
Up-Regulation of Claudin-6 in the Distal Lung Impacts Secondhand Smoke-Induced Inflammation

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Secondhand Smoke Exposure
2.3. Histology and Immunohistochemistry
2.4. Immunoblotting
2.5. qRT-PCR
2.6. ELISAs
2.7. Statistical Analysis
3. Results
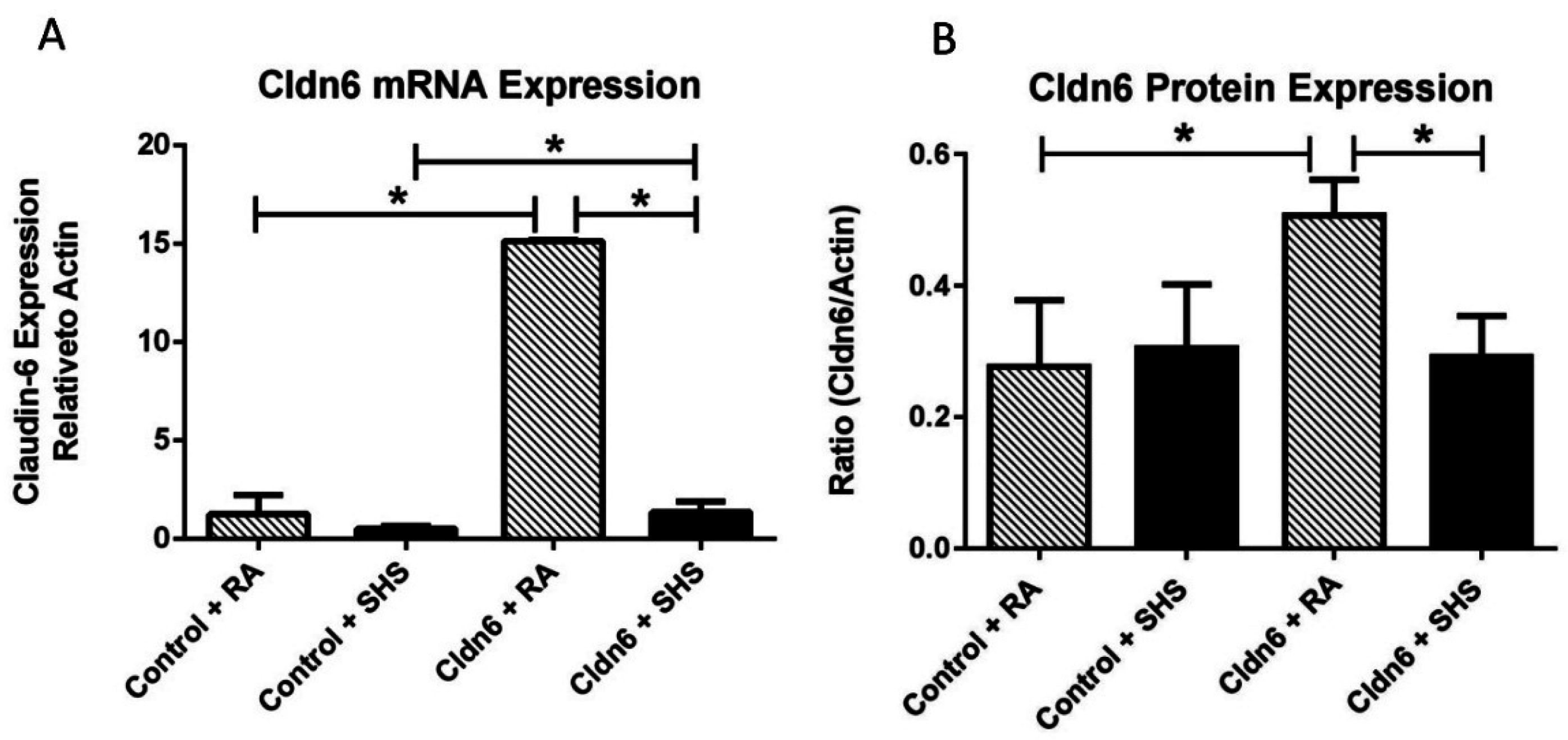
3.1. Cldn6 Expression
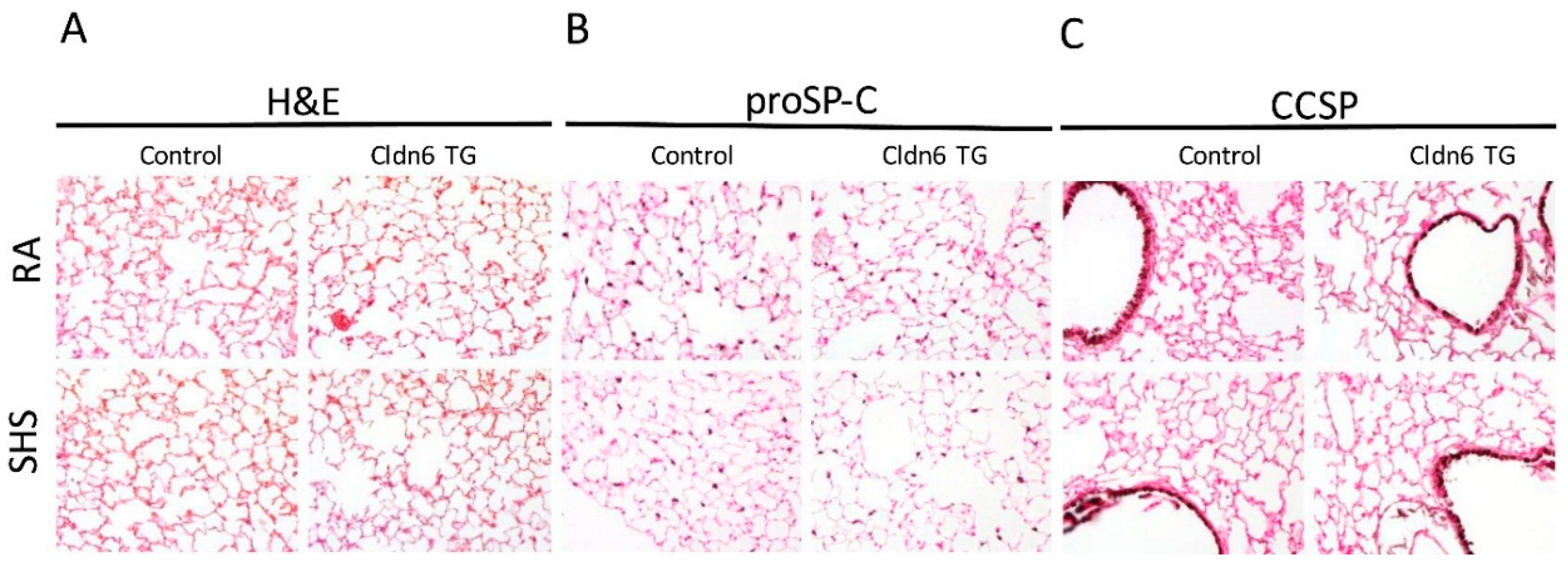
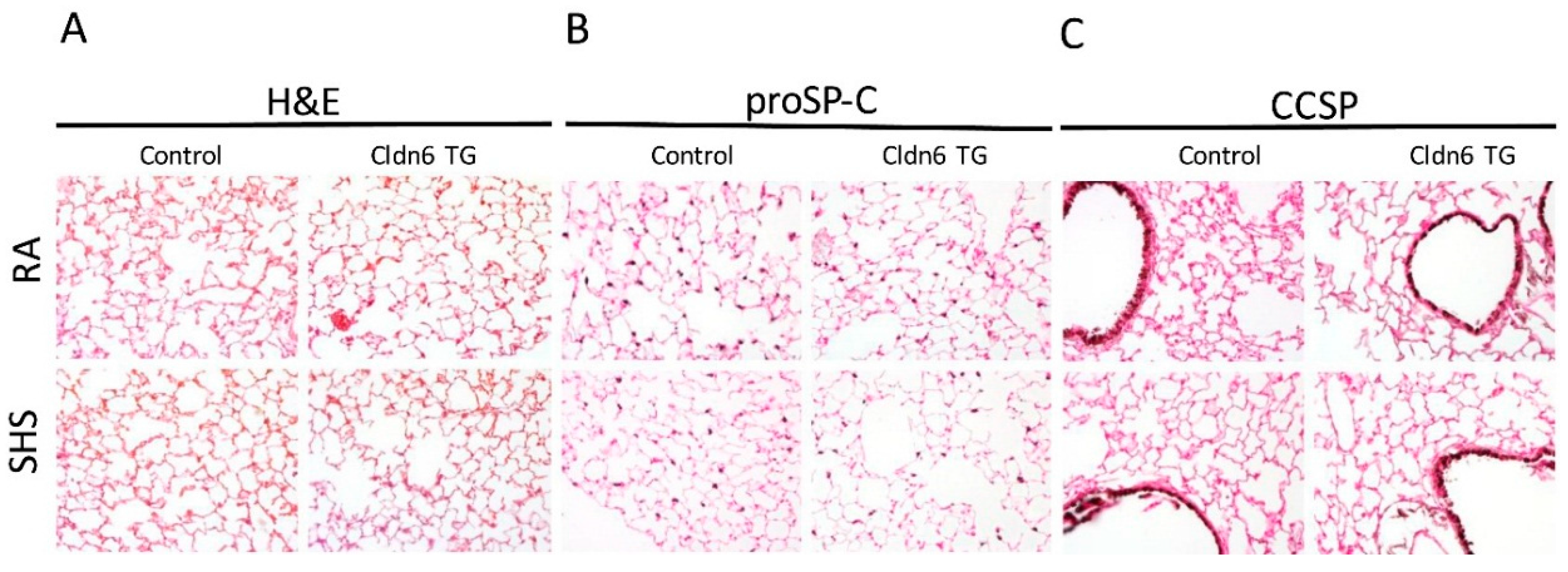
3.2. Lung Morphology
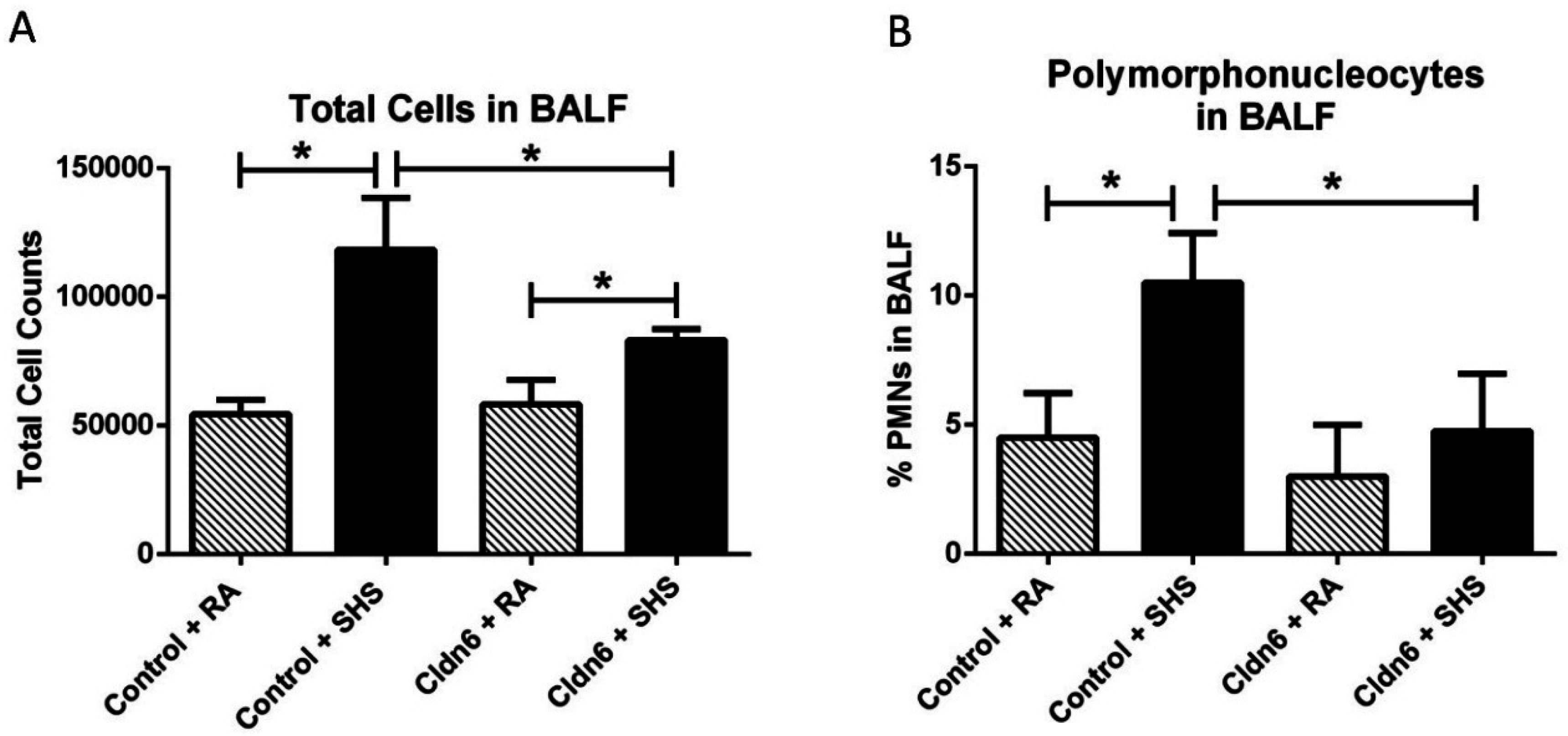
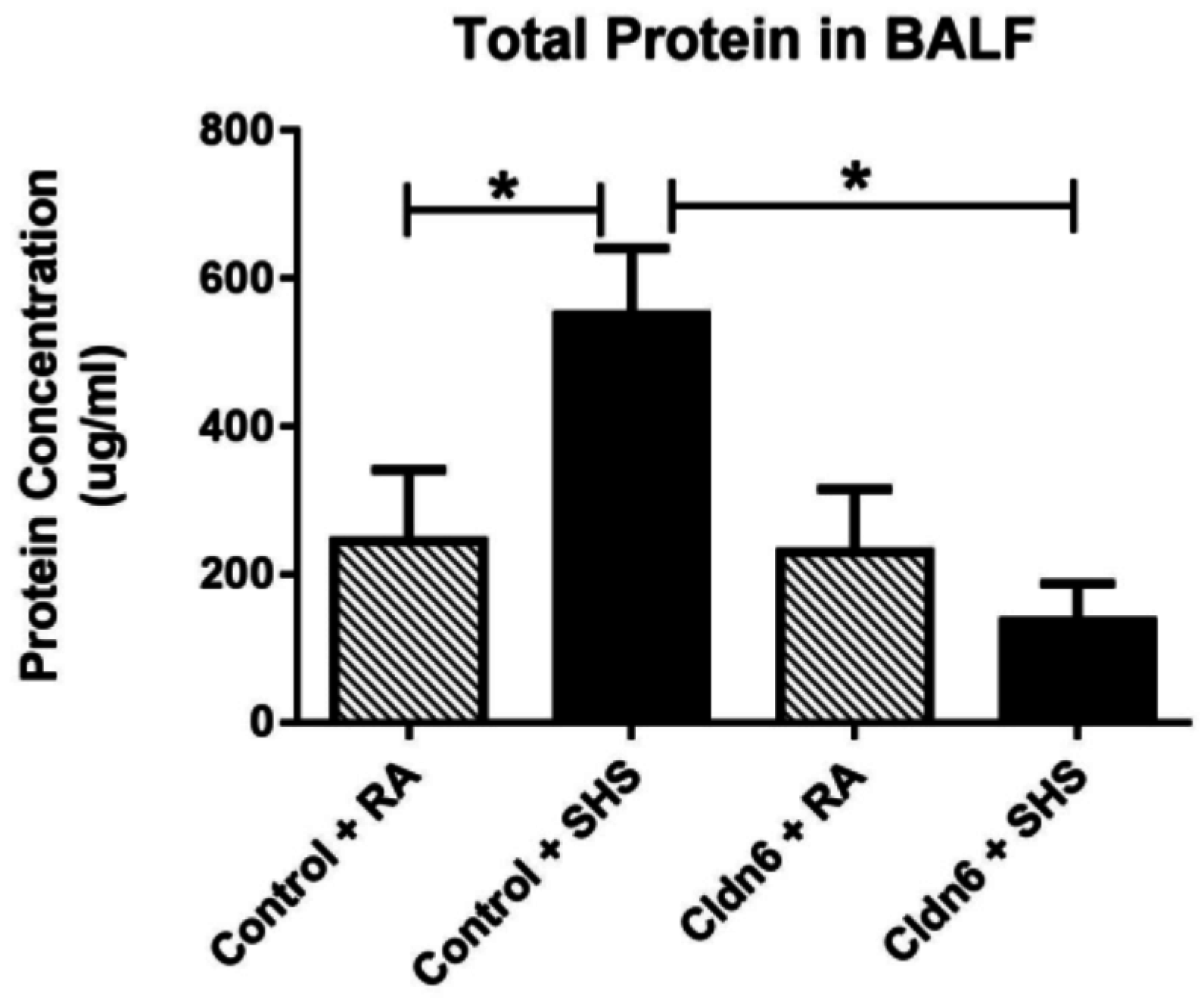
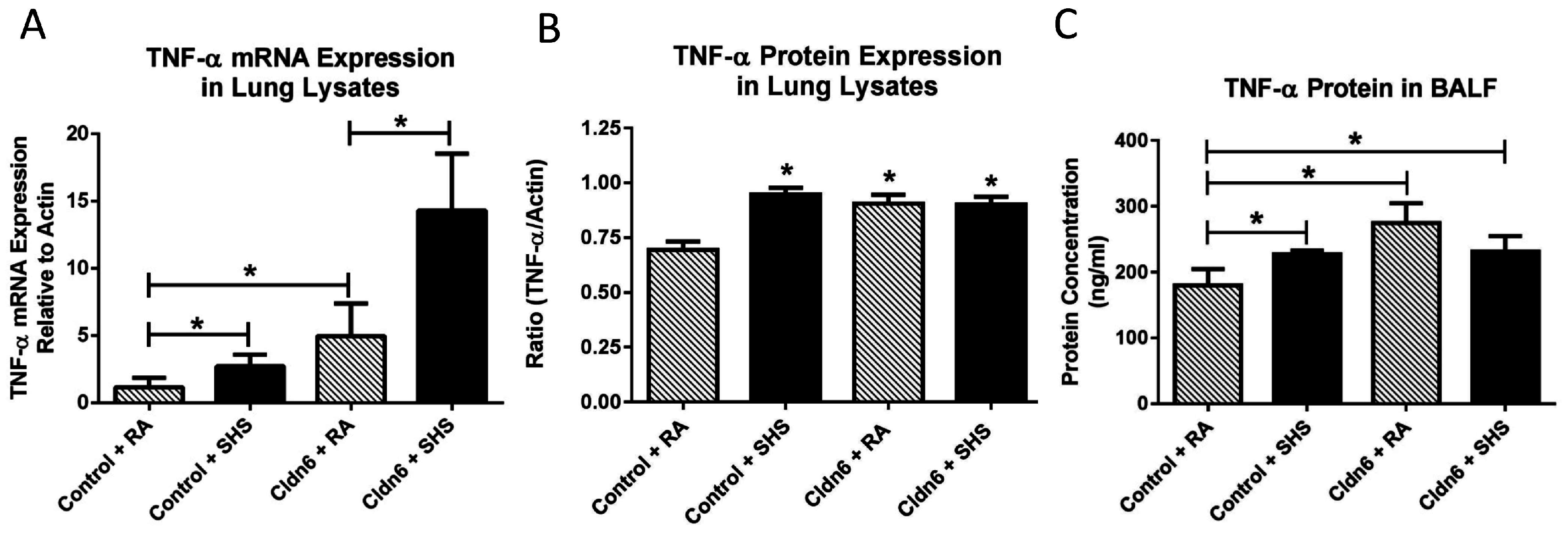
3.3. Pro-Inflammatory Conditions in the Lung
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kirschner, N.; Brandner, J.M. Barriers and more: Functions of tight junction proteins in the skin. Ann. N. Y. Acad. Sci. 2012, 1257, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Matter, K.; Balda, M.S. Epithelial tight junctions, gene expression and nucleo-junctional interplay. J. Cell Sci. 2007, 120, 1505–1511. [Google Scholar] [CrossRef] [PubMed]
- Georas, S.N.; Rezaee, F. Epithelial barrier function: At the front line of asthma immunology and allergic airway inflammation. J. Allergy Clin. Immunol. 2014, 134, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Strengert, M.; Knaus, U.G. Analysis of epithelial barrier integrity in polarized lung epithelial cells. Methods Mol. Biol. 2011, 763, 195–206. [Google Scholar] [PubMed]
- Szaszi, K.; Amoozadeh, Y. New insights into functions, regulation, and pathological roles of tight junctions in kidney tubular epithelium. Int. Rev. Cell Mol. Biol. 2014, 308, 205–271. [Google Scholar] [PubMed]
- Shaykhiev, R.; Otaki, F.; Bonsu, P.; Dang, D.T.; Teater, M.; Strulovici-Barel, Y.; Salit, J.; Harvey, B.G.; Crystal, R.G. Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo. Cell Mol. Life Sci. 2011, 68, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Schamberger, A.C.; Mise, N.; Jia, J.; Genoyer, E.; Yildirim, A.O.; Meiners, S.; Eickelberg, O. Cigarette smoke-induced disruption of bronchial epithelial tight junctions is prevented by transforming growth factor-β. Am. J. Respir. Cell Mol. Biol. 2014, 50, 1040–1052. [Google Scholar] [CrossRef] [PubMed]
