Exploring the Impacts of Anthropogenic Disturbance on Seawater and Sediment Microbial Communities in Korean Coastal Waters Using Metagenomics Analysis


Abstract
:1. Introduction
2. Materials and Methods
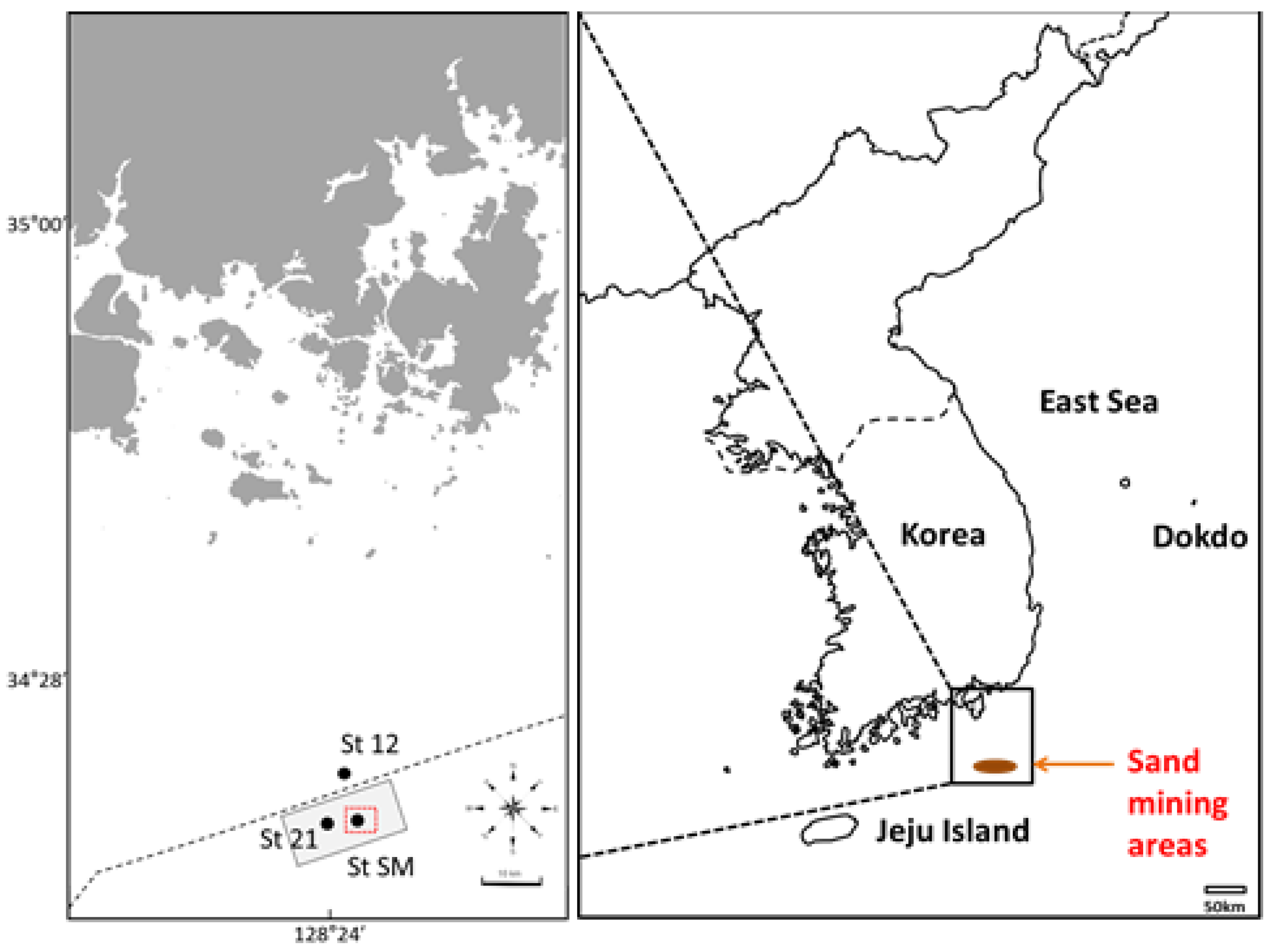
2.1. Sample Collection, Processing, and eDNA Isolation
2.2. Sequencing and Prediction of 16S rRNA Data
2.3. Metagenome Predictions from 16S rRNA Data
2.4. 16S rRNA Sequence Assembly and Statistical Analysis
3. Results
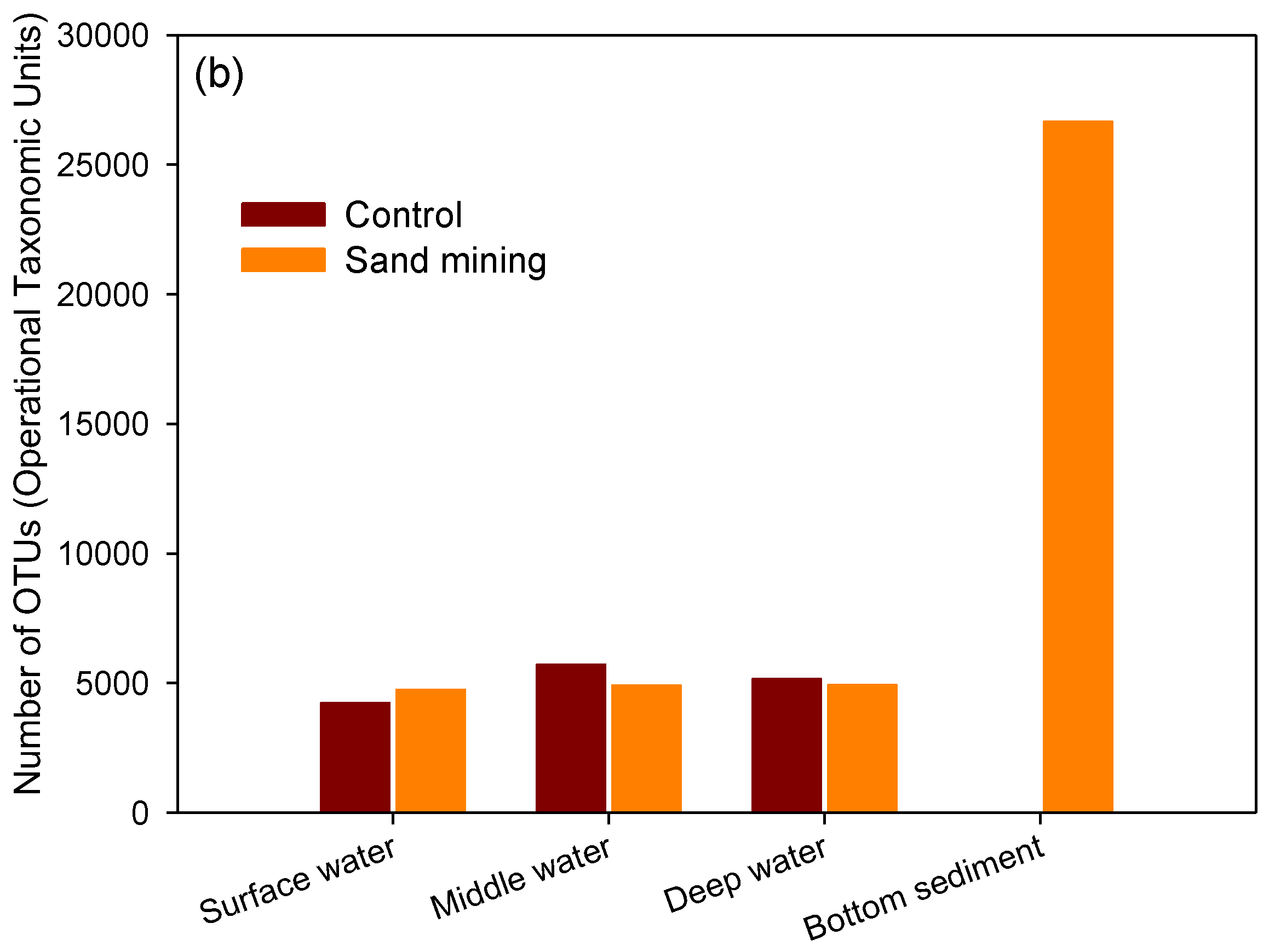
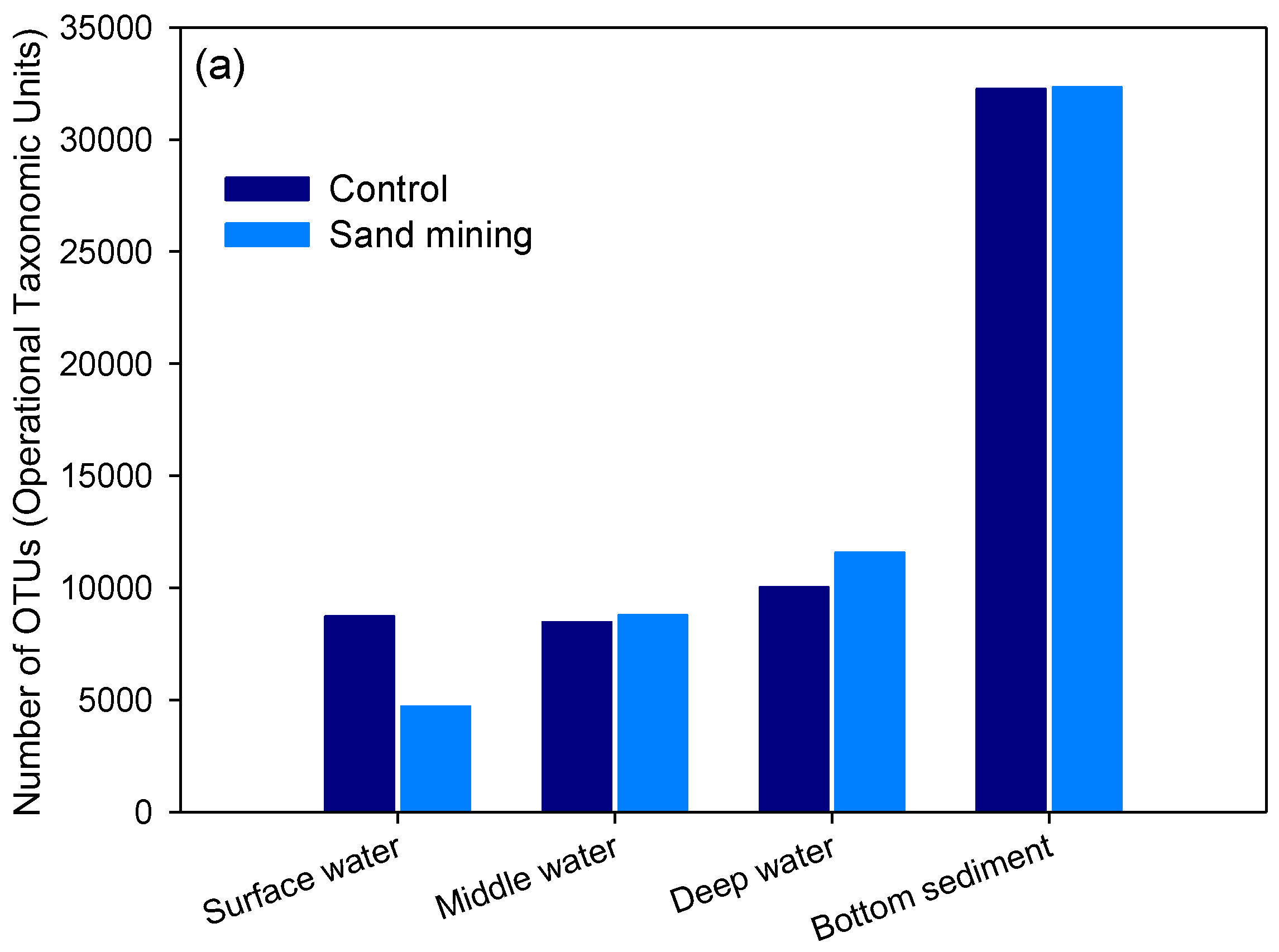
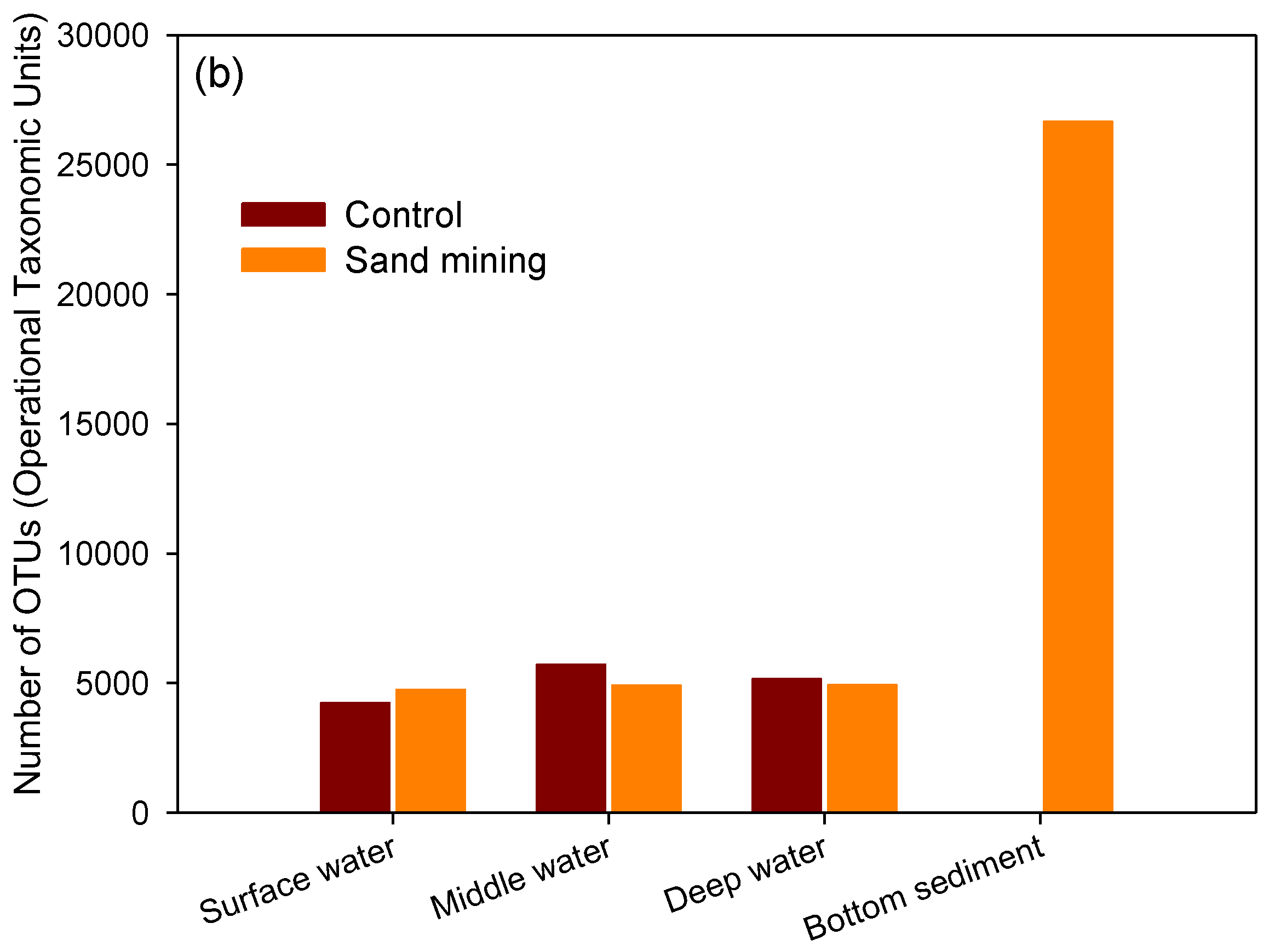
3.1. Differences in Taxonomic Richness between Sand Mining vs. Control Microbial Communities
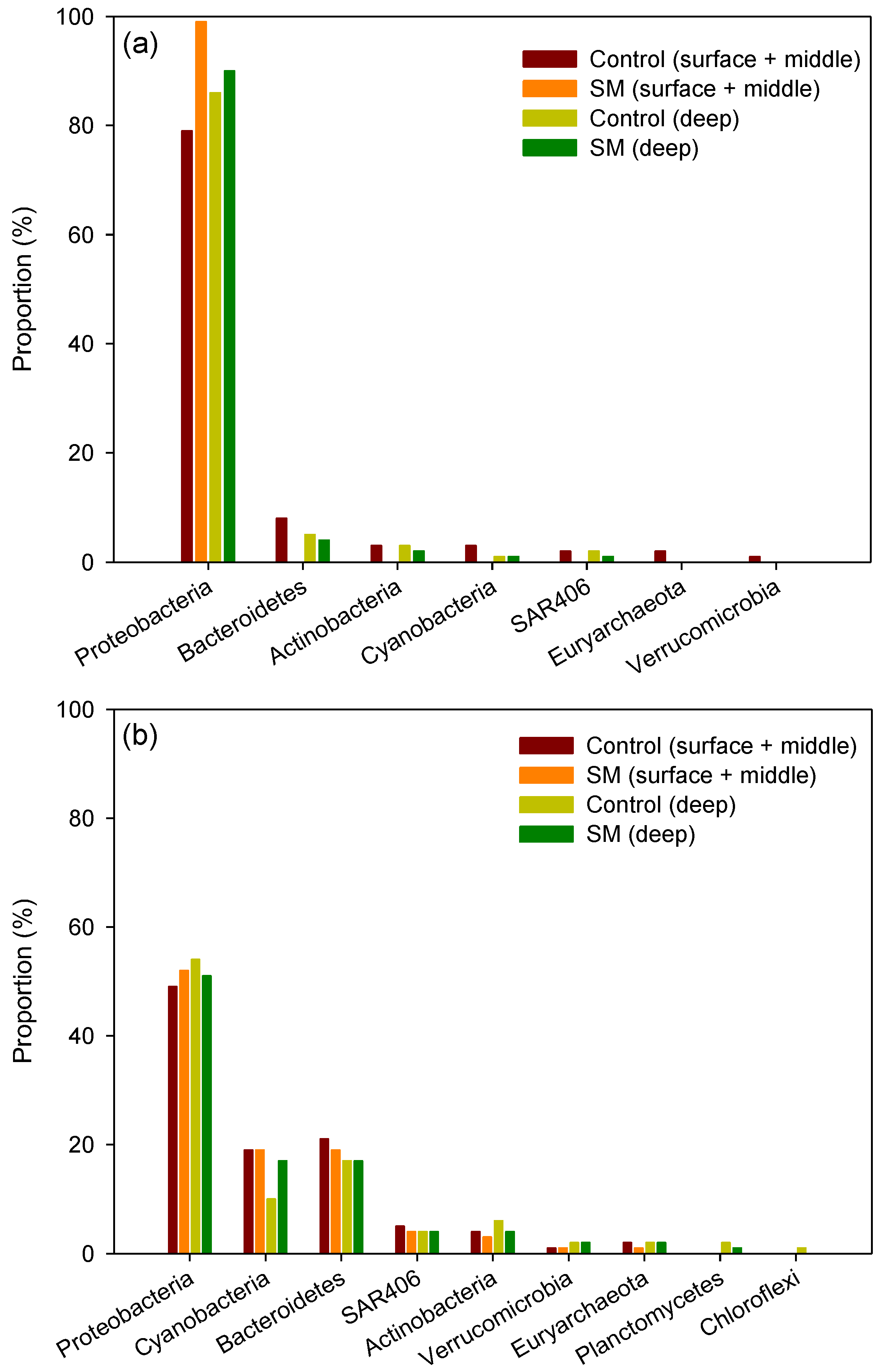
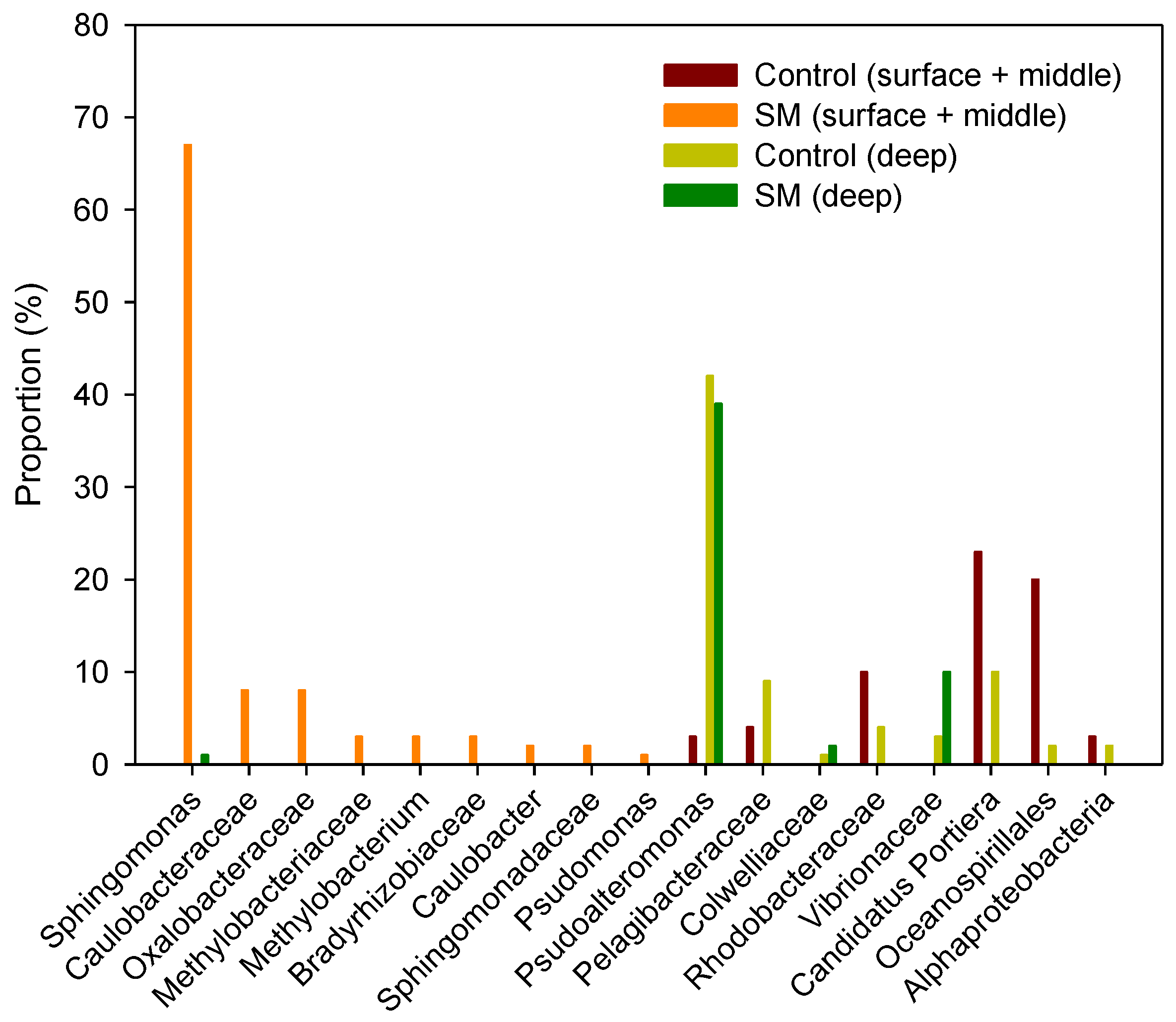
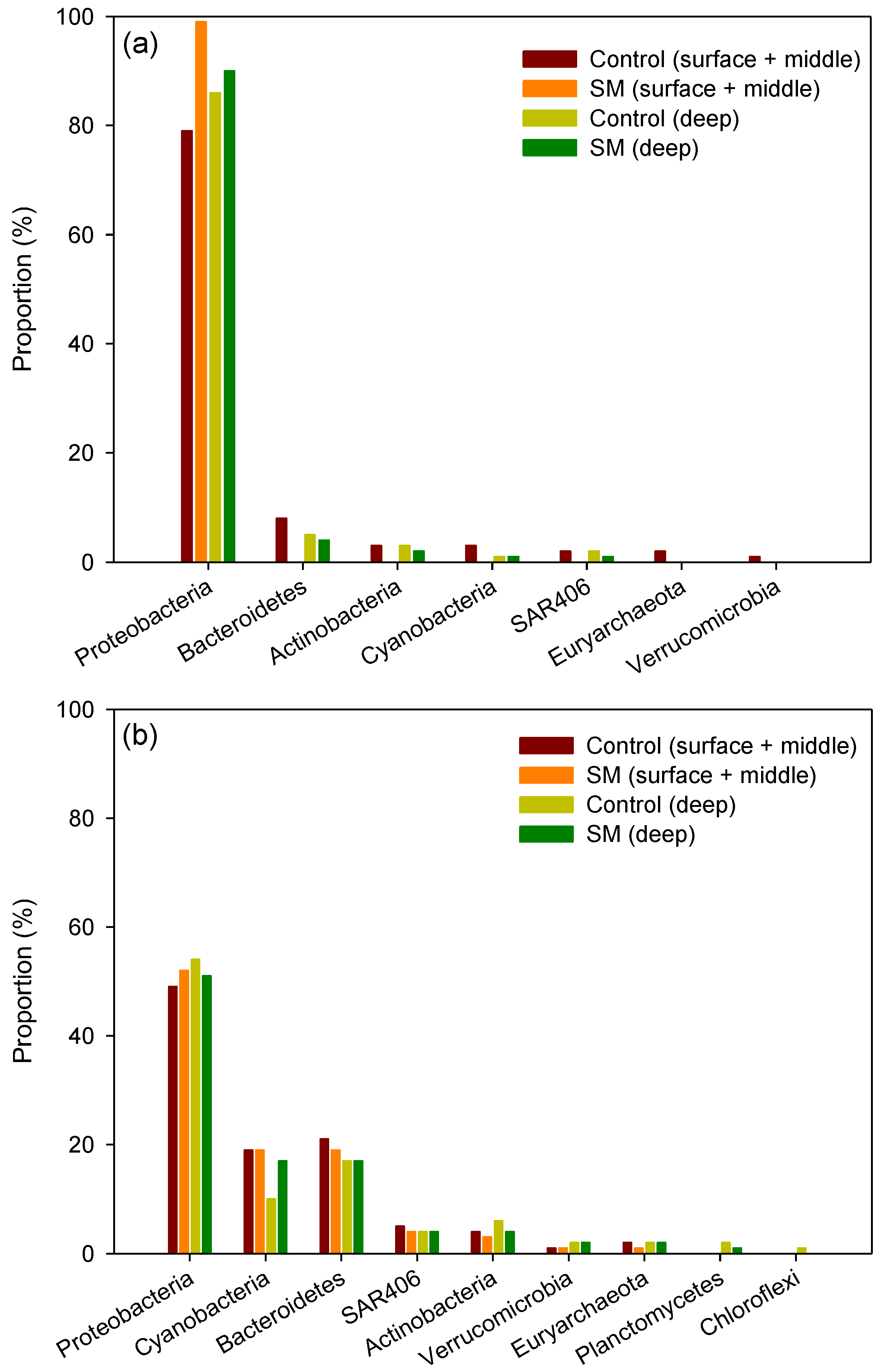
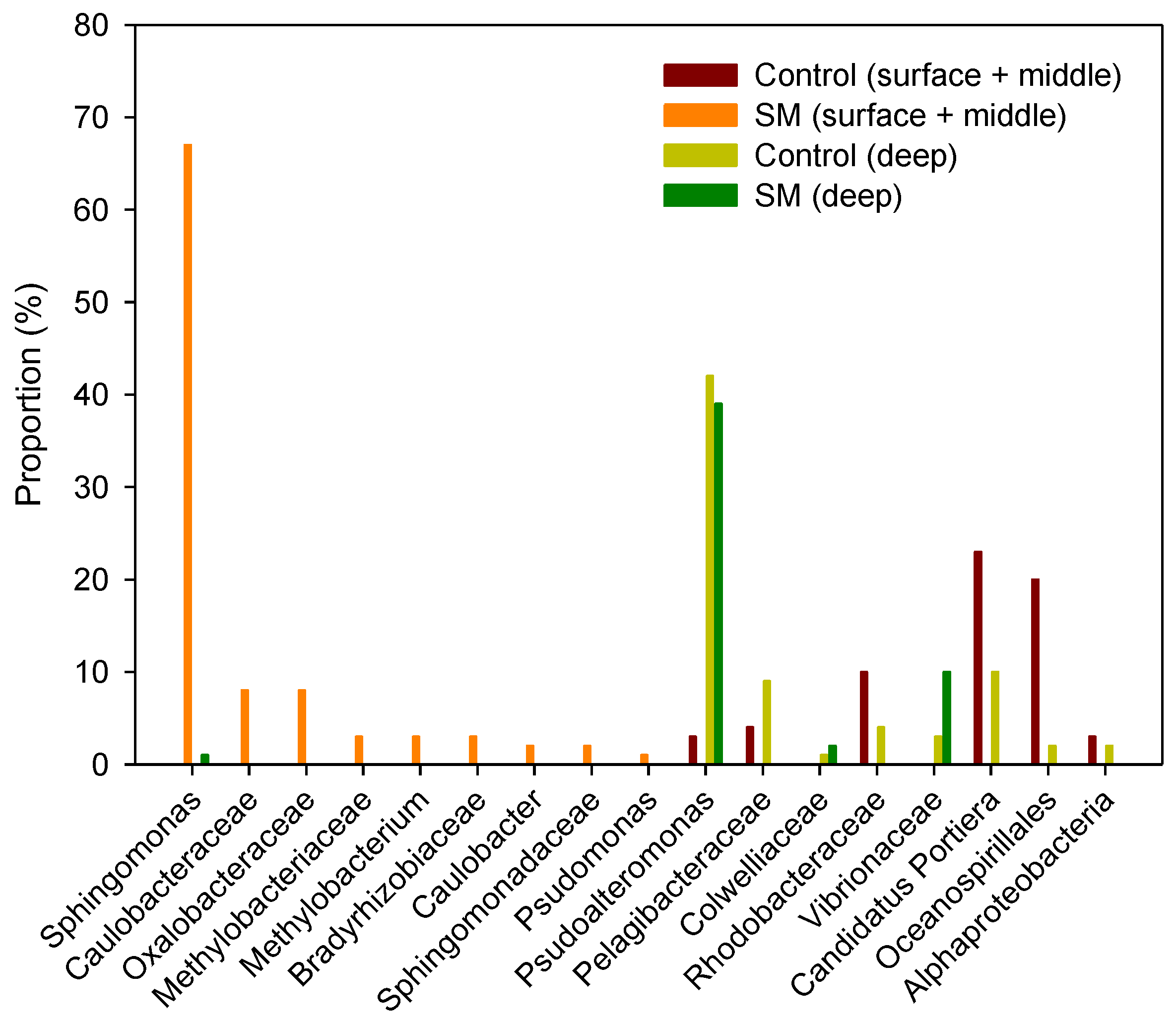
3.2. Differences in Composition and Diversity of Microbial Seawater Community between Sand Mining vs. Control Sites
