Cross-Talk between Wnt and Hh Signaling Pathways in the Pathology of Basal Cell Carcinoma

 , ,
, ,
Abstract
:1. Introduction
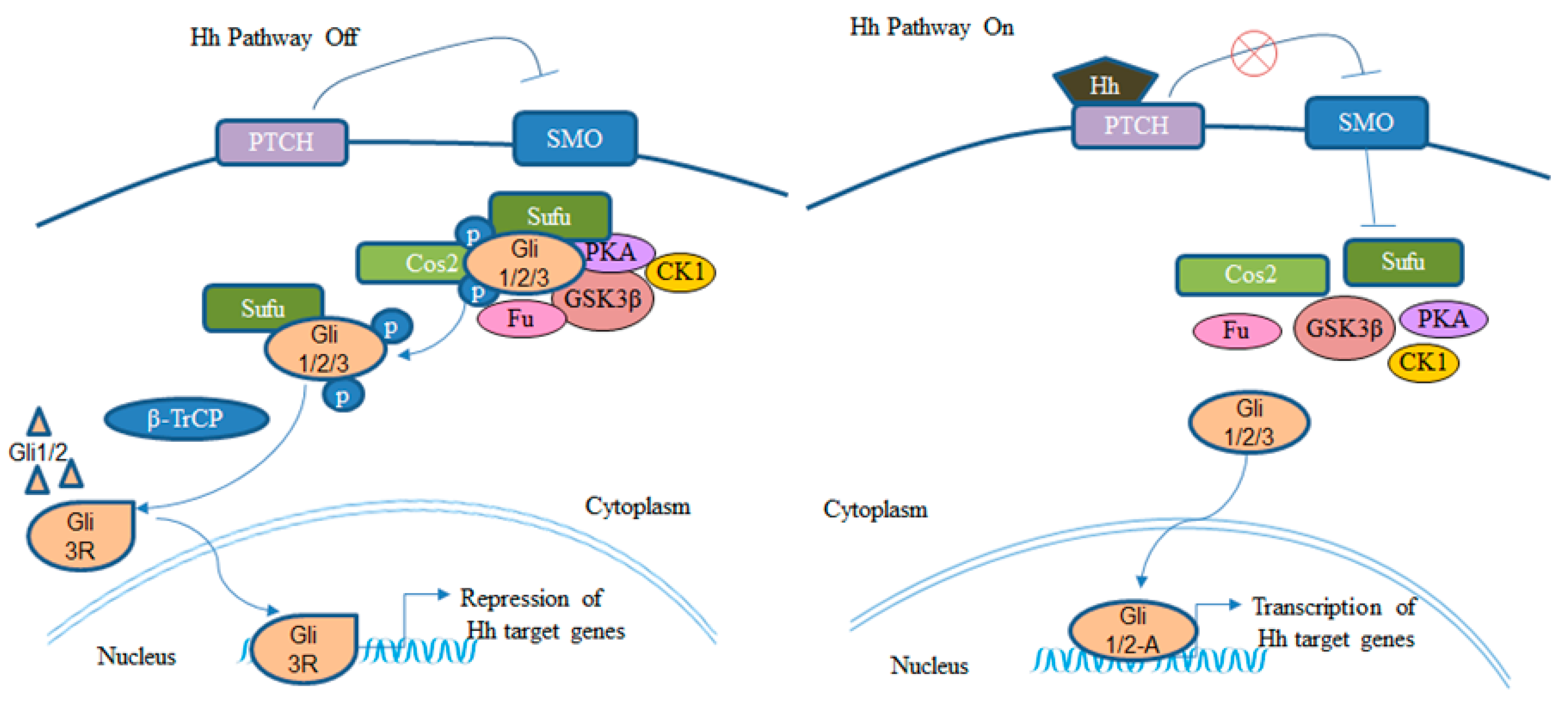
2. BCC and Hh Signaling Pathway
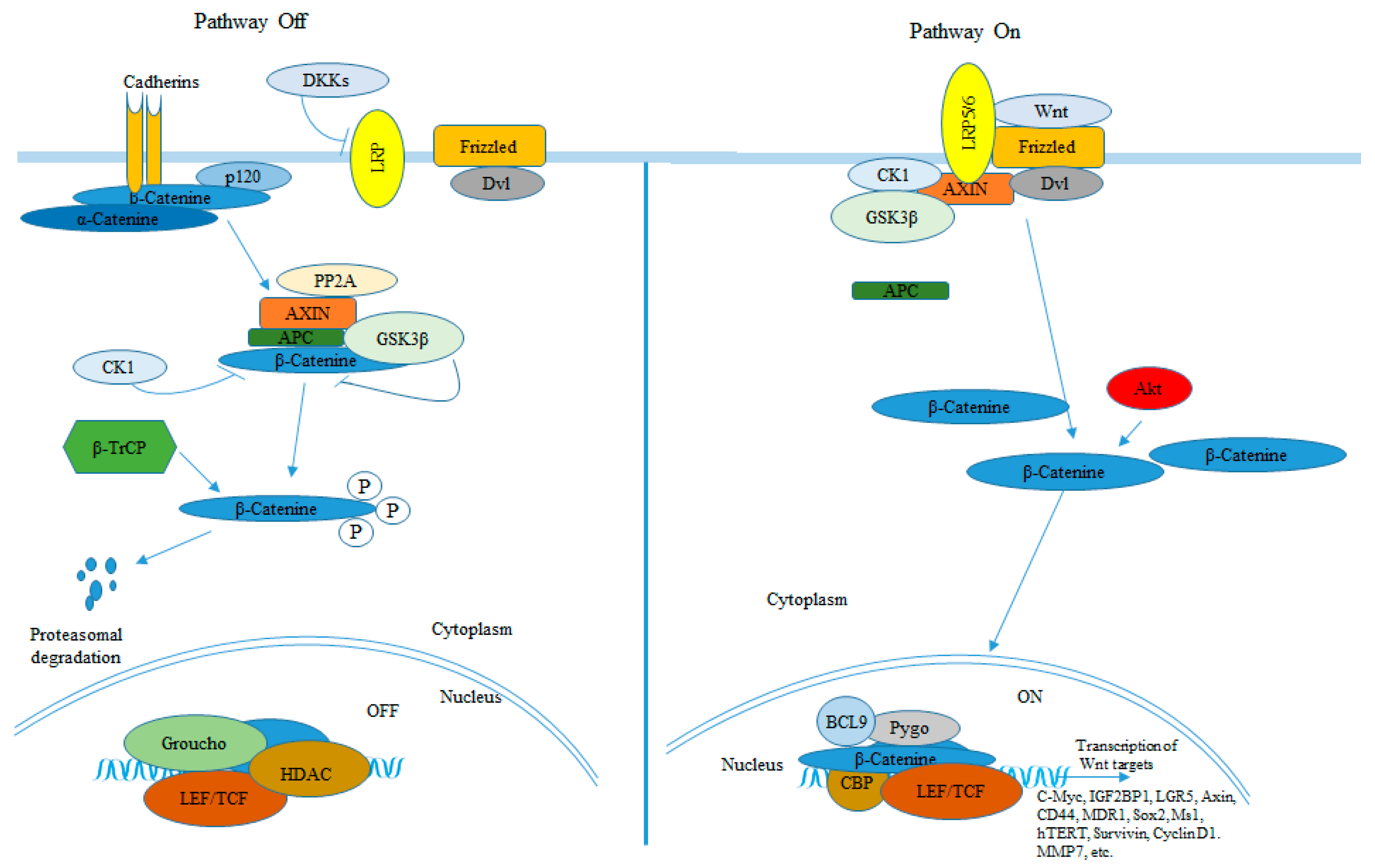
3. BCC and Wnt/β-Catenin Signaling Pathway
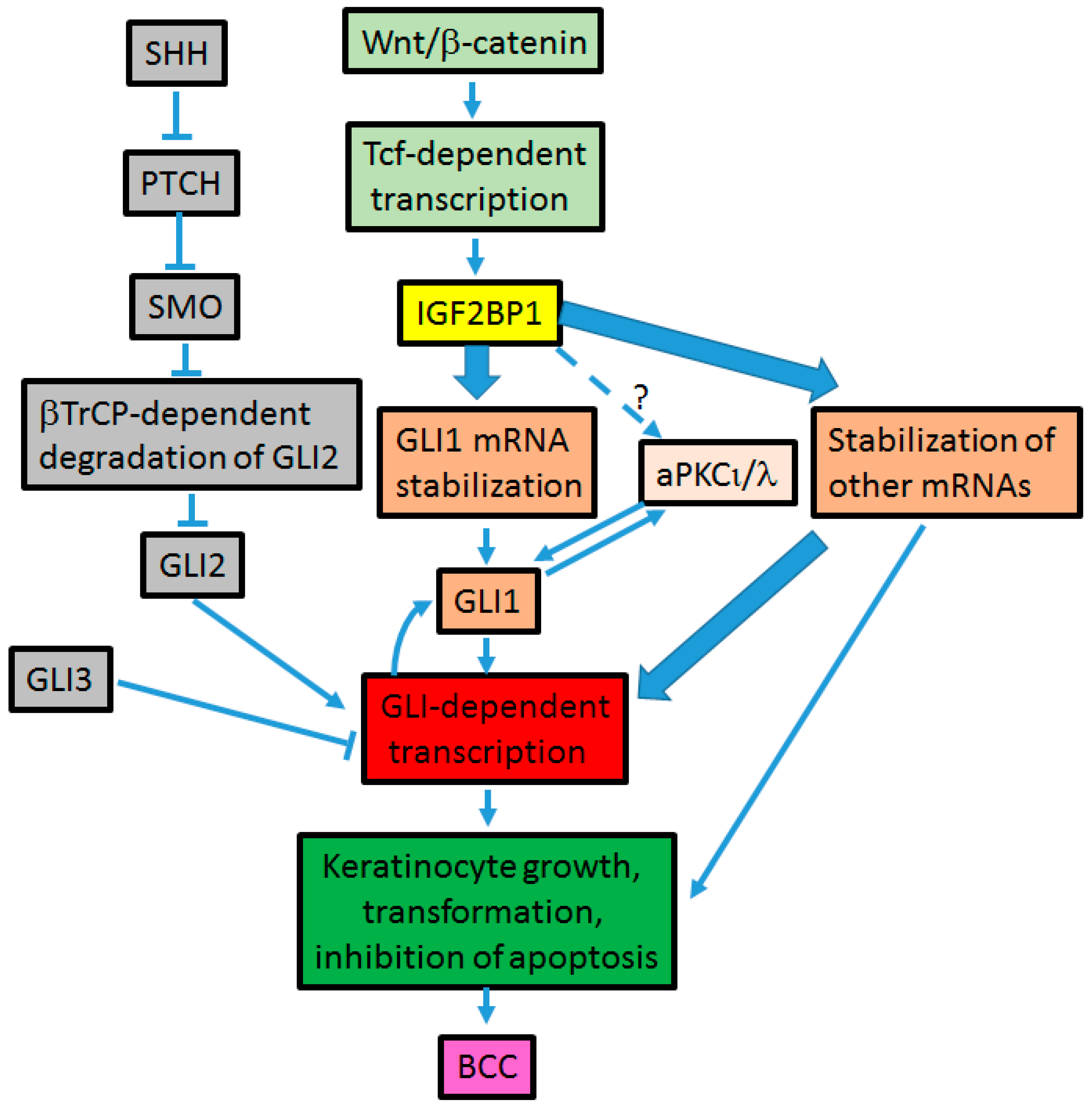
4. Cross-Talk between Hh and Wnt Signaling Pathways in BCC Development (Figure 3)
5. BCC Treatment
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Milroy, C.J.; Horlock, N.; Wilson, G.D.; Sanders, R. Aggressive basal cell carcinoma in young patients: Fact or fiction? Br. J. Plast. Surg. 2000, 53, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Leffell, D.J.; Headington, J.T.; Wong, D.S.; Swanson, N.A. Aggressive-growth basal cell carcinoma in young adults. Arch. Dermatol. 1991, 127, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Christenson, L.J.; Borrowman, T.A.; Vachon, C.M.; Tollefson, M.M.; Otley, C.C.; Weaver, A.L.; Roenigk, R.K. Incidence of basal cell and squamous cell carcinomas in a population younger than 40 years. JAMA 2005, 294, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Rippey, J.J. Why classify basal cell carcinomas? Histopathology 1998, 32, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Rubin, A.I.; Chen, E.H.; Ratner, D. Basal-cell carcinoma. N. Engl. J. Med. 2005, 353, 2262–2269. [Google Scholar] [CrossRef] [PubMed]
- Lomas, A.; Leonardi-Bee, J.; Bath-Hextall, F. A systematic review of worldwide incidence of nonmelanoma skin cancer. Br. J. Dermatol. 2012, 166, 1069–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karagas, M.R.; Greenberg, E.R.; Spencer, S.K.; Stukel, T.A.; Mott, L.A. Increase in incidence rates of basal cell and squamous cell skin cancer in New Hampshire, USA. New Hampshire Skin Cancer Study Group. Int. J. Cancer 1999, 81, 555–559. [Google Scholar] [CrossRef]
- Euvrard, S.; Kanitakis, J.; Claudy, A. Skin cancers after organ transplantation. N. Engl. J. Med. 2003, 348, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Yanik, E.L.; Pfeiffer, R.M.; Freedman, D.M.; Weinstock, M.A.; Cahoon, E.K.; Arron, S.T.; Chaloux, M.; Connolly, M.K.; Nagarajan, P.; Engels, E.A. Spectrum of immune-related conditions associated with risk of keratinocyte cancers among elderly adults in the United States. Cancer Epidemiol. Biomarkers Prev. 2017, 26, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Oram, Y.; Orengo, I.; Griego, R.D.; Rosen, T.; Thornby, J. Histologic patterns of basal cell carcinoma based upon patient immunostatus. Dermatol. Surg. 1995, 21, 611–614. [Google Scholar] [CrossRef] [PubMed]
- Gulshan, S.; Rahman, M.J.; Sarkar, R.; Ghosh, S.; Hazra, R. An Interesting Case of Basal Cell Carcinoma with Raynaud’s Phenomenon Following Chronic Arsenic Exposure. JNMA J. Nepal Med. Assoc. 2016, 55, 100–102. [Google Scholar] [PubMed]
- Cheng, P.S.; Weng, S.F.; Chiang, C.H.; Lai, F.J. Relationship between arsenic-containing drinking water and skin cancers in the arseniasis endemic areas in Taiwan. J. Dermatol. 2016, 43, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Kile, M.L.; Hoffman, E.; Rodrigues, E.G.; Breton, C.V.; Quamruzzaman, Q.; Rahman, M.; Mahiuddin, G.; Hsueh, Y.M.; Christiani, D.C. A pathway-based analysis of urinary arsenic metabolites and skin lesions. Am. J. Epidemiol. 2011, 173, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Nahhas, A.F.; Scarbrough, C.A.; Trotter, S. A Review of the Global Guidelines on Surgical Margins for Nonmelanoma Skin Cancers. J. Clin. Aesthet. Dermatol. 2017, 10, 37–46. [Google Scholar] [PubMed]
