Sanger and Next Generation Sequencing Approaches to Evaluate HIV-1 Virus in Blood Compartments


Abstract
:1. Introduction
2. Methods
2.1. Cell Sorting
2.2. Nucleotide Acid Purification and PCR Amplification
2.3. Sequencing and Phylogenetic Analyses
3. Results and Discussion
4. Conclusion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Svicher, V.; Ceccherini-Silberstein, F.; Antinori, A.; Aquaro, S.; Perno, C.F. Understanding HIV compartments and reservoirs. Curr. HIV/AIDS Rep. 2014, 11, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Fauci, A.S. HIV reservoirs: Pathogenesis and obstacles to viral eradication and cure. AIDS 2012, 26, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gamez, S.; Hill, A.L.; Rosenbloom, D.I.; Petrov, D.A.; Nowak, M.A.; Pennings, P.S. Imperfect drug penetration leads to spatial monotherapy and rapid evolution of multidrug resistance. Proc. Natl. Acad. Sci. USA 2015, 112. [Google Scholar] [CrossRef] [PubMed]
- Potter, S.J.; Lemey, P.; Achaz, G.; Chew, C.B.; Vandamme, A.M.; Dwyer, D.E.; Dwyer, D.E.; Saksena, N.K. HIV-1 compartmentalization in diverse leukocyte populations during antiretroviral therapy. J. Leukoc. Biol. 2004, 76, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Ding, M.; Craigo, J.K.; Tarwater, P.; Chatterjee, R.; Roy, P.; Pratima, R.; Subhasish, K.G.; Bibhuti, S.; Dolonchapa, M.; et al. Genetic characterization of HIV-1 from semen and blood from clade C-infected subjects from India and effect of therapy in these body compartments. Virology 2010, 401, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.J.; Peters, P.J.; Caron, C.; Gonzalez-Perez, M.P.; Stones, L.; Ankghuambom, C.; Kemebradikumo, P.; McClure, C.P.; George, A.; Stephen, T.; et al. Intercompartmental recombination of HIV-1 contributes to env intrahost diversity and modulates viral tropism and sensitivity to entry inhibitors. J. Virol. 2011, 85, 6024–6037. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Picado, J.; Deeks, S.G. Persistent HIV-1 replication during antiretroviral therapy. Curr. Opin. HIV AIDS 2016, 11, 417–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.K.; Hezareh, M.; Gunthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.Q.; Lichterfeld, M. Diversity of HIV-1 reservoirs in CD4+ T-cell subpopulations. Curr. Opin. HIV AIDS 2016, 11, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Paquet, A.C.; Baxter, J.; Weidler, J.; Lie, Y.; Lawrence, J.; Kim, R.; Michael, B.; Eoin, C.; Colombe, C. Differences in reversion of resistance mutations to wild-type under structured treatment interruption and related increase in replication capacity. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.F.; Shafer, R.W. Web resources for HIV type 1 genotypic-resistance test interpretation. Clin. Infect. Dis. 2006, 42, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zou, X.; He, J.; Zheng, J.; Chiarella, J.; Kozal, M.J. HIV Drug Resistance Mutations (DRMs) Detected by Deep Sequencing in Virologic Failure Subjects on Therapy from Hunan Province, China. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Kearney, M.; Maldarelli, F.; Halvas, E.K.; Bixby, C.J.; Bazmi, H.; Rock, D.; Falloon, J.; Davey, R.T.J.; Dewar, R.L.; et al. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 2005, 43, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Parikh, U.M.; McCormick, K.; van Zyl, G.; Mellors, J.W. Future technologies for monitoring HIV drug resistance and cure. Curr. Opin. HIV AIDS 2017, 12, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Gibson, R.M.; Schmotzer, C.L.; Quinones-Mateu, M.E. Next-Generation Sequencing to Help Monitor Patients Infected with HIV: Ready for Clinical Use? Curr. Infect. Dis. Rep. 2014, 16. [Google Scholar] [CrossRef] [PubMed]
- Dudley, D.M.; Chin, E.N.; Bimber, B.N.; Sanabani, S.S.; Tarosso, L.F.; Costa, P.R.; Sauer, M.M.; Kallas, E.G.; O’Connor, D.H. Low-cost ultra-wide genotyping using Roche/454 pyrosequencing for surveillance of HIV drug resistance. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, C.K.; Brumme, C.J.; Liu, T.F.; Chui, C.K.; Chu, A.L.; Wynhoven, B.; Hall, T.A.; Trevino, C.; Shafer, R.W.; Harrigan, P.R. Automating HIV drug resistance genotyping with RECall, a freely accessible sequence analysis tool. J. Clin. Microbiol. 2012, 50, 1936–1942. [Google Scholar] [CrossRef] [PubMed]
- Shafer, R.W. Rationale and uses of a public HIV drug-resistance database. J. Infect. Dis. 2006, 194 (Suppl. 1), S51–S58. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Illumina. Available online: https://www.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 1 April 2018).
