The Occurrence of 275 Rare Diseases and 47 Rare Disease Groups in Italy. Results from the National Registry of Rare Diseases


Abstract
:1. Introduction
2. Materials and Methods
2.1. Set of Data Communicated to RNMR and Collection Procedures
- Personal identifiers of the patient: name, surname, place and date of birth, sex, and national ID code (represented by the Fiscal Code and by an Encrypted Univocal Patient Code derived from it)
- Patient place of residence
- Disease diagnosed (defined according to the exemption codes listed in MD 279/2001 [15])
- Center making the diagnosis, with geographical location (coded according to the National Health Service Information System [19])
- Date of disease onset
- Date of diagnosis
2.2. Aims, Procedures and Limitations of Data Curation
2.3. Symptoms Onset Age and Distribution between Sexes
2.4. Birth Prevalence and Incidence of Rare Diseases
2.5. Internal Consistency of the Incidence and Birth Prevalence Data and Comparison with Literature Data
2.6. Statistical Methods
3. Results and Discussion
3.1. General Description of the National Database
3.2. Data Quality: Missing, Inaccurate or Inconsistent Values
3.3. Data Quality: Selection of the Regions and Reference Period for Frequency Rates Assessment
3.4. Symptoms Onset Age and Sex Distribution
3.5. Incidence in the General Population and Prevalence in Live Births
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- ORPHANET. Rare Disease Registries in EU. ORPHANET Report Series. May 2017. Available online: http://www.orpha.net/orphacom/cahiers/docs/GB/Registries.pdf (accessed on 15 November 2017).
- Taruscio, D.; Gainotti, S.; Mollo, E.; Vittozzi, L.; Bianchi, F.; Ensini, M.; Posada, M. The current situation and needs of rare disease registries in Europe. Public Health Genom. 2013, 16, 288–298. [Google Scholar] [CrossRef] [PubMed]
- ORPHANET. Prevalence and Incidence of Rare Diseases: Bibliographic Data Prevalence, Incidence or Number of Published Cases Listed by Diseases (in Alphabetical Order). ORPHANET Report Series N.1. June 2017. Available online: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf (accessed on 15 July 2017).
- Rubinstein, Y.R.; Groft, S.C.; Bartek, R.; Brown, K.; Christensen, R.A.; Collier, E.; Farber, A.; Farmer, J.; Ferguson, J.H.; Forrest, C.B.; et al. Creating a global rare disease patient registry linked to a rare diseases biorepository database: Rare Disease-HUB (RD-HUB). Contemp. Clin. Trials 2010, 31, 394–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Union. Council Recommendation of 8 June 2009 on an Action in the Field of Rare Diseases. The Council of the European Union. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:C:2009:151:0007:0010:EN:PDF (accessed on 23 August 2013).
- European Project for Rare Diseases National Plans Development (EUROPLAN). Selecting Indicators to Evaluate the Achievements of RD Initiatives. Available online: http://www.europlanproject.eu/_europlanproject/Resources/docs/2008-2011_3.EuroplanIndicators.pdf (accessed on 22 November 2017).
- European Committee of Experts on Rare Diseases (EUCERD). Core Recommendations on Rare Disease Patient Registration and Data Collection. June 2013. Available online: http://www.eucerd.eu/wp-content/uploads/2013/06/EUCERD_Recommendations_RDRegistryDataCollection_adopted.pdf (accessed on 15 July 2017).
- EU Patient Registries Initiative (PARENT Joint Action). Available online: www.patientregistries.eu (accessed on 3 April 2017).
- Building Consensus and Synergies for the Registration of Rare Disease Patients in the European Union (EPIRARE Project). Available online: www.epirare.eu (accessed on 3 April 2017).
- An Integrated Platform Connecting Databases, Registries, Biobanks and Clinical Bioinformatics for Rare Disease Research (RD-Connect Project). Available online: www.rd-connect.eu (accessed on 3 April 2017).
- Martin, S.; Kinsner-Ovaskainen, A.; Lanzoni, M.; Papadopoulou, A. European Platform on Rare Diseases Registration. Oral Presentation at 3rd European Reference Networks Conference (Vilnius, 9 March 2017). Available online: https://ec.europa.eu/health/sites/health/files/ern/docs/20170309_rt2_05_martin_pres_en.pdf (accessed on 3 April 2017).
- International Rare Disease Research Consortium (IRDiRC). Available online: http://www.irdirc.org/ (accessed on 3 April 2017).
- European Union. European Reference Networks: Working for Patients with Rare, Low-Prevalence and Complex Diseases. Available online: https://ec.europa.eu/health/sites/health/files/ern/docs/2017_brochure_en.pdf (accessed on 3 April 2017).
- Taruscio, D.; Vittozzi, L.; Choquet, R.; Heimdal, K.; Iskrov, G.; Kodra, Y.; Landais, P.; Posada, M.; Stefanov, R.; Steinmueller, C.; et al. National Registries of Rare Diseases in Europe: An Overview of the Current Situation and Experiences. Public Health Genom. 2015, 18, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Ministero della Salute. Decreto 18 Maggio 2001, n. 279—Regolamento di Istituzione della Rete Nazionale delle Malattie Rare e di Esenzione dalla Partecipazione al Costo delle Relative Prestazioni Sanitarie, ai Sensi dell’Articolo 5, Comma 1, Lettera b), del Decreto Legislativo 29 Aprile 1998, n. 124. (Gazzetta Ufficiale n. 160 of 12/07/2001 Suppl. Ordinario n.180/L). Available online: http://www.salute.gov.it/imgs/C_17_normativa_194_allegato.pdf (accessed on 3 April 2017).
