- Zalewski, PD; Forbes, IJ; Giannakis, C. Physiological role for zinc in prevention of apoptosis (gene-directed death). Biochem. Int 1991, 24, 1093–1101. [Google Scholar]
- Zalewski, PD; Forbes, IJ; Betts, WH. Correlation of apoptosis with change in intracellular labile Zn(II) using zinquin [(2-methyl-8-p-toluenesulphonamido-6-quinolyloxy)acetic acid], a new specific fluorescent probe for Zn(II). Biochem. J 1993, 296, 403–408. [Google Scholar]
- Murphy, JV. Intoxication following ingestion of elemental zinc. JAMA 1970, 212, 2119–2120. [Google Scholar]
- Colvin, RA; Fontaine, CP; Laskowski, M; Thomas, D. Zn2+ transporters and Zn2+ homeostasis in neurons. Eur. J. Pharmacol 2003, 479, 171–185. [Google Scholar]
- Frederickson, CJ; Bush, AI. Synaptically released zinc: physiological functions and pathological effects. Biometals 2001, 14, 353–366. [Google Scholar]
- Takeda, A. Movement of zinc and its functional significance in the brain. Brain. Res. Rev 2000, 34, 137–148. [Google Scholar]
- Vogt, K; Mellor, J; Tong, G; Nicoll, R. The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 2000, 26, 187–196. [Google Scholar]
- Cuajungco, MP; Lees, GJ. Zinc and Alzheimer’s disease: is there a direct link? Brain. Res. Rev 1997, 23, 219–236. [Google Scholar]
- Cuajungco, MP; Lees, GJ. Zinc metabolism in the brain: relevance to human neurodegenerative disorders. Neurobiol. Disease 1997, 4, 137–169. [Google Scholar]
- Frederickson, CJ; Suh, SW; Silva, D; Thompson, RB. Importance of zinc in the central nervous system: the zinc-containing neuron. J. Nutr 2000, 130, 1471S–1483S. [Google Scholar]
- Duncan, MW; Marini, AM; Watters, R; Kopin, IJ; Markey, SP. Zinc, a neurotoxin to cultured neurons, contaminates cycad flour prepared by traditional guamanian methods. J. Neurosci 1992, 12, 1523–1537. [Google Scholar]
- Choi, DW; Yokoyama, M; Koh, J. Zinc neurotoxicity in cortical cell culture. Neuroscience 1988, 24, 67–79. [Google Scholar]
- Frederickson, CJ. Neurobiology of zinc and zinc-containing neurons. Int. Rev. Neurobiol 1989, 31, 145–238. [Google Scholar]
- Koh, JY; Choi, DW. Zinc toxicity on cultured cortical neurons: involvement of N-methyl-D-aspartate receptors. Neuroscience 1994, 60, 1049–1057. [Google Scholar]
- Wang, YX; Quastel, DM. Multiple actions of zinc on transmitter release at mouse end-plates. Pflugers. Arch.-Eur. J. Physiol 1990, 415, 582–587. [Google Scholar]
- Colvin, RA; Davis, N; Nipper, RW; Carter, PA. Zinc transport in the brain: routes of zinc influx and efflux in neurons. J. Nutr 2000, 130, 1484S–1487S. [Google Scholar]
- Weiss, JH; Hartley, DM; Koh, JY; Choi, DW. AMPA receptor activation potentiates zinc neurotoxicity. Neuron 1993, 10, 43–49. [Google Scholar]
- Yin, HZ; Weiss, JH. Zn(2+) permeates Ca(2+) permeable AMPA/kainate channels and triggers selective neural injury. Neuroreport 1995, 6, 2553–2556. [Google Scholar]
- Sensi, SL; Ton-That, D; Weiss, JH. Mitochondrial sequestration and Ca(2+)-dependent release of cytosolic Zn(2+) loads in cortical neurons. Neurobiol. Disease 2002, 10, 100–108. [Google Scholar]
- Masters, BA; Quaife, CJ; Erickson, JC; Kelly, EJ; Froelick, GJ; Zambrowicz, BP; Brinster, RL; Palmiter, RD. Metallothionein III is expressed in neurons that sequester zinc in synaptic vesicles. J. Neurosci 1994, 14, 5844–5857. [Google Scholar]