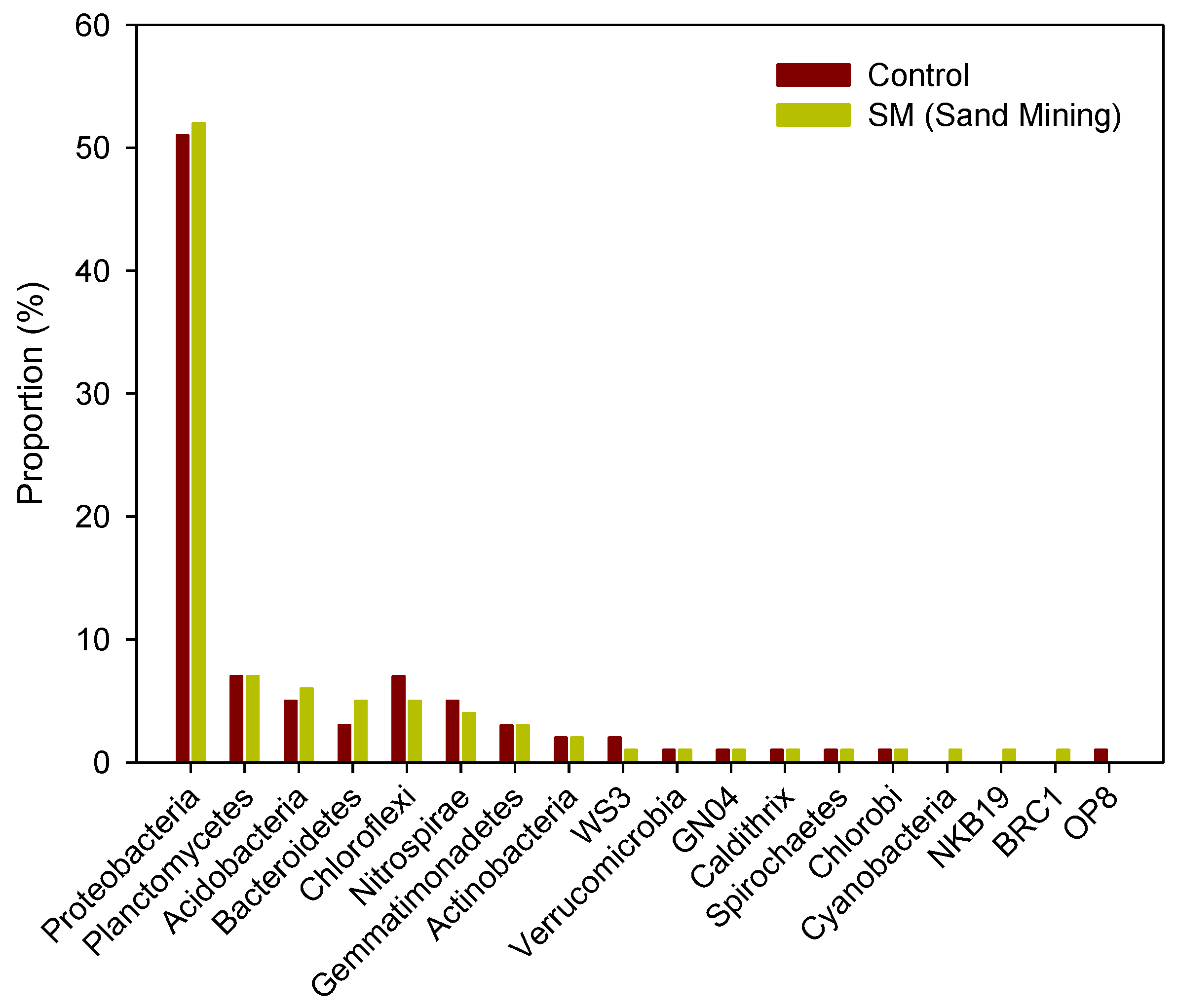
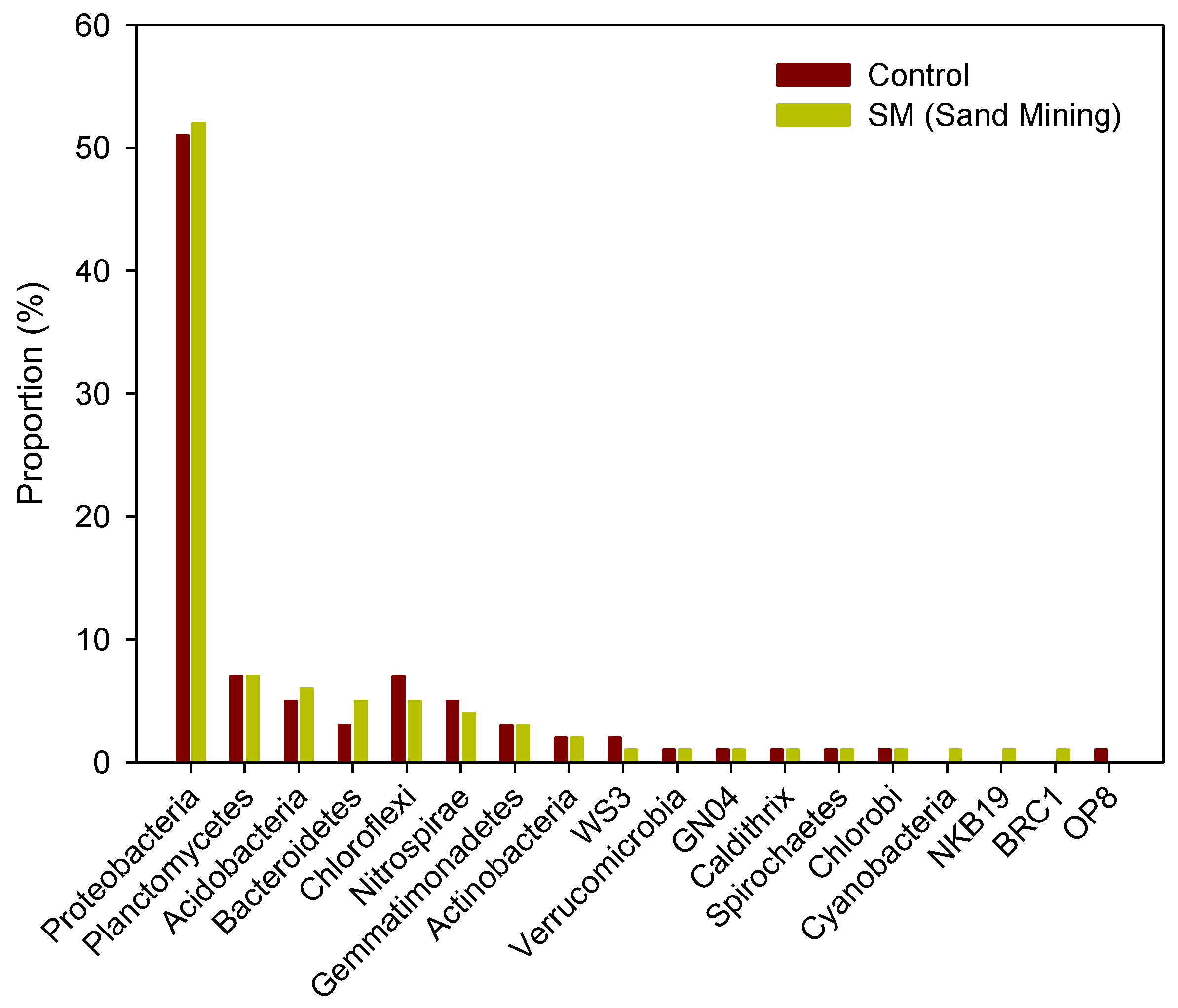
3.3. Differences in Composition and Diversity of Microbial Sediment Community between Sand Mining vs. Control Sites
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Palumbi, S.R.; Sandifer, P.A.; Allan, J.D.; Beck, M.W.; Fautin, D.G.; Fogarty, M.J.; Halpern, B.S.; Incze, L.S.; Leong, J.A.; Norse, E.; et al. Managing for ocean biodiversity to sustain marine ecosystem services. Front. Ecol. Environ. 2009, 7, 204–211. [Google Scholar] [CrossRef]
- UNEP-WCMC. Marine and Coastal Ecosystem Services: Valuation Methods and Their Application; UNEP-WCMC Biodiversity: Cambridge, UK, 2011. [Google Scholar]
- Halpern, B.S.; Longo, C.; Hardy, D.; McLeod, K.L.; Samhouri, J.F.; Katona, S.K.; Kleisner, K.; Lester, S.E.; O’Leary, J.; Ranelletti, M.; et al. An index to assess the health and benefits of the global ocean. Nature 2012, 488, 