- Cuzic, S.; Bosnar, M.; Kramaric, M.D.; Ferencic, Z.; Markovic, D.; Glojnaric, I.; Erakovic Haber, V. Claudin-3 and Clara cell 10 kDa protein as early signals of cigarette smoke-induced epithelial injury along alveolar ducts. Toxicol. Pathol. 2012, 40, 1169–1187. [Google Scholar] [CrossRef] [PubMed]
- Bodas, M.; Min, T.; Vij, N. Critical role of CFTR-dependent lipid rafts in cigarette smoke-induced lung epithelial injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L811–L820. [Google Scholar] [CrossRef] [PubMed]
- Lappi-Blanco, E.; Lehtonen, S.T.; Sormunen, R.; Merikallio, H.M.; Soini, Y.; Kaarteenaho, R.L. Divergence of tight and adherens junction factors in alveolar epithelium in pulmonary fibrosis. Hum. Pathol. 2013, 44, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Ohta, H.; Chiba, S.; Ebina, M.; Furuse, M.; Nukiwa, T. Altered expression of tight junction molecules in alveolar septa in lung injury and fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 302, L193–L205. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Liu, M.; Zhao, T.; Wang, D.; Wang, Y. Protective role of p120-catenin in maintaining the integrity of adherens and tight junctions in ventilator-induced lung injury. Respir. Res. 2015, 16, 58. [Google Scholar] [CrossRef] [PubMed]
- Overgaard, C.E.; Schlingmann, B.; Dorsainvil White, S.; Ward, C.; Fan, X.; Swarnakar, S.; Brown, L.A.; Guidot, D.M.; Koval, M. The relative balance of GM-CSF and TGF-β1 regulates lung epithelial barrier function. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L1212–L1223. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.; Schlingmann, B.L.; Stecenko, A.A.; Guidot, D.M.; Koval, M. NF-kappaB inhibitors impair lung epithelial tight junctions in the absence of inflammation. Tissue Barriers 2015, 3, e982424. [Google Scholar] [CrossRef] [PubMed]