- McCusker, M.; Basset-Seguin, N.; Dummer, R.; Lewis, K.; Schadendorf, D.; Sekulic, A.; Hou, J.; Wang, L.; Yue, H.; Hauschild, A. Metastatic basal cell carcinoma: Prognosis dependent on anatomic site and spread of disease. Eur. J. Cancer 2014, 50, 774–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, N.M.; Holliday, A.C.; Luyimbazi, D.T.; Phillips, M.A.; Collins, G.R.; Grider, D.J. Metastatic basal cell carcinoma with loss of p63 and mismatch repair proteins. JAAD Case Rep. 2017, 3, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Wysong, A.; Aasi, S.Z.; Tang, J.Y. Update on metastatic basal cell carcinoma: A summary of published cases from 1981 through 2011. JAMA Dermatol. 2013, 149, 615–616. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.A.; Kelly, D.; Prior, L.; Stanley, E.; MacEneaney, O.; Walsh, T.; Kelly, C.M. An unusual case of basal cell carcinoma of the vulva with lung metastases. Gynecol. Oncol. Rep. 2016, 18, 32–35. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.S.; Strange, R.C.; Lear, J.T. Basal cell carcinoma. BMJ 2003, 327, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Reifenberger, J.; Wolter, M.; Knobbe, C.B.; Kohler, B.; Schonicke, A.; Scharwachter, C.; Kumar, K.; Blaschke, B.; Ruzicka, T.; Reifenberger, G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br. J. Dermatol. 2005, 152, 43–51. [Google Scholar] [CrossRef] [PubMed]
- De Zwaan, S.E.; Haass, N.K. Genetics of basal cell carcinoma. Australas. J. Dermatol. 2010, 51, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef] [PubMed]
- Axelson, M.; Liu, K.; Jiang, X.; He, K.; Wang, J.; Zhao, H.; Kufrin, D.; Palmby, T.; Dong, Z.; Russell, A.M.; et al. U.S. Food and Drug Administration approval: Vismodegib for recurrent, locally advanced, or metastatic basal cell carcinoma. Clin. Cancer Res. 2013, 19, 2289–2293. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.Y.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Yauch, R.L.; Lindgren, J.; Chang, K.; Coppola, C.; Chanana, A.M.; Marji, J.; Bickers, D.R.; et al. Inhibiting the hedgehog pathway in patients with the basal-cell nevus syndrome. N. Engl. J. Med. 2012, 366, 2180–2188. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, S.C.; Tang, X.; Arumugam, A.; Li, C.; Srivastava, R.K.; Weng, Z.; Xu, J.; Zhang, X.; Kim, A.L.; McKay, K.; et al. Shh and p50/Bcl3 signaling crosstalk drives pathogenesis of BCCs in Gorlin syndrome. Oncotarget 2015, 6, 36789–36814. [Google Scholar] [CrossRef] [PubMed]
- Noubissi, F.K.; Kim, T.; Kawahara, T.N.; Aughenbaugh, W.D.; Berg, E.; Longley, B.J.; Athar, M.; Spiegelman, V.S. Role of CRD-BP in the growth of human basal cell carcinoma cells. J. Investig. Dermatol. 2014, 134, 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Liu, G.J.; Liu, J. Genetic association between TNF-alpha promoter polymorphism and susceptibility to squamous cell carcinoma, basal cell carcinoma, and melanoma: A meta-analysis. Oncotarget 2017. [Google Scholar] [CrossRef]
- Kim, A.L.; Back, J.H.; Zhu, Y.; Tang, X.; Yardley, N.P.; Kim, K.J.; Athar, M.; Bickers, D.R. AKT1 Activation is Obligatory for Spontaneous BCC Tumor Growth in a Murine Model that Mimics Some Features of Basal Cell Nevus Syndrome. Cancer Prev. Res. 2016, 9, 794–802. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Oro, A.E. “Atypical” regulation of Hedgehog-dependent cancers. Cancer Cell 2014, 25, 133–134. [Google Scholar] [CrossRef] [PubMed]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.; et al. Smoothened variants explain the majority of drug resistance in basal cell carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [PubMed]