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Babraham Bioinformatics. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 1 May 2018).
- Armin, T.; Beerenwinkel, N. Available online: https://github.com/cbg-ethz/InDelFixer (accessed on 1 May 2018).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnett, D. Available online: https://github.com/pezmaster31/bamtools (accessed on 1 May 2018).
- Tzou, P.L.; Ariyaratne, P.; Varghese, V.; Lee, C.; Rakhmanaliev, E.; Villy, C.; Yee, M.; Tan, K.; Michel, G.; Pinsky, B.A.; et al. Comparison of an in vitro Diagnostic Next-Generation Sequencing Assay with Sanger Sequencing for HIV-1 Genotypic Resistance Testing. J. Clin. Microbiol. 2018, 56. [Google Scholar] [CrossRef] [PubMed]
- Alidjinou, E.K.; Deldalle, J.; Hallaert, C.; Robineau, O.; Ajana, F.; Choisy, P.; Hober, D.; Bocket, L. RNA and DNA Sanger sequencing versus next-generation sequencing for HIV-1 drug resistance testing in treatment-naive patients. J. Antimicrob. Chemother. 2017, 72, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Huber, M.; Metzner, K.J.; Geissberger, F.D.; Shah, C.; Leemann, C.; Klimkait, T.; Böni, J.; Trkola, A.; Zagordi, O. MinVar: A rapid and versatile tool for HIV-1 drug resistance genotyping by deep sequencing. J. Virol. Methods 2017, 240, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Nykoluk, M.; Taylor, T. HyDRA Web User Guide 2016. Available online: https://hydra.canada.ca/HyDRA_Web_User_Guide_Final_6Sept2016.pdf. (accessed on 1 May 2018).
- Wirden, M.; Soulie, C.; Valantin, M.A.; Fourati, S.; Simon, A.; Lambert-Niclot, S.; Bonmarchand, M.; Clavel-Osorio, C.; Marcelin, A.G.; Katlama, C.; et al. Historical HIV-RNA resistance test results are more informative than proviral DNA genotyping in cases of suppressed or residual viraemia. J. Antimicrob. Chemother. 2011, 66, 709–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaugerre, C.; Braun, J.; Charreau, I.; Delarue, S.; Nere, M.L.; de Castro, N.; May, T.; Marchou, B.; Simon, F.; Molina, J.M.; et al. Comparison of resistance mutation patterns in historical plasma HIV RNA genotypes with those in current proviral HIV DNA genotypes among extensively treated patients with suppressed replication. HIV Med. 2012, 13, 517–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derache, A.; Shin, H.S.; Balamane, M.; White, E.; Israelski, D.; Klausner, J.D.; Freeman, A.H.; Katzenstein, D. HIV drug resistance mutations in proviral DNA from a community treatment program. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Keulen, W.; Boucher, C.; Berkhout, B. Nucleotide substitution patterns can predict the requirements for drug-resistance of HIV-1 proteins. Antivir. Res. 1996, 31, 45–57. [Google Scholar] [CrossRef]
- Frost, S.D.; Nijhuis, M.; Schuurman, R.; Boucher, C.A.; Brown, A.J. Evolution of lamivudine resistance in human immunodeficiency virus type 1-infected individuals: The relative roles of drift and selection. J. Virol. 2000, 74, 6262–6268. [Google Scholar] [CrossRef] [PubMed]
- Rimsky, L.; Vingerhoets, J.; Van Eygen, V.; Eron, J.; Clotet, B.; Hoogstoel, A.; Boven, K.; Picchio, G. Genotypic and phenotypic characterization of HIV-1 isolates obtained from patients on rilpivirine therapy experiencing virologic failure in the phase 3 ECHO and THRIVE studies: 48-week analysis. J. Acquir. Immune Defic. Syndr. 2012, 59, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Nijhuis, M.; Schuurman, R.; de Jong, D.; Erickson, J.; Gustchina, E.; Albert, J.; Schipper, P.; Gulnik, S.; Boucher, C.A. Increased fitness of drug resistant HIV-1 protease as a result of acquisition of compensatory mutations during suboptimal therapy. AIDS 1999, 13, 2349–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cane, P.A.; Kaye, S.; Smit, E.; Tilston, P.; Kirk, S.; Shepherd, J.; Hopkins, M.; Zhang, H.; Geretti, A.M. Genotypic antiretroviral drug resistance testing at low viral loads in the UK. HIV Med. 2008, 9, 673–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monie, D.; Simmons, R.P.; Nettles, R.E.; Kieffer, T.L.; Zhou, Y.; Zhang, H.; Karmon, S.; Ingersoll, R.; Chadwick, K.; Zhang, H.; et al. A novel assay allows genotyping of the latent reservoir for human immunodeficiency virus type 1 in the resting CD4+ T cells of viremic patients. J. Virol. 2005, 79, 5185–5202. [Google Scholar] [CrossRef] [PubMed]