- Ehteshami-Afshar, S.; FitzGerald, J.M.; Doyle-Waters, M.M.; Sadatsafavi, M. The global economic burden of asthma and chronic obstructive pulmonary disease. Int. J. Tuberc. Lung Dis. 2016, 20, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Campos, J.L.; Tan, W.; Soriano, J.B. Global burden of COPD. Respirology 2016, 21, 14–23. [Google Scholar] [CrossRef] [PubMed]
- Mannino, D.M.; Buist, A.S. Global burden of COPD: Risk factors, prevalence, and future trends. Lancet 2007, 370, 765–773. [Google Scholar] [CrossRef]
- Zuo, L.; He, F.; Sergakis, G.G.; Koozehchian, M.S.; Stimpfl, J.N.; Rong, Y.; Diaz, P.T.; Best, T.M. Interrelated role of cigarette smoking, oxidative stress, and immune response in COPD and corresponding treatments. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L205–L218. [Google Scholar] [CrossRef] [PubMed]
- Tsukita, S.; Furuse, M. Claudin-based barrier in simple and stratified cellular sheets. Curr. Opin. Cell Biol. 2002, 14, 531–536. [Google Scholar] [CrossRef]
- Kielgast, F.; Schmidt, H.; Braubach, P.; Winkelmann, V.E.; Thompson, K.E.; Frick, M.; Dietl, P.; Wittekindt, O.H. Glucocorticoids regulate tight junction permeability of lung epithelia by modulating Claudin 8. Am. J. Respir. Cell Mol. Biol. 2016, 54, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Krause, G.; Winkler, L.; Mueller, S.L.; Haseloff, R.F.; Piontek, J.; Blasig, I.E. Structure and function of claudins. Biochim. Biophys. Acta 2008, 1778, 631–645. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, B.L.; Mateescu, M.; Patel, A.S.; Wade, K.; Kimura, S.; Gonzales, L.W.; Guttentag, S.; Ballard, P.L.; Koval, M. Developmental regulation of claudin localization by fetal alveolar epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L1266–L1273. [Google Scholar] [CrossRef] [PubMed]
