
Altered Expression of Genes in Signaling Pathways Regulating Proliferation of Hematopoietic Stem and Progenitor Cells in Mice with Subchronic Benzene Exposure


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Animals and Treatments
2.3. Blood Parameters, Relative Organ Weight and Bone Marrow Smears
2.4. Collection of Hematopoietic Stem cells
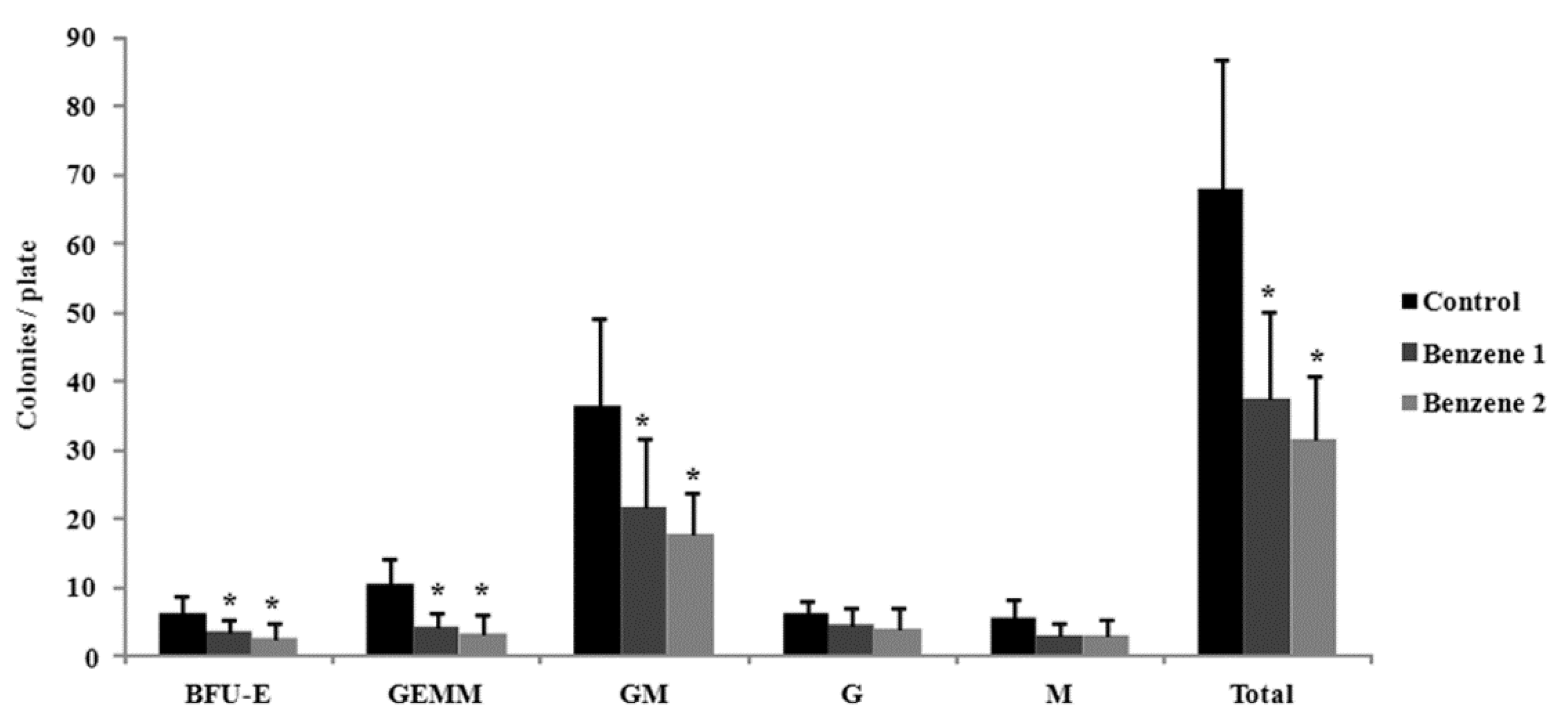
2.5. Colony Forming Cells Assay
2.6. Real-time PCR Assay
2.7. Statistical Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Primer Set | Sequence (5′-3′) |
|---|---|---|
| β-actin | Outer | Forward: CGAGCGGTTCCGATGCCCTG; Reverse: ACGCAGCTCAGTAACAGTCCGC |
| Inner | Forward: ATCCGCAAAGACCTGT; Reverse: GGGTGTAACGCAACTAAG | |