- Palmiter, RD; Findley, SD; Whitmore, TE; Durnam, DM. MT-III, a brain-specific member of the metallothionein gene family. Proc. Nat. Acad. Sci. USA 1992, 89, 6333–6337. [Google Scholar]
- Yokoyama, M; Koh, J; Choi, DW. Brief exposure to zinc is toxic to cortical neurons. Neurosci. Lett 1986, 71, 351–355. [Google Scholar]
- Frederickson, CJ; Klitenick, MA; Manton, WI; Kirkpatrick, JB. Cytoarchitectonic distribution of zinc in the hippocampus of man and the rat. Brain. Res 1983, 273, 335–339. [Google Scholar]
- Assaf, SY; Chung, SH. Release of endogenous Zn2+ from brain tissue during activity. Nature 1984, 308, 734–736. [Google Scholar]
- Sloviter, RS. A selective loss of hippocampal mossy fiber Timm stain accompanies granule cell seizure activity induced by perforant path stimulation. Brain. Res 1985, 330, 150–153. [Google Scholar]
- Siesjo, BK. Basic mechanisms of traumatic brain damage. Ann. Emerg. Med 1993, 22, 959–969. [Google Scholar]
- Frederickson, CJ; Koh, JY; Bush, AI. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci 2005, 6, 449–462. [Google Scholar]
- Choi, DW; Koh, JY. Zinc and brain injury. Annu. Rev. Neurosci 1998, 21, 347–375. [Google Scholar]
- Weiss, JH; Sensi, SL; Koh, JY. Zn(2+): a novel ionic mediator of neural injury in brain disease. Trends. Pharmacol. Sci 2000, 21, 395–401. [Google Scholar]
- Tonder, N; Johansen, FF; Frederickson, CJ; Zimmer, J; Diemer, NH. Possible role of zinc in the selective degeneration of dentate hilar neurons after cerebral ischemia in the adult rat. Neurosci. Lett 1990, 109, 247–252. [Google Scholar]
- Koh, JY; Suh, SW; Gwag, BJ; He, YY; Hsu, CY; Choi, DW. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 1996, 272, 1013–1016. [Google Scholar]
- Suh, SW; Chen, JW; Motamedi, M; Bell, B; Listiak, K; Pons, NF; Danscher, G; Frederickson, CJ. Evidence that synaptically-released zinc contributes to neuronal injury after traumatic brain injury. Brain. Res 2000, 852, 268–273. [Google Scholar]
- Lee, JY; Cole, TB; Palmiter, RD; Koh, JY. Accumulation of zinc in degenerating hippocampal neurons of ZnT3-null mice after seizures: evidence against synaptic vesicle origin. J Neurosci 2000, 20, RC79. [Google Scholar]
- Jiang, D; Sullivan, PG; Sensi, SL; Steward, O; Weiss, JH. Zn(2+) induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J. Biol. Chem 2001, 276, 47524–47529. [Google Scholar]
- Sensi, SL; Ton-That, D; Sullivan, PG; Jonas, EA; Gee, KR; Kaczmarek, LK; Weiss, JH. Modulation of mitochondrial function by endogenous Zn2+ pools. Proc. Nat. Acad. Sci. USA 2003, 100, 6157–6162. [Google Scholar]
- Kim, EY; Koh, JY; Kim, YH; Sohn, S; Joe, E; Gwag, BJ. Zn2+ entry produces oxidative neuronal necrosis in cortical cell cultures. Eur. J. Neurosci 1999, 11, 327–334. [Google Scholar]
- Sensi, SL; Yin, HZ; Weiss, JH. Glutamate triggers preferential Zn2+ flux through Ca2+ permeable AMPA channels and consequent ROS production. Neuroreport 1999, 10, 1723–1727. [Google Scholar]
- Noh, KM; Koh, JY. Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J Neurosci 2000, 20, RC111. [Google Scholar]
- Seo, SR; Chong, SA; Lee, SI; Sung, JY; Ahn, YS; Chung, KC; Seo, JT. Zn2+-induced ERK activation mediated by reactive oxygen species causes cell death in differentiated PC12 cells. J. Neurochem 2001, 78, 600–610. [Google Scholar]