- Kaarteenaho, R.; Merikallio, H.; Lehtonen, S.; Harju, T.; Soini, Y. Divergent expression of Claudin -1, -3, -4, -5 and -7 in developing human lung. Respir. Res. 2010, 11, 59. [Google Scholar] [CrossRef] [PubMed]
- Kaarteenaho-Wiik, R.; Soini, Y. Claudin-1, -2, -3, -4, -5, and -7 in usual interstitial pneumonia and sarcoidosis. J. Histochem. Cytochem. 2009, 57, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Zhang, T.; Han, Z.G.; Shan, L. Low Claudin-6 expression correlates with poor prognosis in patients with non-small cell lung cancer. Onco Targets Ther. 2015, 8, 1971–1977. [Google Scholar] [PubMed]
- Jimenez, F.R.; Lewis, J.B.; Belgique, S.T.; Wood, T.T.; Reynolds, P.R. Developmental lung expression and transcriptional regulation of Claudin-6 by TTF-1, Gata-6, and FoxA2. Respir. Res. 2014, 15, 70. [Google Scholar] [CrossRef] [PubMed]
- Bohinski, R.J.; Di Lauro, R.; Whitsett, J.A. The lung-specific surfactant protein B gene promoter is a target for thyroid transcription factor 1 and hepatocyte nuclear factor 3, indicating common factors for organ-specific gene expression along the foregut axis. Mol. Cell Biol. 1994, 14, 5671–5681. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.H.; Kalinichenko, V.V.; Lim, L. Transcription factors in mouse lung development and function. Am. J. Physiol. Lung Cell Mol. Physiol. 2001, 280, L823–L838. [Google Scholar] [PubMed]
- Jimenez, F.R.; Belgique, S.T.; Lewis, J.B.; Albright, S.A.; Jones, C.M.; Howell, B.M.; Mika, A.P.; Jergensen, T.R.; Gassman, J.R.; Morris, R.J.; et al. Conditional pulmonary over-expression of Claudin 6 during embryogenesis delays lung morphogenesis. Int. J. Dev. Biol. 2015, 59, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ruan, Y.; Li, Y.; Lin, D.; Liu, Z.; Quan, C. Expression of apoptosis signal-regulating kinase 1 is associated with tight junction protein Claudin-6 in cervical carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 5535–5541. [Google Scholar] [PubMed]