| β-catenin | Outer | Forward: GACACCTCCCAAGTCCTTT; Reverse: CCTCTGAGCCCTAGTCATT |
| Inner | Forward: ACACCTCCCAAGTCCTTTATGAAT; Reverse: CCCGTCAATATCAGCTACTTGCT | |
| c-Myc | Outer | Forward: GCGCTCTCCTTCCTCGGACTCGC; Reverse: GGTTTGCCTCTTCTCCACAG; |
| Inner | Forward: GCCCCTAGTGCTGCATGAG; Reverse: CCACAGACACCACATCAATTTCTT | |
| Notch1 | Outer | Forward: GGCCTTGAGACAGGCAACA; Reverse: CTGCACTGGCCTCCAGCAA |
| Inner | Forward: CCTTGAGACAGGCAACAGTGAAG; Reverse: GAGAGTATCGGGCAGCCAAGT | |
| cyclinD1 | Outer | Forward: GCGCTTCCAACCCACCCTCCATG; Reverse: GCGCCGCAGGCTTGACTCCAGAA |
| Inner | Forward: GGGCAGCCCCAACAACTT; Reverse: GCAGTCCGGGTC ACACTTG | |
| Bmi1 | Outer | Forward: CAGAGGGATGGACTGACGA; Reverse: GCGTGAGGGAACTGTGGGTGA |
| Inner | Forward: GAAAGTGACTCTGGGAGTGACAAG; Reverse: GGGACTGGGCAAACAGGAA | |