- Bakshi, A.; Chaudhary, S.C.; Rana, M.; Elmets, C.A.; Athar, M. Basal cell carcinoma pathogenesis and therapy involving hedgehog signaling and beyond. Mol. Carcinog. 2017, 56, 2543–2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wicking, C.; Smyth, I.; Bale, A. The hedgehog signalling pathway in tumorigenesis and development. Oncogene 1999, 18, 7844–7851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Athar, M.; Li, C.; Kim, A.L.; Spiegelman, V.S.; Bickers, D.R. Sonic hedgehog signaling in Basal cell nevus syndrome. Cancer Res. 2014, 74, 4967–4975. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Scott, M.P. Patching the gaps in Hedgehog signalling. Nat. Cell Biol. 2007, 9, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Lum, L.; Beachy, P.A. The Hedgehog response network: Sensors, switches, and routers. Science 2004, 304, 1755–1759. [Google Scholar] [CrossRef] [PubMed]
- Oro, A.E.; Higgins, K.M.; Hu, Z.; Bonifas, J.M.; Epstein, E.H., Jr.; Scott, M.P. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science 1997, 276, 817–821. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wicking, C.; Zaphiropoulous, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Nilsson, M.; Unden, A.B.; Krause, D.; Malmqwist, U.; Raza, K.; Zaphiropoulos, P.G.; Toftgard, R. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1 [In Process Citation]. Proc. Natl. Acad. Sci. USA 2000, 97, 3438–3443. [Google Scholar] [CrossRef] [PubMed]
- Green, J.; Leigh, I.M.; Poulsom, R.; Quinn, A.G. Basal cell carcinoma development is associated with induction of the expression of the transcription factor Gli-1. Br. J. Dermatol. 1998, 139, 911–915. [Google Scholar] [CrossRef] [PubMed]
- Noubissi, F.K.; Goswami, S.; Sanek, N.A.; Kawakami, K.; Minamoto, T.; Moser, A.; Grinblat, Y.; Spiegelman, V.S. Wnt signaling stimulates transcriptional outcome of the Hedgehog pathway by stabilizing GLI1 mRNA. Cancer Res. 2009. [Google Scholar] [CrossRef] [PubMed]
- Justilien, V.; Fields, A.P. Molecular pathways: Novel approaches for improved therapeutic targeting of Hedgehog signaling in cancer stem cells. Clin. Cancer Res. 2015, 21, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. The oncogenic activation of beta-catenin. Curr. Opin. Genet. Dev. 1999, 9, 15–21. [Google Scholar] [CrossRef]
- Nusse, R. Wnt signaling in disease and in development. Cell Res. 2005, 15, 28–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt signalling pathways in cancer. Nature 2001, 411, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Warren, R.Q.; Ivy, S.P. Breast cancer growth and metastasis: Interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res. BCR 2011, 13, 211. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Havighurst, T.; Kim, K.; Albertini, M.; Xu, Y.G.; Spiegelman, V.S. Targeting insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) in metastatic melanoma to increase efficacy of BRAF(V600E) inhibitors. Mol. Carcinog. 2018, 57, 678–683. [Google Scholar] [CrossRef] [PubMed]
- Elcheva, I.; Tarapore, R.S.; Bhatia, N.; Spiegelman, V.S. Overexpression of mRNA-binding protein CRD-BP in malignant melanomas. Oncogene 2008, 27, 5069–5074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullor, J.L.; Dahmane, N.; Sun, T.; Ruiz i Altaba, A. Wnt signals are targets and mediators of Gli function. Curr. Biol. 2001, 11, 769–773. [Google Scholar] [CrossRef]