- Brennan, T.P.; Woods, J.O.; Sedaghat, A.R.; Siliciano, J.D.; Siliciano, R.F.; Wilke, C.O. Analysis of human immunodeficiency virus type 1 viremia and provirus in resting CD4+ T cells reveals a novel source of residual viremia in patients on antiretroviral therapy. J. Virol. 2009, 83, 8470–8481. [Google Scholar] [CrossRef] [PubMed]
- Jung, A.; Maier, R.; Vartanian, J.P.; Bocharov, G.; Jung, V.; Fischer, U.; Meese, E.; Wain-Hobson, S.; Meyerhans, A. Recombination: Multiply infected spleen cells in HIV patients. Nature 2002, 418. [Google Scholar] [CrossRef] [PubMed]
- Fraser, C. HIV recombination: What is the impact on antiretroviral therapy? J. R. Soc. Interface 2005, 2, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Geneviève, B.; Mohamed-Rachid, B.; Georges, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.M.; Dyer, W.B.; Workman, C.; Wang, B.; Peng, N.K.; Lachireddy, K.; Beng, C.C.; Sullivan, J.; Saksena, N.K. Drug resistance and viral evolution in plasma and peripheral blood cells during structured treatment interruption (STI) and non-interrupted HAART. Curr. HIV Res. 2007, 5, 235–250. [Google Scholar] [CrossRef] [PubMed]
- Turriziani, O.; Andreoni, M.; Antonelli, G. Resistant viral variants in cellular reservoirs of human immunodeficiency virus infection. Clin. Microbiol. Infect. 2010, 16, 1518–1524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nottet, H.S.; van Dijk, S.J.; Fanoy, E.B.; Goedegebuure, I.W.; de Jong, D.; Vrisekoop, N.; van Baarle, D.; Boltz, V.; Palmer, S.; Borleffs, J.C.; et al. HIV-1 can persist in aged memory CD4+ T lymphocytes with minimal signs of evolution after 8.3 years of effective highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2009, 50, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Chew, C.B.; Potter, S.J.; Wang, B.; Wang, Y.M.; Shaw, C.O.; Dwyer, D.E.; Saksena, N.K. Assessment of drug resistance mutations in plasma and peripheral blood mononuclear cells at different plasma viral loads in patients receiving HAART. J. Clin. Virol. 2005, 33, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Miller, M.D.; Danovich, R.M.; Vandergrift, N.; Cai, F.; Hicks, C.B.; Daria, J.H.; Feng, G. Analysis of low-frequency mutations associated with drug resistance to raltegravir before antiretroviral treatment. Antimicrob. Agents Chemother. 