- Taruscio, D.; Mollo, E.; Gainotti, S.; Posada, M.; Bianchi, F.; Vittozzi, L. The EPIRARE proposal of a set of indicators and common data elements for the European platform for rare disease registration. Arch. Public Health 2014, 72, 35. [Google Scholar] [CrossRef] [PubMed]
- Fregonese, L.; Rodwell, C.; Aymé, S. RDTF Report on Health Indicators for Rare Diseases: II—Conceptual Framework for the Use of Health Indicators for the Monitoring of Quality of Care. September 2011. Available online: http://nestor.orpha.net/EUCERD/upload/file/RDTFIndicators2011.pdf (accessed on 15 April 2013).
- Mazzucato, M.; Visonà Dalla Pozza, L.; Manea, S.; Minichiello, C.; Facchin, P. A population-based registry as a source of health indicators for rare diseases: The ten-year experience of the Veneto Region’s rare diseases registry. Orphanet J. Rare Dis. 2014, 9, 37–49. Available online: http://www.ojrd.com/content/9/1/37 (accessed on 3 April 2017). [CrossRef] [PubMed]
- Ministero della Salute. Elenco delle Strutture di Ricovero Attive al 30 Giugno 2014. Available online: http://www.salute.gov.it/portale/documentazione/p6_2_8_1_1.jsp?id=13 (accessed on 15 June 2015).
- Conferenza Permanente per i Rapporti tra lo Stato, le Regioni e le Province Autonome di Trento e di Bolzano. Accordo, ai Sensi dell’Articolo 4 del Decreto Legislativo 28 Agosto 1997, n.281, tra il Governo, le Regioni e le Province Autonome di Trento e di Bolzano sul Riconoscimento di Centri di Coordinamento Regionali e/o Interregionali, di Presidi Assistenziali Sovraregionali per Patologie a Bassa Prevalenza e sull’Attivazione dei Registri Regionali e/o Interregionali delle Malattie Rare. Rep. 103/CSR del 10 Maggio 2007. Available online: http://www.regione.lazio.it/malattierare/allegati/ASR-10-5-2007.pdf (accessed on 3 April 2017).
- Taruscio, D.; Kodra, Y.; Ferrari, G.; Vittozzi, L.; National Rare Diseases Registry Collaborating Group. The Italian National Rare Diseases Registry. Blood Transfus. 2014, 12 (Suppl. 3), 606–613. [Google Scholar]
- ISTAT Demographic Tables. Available online: http://www.istat.it/it/archivio/bilancio+demografico (accessed on 10 May 2016).
- ISTAT Natality Tables. Available online: http://dati.istat.it/Index.aspx (accessed on 10 May 2016).
- ORPHANET. Disease Data Sheets. Available online: http://www.orpha.net/consor/cgi-bin/Disease_Search_Simple.php?lng=EN (accessed on 3 April 2017).
- Kodra, Y.; Minelli, G.; Rocchetti, A.; Manno, V.; Carinci, A.; Conti, S.; Taruscio, D.; on behalf of the National Rare Diseases Registry Collaborating Group. The Italian National Rare Diseases Registry: A Model of Comparison and Integration with Hospital Discharge Data. J. Public Health 2017. [Google Scholar] [CrossRef]
- Dutly, F.; Altwegg, M. Whipple’s Disease and “Tropheryma whippelii”. Clin. Microbiol. Rev. 2001, 14, 561–583. [Google Scholar] [CrossRef] [PubMed]
- Pedrini, E.; Jennes, I.; Tremosini, M.; Milanesi, A.; Mordenti, M.; Parra, A.; Sgariglia, F.; Zuntini, M.; Campanacci, L.; Fabbri, N.; et al. Genotype-phenotype correlation study in 529 patients with multiple hereditary exostoses: Identification of “protective” and “risk” factors. J. Bone Jt. Surg. Am. 2011, 93, 2294–2302. [Google Scholar] [CrossRef] [PubMed]
- Hausmanowa-Petrusewicz, I.; Zaremba, J.; Borkowska, J.; Szirkowiec, W. Chronic proximal spinal muscular atrophy of childhood and adolescence: Sex influence. J. Med. Genet. 1984, 21, 447–450. [Google Scholar] [CrossRef] [PubMed]
- NIH-NCATS Best Vitelliform Macular Dystrophy. Available online: https://rarediseases.info.nih.gov/diseases/182/best-vitelliform-macular-dystrophy (accessed on 10 November 2017).
- OMIM Entry #162500 Neuropathy, Hereditary, with Liability to Pressure Palsies; HNPP. Available online: https://www.omim.org/entry/162500#clinicalFeatures (accessed on 5 November 2017).
- Roos, D.; Thygesen, P. Familial recurrent polyneuropathy: A family and a survey. Brain 1972, 95, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, V.; Beldjord, C.; Taillandier, A.; Szpiro-Tapia, S.; Cusin, V.; Gerson, F.; Philippe, C.; Mandel, J.-L.; French National Working Group on Fragile X Syndrome. Five years of molecular diagnosis of fragile X syndrome (1997–2001): A collaborative study reporting 95% of the activity in France. Am. J. Med. Genet. 2004, 129A, 218–224. [Google Scholar] [CrossRef] [PubMed]
- ISTAT. Popolazione per Età, Sesso e Stato Civile 2013. Available online: http://www.tuttitalia.it/statistiche/popolazione-eta-sesso-stato-civile-2013/ (accessed on 10 November 2017).