- Liu, Y.; Jin, X.; Li, Y.; Ruan, Y.; Lu, Y.; Yang, M.; Lin, D.; Song, P.; Guo, Y.; Zhao, S.; et al. DNA methylation of Claudin-6 promotes breast cancer cell migration and invasion by recruiting MeCP2 and deacetylating H3Ac and H4Ac. J. Exp. Clin. Cancer Res. 2016, 35, 120. [Google Scholar] [CrossRef] [PubMed]
- Stadler, C.R.; Bahr-Mahmud, H.; Plum, L.M.; Schmoldt, K.; Kolsch, A.C.; Tureci, O.; Sahin, U. Characterization of the first-in-class T-cell-engaging bispecific single-chain antibody for targeted immunotherapy of solid tumors expressing the oncofetal protein Claudin 6. Oncoimmunology 2016, 5, e1091555. [Google Scholar] [CrossRef] [PubMed]
- Micke, P.; Mattsson, J.S.; Edlund, K.; Lohr, M.; Jirstrom, K.; Berglund, A.; Botling, J.; Rahnenfuehrer, J.; Marincevic, M.; Ponten, F.; et al. Aberrantly activated Claudin 6 and 18.2 as potential therapy targets in non-small-cell lung cancer. Int. J. Cancer 2014, 135, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Stogsdill, J.A.; Stogsdill, M.P.; Porter, J.L.; Hancock, J.M.; Robinson, A.B.; Reynolds, P.R. Embryonic overexpression of receptors for advanced glycation end-products by alveolar epithelium induces an imbalance between proliferation and apoptosis. Am. J. Respir. Cell Mol. Biol. 2012, 47, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Stogsdill, J.A.; Stogsdill, M.P.; Heimann, N.B. Up-regulation of receptors for advanced glycation end-products by alveolar epithelium influences cytodifferentiation and causes severe lung hypoplasia. Am. J. Respir. Cell Mol. Biol. 2011, 45, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Stogsdill, M.P.; Stogsdill, J.A.; Bodine, B.G.; Fredrickson, A.C.; Sefcik, T.L.; Wood, T.T.; Kasteler, S.D.; Reynolds, P.R. Conditional overexpression of receptors for advanced glycation end-products