- Lo Muzio, L.; Pannone, G.; Staibano, S.; Mignogna, M.D.; Grieco, M.; Ramires, P.; Romito, A.M.; De Rosa, G.; Piattelli, A. WNT-1 expression in basal cell carcinoma of head and neck. An immunohistochemical and confocal study with regard to the intracellular distribution of beta-catenin. Anticancer Res. 2002, 22, 565–576. [Google Scholar] [PubMed]
- Doglioni, C.; Piccinin, S.; Demontis, S.; Cangi, M.G.; Pecciarini, L.; Chiarelli, C.; Armellin, M.; Vukosavljevic, T.; Boiocchi, M.; Maestro, R. Alterations of beta-catenin pathway in non-melanoma skin tumors: Loss of alpha-ABC nuclear reactivity correlates with the presence of beta-catenin gene mutation. Am. J. Pathol. 2003, 163, 2277–2287. [Google Scholar] [CrossRef]
- Yamazaki, F.; Aragane, Y.; Kawada, A.; Tezuka, T. Immunohistochemical detection for nuclear beta-catenin in sporadic basal cell carcinoma. Br. J. Dermatol. 2001, 145, 771–777. [Google Scholar] [CrossRef] [PubMed]
- El-Bahrawy, M.; El-Masry, N.; Alison, M.; Poulsom, R.; Fallowfield, M. Expression of beta-catenin in basal cell carcinoma. Br. J. Dermatol. 2003, 148, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Saldanha, G.; Ghura, V.; Potter, L.; Fletcher, A. Nuclear beta-catenin in basal cell carcinoma correlates with increased proliferation. Br. J. Dermatol. 2004, 151, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Jaitner, S.; Reiche, J.A.; Schaffauer, A.J.; Hiendlmeyer, E.; Herbst, H.; Brabletz, T.; Kirchner, T.; Jung, A. Human telomerase reverse transcriptase (hTERT) is a target gene of beta-catenin in human colorectal tumors. Cell Cycle 2012, 11, 3331–3338. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Toh, L.; Lau, P.; Wang, X. Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/beta-catenin pathway in human cancer. J. Biol. Chem. 2012, 287, 32494–32511. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Xie, R.; Qin, Y.; Xiao, Y.F.; Yong, X.; Zheng, L.; Dong, H.; Yang, S.M. Human telomerase reverse transcriptase (hTERT) promotes gastric cancer invasion through cooperating with c-Myc to upregulate heparanase expression. Oncotarget 2016, 7, 11364–11379. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Duan, N.; Zhang, C.; Zhang, W. Survivin and Tumorigenesis: Molecular Mechanisms and Therapeutic Strategies. J. Cancer 2016, 7, 314–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Lyu, H.; Wang, J.; Liu, B. MicroRNA regulation and therapeutic targeting of survivin in cancer. Am. J. Cancer Res. 2015, 5, 20–31. [Google Scholar] [PubMed]
- Rezza, A.; Skah, S.; Roche, C.; Nadjar, J.; Samarut, J.; Plateroti, M. The overexpression of the putative gut stem cell marker Musashi-1 induces tumorigenesis through Wnt and Notch activation. J. Cell Sci. 2010, 123, 3256–3265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.R.; Xi, S.; Shukla, V.; Hong, J.A.; Chen, H.; Xiong, Y.; Ripley, R.T.; Hoang, C.D.; Schrump, D.S. The Pluripotency Factor Musashi-2 Is a Novel Target for Lung Cancer Therapy. Ann. Am. Thorac. Soc. 2018, 15, S124. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, P.; Rustgi, A.K. RNA Binding Proteins in Intestinal Epithelial Biology and Colorectal Cancer. Trends Mol. Med. 2018, 24, 490–506. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fan, Y.; Wang, X.; Huang, Z.; Shi, K.; Zhou, B. Musashi-2 is a prognostic marker for the survival of patients with cervical cancer. Oncol. Lett. 2018, 15, 5425–5432. [Google Scholar] [CrossRef] [PubMed]