- Hazra, N.; Dregan, A.; Charlton, J.; Gulliford, M.C.; D’Cruz, D.P. Incidence and mortality of relapsing polychondritis in the UK: A population-based cohort study. Rheumatology 2015, 54, 2181–2187. [Google Scholar] [CrossRef] [PubMed]
- Groth, K.A.; Hove, H.; Kyhl, K.; Folkestad, L.; Gaustadnes, M.; Vejlstrup, N.; Stochholm, K.; Østergaard, J.R.; Andersen, N.H.; Gravholt, C.H. Prevalence, incidence, and age at diagnosis in Marfan Syndrome. Orphanet J. Rare Dis. 2015, 10, 153. [Google Scholar] [CrossRef] [PubMed]
- Kleijer, W.J.; Laugel, V.; Berneburg, M.; Nardo, T.; Fawcett, H.; Gratchev, A.; Jaspers, N.G.; Sarasin, A.; Stefanini, M.; Lehmann, A.R. Incidence of DNA repair deficiency disorders in western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair 2008, 7, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Kubota, M.; Ohta, S.; Ando, A.; Koyama, A.; Terashima, H.; Kashii, H.; Hoshino, H.; Sugita, K.; Hayashi, M. Nationwide survey of Cockayne syndrome in Japan: Incidence, clinical course and prognosis. Pediatr. Int. 2015, 57, 339–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual-Castroviejo, I.; Lopez-Rodriguez, L.; de la Cruz Medina, M.; Salamanca-Maesso, C.; Roche Herrero, C. Hypomelanosis of Ito: Neurological complications in 34 cases. Can. J. Neurol. Sci. 1988, 15, 124–129. [Google Scholar] [CrossRef] [PubMed]
- EUROCAT. Prevalence Tables. Available online: http://www.eurocat-network.eu/accessprevalencedata/prevalencetables (accessed on 10 November 2017).
- Zenteno-Ruiz, J.C.; Kofman-Alfaro, S.; Méndez, J.P. 46,XX sex reversal. Arch. Med. Res. 2001, 32, 559–566. [Google Scholar] [CrossRef]
- Shannon, N.L.; Maltby, E.L.; Rigby, A.S.; Quarrell, O.W.J. An epidemiological study of Wolf-Hirschhorn syndrome: Life expectancy and cause of mortality. J. Med. Genet. 2001, 38, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Szabó, N.Z. Epidemiology of Central Nervous System Malformations in South-Eastern Hungary. Ph.D. Thesis, Department of Paediatrics, Faculty of Medicine, University of Szeged, Szeged, Hungary, 2012. Available online: http://doktori.bibl.u-szeged.hu/1485/1/SZAB%C3%93_N%C3%93RA_PhD_ertekezes.pdf (accessed on 15 November 2017).
- Barisic, I.; Boban, L.; Greenlees, R.; Garne, E.; Wellesley, D.; Calzolari, E.; Addor, M.-C.; Arriola, L.; Bergman, J.E.H.; Braz, P.; et al. Holt Oram syndrome: A registry-based study in Europe. Orphanet J. Rare Dis. 2014, 9, 156. Available online: https://ojrd.biomedcentral.com/track/pdf/10.1186/s13023-014-0156-y?site=ojrd.biomedcentral.com (accessed on 10 November 2017). [CrossRef] [PubMed] [Green Version]
- Noonan, J.A. Noonan syndrome revisited. J. Pediatr. 1999, 135, 667–668. [Google Scholar] [CrossRef]
- Stoll, C.; Dott, B.; Roth, M.-P.; Alembik, Y. Birth prevalence rates of skeletal dysplasias. Clin. Genet. 1989, 35, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Holmes, L.B. Common Malformations; Oxford University Press: Oxford, UK, 2012; pp. 277–283. [Google Scholar]
- Blyth, M.; Maloney, V.; Beal, S.; Collinson, M.; Huang, S.; Crolla, J.; Temple, I.K.; Baralle, D. Pallister-Killian syndrome: A study of 22 British patients. J. Med. Genet. 2015, 52, 454–464. [Google Scholar] [CrossRef] [PubMed]


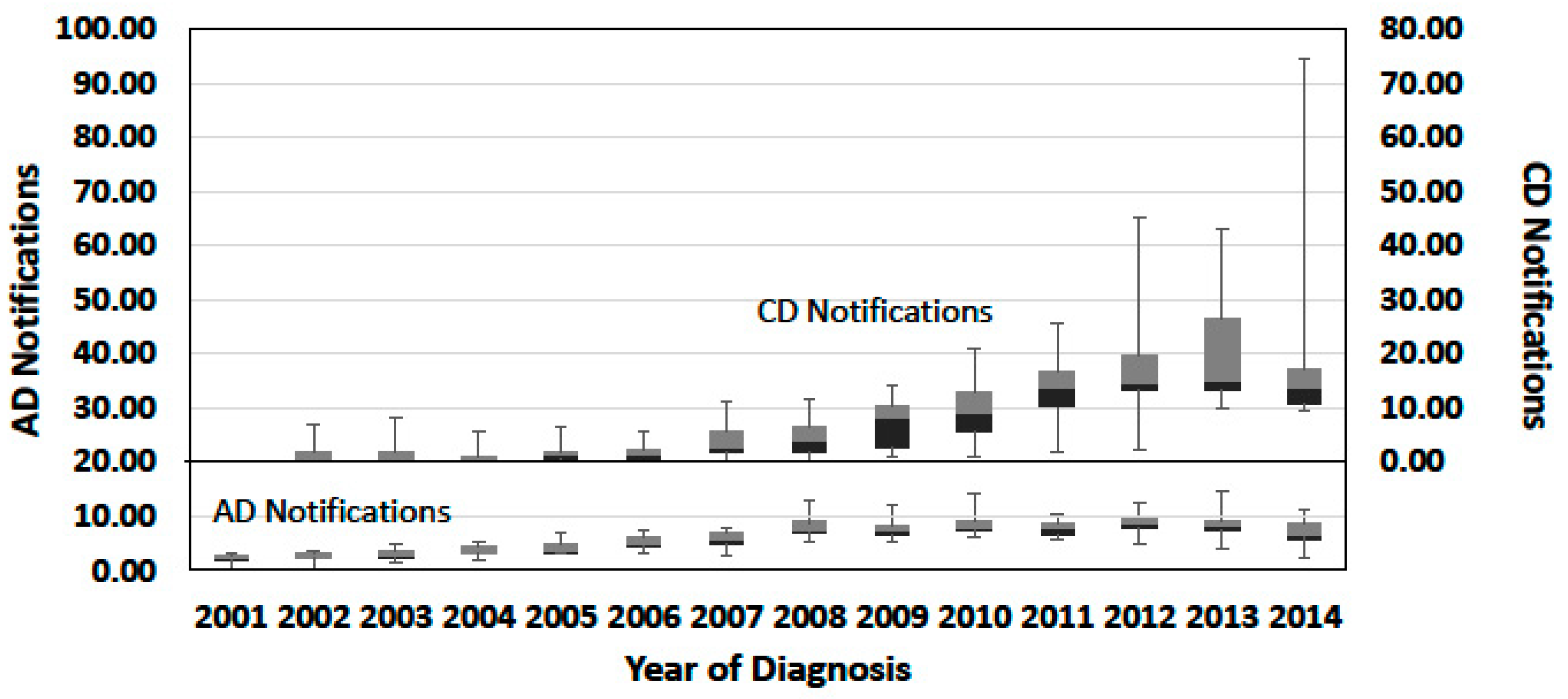{kind=link}
| RNMR Code | ORPHA Code | Disease | Records (N) | Median Onset Age (Years) | Life Stages Reported by ORPHANET [24] |
|---|---|---|---|---|---|