in the adult murine lung causes airspace enlargement and induces inflammation. Am. J. Respir. Cell Mol. Biol. 2013, 49, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Rao, N.V.; Argyle, B.; Xu, X.; Reynolds, P.R.; Walenga, J.M.; Prechel, M.; Prestwich, G.D.; MacArthur, R.B.; Walters, B.B.; Hoidal, J.R.; et al. Low anticoagulant heparin targets multiple sites of inflammation, suppresses heparin-induced thrombocytopenia, and inhibits interaction of rage with its ligands. Am. J. Physiol. Cell Physiol. 2010, 299, C97–C110. [Google Scholar] [CrossRef] [PubMed]
- Wood, T.T.; Winden, D.R.; Marlor, D.R.; Wright, A.J.; Jones, C.M.; Chavarria, M.; Rogers, G.D.; Reynolds, P.R. Acute secondhand smoke-induced pulmonary inflammation is diminished in rage knockout mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L758–L764. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, M.; Maes, K.; De Vleeschauwer, S.; Thomas, D.; Verbeken, E.K.; Decramer, M.; Janssens, W.; Gayan-Ramirez, G.N. Long-term nose-only cigarette smoke exposure induces emphysema and mild skeletal muscle dysfunction in mice. Dis. Models Mech. 2012, 5, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Bozinovski, S.; Chan, S.P.; Ivanov, S.; Linden, A.; Hamilton, J.A.; Anderson, G.P. Neutralizing granulocyte/macrophage colony-stimulating factor inhibits cigarette smoke-induced lung inflammation. Am. J. Respir. Crit. Care Med. 2010, 182, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.L.; Cosio, M.; Churg, A. Animal models of chronic obstructive pulmonary disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L1–L15. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.R.; Mucenski, M.L.; Le Cras, T.D.; Nichols, W.C.; Whitsett, J.A. Midkine is regulated by hypoxia and causes pulmonary vascular remodeling. J. Biol. Chem. 2004, 279, 37124–37132. [Google Scholar] [CrossRef] [PubMed]