2011, 55, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- El Bouzidi, K.; White, E.; Mbisa, J.L.; Sabin, C.A.; Phillips, A.N.; Mackie, N.; Pozniak, A.L.; Tostevin, A.; Pillay, D.; Dunn, D.T.; et al. HIV-1 drug resistance mutations emerging on darunavir therapy in PI-naive and -experienced patients in the UK. J. Antimicrob. Chemother. 2016, 71, 3487–3494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyeyune, F.; Gibson, R.M.; Nankya, I.; Venner, C.; Metha, S.; Akao, J.; Ndashimye, E.; Kityo, C.M.; Salata, R.A.; Mugyenyi, P.; et al. Low-Frequency Drug Resistance in HIV-Infected Ugandans on Antiretroviral Treatment Is Associated with Regimen Failure. Antimicrob. Agents Chemother. 2016, 60, 3380–3397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Messiaen, P.; Verhofstede, C.; Vandenbroucke, I.; Dinakis, S.; Van Eygen, V.; Thys, K.; Winters, B.; Aerssens, J.; Vogelaers, D.; Stuyver, L.J.; et al. Ultra-deep sequencing of HIV-1 reverse transcriptase before start of an NNRTI-based regimen in treatment-naive patients. Virology 2012, 426, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.; van Zyl, G.U.; Travers, S.A.; Kosakovsky Pond, S.L.; Engelbrech, S.; Murrell, B.; Scheffler, K.; Smith, D. Deep sequencing reveals minor protease resistance mutations in patients failing a protease inhibitor regimen. J. Virol. 2012, 86, 6231–6237. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Maggie, L. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Casadella, M.; Paredes, R. Deep sequencing for HIV-1 clinical management. Virus Res. 2017, 239, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, I.; Van Marck, H.; Mostmans, W.; Van Eygen, V.; Rondelez, E.; Thys, K.; Van Baelen, K.; Fransen, K.; Vaira, D.; Kabeya, K.; et al. HIV-1 V3 envelope deep sequencing for clinical plasma specimens failing in phenotypic tropism assays. AIDS Res. Ther. 2010, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]



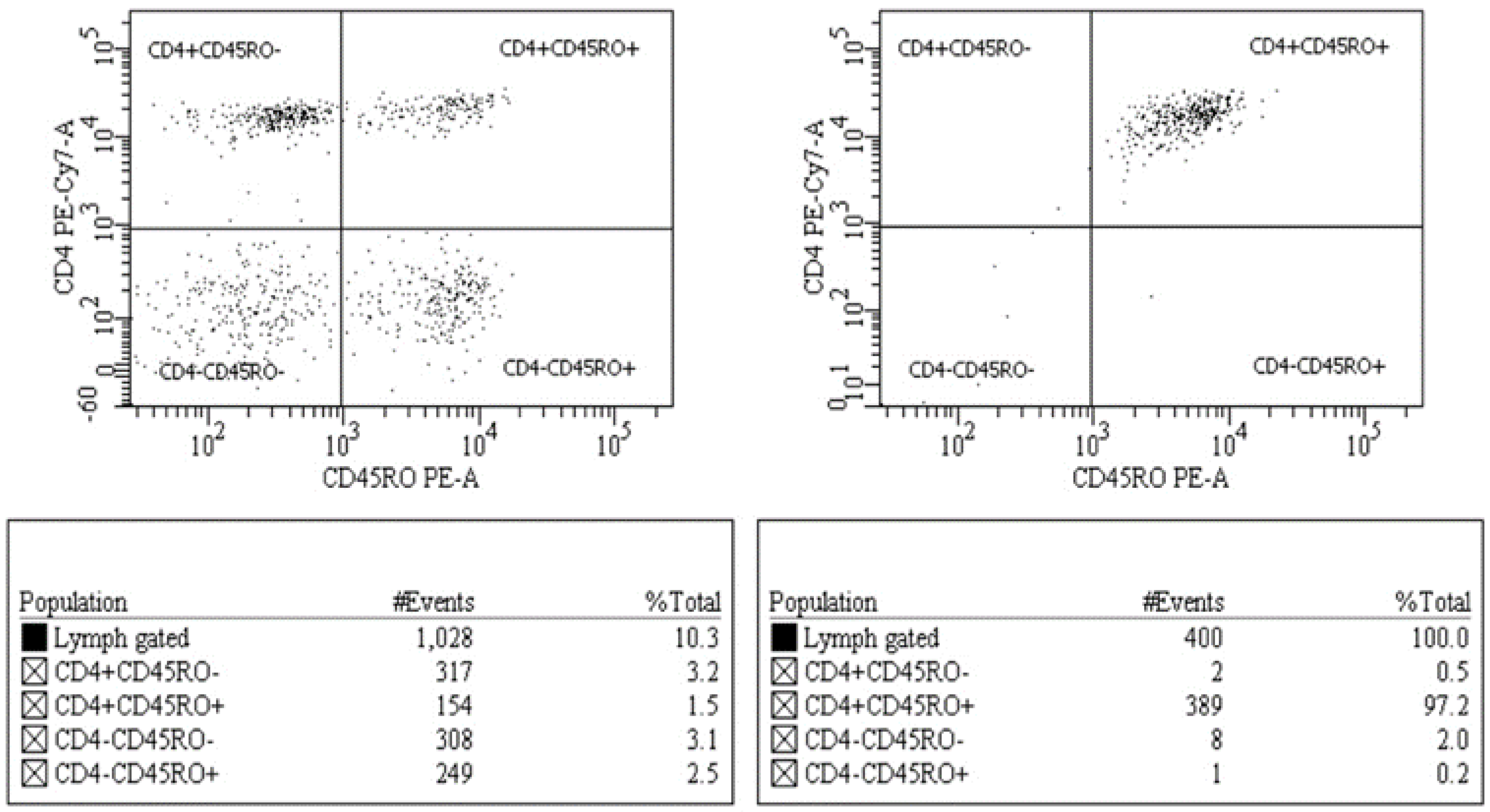{kind=link}
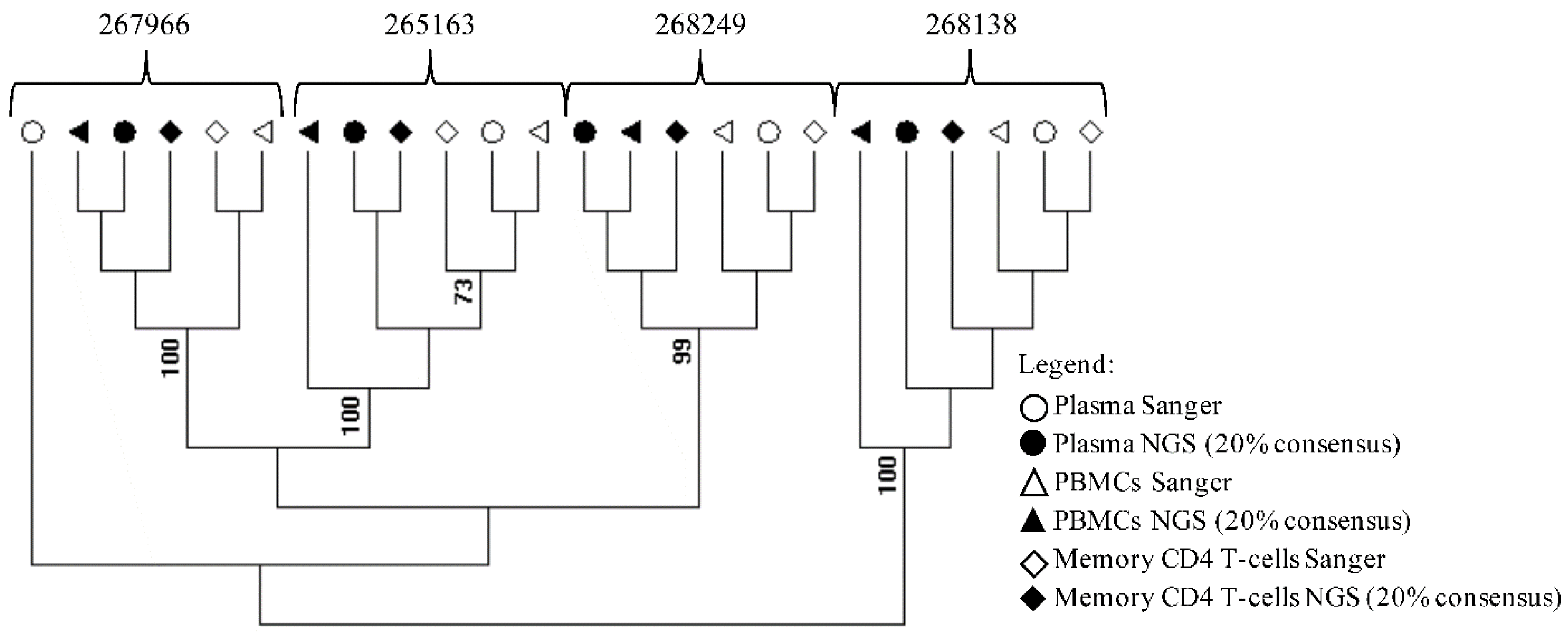{kind=link}
| Sanger |
|---|
| First Round Forward (Protease):5′-TGAARGAITGYACTGARAGRCAGGCTAAT-3′ Reverse (Protease): 5′-AYCTIATYCCTGGTGTYTCATTRTT-3′ Forward (RT): 5′-TTTYAGRGARCTYAATAARAGAACTCA-3′ Reverse (RT): 5′-CCTCITTYTTGCATAYTTYCCTGTT-3′ |