| RNG040 | 1791 | Frontofacionasal dysplasia | 7 | −0.28 | Neonatal |
| RN0490 | 3447 | Weaver syndrome | 7 | −0.23 | Neonatal, Antenatal |
| RNG030 | 87 | Apert syndrome | 13 | −0.18 | Antenatal, Neonatal |
| RNG040 | 2108 | Hallermann-Streiff syndrome | 9 | −0.17 | Neonatal, Infancy |
| RP0040 | 1915 | Fetal alcohol syndrome | 56 | −0.14 | Antenatal, Neonatal |
| RNG070 | 313 | Lamellar ichthyosis | 60 | −0.11 | Neonatal |
| RN0400 | 1540 | Jackson-Weiss syndrome | 12 | −0.10 | Neonatal |
| RNG040 | 207 | Crouzon disease | 43 | −0.09 | Infancy, Neonatal |
| RN1150 | 1340 | Cardiofaciocutaneous syndrome | 54 | −0.06 | Antenatal, Neonatal |
| RN0640 | 1114 | Aplasia cutis congenita | 14 | −0.06 | Antenatal, Neonatal |
| RFG010 | 141 | Canavan disease | 6 | −0.06 | Neonatal, infancy (severe form); childhood (mild form) |
| RNG040 | 1452 | Cleidocranial dysostosis | 7 | −0.05 | Neonatal |
| RCG060 | 352 | Galactosemia | 58 | −0.05 | Infancy, Neonatal, Childhood |
| RN0130 | 35,737 | Morning glory syndrome | 10 | −0.05 | Childhood |
| RNG050 | 429 | Hypochondroplasia | 7 | −0.05 | Childhood |
| RP0010 | 290 | Congenital rubella syndrome | 31 | −0.05 | Antenatal, Neonatal |
| RN0390 | 380 | Greig cephalopolysyndactyly syndrome | 15 | −0.04 | Neonatal |
| RDG020 | 326 | Factor V deficiency | 24 | −0.04 | All ages |
| RN0100 | 708 | Peters anomaly | 12 | −0.04 | Infancy, Neonatal |
| RCG040 | 238,583 | Hyperphenylalaninemia | 673 | −0.04 | Neonatal, Infancy |
| RN1040 | 710 | Pfeiffer syndrome | 10 | −0.03 | Antenatal, Neonatal |
| RN1530 | 500 | LEOPARD syndrome | 37 | −0.03 | Childhood |
| RCG040 | 407 | Non-ketotic hyperglycinemia | 6 | −0.03 | Infancy, Neonatal |
| RCG060 | 348 | Fructose-1,6-bisphosphatase deficiency | 4 | −0.03 | All ages |
| RN0510 | 464 | Incontinentia pigmenti | 74 | −0.03 | Neonatal |
| RN1640 | 1466 | COFS syndrome | 6 | −0.03 | Neonatal, Antenatal |
| RN1760 | 912 | Zellweger syndrome | 13 | −0.02 | Neonatal |
| RNG070 | 461 | Recessive X-linked ichthyosis | 60 | −0.02 | Neonatal |
| RP0060 | 415,286 | Bilirubin encephalopathy | 7 | −0.02 | Neonatal |
| RCG050 | 23 | Argininosuccinatelyase deficiency | 12 | −0.02 | Neonatal, All ages |
| RN0910 | 374 | Goldenhar syndrome | 187 | −0.02 | Neonatal, Antenatal |
| RN0600 | 312 | Epidermolytic ichthyosis | 32 | −0.02 | Neonatal |
| RDG020 | 327 | Factor VII deficiency | 98 | −0.02 | All ages |
| RN0990 | 570 | Moebius syndrome | 52 | −0.02 | Neonatal |
| RN0320 | 2368 | Gastroschisis | 57 | −0.02 | Neonatal, Antenatal |
| RDG020 | 328 | Factor X deficiency | 9 | −0.02 | All ages |
| RN0700 | 280 | Wolf-Hirschhorn syndrome | 64 | −0.02 | Neonatal, Antenatal |
| RN1080 | 813 | Silver-Russell syndrome | 85 | −0.02 | Neonatal, Antenatal |
| RN0180 | 1203 | Duodenal atresia | 75 | −0.02 | Antenatal, Neonatal, Infancy, Childhood |
| RN0540 | 1556 | Cutis marmorata telangiectatica congenita | 21 | −0.02 | Neonatal |
| RCG040 | 511 | Maple syrup urine disease | 26 | −0.02 | Infancy, Neonatal, Childhood |
| RNG070 | 634 | Netherton syndrome | 17 | −0.01 | Infancy, Neonatal |
| RN1100 | 808 | Seckel syndrome | 11 | −0.01 | Neonatal, Antenatal |
| RN1310 | 739 | Prader-Willi syndrome | 388 | −0.01 | Neonatal, Antenatal |
| RCG040 | 35 | Propionic acidemia | 4 | −0.01 | Infancy, Neonatal |
| RN0080 | 1764 | Familial dysautonomia | 6 | −0.01 | Neonatal, Antenatal |
| RDG020 | 331 | Factor XIII deficiency | 5 | −0.01 | All ages |