- Winden, D.R.; Barton, D.B.; Betteridge, B.C.; Bodine, J.S.; Jones, C.M.; Rogers, G.D.; Chavarria, M.; Wright, A.J.; Jergensen, Z.R.; Jimenez, F.R.; et al. Antenatal exposure of maternal secondhand smoke (SHS) increases fetal lung expression of RAGE and induces RAGE-mediated pulmonary inflammation. Respir. Res. 2014, 15, 129. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Roth, G.A.; Naghavi, M.; Parmar, P.; Krishnamurthi, R.; Chugh, S.; Mensah, G.A.; Norrving, B.; Shiue, I.; Ng, M.; et al. Global burden of stroke and risk factors in 188 countries, during 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet Neurol. 2016, 15, 913–924. [Google Scholar] [CrossRef]
- Mbulo, L.; Palipudi, K.M.; Andes, L.; Morton, J.; Bashir, R.; Fouad, H.; Ramanandraibe, N.; Caixeta, R.; Dias, R.C.; Wijnhoven, T.M.; et al. Secondhand smoke exposure at home among one billion children in 21 countries: Findings from the Global Adult Tobacco Survey (GATS). Tob. Control 2016. [Google Scholar] [CrossRef] [PubMed]
- Olivera, D.S.; Boggs, S.E.; Beenhouwer, C.; Aden, J.; Knall, C. Cellular mechanisms of mainstream cigarette smoke-induced lung epithelial tight junction permeability changes in vitro. Inhal. Toxicol. 2007, 19, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, F.; Lewis, J.; Wood, T.; Reynolds, P. Pulmonary expression and regulation of Cldn6 by tobacco smoke (834.3). FASEB J. 2014, 28. [Google Scholar]
- Van der Toorn, M.; Slebos, D.J.; de Bruin, H.G.; Gras, R.; Rezayat, D.; Jorge, L.; Sandra, K.; van Oosterhout, A.J. Critical role of aldehydes in cigarette smoke-induced acute airway inflammation. Respir. Res. 2013, 14, 45. [Google Scholar] [CrossRef] [PubMed]
- Stockley, R.A. Neutrophils and the pathogenesis of COPD. Chest 2002, 121, 151S–155S. [Google Scholar] [CrossRef] [PubMed]
- Caramori, G.; Adcock, I.M.; Di Stefano, A.; Chung, K.F. Cytokine inhibition in the treatment of COPD. Int. J. Chron. Obstruct. Pulm. Dis. 2014, 9, 397–412. [Google Scholar]