- Piva, M.; Domenici, G.; Iriondo, O.; Rabano, M.; Simoes, B.M.; Comaills, V.; Barredo, I.; Lopez-Ruiz, J.A.; Zabalza, I.; Kypta, R.; et al. Sox2 promotes tamoxifen resistance in breast cancer cells. EMBO Mol. Med. 2014, 6, 66–79. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Dong, J.; Li, D.; Lai, L.; Siwko, S.; Li, Y.; Liu, M. Lgr4 regulates mammary gland development and stem cell activity through the pluripotency transcription factor Sox2. Stem Cells 2013, 31, 1921–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhang, X.; Wu, X.; Li, W.; Su, P.; Cheng, H.; Xiang, L.; Gao, P.; Zhou, G. Interference of Frizzled 1 (FZD1) reverses multidrug resistance in breast cancer cells through the Wnt/beta-catenin pathway. Cancer Lett. 2012, 323, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T. Studies on molecular mechanism of drug resistance of mycobacteria and recombinant BCG to prevent infection of intracellular pathogens. Nihon Saikingaku Zasshi 2000, 55, 613–627. [Google Scholar] [CrossRef] [PubMed]
- Duchartre, Y.; Kim, Y.M.; Kahn, M. The Wnt signaling pathway in cancer. Crit. Rev. Oncol./Hematol. 2016, 99, 141–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaguchi, T.; Goto, Y.; Kido, K.; Mochimaru, H.; Sakurai, T.; Tsukamoto, N.; Kudo-Saito, C.; Fujita, T.; Sumimoto, H.; Kawakami, Y. Immune suppression and resistance mediated by constitutive activation of Wnt/beta-catenin signaling in human melanoma cells. J. Immunol. 2012, 189, 2110–2117. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, J.; Wang, H.; Wu, L.; Yuan, W.; Du, J.; Cai, S. Trichostatin A, a histone deacetylase inhibitor, reverses epithelial-mesenchymal transition in colorectal cancer SW480 and prostate cancer PC3 cells. Biochem. Biophys. Res. Commun. 2015, 456, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Wickstrom, M.; Dyberg, C.; Milosevic, J.; Einvik, C.; Calero, R.; Sveinbjornsson, B.; Sanden, E.; Darabi, A.; Siesjo, P.; Kool, M.; et al. Wnt/beta-catenin pathway regulates MGMT gene expression in cancer and inhibition of Wnt signalling prevents chemoresistance. Nat. Commun. 2015, 6, 8904. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef] [PubMed]
- Gattinoni, L.; Ji, Y.; Restifo, N.P. Wnt/beta-catenin signaling in T-cell immunity and cancer immunotherapy. Clin. Cancer Res. 2010, 16, 4695–4701. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Huelsken, J.; Vogel, R.; Erdmann, B.; Cotsarelis, G.; Birchmeier, W. beta-Catenin controls hair follicle morphogenesis and stem cell differentiation in the skin. Cell 2001, 105, 533–545. [Google Scholar] [CrossRef]
- Huelsken, J.; Birchmeier, W. New aspects of Wnt signaling pathways in higher vertebrates. Curr. Opin. Genetics Dev. 2001, 11, 547–553. [Google Scholar] [CrossRef]
- Andl, T.; Reddy, S.T.; Gaddapara, T.; Millar, S.E. WNT signals are required for the initiation of hair follicle development. Dev. Cell 2002, 2, 643–653. [Google Scholar] [CrossRef]
- St-Jacques, B.; Dassule, H.R.; Karavanova, I.; Botchkarev, V.A.; Li, J.; Danielian, P.S.; McMahon, J.A.; Lewis, P.M.; Paus, R.; McMahon, A.P. Sonic hedgehog signaling is essential for hair development. Curr. Biol. CB 1998, 8, 1058–1068. [Google Scholar] [CrossRef]