| RN0850 | 138 | CHARGE syndrome | 116 | −0.01 | Neonatal |
| RCG040 | 293,355 | Methylmalonic acidemia | 11 | −0.01 | All ages |
| RN1170 | 744 | Proteus syndrome | 19 | −0.01 | Infancy |
| RN0170 | 1201 | Atresia of small intestine | 77 | −0.01 | Neonatal |
| RN1140 | 107 | Branchio-oto-renal syndrome | 35 | −0.01 | All ages |
| RN1250 | 887 | VACTERL/VATER association | 80 | −0.01 | Neonatal |
| RN1410 | 199 | Cornelia de Lange syndrome | 74 | −0.01 | Neonatal, Antenatal |
| RN0930 | 392 | Holt-Oram syndrome | 23 | −0.01 | Neonatal |
| RDG020 | 98,878 | Hemophilia A | 1209 | −0.01 | Infancy, Neonatal |
| RDG020 | 98,879 | Hemophilia B | 202 | 0.00 | Infancy, Neonatal |
| RN1740 | 899 | Walker-Warburg syndrome | 6 | 0.00 | Infancy, Neonatal |
| RN1010 | 648 | Noonan syndrome | 459 | 0.00 | Neonatal |
| RN0430 | 2911 | Poland syndrome | 299 | 0.00 | Infancy, Neonatal |
| RDG020 | 745 | Protein C deficiency | 233 | 0.00 | Childhood |
| RN0200 | 388 | Hirschsprung disease | 244 | 0.00 | Infancy, Neonatal |
| RN0060 | 2162 | Holoprosencephaly | 46 | 0.00 | Neonatal, Antenatal |
| RN1240 | 857 | Townes-Brocks syndrome | 8 | 0.00 | All ages |
| RN1130 | 1297 | Branchio-oculo-facial syndrome | 7 | 0.00 | Neonatal |
| RNG060 | 1522 | Craniometaphyseal dysplasia | 11 | 0.00 | Childhood |
| RN1210 | 819 | Smith-Magenis syndrome | 45 | 0.00 | Infancy, Neonatal |
| RN0890 | 2053 | Freeman-Sheldon syndrome | 9 | 0.00 | Neonatal |
| RCG050 | 187 | Citrullinemia | 10 | 0.00 | Neonatal, Adult |
| RN0360 | 1465 | Coffin-Siris syndrome | 13 | 0.00 | Neonatal |
| RN0410 | 2311 | Jarcho-Levin syndrome | 11 | 0.00 | Neonatal, Antenatal |
| RN0280 | 950 | Acrodysostosis | 12 | 0.00 | Neonatal, Antenatal |
| RN1660 | 35,125 | Epidermal nevus syndrome | 15 | 0.00 | Childhood, Adolescent, Adult |
| RNG040 | 861 | Treacher-Collins syndrome | 16 | 0.00 | Neonatal |
| RN1450 | 94,068 | Congenital spondyloepiphyseal dysplasia | 14 | 0.00 | Neonatal |
| RNG060 | 289 | Ellis-Van Creveld syndrome | 8 | 0.00 | Neonatal, Antenatal |
| RN1590 | 884 | Pallister-Killian syndrome | 16 | 0.00 | Neonatal, Antenatal |
| RN0670 | 281 | Cri du chat syndrome | 46 | 0.00 | Neonatal |
| RN0790 | 915 | Aarskog-Scott syndrome | 28 | 0.00 | Childhood |
| RN0940 | 2322 | Kabuki make-up syndrome | 112 | 0.00 | Infancy, Neonatal |
| RN0040 | 475 | Joubert syndrome | 96 | 0.00 | Neonatal, Antenatal |
| RN1270 | 904 | Williams syndrome | 316 | 0.00 | Neonatal, Antenatal |
| RCG160 | 567 | Di George syndrome | 359 | 0.00 | Neonatal |
| RN1620 | 783 | Rubinstein-Taybi syndrome | 63 | 0.00 | All ages |
| RN1380 | 110 | Bardet-Biedl syndrome | 51 | 0.00 | Infancy, Neonatal, Antenatal |
| RN0660 | 870 | Down syndrome | 2283 | 0.00 | Antenatal, Neonatal |
| RN0680 | 881 | Turner syndrome | 947 | 0.00 | Infancy, Neonatal, Antenatal, Childhood |
| RN0770 | 3205 | Sturge-Weber syndrome | 134 | 0.00 | Infancy, Neonatal, Childhood, Adolescent |
| RN1330 | 908 | Fragile X syndrome | 288 | 0.00 | Neonatal, Infancy, Childhood |
| RN1510 | 90,308 | Klippel-Trénaunay syndrome | 170 | 0.00 | Infancy, Childhood, Adolescent |
| RN0110 | 77 | Aniridia | 69 | 0.00 | Infancy, Neonatal |
| RFG050 | 83,330 | Werdnig-Hoffman disease | 13 | 0.00 | Infancy, Neonatal |
| RCG040 | 26 | Methylmalonic acidemia with homocystinuria | 16 | 0.00 | All ages |
| RN0860 | 3157 | De Morsier syndrome | 46 | 0.00 | Infancy, Neonatal |
| RN1400 | 191 | Cockayne syndrome | 6 | 0.01 | All ages |