- Runkle, E.A.; Rice, S.J.; Qi, J.; Masser, D.; Antonetti, D.A.; Winslow, M.M.; Mu, D. Occludin is a direct target of thyroid transcription factor-1 (TTF-1/NKX2-1). J. Biol. Chem. 2012, 287, 28790–28801. [Google Scholar] [CrossRef] [PubMed]
- You, K.; Xu, X.; Fu, J.; Xu, S.; Yue, X.; Yu, Z.; Xue, X. Hyperoxia disrupts pulmonary epithelial barrier in newborn rats via the deterioration of occludin and ZO-1. Respir. Res. 2012, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, D.R.; Marchiando, A.M.; Zhang, Y.; Shen, L.; Sasaki, H.; Wang, Y.; Long, M.; Turner, J.R. Tight junction-associated MARVEL proteins marveld3, tricellulin, and occludin have distinct but overlapping functions. Mol. Biol. Cell 2010, 21, 1200–1213. [Google Scholar] [CrossRef] [PubMed]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lewis, J.B.; Milner, D.C.; Lewis, A.L.; Dunaway, T.M.; Egbert, K.M.; Albright, S.C.; Merrell, B.J.; Monson, T.D.; Broberg, D.S.; Gassman, J.R.; et al. Up-Regulation of Claudin-6 in the Distal Lung Impacts Secondhand Smoke-Induced Inflammation. Int. J. Environ. Res. Public Health 2016, 13, 1018. https://doi.org/10.3390/ijerph13101018
Lewis JB, Milner DC, Lewis AL, Dunaway TM, Egbert KM, Albright SC, Merrell BJ, Monson TD, Broberg DS, Gassman JR, et al. Up-Regulation of Claudin-6 in the Distal Lung Impacts Secondhand Smoke-Induced Inflammation. International Journal of Environmental Research and Public Health. 2016; 13(10):1018. https://doi.org/10.3390/ijerph13101018
Chicago/Turabian StyleLewis, Joshua B., Dallin C. Milner, Adam L. Lewis, Todd M. Dunaway, Kaleb M. Egbert, Scott C. Albright, Brigham J. Merrell, Troy D. Monson, Dallin S. Broberg, Jason R. Gassman, and et al. 2016. "Up-Regulation of Claudin-6 in the Distal Lung Impacts Secondhand Smoke-Induced Inflammation" International Journal of Environmental Research and Public Health 13, no. 10: 1018. https://doi.org/10.3390/ijerph13101018