615–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halpern, B.S.; Longo, C.; Stewart Lowndes, J.S.; Best, B.D.; Frazier, M.; Katona, S.K.; Kleisner, K.M.; Rosenberg, A.A.; Scarborough, C.; Selig, E.R. Patterns and emerging trends in global ocean health. PLoS ONE 2015, 10, e0117863. [Google Scholar] [CrossRef] [PubMed]
- Oliver, T.H.; Isaac, N.J.B.; August, T.A.; Woodcock, B.A.; Roy, D.B.; Bullock, J.M. Declining resilience of ecosystem functions under biodiversity loss. Nat. Commun. 2015, 6, 10122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glöckner, F.O.; Stal, L.J.; Sandaa, R.A.; Gasol, J.M.; O’Gara, F.; Hernandez, F.; Labrenz, M.; Stoica, E.; Varela, M.M.; Bordalo, A.; et al. Marine Microbial Diversity and Its Role in Ecosystem Functioning and Environmental Chang; Marine Board Position Paper 17; Calewaert, J.B., McDonough, N., Eds.; Marine Board-ESF: Ostend, Belgium, 2012. [Google Scholar]
- Cardinale, B.J.; Duffy, J.E.; Gonzalez, A.; Hooper, D.U.; Perrings, C.; Venail, P.; Narwani, A.; Mace, G.M.; Tilman, D.; Wardle, D.A.; et al. Biodiversity loss and its impact on humanity. Nature 2012, 486, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Marcus, N.H.; Boero, F. Minireview: The importance of benthic-pelagic coupling and the forgotten role of life cycles in coastal aquatic systems. Limnol. Oceanogr. 1998, 43, 763–768. [Google Scholar] [CrossRef]
- Jeffries, T.C.; Schmitz Fontes, M.L.; Harrison, D.P.; Van-Dongen-Vogels, V.; Eyre, B.D.; Ralph, P.J.; Seymour, J.R. Bacterioplankton dynamics within a large anthropogenically impacted urban estuary. Front. Microbiol. 2016, 6, 1438. [Google Scholar] [CrossRef] [PubMed]
- Meziti, A.; Tsementzi, D.; Kormas, K.A.; Karayanni, H.; Konstantinidis, K.T. Anthropogenic effects on bacterial diversity and function along a river-to-estuary gradient in Northwest Greece revealed by metagenomics. Environ. Microbiol. 2016, 18, 4640–4652. [Google Scholar] [CrossRef] [PubMed]