| RN1430 | 220 | Denys-Drash syndrome | 5 | 0.02 | Infancy, Neonatal |
| RN0240 | 2138 | True hermaphroditism | 17 | 0.02 | Infancy, Neonatal |
| RN1190 | 2614 | Nail-patella syndrome | 33 | 0.02 | Neonatal, Infancy, Childhood |
| RN0340 | 974 | Adams-Oliver syndrome | 11 | 0.02 | Neonatal |
| RDG020 | 903 | Von Willebrand disease | 800 | 0.02 | All ages |
| RC0180 | 205 | Crigler-Najjar syndrome | 27 | 0.04 | Neonatal |
| RN1730 | 893 | WAGR syndrome | 8 | 0.07 | Neonatal |
| RN0950 | 98,861 | Primary ciliary dyskinesia, Kartagener type | 345 | 0.08 | Neonatal, infancy |
| ORPHA Code | Disease | Records (N) | Females (%) | Males (%) | F:M Ratio and Other Literature Information Reported in ORPHANET Data Sheets [24] |
|---|---|---|---|---|---|
| 1791 | Frontofacionasal dysplasia | 7 | 100 | 0 | - |
| 209,981 | Iron refractory iron deficiency anemia | 4 | 100 | 0 | - |
| 881 | Turner syndrome | 1754 | 99 | 1 | Almost exclusively in F |
| 778 | Rett syndrome | 603 | 97 | 3 | Predominant in F |
| 538 | Lymphangioleiomyomatosis | 180 | 97 | 3 | Almost exclusively in F |
| 464 | Incontinentia pigmenti | 95 | 97 | 3 | 20:1 |
| 37,202 | Interstitial cystitis | 1192 | 95 | 5 | 9:1 |
| - | Storage pool deficiency | 11 | 91 | 9 | - |
| 816 | Sjögren-Larsson syndrome | 93 | 90 | 9 | - |
| - | Thrombocytopenic thrombotic purpura-hemolytic-uremic syndrome | 10 | 90 | 10 | - |
| 559 | Marinesco-Sjögren syndrome | 30 | 90 | 10 | - |
| 169,615 | Idiopathic central precocious puberty | 3686 | 89 | 11 | 10:1 |
| 98,973 | Posterior polymorphous corneal dystrophy | 8 | 88 | 13 | - |
| 79,473 | Porphyria variegata | 8 | 88 | 13 | Predominant in F |
| 1297 | Branchio-oculo-facial syndrome | 8 | 88 | 13 | - |
| 3143 | Schmidt syndrome | 72 | 86 | 14 | - |
| 1452 | Cleidocranial dysostosis | 7 | 86 | 14 | - |
| 429 | Hypochondroplasia | 7 | 86 | 14 | - |
| 809 | Mixed connective tissue disease | 1092 | 86 | 14 | 10:1 |
| 3287 | Takayasu arteritis | 346 | 85 | 14 | Predominant in F |
| 2483 | Melkersson-Rosenthal syndrome | 25 | 84 | 16 | - |
| 317 | Erythrokeratodermia variabilis | 6 | 83 | 17 | - |
| 98,958 | Droplet cornea | 17 | 82 | 18 | - |
| 436 | Hypophosphatasia | 11 | 82 | 18 | - |
| - | Secretion deficiency thrombocytopathy | 11 | 82 | 18 | - |
| 125 | Bloom syndrome | 5 | 80 | 20 | - |
| 79,273 | Hereditary coproporphyria | 5 | 80 | 20 | Predominant in F |
| 98,974 | Fuchs endothelial dystrophy | 30 | 80 | 20 | 3–4:1 |
| 581 | Mucopolysaccharidosis type 3 | 5 | 80 | 20 | - |
| 2059 | Fryns syndrome | 5 | 80 | 20 | - |
| 65,684 | Hirayama disease | 5 | 20 | 80 | 1:20 |
| 3447 | Weaver syndrome | 10 | 20 | 80 | - |
| 98,895 | Becker muscular dystrophy | 221 | 19 | 81 | Predominant in M |
| 379 | Chronic granulomatous disease | 87 | 18 | 82 | - |
| 221,117 | Gerstmann syndrome | 6 | 17 | 83 | - |
| 97,360 | Robinow syndrome | 6 | 17 | 83 | - |
| 478 | Kallmann syndrome | 509 | 16 | 84 | 1:5 |
| 101,330 | Porphyria cutanea tarda | 53 | 15 | 85 | Predominant in M |
| - | Opitz syndrome | 20 | 15 | 85 | - |
| 1466 | COFS syndrome | 7 | 14 | 86 | - |
| - | Simpson-Golabi-Behmel syndrome | 7 | 14 | 86 | - |
| 2796 | Pachydermoperiostosis | 15 | 13 | 87 | 1:7 |
| 481 | Kennedy disease | 51 | 12 | 88 | - |
| 98,879 | Hemophilia B | 271 | 11 | 89 | Predominant in M |
| 223 | Nephrogenic diabetes insipidus | 66 | 11 | 89 | - |
| 915 | Aarskog-Scott syndrome | 38 | 8 | 92 | Predominant in M |