- Aizenman, E; Stout, AK; Hartnett, KA; Dineley, KE; McLaughlin, B; Reynolds, IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J. Neurochem 2000, 75, 1878–1888. [Google Scholar]
- Wei, G; Hough, CJ; Li, Y; Sarvey, JM. Characterization of extracellular accumulation of Zn2+ during ischemia and reperfusion of hippocampus slices in rat. Neuroscience 2004, 125, 867–877. [Google Scholar]
- Frederickson, CJ; Cuajungco, MP; LaBuda, CJ; Suh, SW. Nitric oxide causes apparent release of zinc from presynaptic boutons. Neuroscience 2002, 115, 471–474. [Google Scholar]
- Park, JA; Lee, JY; Sato, TA; Koh, JY. Co-induction of p75NTR and p75NTR-associated death executor in neurons after zinc exposure in cortical culture or transient ischemia in the rat. J. Neurosci 2000, 20, 9096–9103. [Google Scholar]
- Mukai, J; Hachiya, T; Shoji-Hoshino, S; Kimura, MT; Nadano, D; Suvanto, P; Hanaoka, T; Li, Y; Irie, S; Greene, LA; Sato, TA. NADE, a p75NTR-associated cell death executor, is involved in signal transduction mediated by the common neurotrophin receptor p75NTR. J. Biol. Chem 2000, 275, 17566–17570. [Google Scholar]
- Lobner, D; Canzoniero, LM; Manzerra, P; Gottron, F; Ying, H; Knudson, M; Tian, M; Dugan, LL; Kerchner, GA; Sheline, CT; Korsmeyer, SJ; Choi, DW. Zinc-induced neuronal death in cortical neurons. Cell. Mol. Biol 2000, 46, 797–806. [Google Scholar]
- Manev, H; Kharlamov, E; Uz, T; Mason, RP; Cagnoli, CM. Characterization of zinc-induced neuronal death in primary cultures of rat cerebellar granule cells. Exp. Neurol 1997, 146, 171–178. [Google Scholar]
- Barkalifa, R; Hershfinkel, M; Friedman, JE; Kozak, A; Sekler, I. The lipophilic zinc chelator DP-b99 prevents zinc induced neuronal death. Eur. J. Pharmacol 2009, 618, 15–21. [Google Scholar]
- Devirgiliis, C; Zalewski, PD; Perozzi, G; Murgia, C. Zinc fluxes and zinc transporter genes in chronic diseases. Mutat. Res 2007, 622, 84–93. [Google Scholar]
- Sensi, SL; Paoletti, P; Bush, AI; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci 2009, 10, 780–791. [Google Scholar]
- Ackland, ML; Michalczyk, A. Zinc deficiency and its inherited disorders: a review. Genes. Nutr 2006, 1, 41–49. [Google Scholar]
- Prasad, AS. Clinical manifestations of zinc deficiency. Annu. Rev. Nutr 1985, 5, 341–363. [Google Scholar]
- Prasad, AS; Halsted, JA; Nadimi, M. Syndrome of iron deficiency anemia, hepatosplenomegaly, hypogonadism, dwarfism and geophagia. Am. J. Med 1961, 31, 532–546. [Google Scholar]
- Prasad, AS; Miale, AJ; Farid, Z; Sandstead, HH; Schulert, AR. Zinc metabolism in patients with the symptoms of iron deficiency, anaemia, hepatosplenomegaly, dwarfism and hypogonadism. J. Lab. Clin. Med 1963, 61, 537–549. [Google Scholar]
- Sandstead, HH. Zinc deficiency. A public health problem? Am. J. Dis. Child 1991, 145, 853–859. [Google Scholar]
- Wang, K; Zhou, B; Kuo, YM; Zemansky, J; Gitschier, J. A novel member of a zinc transporter family is defective in acrodermatitis enteropathica. Am. J. Hum. Genet 2002, 71, 66–73. [Google Scholar]
- Aggett, PJ. Acrodermatitis enteropathica. J. Inherit. Metab. Dis 1983, 6, 39–43. [Google Scholar]
- Failla, ML; van de Veerdonk, M; Morgan, WT; Smith, JC, Jr. Characterization of zinc-binding proteins of plasma in familial hyperzincemia. J. Lab. Clin. Med 1982, 100, 943–952. [Google Scholar]