- Chiang, C.; Swan, R.Z.; Grachtchouk, M.; Bolinger, M.; Litingtung, Y.; Robertson, E.K.; Cooper, M.K.; Gaffield, W.; Westphal, H.; Beachy, P.A.; et al. Essential role for Sonic hedgehog during hair follicle morphogenesis. Dev. Biol. 1999, 205, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Andl, T.; Grachtchouk, V.; Wang, A.; Liu, J.; Syu, L.J.; Ferris, J.; Wang, T.S.; Glick, A.B.; Millar, S.E.; et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/beta3-catenin signaling. Nat. Genet. 2008, 40, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Akiyoshi, T.; Nakamura, M.; Koga, K.; Nakashima, H.; Yao, T.; Tsuneyoshi, M.; Tanaka, M.; Katano, M. Gli1, downregulated in colorectal cancers, inhibits proliferation of colon cancer cells involving Wnt signalling activation. Gut 2006, 55, 991–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borycki, A.; Brown, A.M.; Emerson, C.P., Jr. Shh and Wnt signaling pathways converge to control Gli gene activation in avian somites. Development 2000, 127, 2075–2087. [Google Scholar] [PubMed]
- Iwatsuki, K.; Liu, H.X.; Gronder, A.; Singer, M.A.; Lane, T.F.; Grosschedl, R.; Mistretta, C.M.; Margolskee, R.F. Wnt signaling interacts with Shh to regulate taste papilla development. Proc. Natl. Acad. Sci. USA 2007, 104, 2253–2258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.S.; Buttitta, L.A.; May, N.R.; Kispert, A.; Fan, C.M. SHH-N upregulates Sfrp2 to mediate its competitive interaction with WNT1 and WNT4 in the somitic mesoderm. Development 2000, 127, 109–118. [Google Scholar] [PubMed]
- Li, X.; Deng, W.; Lobo-Ruppert, S.M.; Ruppert, J.M. Gli1 acts through Snail and E-cadherin to promote nuclear signaling by beta-catenin. Oncogene 2007, 26, 4489–4498. [Google Scholar] [CrossRef] [PubMed]
- Marcelle, C.; Stark, M.R.; Bronner-Fraser, M. Coordinate actions of BMPs, Wnts, Shh and noggin mediate patterning of the dorsal somite. Development 1997, 124, 3955–3963. [Google Scholar] [PubMed]
- Maeda, O.; Kondo, M.; Fujita, T.; Usami, N.; Fukui, T.; Shimokata, K.; Ando, T.; Goto, H.; Sekido, Y. Enhancement of GLI1-transcriptional activity by beta-catenin in human cancer cells. Oncol. Rep. 2006, 16, 91–96. [Google Scholar] [PubMed]
- Bhatia, N.; Thiyagarajan, S.; Elcheva, I.; Saleem, M.; Dlugosz, A.; Mukhtar, H.; Spiegelman, V.S. Gli2 is targeted for ubiquitination and degradation by beta-TrCP ubiquitin ligase. J. Biol. Chem. 2006, 281, 19320–19326. [Google Scholar] [CrossRef] [PubMed]
- Noubissi, F.K.; Elcheva, I.; Bhatia, N.; Shakoori, A.; Ougolkov, A.; Liu, J.; Minamoto, T.; Ross, J.; Fuchs, S.Y.; Spiegelman, V.S. CRD-BP mediates stabilization of betaTrCP1 and c-myc mRNA in response to beta-catenin signalling. Nature 2006, 441, 898–901. [Google Scholar] [CrossRef] [PubMed]
- Mehmood, K.; Akhtar, D.; Mackedenski, S.; Wang, C.; Lee, C.H. Inhibition of GLI1 Expression by Targeting the CRD-BP-GLI1 mRNA Interaction Using a Specific Oligonucleotide. Mol. Pharmacol. 2016, 89, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Elcheva, I.; Goswami, S.; Noubissi, F.K.; Spiegelman, V.S. CRD-BP protects the coding region of betaTrCP1 mRNA from miR-183-mediated degradation. Mol. Cell 2009, 35, 240–246. [Google Scholar] [CrossRef] [PubMed]
- Heffelfinger, C.; Ouyang, Z.; Engberg, A.; Leffell, D.J.; Hanlon, A.M.; Gordon, P.B.; Zheng, W.; Zhao, H.; Snyder, M.P.; Bale, A.E. Correlation of Global MicroRNA Expression With Basal Cell Carcinoma Subtype. G3 2012, 2, 279–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Dinehart, M.S. Triple Hedgehog Pathway Inhibition for Basal Cell Carcinoma. J. Clin. Aesthet. Dermatol. 2017, 10, 47–49. [Google Scholar] [CrossRef] [PubMed]
- Karabulut, G.O.; Kaynak, P.; Ozturker, C.; Fazil, K.; Ocak, O.B.; Taskapili, M. Imiquimod 5% cream for the treatment of large nodular basal cell carcinoma at the medial canthal area. Indian J. Ophthalmol. 2017, 65, 48–51. [Google Scholar] [CrossRef] [PubMed]