| 98,878 | Hemophilia A | 1577 | 7 | 93 | Predominant in M |
| 98,896 | Duchenne muscular dystrophy | 258 | 5 | 95 | Predominant in M |
| 461 | Recessive X-linked ichthyosis | 65 | 3 | 97 | Almost exclusively in M |
| - | Klinefelter syndrome | 2071 | 0 | 100 | - |
| 330 | Factor XII deficiency | 4 | 0 | 100 | - |
| 425 | Familial hypoalphalipoproteinemia | 4 | 0 | 100 | - |
| 510 | Lesch-Nyhan disease | 12 | 0 | 100 | Severe form predominant in M |
| 534 | Lowe syndrome | 5 | 0 | 100 | Almost exclusively in M |
| 649 | Norrie disease | 5 | 0 | 100 | Almost exclusively in M |
| 580 | Mucopolysaccharidosis type 2 | 27 | 0 | 100 | Almost exclusively in M |
| 245 | Nager syndrome | 4 | 0 | 100 | - |
| ORPHA Code | Disease Name | National Incidence (/Million) |
|---|---|---|
| 156,071 | Keratoconus | 25.53 |
| 803 | Amyotrophic lateral sclerosis | 19.15 |
| 870 | Down syndrome | 7.76 |
| 169,615 | Idiopathic central precocious puberty | 7.33 |
| - | Lichen sclerosus | 5.92 |
| - | Bullous pemphigoid | 4.88 |
| 930 | Idiopathic achalasia | 4.63 |
| 117 | Behçet disease | 4.57 |
| - | Arnold-Chiari malformation | 4.19 |
| 2932 | Chronic inflammatory demyelinating polyneuropathy | 3.77 |
| - | Pemphigus vulgaris | 3.75 |
| - | Klinefelter syndrome | 3.75 |
| 397 | Giant cell arteritis | 3.73 |
| 399 | Huntington disease | 3.38 |
| 37,202 | Interstitial cystitis | 3.19 |
| 881 | Turner syndrome | 2.53 |
| 98,249 | Ehlers-Danlos syndrome | 2.43 |
| 774 | Hereditary hemorrhagic telangiectasia | 2.36 |
| 2331 | Kawasaki disease | 2.08 |
| 732 | Polymyositis | 1.95 |
| 733 | Familial adenomatous polyposis | 1.90 |
| 183 | Churg-Strauss syndrome | 1.88 |
| 221 | Dermatomyositis | 1.87 |
| 809 | Mixed connective tissue disease | 1.80 |
| 558 | Marfan syndrome | 1.79 |
| 900 | Wegener granulomatosis | 1.77 |
| - | Mixed cryoglobulinemia | 1.75 |
| - | Idiopathic torsion dystonia | 1.46 |
| - | Rheumatic endocarditis | 1.36 |
| 761 | Henoch-Schönlein purpura | 1.35 |
| 648 | Noonan syndrome | 1.30 |
| 805 | Tuberous sclerosis | 1.18 |
| 908 | Fragile X syndrome | 1.15 |
| 2911 | Poland syndrome | 1.14 |
| 727 | Microscopic polyangiitis | 1.14 |
| 171 | Primary sclerosing cholangitis | 1.03 |
| 904 | Williams syndrome | 0.98 |
| 2073 | Narcolepsy | 0.97 |
| 478 | Kallmann syndrome | 0.94 |
| 96,346 | Anorectal malformation | 0.93 |
| 388 | Hirschsprung disease | 0.88 |
| 790 | Retinoblastoma | 0.83 |
| 91,378 | Hereditary angioedema | 0.81 |
| 104 | Leber hereditary optic neuropathy | 0.79 |
| 739 | Prader-Willi syndrome | 0.78 |
| 70,590 | Infantile apnea | 0.78 |
| 116 | Beckwith-Wiedemann syndrome | 0.76 |
| 550 | MELAS | 0.73 |
| 49,041 | Retroperitoneal fibrosis | 0.72 |
| 3451 | West syndrome | 0.70 |
| Disease Group Name | ||
| - | Hereditary coagulation defects | 25.16 |
| - | Undifferentiated connective tissue syndromes | 17.75 |
| - | Hereditary retinic dystrophies | 17.60 |
| - | Hereditary anemias | 15.04 |
| - | Neurofibromatoses | 10.08 |
| - | Muscular dystrophies | 8.12 |
| - | Congenital alterations of iron metabolism | 8.04 |
| - | Disturbances of aminoacid transport and metabolism | 7.92 |
| - | Chromosomal duplication/deficiency syndromes | 7.82 |
| - | Hereditary neuropathies | 6.61 |
| ORPHA Code | Disease Name | Birth Prevalence (/100,000) |
|---|---|---|
| 870 | Down syndrome | 35.00 |
| - | Esophageal atresia and/or Isolated tracheo-esophageal fistula | 6.67 |