- Lai, X.T.; Zeng, X.F.; Fang, S.; Huang, Y.L.; Cao, L.X.; Zhou, S.N. Denaturing gradient gel electrophoresis (DGGE) analysis of bacterial community composition in deep-sea sediments of the South China Sea. World J. Microb. Biot. 2006, 22, 1337–1345. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Field, D.; Swift, P.; Newbold, L.; Oliver, A.; Smyth, T.; Somerfield, P.J.; Huse, S.; Joint, I. The seasonal structure of microbial communities in the Western English Channel. Environ. Microbiol. 2009, 11, 3132–3139. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Steele, J.A.; Caporaso, J.G.; Steinbrück, L.; Reeder, J.; Temperton, B.; Huse, S.; McHardy, A.C.; Knight, R.; Joint, I.; et al. Defining seasonal marine microbial community dynamics. ISME J. 2012, 6, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.-S.; Park, M.; Hwang, J.; Kil, E.-J.; Jung, S.W.; Lee, S.; Lee, T.-K. Seasonal dynamics of marine microbial community in the south sea of Korea. PLoS ONE 2015, 10, e0131633. [Google Scholar] [CrossRef] [PubMed]
- Meziti, A.; Kormas, K.A.; Moustaka-Gouni, M.; Karayanni, H. Spatially uniform but temporally variable bacterioplankton in a semi-enclosed coastal area. Syst. Appl. Microbiol. 2015, 38, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Fuhrman, J.A.; Cram, J.A.; Needham, D.M. Marine microbial community dynamics and their ecological interpretation. Nat. Rev. Microbiol. 2015, 13, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Nogales, B.; Lanfranconi, M.P.; Piña-Villalonga, J.M.; Bosch, R. Anthropogenic perturbations in marine microbial communities. FEMS Microbiol. Rev. 2011, 358, 275–298. [Google Scholar] [CrossRef] [PubMed]
- Phua, C.; Van den Akker, S.; Baretta, M.; Van Dalfsen, J. Ecological Effects of Sand Extraction in the North Sea. Available online: http://www.vliz.be/imisdocs/publications/154975.pdf (accessed on 4 November 2016).