- Fessatou, S; Fagerhol, MK; Roth, J; Stamoulakatou, A; Kitra, V; Hadarean, M; Paleologos, G; Chandrinou, H; Sampson, B; Papassotiriou, I. Severe anemia and neutropenia associated with hyperzincemia and hypercalprotectinemia. J. Pediatr. Hematol. Oncol 2005, 27, 477–480. [Google Scholar]
- Smith, JC; Zeller, JA; Brown, ED; Ong, SC. Elevated plasmz zinc: a heritable anomaly. Science 1976, 193, 496–498. [Google Scholar]
- Saito, Y; Saito, K; Hirano, Y; Ikeya, K; Suzuki, H; Shishikura, K; Manno, S; Takakuwa, Y; Nakagawa, K; Iwasa, A; Fujikawa, S; Moriya, M; Mizoguchi, N; Golden, BE; Osawa, M. Hyperzincemia with systemic inflammation: a heritable disorder of calprotectin metabolism with rheumatic manifestations? J. Pediatr 2002, 140, 267–269. [Google Scholar]
- Sampson, B; Kovar, IZ; Rauscher, A; Fairweather-Tait, S; Beattie, J; McArdle, HJ; Ahmed, R; Green, C. A case of hyperzincemia with functional zinc depletion: a new disorder? Pediatr. Res 1997, 42, 219–225. [Google Scholar]
- Prasad, AS. Zinc deficiency and effects of zinc supplementation on sickle cell anemia subjects. Prog. Clin. Biol. Res 1981, 55, 99–122. [Google Scholar]
- Prasad, AS. Zinc deficiency in patients with sickle cell disease. Am. J. Clin. Nutr 2002, 75, 181–182. [Google Scholar]
- Dardenne, M; Savino, W; Wade, S; Kaiserlian, D; Lemonnier, D; Bach, JF. In vivo and in vitro studies of thymulin in marginally zinc-deficient mice. Eur. J. Immunol 1984, 14, 454–458. [Google Scholar]
- Wuehler, SE; Peerson, JM; Brown, KH. Use of national food balance data to estimate the adequacy of zinc in national food supplies: methodology and regional estimates. Public Health Nutr 2005, 8, 812–819. [Google Scholar]
- Cavdar, AO; Arcasoy, A; Cin, S; Babacan, E; Gozdasoglu, S. Geophagia in Turkey: iron and zinc deficiency, iron and zinc absorption studies and response to treatment with zinc in geophagia cases. Prog. Clin. Biol. Res 1983, 129, 71–97. [Google Scholar]
- Cavdar, OA. Zinc deficiency and geophagia. J. Pediatr 1982, 100, 1003–1004. [Google Scholar]
- Prasad, AS; Miale, A, Jr; Farid, Z; Sandstead, HH; Schulert, AR. Zinc metabolism in patients with the syndrome of iron deficiency anemia, hepatosplenomegaly, dwarfism, and hypognadism. J. Lab. Clin. Med 1963, 61, 537–549. [Google Scholar]
- Briefel, RR; Bialostosky, K; Kennedy-Stephenson, J; McDowell, MA; Ervin, RB; Wright, JD. Zinc intake of the U.S. population: findings from the third National Health and Nutrition Examination Survey, 1988–1994. J. Nutr 2000, 130, 1367S–1373S. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Plum, L.M.; Rink, L.; Haase, H. The Essential Toxin: Impact of Zinc on Human Health. Int. J. Environ. Res. Public Health 2010, 7, 1342-1365. https://doi.org/10.3390/ijerph7041342
Plum LM, Rink L, Haase H. The Essential Toxin: Impact of Zinc on Human Health. International Journal of Environmental Research and Public Health. 2010; 7(4):1342-1365. https://doi.org/10.3390/ijerph7041342
Chicago/Turabian StylePlum, Laura M., Lothar Rink, and Hajo Haase. 2010. "The Essential Toxin: Impact of Zinc on Human Health" International Journal of Environmental Research and Public Health 7, no. 4: 1342-1365. https://doi.org/10.3390/ijerph7041342