| Second Round Forward (Protease): 5′-YTCAGRCAGRCCRGARCCAACAGC-3′ Reverse (Protease): 5′-CTGGTGTYTCATTRTTKRTACTAGGT-3′ Forward (RT): 5′- TTYTGGGARGTYCARYTAGGRATACC-3′ Reverse (RT): 5′- GGYTCTTGRTAAATTTGRTATGTCCA-3′ |
| NGS |
| PR-INNER_F 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCTTTAACTTCCCTCAGGTCACTCT-3′ RT-1_R 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGTCAATGGCCATTGTTTAACTTTTGG-3′ RT-1_F 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCAAAAGTTAAACAATGGCCATTGAC-3′ PRNEWIN_R 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGCTGGTGTYTCATTRTTKRTACTAGGT-3′ 5FP127_F 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGATACTGCATTTACCATACCTAG-3′ 3F262_R 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGTCCCACTAACTTCTGTATGTC-3′ |
| Samples | Protease Major Resistance Mutations | RT Major Resistance Mutations |
|---|---|---|
| 267966 (Female) Viral load: 1450 copies/mL CD4+ T cell counts: 649 cells/µL Antiretroviral therapy: Yes | ||
| Plasma Sanger | none | none |
| Plasma NGS (consensus 20%) | none | M184V, E138EA |
| PBMCs Sanger | none | M184V, E138A |
| PBMC’s NGS (consensus 20%) | none | M184MV, E138EA |
| CD4+ memory T-cells Sanger | none | M184V, E138A |
| CD4+ memory T-cells NGS (consensus 20%) | none | M184V, E138A |
| 268138 (Male) Viral load: 12,500 copies/mL CD4+ T cell counts: 937 cells/µL Antiretroviral therapy: Not available | ||
| Plasma Sanger | none | none |
| Plasma NGS (consensus 20%) | none | none |
| PBMCs Sanger | none | none |
| PBMCs NGS (consensus 20%) | none | none |
| CD4+ memory T-cells Sanger | none | none |
| CD4+ memory T-cells NGS (consensus 20%) | none | none |
| 275163 (Female) Viral load: 16,105 copies/mL CD4+ T cell counts: 369 cells/µL Antiretroviral therapy: Yes | ||
| Plasma Sanger | none | none |
| Plasma NGS (consensus 20%) | none | none |
| PBMCs Sanger | none | none |
| PBMCs NGS (consensus 20%) | none | none |
| CD4+ memory T-cells Sanger | none | none |
| CD4+ memory T-cells NGS (consensus 20%) | none | none |
| 268249 (Female) Viral load: 32,468 copies/mL CD4+ T cell counts: 511 cells/µL Antiretroviral therapy: Yes | ||
| Plasma Sanger | none | none |
| Plasma NGS (consensus 20%) | none | none |
| PBMCs Sanger | none | none |