- Edlund, A.; Jansson, J.K. Changes in active bacterial communities before and after dredging of highly polluted Baltic Sea sediments. Appl. Environ. Microbiol. 2006, 72, 6800–6807. [Google Scholar] [CrossRef] [PubMed]
- De Jong, M.F.; Baptist, M.J.; van Hal, R.; de Boois, I.J.; Lindeboom, H.J.; Hoekstra, P. Impact on demersal fish of a large-scale and deep sand extraction site with ecosystem-based landscaped sandbars. Estuar. Coast. Shelf Sci. 2014, 146, 83–94. [Google Scholar] [CrossRef]
- Bohmann, K.; Evans, A.; Gilbert, M.T.P.; Carvalho, G.R.; Creer, S.; Knapp, M.; Yu, D.W.; de Bruyn, M. Environmental DNA for wildlife biology and biodiversity monitoring. Trends Ecol. Evol. 2015, 29, 358–367. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.F.; Preston, C.M.; Mincer, T.; Rich, V.; Hallam, S.J.; Friggard, N.U.; Martinez, A.; Sullivan, M.B.; Edwards, R.; Brito, B.R.; et al. Community genomics among stratified microbial assemblages in the ocean’s interior. Science 2006, 5760, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Field, D.; Huang, Y.; Edwards, R.; Li, W.; Gilna, P.; Joint, L. Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. PLoS ONE 2008, 3, e3042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, J.A.; Dupont, C.L. Microbial metagenomics: beyond the genome. Annu. Rev. Mar. Sci. 2011, 3, 347–371. [Google Scholar] [CrossRef] [PubMed]
- Zinger, L.; Amaral-Zettler, L.A.; Fuhrman, J.A.; Horner-Devine, M.C.; Huse, S.M.; Mark Welch, D.B.; Martiny, J.B.H.; Sogin, M.; Boetius, A.; Ramette, A. Global patterns of bacterial beta-diversity in seafloor and seawater ecosystems. PLoS ONE 2011, 6, e24570. [Google Scholar] [CrossRef] [PubMed]
- Kisand, V.; Valente, A.; Lahm, A.; Tanet, G.; Lettieri, T. Phylogenetic and functional metagenomic profiling for assessing microbial biodiversity in environmental monitoring. PLoS ONE 2012, 7, e43630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, X.Y.; Arumugam, R.; Choo, S.W.; Yin, W.F.; Chan, K.G. Metagenomic sequencing of prokaryotic microbiota from tropical surface seawater. Genome Announc. 2013, 1, e00540-13. [Google Scholar] [CrossRef] [PubMed]
- Boulat, S.J.; Borja, A.; Gilbert, J.; Taylor, M.I.; Davies, N.; Weisberg, S.B.; Griffith, J.F.; Lettieri, T.; Field, D.; Benzie, J.; et al. Genomics in marine monitoring: New opportunities for assessing marine health status. Mar. Poll. Bull. 2013, 74, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 04 March 2016 ).
- Magoč, T.; Salzberg, S.L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Nilgiriwala, K.S.; Alahari, A.; Rao, A.S.; Apte, S.K. Cloning and overexpression of alkaline phasphatase phoK from Sphingomonas sp. stran BSAR-1 for bioprecipitation of uranium from alkaline solutions. Appl. Environ. Microbiol. 2008, 74, 5516–5523. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.-W.; Li, X.-R.; Wang, J-H.; Hu, Z.-Y.; Meng, H.; Xiang, L.-Y.; Quan, Z.-X. Bacterial diversity of water and sediment in the Changjiang estuary and coastal area of the East China Sea. FEMS Microbiol. Ecol. 2009, 70, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.A.; Kirkpatrick, J.B.; Rutherford, S.D.; Smith, D.C.; Sogin, M.; D’Hondt, S. Bacterial diversity and community composition from sea surface to subseafloor. ISME J. 2016, 10, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.H.; Lee, H.S.; Kim, J.T.; Kim, S.-J.; Choi, S.H.; Kang, S.G.; Lee, J.-H. Identification of a new subfamily of salt-tolerant esterases from a metagenomic library of tidal flat sediment. Appl. Microbiol. Biotech. 2012, 93, 623–631. [Google Scholar] [CrossRef]
- Oh, K.H.; Nguyen, G.S.; Kim, E.Y.; Kourist, R.; Bornscheuer, U.; Oh, T.K.; Yoon, J.H. Characterization of a novel esterase isolated from intertidal flat metagenome and its tertiary alcohols synthesis. J. Mol. Catal. B 2012, 80, 67–73. [Google Scholar] [CrossRef]
- Bowman, J.P.; McCuaig, R.D. Biodiversity, community structural shifts, and biogeography of prokaryotes within Antarctic continental shelf sediment. Appl. Environ. Microb. 2003, 69, 2463–2483. [Google Scholar] [CrossRef]
- Jones, S.E.; Chiu, C.-Y.; Kratz, T.K.; Wu, J.-T.; Shade, A.; McMahon, K.D. Typhoons initiate predictable change in aquatic bacterial communities. Limnol. Oceanogr. 2008, 53, 1319–1326. [Google Scholar] [CrossRef]
- Brito, E.M.; Guyoneaud, R.; Goñi-Urriza, M.; Ranchou-Peyruse, A.; Verbaere, A.; Crapez, M.A.; Wasserman, J.C.; Duran, R. Characterization of hydrocarbonoclastic bacterial communites from mangrove sediments in Guanabara Bay. Res. Microbiol. 2006, 157, 752–762. [Google Scholar] [CrossRef] [PubMed]
- Kasai, Y.; Kishira, H.; Sasaki, T.; Syutsubo, K.; Watanabe, K.; Harayama, S. Predominant growth of Alcanivorax strains in oil-contaminated and nutrient-supplemented sea water. Environ. Microbiol. 2002, 4, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.P.; Wang, B.J.; Liu, Y.H.; Liu, S.J. Novosphingobium taihuense sp. nov., a novel aromatic-compund-degrading bacterium isolated from Taihu Lake, China. Int. J. Syst. Evol. Microbiol. 2005, 55, 1229–1232. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site ID | Latitude; Longitude | Sample Type | Samples | Sampling Period | Read Counts | Total Base Pair Counts |
|---|---|---|---|---|---|---|
| Site12 | 34°15.05′ N; 128°23.68′ E | Control | Surface | April 2015 (Spring) | 876,484 | 263,821,684 |
| Middle | 904,794 | 272,342,994 | ||||
| Deep | 865,924 | 260,643,124 | ||||
| Site21 | 34°11.55′ N; 128°22.55′ E | Sand mining | Surface | 887,080 | 267,011,080 | |
| Middle | 574,816 | 173,019,616 | ||||
| Deep | 511,724 | 154,028,924 | ||||
| Site12 | 34°15.05′ N; 128°23.68′ E | Control | Sediment | 845,526 | 254,503,326 | |