| 96,346 | Anorectal malformation | 6.37 |
| 3451 | West syndrome | 5.69 |
| 388 | Hirschsprung disease | 4.22 |
| 739 | Prader-Willi syndrome | 3.63 |
| - | Biliary atresia | 3.14 |
| 805 | Tuberous sclerosis | 2.84 |
| 116 | Beckwith-Wiedemann syndrome | 2.65 |
| 648 | Noonan syndrome | 2.45 |
| 881 | Turner syndrome | 1.96 |
| 1203 | Duodenal atresia | 1.86 |
| 374 | Goldenhar syndrome | 1.67 |
| 1201 | Atresia of small intestine | 1.67 |
| 290 | Congenital rubella syndrome | 1.47 |
| 904 | Williams syndrome | 1.47 |
| - | Epidermolysis bullosa | 1.37 |
| 2911 | Poland syndrome | 1.27 |
| 2368 | Gastroschisis | 1.18 |
| 138 | CHARGE syndrome | 1.18 |
| - | Lissencephaly | 1.08 |
| 98,861 | Primary ciliary dyskinesia, Kartagener type | 1.08 |
| 464 | Incontinentia pigmenti | 0.78 |
| 887 | VACTERL/VATER association | 0.78 |
| - | Congenital colobomatous optic disc | 0.69 |
| 3205 | Sturge-Weber syndrome | 0.69 |
| - | Alagille syndrome | 0.69 |
| 2162 | Holoprosencephaly | 0.59 |
| 1896 | EEC syndrome | 0.59 |
| 205 | Crigler-Najjar syndrome | 0.59 |
| 77 | Aniridia | 0.49 |
| 70,590 | Infantile apnea | 0.49 |
| 1915 | Fetal alcohol syndrome | 0.39 |
| 199 | Cornelia de Lange syndrome | 0.39 |
| 908 | Fragile X syndrome | 0.39 |
| 90,308 | Klippel-Trénaunay syndrome | 0.39 |
| 912 | Zellweger syndrome | 0.29 |
| 570 | Moebius syndrome | 0.29 |
| 199,642 | Microcephaly | 0.29 |
| 281 | Cri du chat syndrome | 0.29 |
| 783 | Rubinstein-Taybi syndrome | 0.29 |
| 380 | Greig cephalopolysyndactyly syndrome | 0.20 |
| 280 | Wolf-Hirschhorn syndrome | 0.20 |
| 813 | Silver-Russell syndrome | 0.20 |
| 475 | Joubert syndrome | 0.20 |
| 884 | Pallister-Killian syndrome | 0.20 |
| 3157 | De Morsier syndrome | 0.20 |
| 974 | Adams-Oliver syndrome | 0.20 |
| 726 | Alpers syndrome | 0.20 |
| 435 | Ito hypomelanosis | 0.20 |
| Disease Group Name | ||
| - | Disturbances of aminoacid transport and metabolism | 30.10 |
| - | Congenital craniofacial anomalies | 21.57 |
| - | Chromosomal duplication/deficiency syndromes | 8.73 |
| - | Disturbances of carbohydrate transport and metabolism, excluded diabetes mellitus | 5.39 |
| - | Neurofibromatoses | 3.92 |
| - | Congenital chondrodystrophies | 3.53 |
| - | Congenital ichthyoses | 2.94 |
| - | Other congenital anomalies with intellectual disability | 2.84 |
| - | Chromosomal aneuploidy syndromes | 2.55 |
| - | Urea cycle disturbances | 1.18 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taruscio, D.; Vittozzi, L.; Rocchetti, A.; Torreri, P.; Ferrari, L. The Occurrence of 275 Rare Diseases and 47 Rare Disease Groups in Italy. Results from the National Registry of Rare Diseases. Int. J. Environ. Res. Public Health 2018, 15, 1470. https://doi.org/10.3390/ijerph15071470
Taruscio D, Vittozzi L, Rocchetti A, Torreri P, Ferrari L. The Occurrence of 275 Rare Diseases and 47 Rare Disease Groups in Italy. Results from the National Registry of Rare Diseases. International Journal of Environmental Research and Public Health. 2018; 15(7):1470. https://doi.org/10.3390/ijerph15071470
Chicago/Turabian StyleTaruscio, Domenica, Luciano Vittozzi, Adele Rocchetti, Paola Torreri, and Luca Ferrari. 2018. "The Occurrence of 275 Rare Diseases and 47 Rare Disease Groups in Italy. Results from the National Registry of Rare Diseases" International Journal of Environmental Research and Public Health 15, no. 7: 1470. https://doi.org/10.3390/ijerph15071470