| PBMCs NGS (consensus 20%) | none | none |
| CD4+ memory T-cells Sanger | none | none |
| CD4+ memory T-cells NGS (consensus 20%) | none | none |
| Sample | Gene | Classification | Surveillance | WT | Position | Mutation | Frequency | Coverage |
|---|---|---|---|---|---|---|---|---|
| 267966 | ||||||||
| Plasma | RT | NRTI | Yes | M | 184 | V | 99.4 | 46,490 |
| RT | NNRTI | No | E | 138 | A | 99.29 | 55,090 | |
| PBMCs | RT | NRTI | Yes | M | 184 | V | 42.23 | 69,897 |
| RT | NNRTI | No | E | 138 | A | 42.29 | 69,891 | |
| RT | Other | No | V | 179 | I | 41.47 | 69,896 | |
| CD4+ memory T-cells | RT | NRTI | Yes | M | 184 | V | 99.12 | 54,457 |
| RT | NNRTI | No | E | 138 | A | 99.38 | 54,452 | |
| 268249 | ||||||||
| Plasma | PR/RT | - | - | WT | - | NP | - | - |
| PBMCs | PR/RT | - | - | WT | - | NP | - | - |
| CD4+ memory T-cells | PR | Other | No | L | 10 | V | 12.69 | 9382 |
| 268138 | ||||||||
| Plasma | PR | Other | No | A | 71 | V | 99.17 | 20,609 |
| PBMCs | PR | Other | No | A | 71 | V | 98.64 | 66,474 |
| CD4+ memory T-cells | PR | Other | No | A | 71 | V | 98.49 | 79,020 |
| 275163 | ||||||||
| Plasma | PR | Other | No | K | 20 | R | 23.13 | 49,598 |
| PBMCs | PR | Other | No | K | 20 | R | 97.03 | 46,084 |
| CD4+ memory T-cells | PR | Other | No | K | 20 | R | 96.9 | 33,222 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arias, A.; López, P.; Sánchez, R.; Yamamura, Y.; Rivera-Amill, V. Sanger and Next Generation Sequencing Approaches to Evaluate HIV-1 Virus in Blood Compartments. Int. J. Environ. Res. Public Health 2018, 15, 1697. https://doi.org/10.3390/ijerph15081697
Arias A, López P, Sánchez R, Yamamura Y, Rivera-Amill V. Sanger and Next Generation Sequencing Approaches to Evaluate HIV-1 Virus in Blood Compartments. International Journal of Environmental Research and Public Health. 2018; 15(8):1697. https://doi.org/10.3390/ijerph15081697
Chicago/Turabian StyleArias, Andrea, Pablo López, Raphael Sánchez, Yasuhiro Yamamura, and Vanessa Rivera-Amill. 2018. "Sanger and Next Generation Sequencing Approaches to Evaluate HIV-1 Virus in Blood Compartments" International Journal of Environmental Research and Public Health 15, no. 8: 1697. https://doi.org/10.3390/ijerph15081697