| Site21 | 34°11.55′ N; 128°22.55′ E | Sand mining | Sediment | 741,150 | 223,086,150 | |
| Site12 | 34°15.05′ N; 128°23.68′ E | Control | Surface | October 2015 (Autumn) | 845,526 | 254,503,326 |
| Middle | 1,260,066 | 379,279,866 | ||||
| Deep | 1,039,742 | 312,962,342 | ||||
| Site SM | 34°11.17′ N; 128°24.50′ E | Sand mining | Surface | 1,135,076 | 341,657,876 | |
| Middle | 1,052,748 | 316,877,148 | ||||
| Deep | 1,133,566 | 341,203,366 | ||||
| Site SM | Sand mining | Sediment | 1,090,470 | 328,231,470 | ||
| Sediment | 1,496,222 | 450,362,822 | ||||
| Sediment | 1,955,034 | 588,465,234 |
| Sample | Class | Family | Genus |
|---|---|---|---|
| Surface water | Cenarchaeales | Cenarchaeaceae | - |
| Flavobacteriales | Flavobacteriaceae | Mesonia | |
| Ignavibacteriales | Ignavibacteriaceae | - | |
| Kordiimonadales | Kordiimonadaceae | - | |
| Burkholderiales | Alcaligenaceae | - | |
| Alteromonadales | Alteromonadaceae | Marinobacter | |
| Alteromonadales | Idiomarinaceae | Pseudidiomarina | |
| Oceanospirillales | Alcanivoracaceae | Alcanivorax | |
| Oceanospirillales | Halomonadaceae | Cobetia | |
| Marinicellales | Marinicellaceae | Marinicella | |
| Middle water | Bacteroidia | Bacteroidaceae | Bacteroides |
| Bacteroidia | Porphyromonadaceae | Parabacteroides | |
| Flavobacteriia | Flavobacteriaceae | Cellulophaga | |
| Flavobacteriia | Weeksellaceae | - | |
| Clostridia | Lachnospiraceae | Lachnospira | |
| Clostridia | Veillonellaceae | Succiniclasticum | |
| Clostridia | Acidaminobacteraceae | Fusibacter | |
| Fusobacteriia | Fusobacteriaceae | Propionigenium | |
| Alphaproteobacteria | Erythrobacteraceae | - | |
| Alphaproteobacteria | Sphingomonadaceae | Novosphingobium | |
| Deltaproteobacteria | Desulfarculaceae | - | |
| Deltaproteobacteria | Desulfobacteraceae | Desulfofaba | |
| Deep water | Actinobacteria | Micrococcaceae | Micrococcus |
| Cytophagia | Amoebophilaceae | Ucs1325 | |
| Chlamydiia | Simkaniaceae | - | |
| Ignavibacteria | lheB3-7 | - | |
| Synechococcophycideae | Synechococcaceae | - | |
| Bacilli | Streptococcaceae | Streptococcus | |
| Alphaproteobacteria | Kiloniellaceae | Thalassospira | |
| Alphaproteobacteria | Rhodospirillaceae | - | |
| Alphaproteobacteria | Erythrobacteraceae | Erythrobacter | |
| Deltaproteobacteria | Desulfarculaceae | - | |
| Deltaproteobacteria | Desulfobacteraceae | Desulfosarcina | |
| Deltaproteobacteria | Syntrophaceae | Desulfobacca | |
| Gammaproteobacteria | Coxiellaceae | Coxiella | |
| Gammaproteobacteria | Halomonadaceae | Haererehalobacter | |
| Gammaproteobacteria | Oceanospirillaceae | Oleibacter | |
| PRR-12 | KSB4 | - |
| Sample | Class | Family | Genus |
|---|---|---|---|
| Bottom sediments | Actinobacteria | Bifidobacteriaceae | Bifidobacterium |
| Fimbriimonadia | Fimbriimonadaceae | - | |
| Bacteroidia | Bacteroidaceae | Bacteroides | |
| Flavobacteriia | Cryomorphaceae | - | |
| Flavobacteriia | Cryomorphaceae | Cryomorpha | |
| Flavobacteriia | Cryomorphaceae | Owenweeksia | |
| Rhodothermi | Balneolaceae | Balneola | |
| Synechococcophycideae | Synechococcaceae | Prochlorococcus | |
| Bacilli | Planococcaceae | Sporosarcina | |
| Bacilli | Thermoactinomycetaceae | - | |
| Clostridia | Clostridiaceae | Alkaliphilus | |
| Clostridia | Clostridiaceae | Geosporobacter | |
| Clostridia | Lachnospiraceae | - | |
| Clostridia | Lachnospiraceae | Coprococcus | |
| Clostridia | Lachnospiraceae | Lachnospira | |
| Clostridia | Peptostreptococcaceae | - | |
| Clostridia | Ruminococcaceae | Faecalibacterium | |
| Clostridia | Ruminococcaceae | Ruminococcus | |
| Clostridia | Veillonellaceae | Dialister | |
| Clostridia | Veillonellaceae | Megamonas | |
| Clostridia | Acidaminobacteraceae | - | |
| Fusobacteriia | Fusobacteriaceae | u114 | |
| Alphaproteobacteria | Methylocystaceae | Pleomorphomonas | |
| Alphaproteobacteria | Rhodobiaceae | Afifella | |
| Alphaproteobacteria | Rhodobacteraceae | Marivita | |
| Alphaproteobacteria | Rhodobacteraceae | Thalassobius | |
| Alphaproteobacteria | Rhodospirillaceae | Novispirillum | |
| Alphaproteobacteria | Rhodospirillaceae | Rhodospirillum | |
| Alphaproteobacteria | AEGEAN_112 | - | |
| Deltaproteobacteria | S25_1238 | - | |
| Deltaproteobacteria | SAR324 | - | |
| Gammaproteobacteria | Psychromonadaceae | - | |
| Gammaproteobacteria | Legionellaceae | Legionella | |
| Gammaproteobacteria | Legionellaceae | Tatlockia | |
| Gammaproteobacteria | Oceanospirillaceae | Oleispira | |
| AB16 | A714017 | SargSea-WGS | |
| Mollicutes | Mycoplasmataceae | Candidatus Hepatoplasma |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Won, N.-I.; Kim, K.-H.; Kang, J.H.; Park, S.R.; Lee, H.J. Exploring the Impacts of Anthropogenic Disturbance on Seawater and Sediment Microbial Communities in Korean Coastal Waters Using Metagenomics Analysis. Int. J. Environ. Res. Public Health 2017, 14, 130. https://doi.org/10.3390/ijerph14020130
Won N-I, Kim K-H, Kang JH, Park SR, Lee HJ. Exploring the Impacts of Anthropogenic Disturbance on Seawater and Sediment Microbial Communities in Korean Coastal Waters Using Metagenomics Analysis. International Journal of Environmental Research and Public Health. 2017; 14(2):130. https://doi.org/10.3390/ijerph14020130
Chicago/Turabian StyleWon, Nam-Il, Ki-Hwan Kim, Ji Hyoun Kang, Sang Rul Park, and Hyuk Je Lee. 2017. "Exploring the Impacts of Anthropogenic Disturbance on Seawater and Sediment Microbial Communities in Korean Coastal Waters Using Metagenomics Analysis" International Journal of Environmental Research and Public Health 14, no. 2: 130. https://doi.org/10.3390/ijerph14020130