- Schulze, H.J.; Cribier, B.; Requena, L.; Reifenberger, J.; Ferrandiz, C.; Garcia Diez, A.; Tebbs, V.; McRae, S. Imiquimod 5% cream for the treatment of superficial basal cell carcinoma: Results from a randomized vehicle-controlled phase III study in Europe. Br. J. Dermatol. 2005, 152, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Neville, J.A.; Williford, P.M.; Jorizzo, J.L. Pilot study using topical imiquimod 5% cream in the treatment of nodular basal cell carcinoma after initial treatment with curettage. J. Drugs Dermatol. JDD 2007, 6, 910–914. [Google Scholar] [PubMed]
- Arits, A.H.; Parren, L.J.; van Marion, A.M.; Sommer, A.; Frank, J.; Kelleners-Smeets, N.W. Basal cell carcinoma and trichoepithelioma: A possible matter of confusion. Int. J. Dermatol. 2008, 47 (Suppl. 1), 13–17. [Google Scholar] [CrossRef] [PubMed]
- Jerjes, W.; Hamdoon, Z.; Abdulkareem, A.A.; Hopper, C. Photodynamic therapy in the management of actinic keratosis: Retrospective evaluation of outcome. Photodiagn. Photodyn. Ther. 2017, 17, 200–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeimies, R.M.; Ibbotson, S.; Murrell, D.F.; Rubel, D.; Frambach, Y.; de Berker, D.; Dummer, R.; Kerrouche, N.; Villemagne, H.; Excilight Study Group. A clinical study comparing methyl aminolevulinate photodynamic therapy and surgery in small superficial basal cell carcinoma (8–20 mm), with a 12-month follow-up. J. Eur. Acad. Dermatol. Venereol. JEADV 2008, 22, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Dummer, R.; Guminski, A.; Gutzmer, R.; Dirix, L.; Lewis, K.D.; Combemale, P.; Herd, R.M.; Kaatz, M.; Loquai, C.; Stratigos, A.J.; et al. The 12-month analysis from Basal Cell Carcinoma Outcomes with LDE225 Treatment (BOLT): A phase II, randomized, double-blind study of sonidegib in patients with advanced basal cell carcinoma. J. Am. Acad. Dermatol. 2016, 75, 113.e5–125.e5. [Google Scholar] [CrossRef] [PubMed]
- Ramelyte, E.; Amann, V.C.; Dummer, R. Sonidegib for the treatment of advanced basal cell carcinoma. Expert Opin. Pharmacother. 2016, 17, 1963–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.J.; Kim, J.; Spaunhurst, K.; Montoya, J.; Khodosh, R.; Chandra, K.; Fu, T.; Gilliam, A.; Molgo, M.; Beachy, P.A.; et al. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J. Clin. Oncol. 2014, 32, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Weiss, G.J.; Tibes, R.; Blaydorn, L.; Jameson, G.; Downhour, M.; White, E.; Caro, I.; Von Hoff, D.D. Long-term safety, tolerability, and efficacy of vismodegib in two patients with metastatic basal cell carcinoma and basal cell nevus syndrome. Dermatol. Rep. 2011, 3, e55. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.L.; Oro, A.E. Initial assessment of tumor regrowth after vismodegib in advanced Basal cell carcinoma. Arch. Dermatol. 2012, 148, 1324–1325. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, H.J.; Pau, G.; Dijkgraaf, G.J.; Basset-Seguin, N.; Modrusan, Z.; Januario, T.; Tsui, V.; Durham, A.B.; Dlugosz, A.A.; Haverty, P.M.; et al. Genomic analysis of smoothened inhibitor resistance in basal cell carcinoma. Cancer Cell 2015, 27, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.V.; Hammer, N.A.; Nielsen, J.; Madsen, M.; Dalbaeck, C.; Wewer, U.M.; Christiansen, J.; Nielsen, F.C. Dwarfism and impaired gut development in insulin-like growth factor II mRNA-binding protein 1-deficient mice. Mol. Cell. Biol. 2004, 24, 4448–4464. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, P.; Kottaridi, C.; Dimitriadis, E.; Courtis, N.; Mahaira, L.; Talieri, M.; Giannopoulos, A.; Iliadis, K.; Papaioannou, D.; Nasioulas, G.; et al. Expression of the RNA-binding protein CRD-BP in brain and non-small cell lung tumors. Cancer Lett. 2004, 209, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Leeds, P.; Kren, B.T.; Boylan, J.M.; Betz, N.A.; Steer, C.J.; Gruppuso, P.A.; Ross, J. Developmental regulation of CRD-BP, an RNA-binding protein that stabilizes c-myc mRNA in vitro. Oncogene 1997, 14, 1279–1286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, G.A.; Betz, N.A.; Leeds, P.F.; Fleisig, A.J.; Prokipcak, R.D.; Ross, J. The c-myc coding region determinant-binding protein: A member of a family of KH domain RNA-binding proteins. Nucleic Acids Res. 1998, 26, 5036–5044. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, P.; Mahaira, L.; Papadopoulou, A.; Teixeira, M.R.; Heim, S.; Andersen, J.A.; Evangelou, E.; Dafni, U.; Pandis, N.; Trangas, T. 8q24 Copy number gains and expression of the c-myc mRNA stabilizing protein CRD-BP in primary breast carcinomas. Int. J. Cancer 2003, 104, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, P.; Trangas, T.; Dimitriadis, E.; Samiotaki, M.; Kyriazoglou, I.; Tsiapalis, C.M.; Kittas, C.; Agnantis, N.; Nielsen, F.C.; Nielsen, J.; et al. C-MYC and IGF-II mRNA-binding protein (CRD-BP/IMP-1) in benign and malignant mesenchymal tumors. Int. J. Cancer 2001, 94, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Lemm, I.; Berberet, B. Overexpression of an mRNA-binding protein in human colorectal cancer. Oncogene 2001, 20, 6544–6550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, L.; Shigemasa, K.; Ohama, K. Increased expression of IGF II mRNA-binding protein 1 mRNA is associated with an advanced clinical stage and poor prognosis in patients with ovarian cancer. Int. J. Oncol. 2004, 24, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Prokipcak, R.D.; Herrick, D.J.; Ross, J. Purification and properties of a protein that binds to the C-terminal coding region of human c-myc mRNA. J. Biol. Chem. 1994, 269, 9261–9269. [Google Scholar] [PubMed]
- Sparanese, D.; Lee, C.H. CRD-BP shields c-myc and MDR-1 RNA from endonucleolytic attack by a mammalian endoribonuclease. Nucleic Acids Res. 2007, 35, 1209–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, D.T.; Barnes, M.; Thomsen, D.; Lee, C.H. Assessing specific oligonucleotides and small molecule antibiotics for the ability to inhibit the CRD-BP-CD44 RNA interaction. PLoS ONE 2014, 9, e91585. [Google Scholar] [CrossRef] [PubMed]
- Faye, M.D.; Beug, S.T.; Graber, T.E.; Earl, N.; Xiang, X.; Wild, B.; Langlois, S.; Michaud, J.; Cowan, K.N.; Korneluk, R.G.; et al. IGF2BP1 controls cell death and drug resistance in rhabdomyosarcomas by regulating translation of cIAP1. Oncogene 2015, 34, 1532–1541. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noubissi, F.K.; Yedjou, C.G.; Spiegelman, V.S.; Tchounwou, P.B. Cross-Talk between Wnt and Hh Signaling Pathways in the Pathology of Basal Cell Carcinoma. Int. J. Environ. Res. Public Health 2018, 15, 1442. https://doi.org/10.3390/ijerph15071442
Noubissi FK, Yedjou CG, Spiegelman VS, Tchounwou PB. Cross-Talk between Wnt and Hh Signaling Pathways in the Pathology of Basal Cell Carcinoma. International Journal of Environmental Research and Public Health. 2018; 15(7):1442. https://doi.org/10.3390/ijerph15071442
Chicago/Turabian StyleNoubissi, Felicite K., Clement G. Yedjou, Vladimir S. Spiegelman, and Paul B. Tchounwou. 2018. "Cross-Talk between Wnt and Hh Signaling Pathways in the Pathology of Basal Cell Carcinoma" International Journal of Environmental Research and Public Health 15, no. 7: 1442. https://doi.org/10.3390/ijerph15071442