| p53 | Outer | Forward: CTTCCACCTGGGCTTCCTG; Reverse: GTGGCGCTGACCCACAACT |
| Inner | Forward: TGGCGCTGACCCACAACT; Reverse: CCAAGTCTGTTATGTGCACGTACTCT |
3. Results
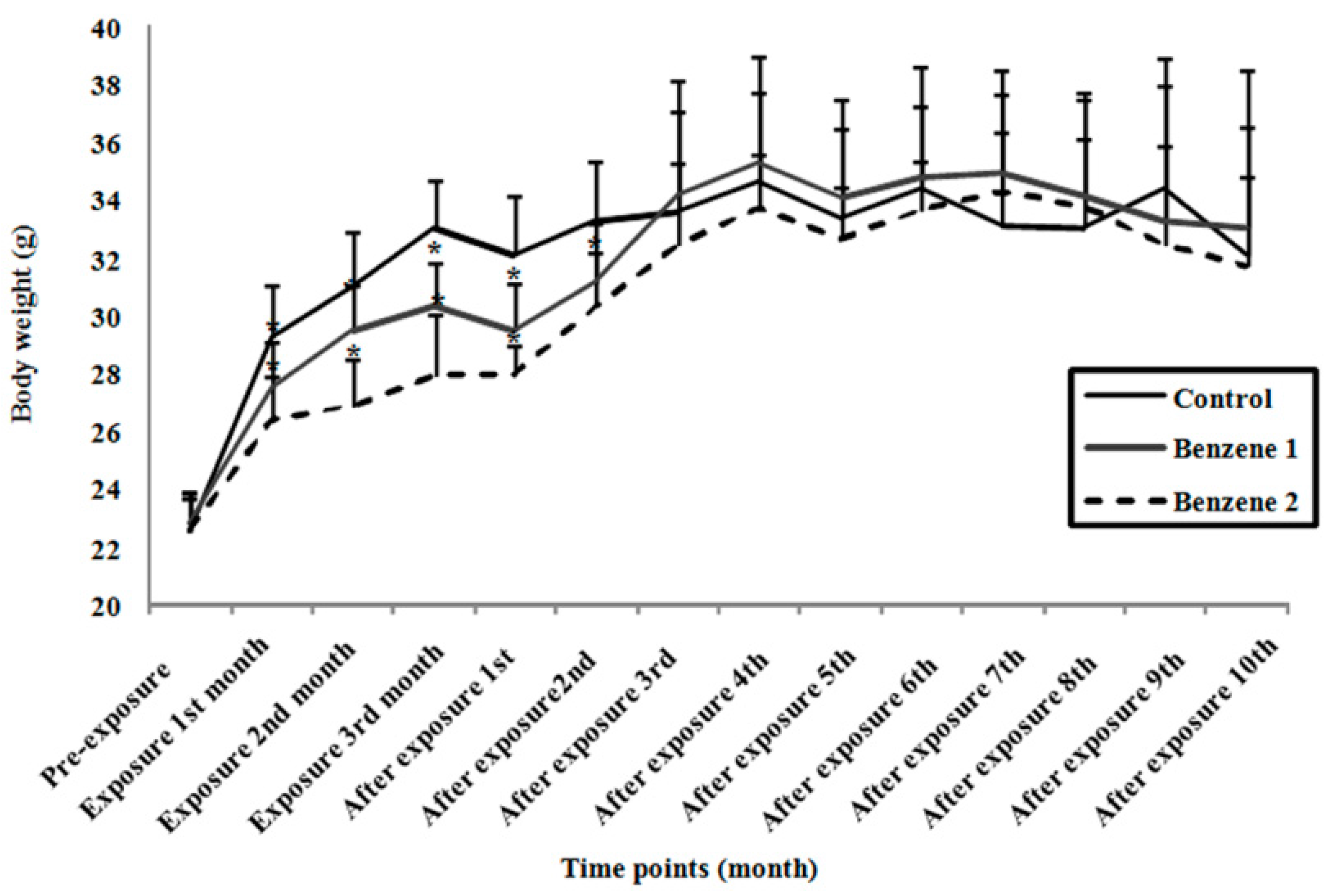
3.1. Body Weight and Relative Organ Weight
| Group | Relative Organ Weight (Mean ± SD) | |||
|---|---|---|---|---|
| Relative Liver Weight | Relative Spleen Weight | Relative Lung Weight | Relative Kidney Weight | |
| Control | 5.31 ± 0.89 | 0.38 ± 0.10 | 0.88 ± 0.44 | 1.93 ± 0.10 |
| Benzene 1 | 5.14 ± 0.39 | 0.41 ± 0.10 | 0.91 ± 0.44 | 1.92 ± 0.17 |
| Benzene 2 | 5.37 ± 0.57 | 0.52 ± 0.14*,# | 0.98 ± 0.43 | 1.89 ± 0.25 |
3.2. Blood Parameters and Bone Marrow Smears
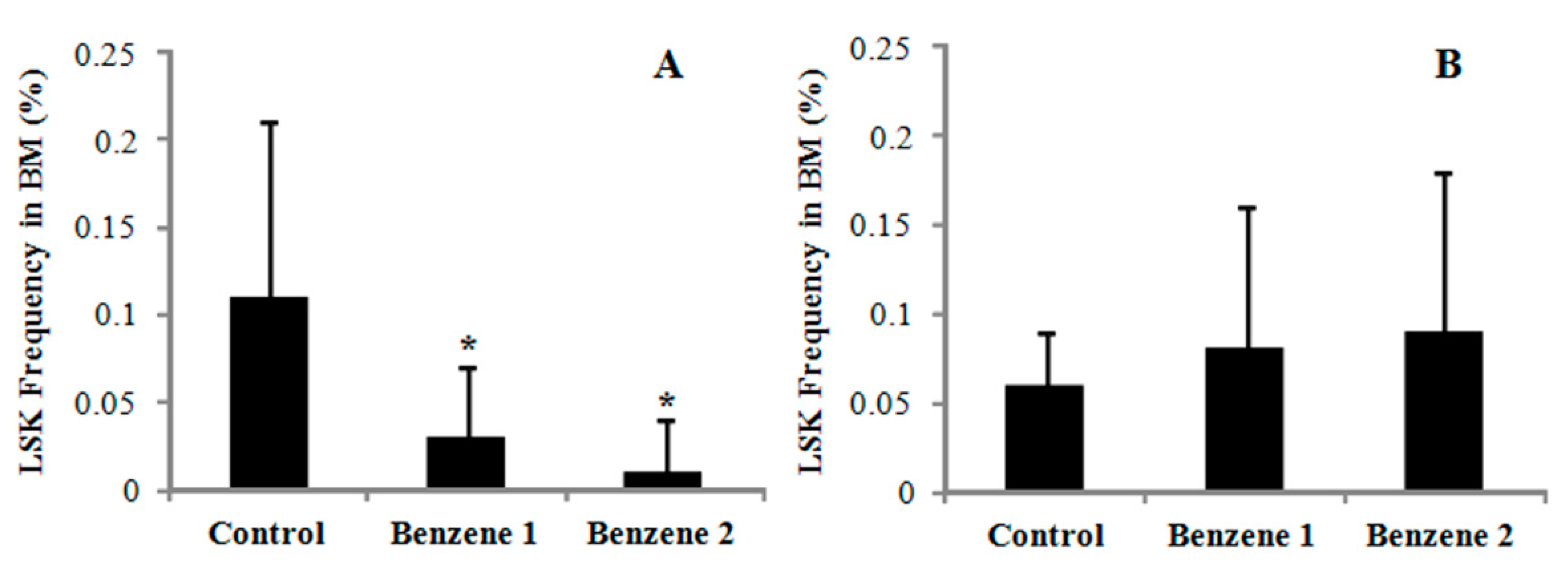
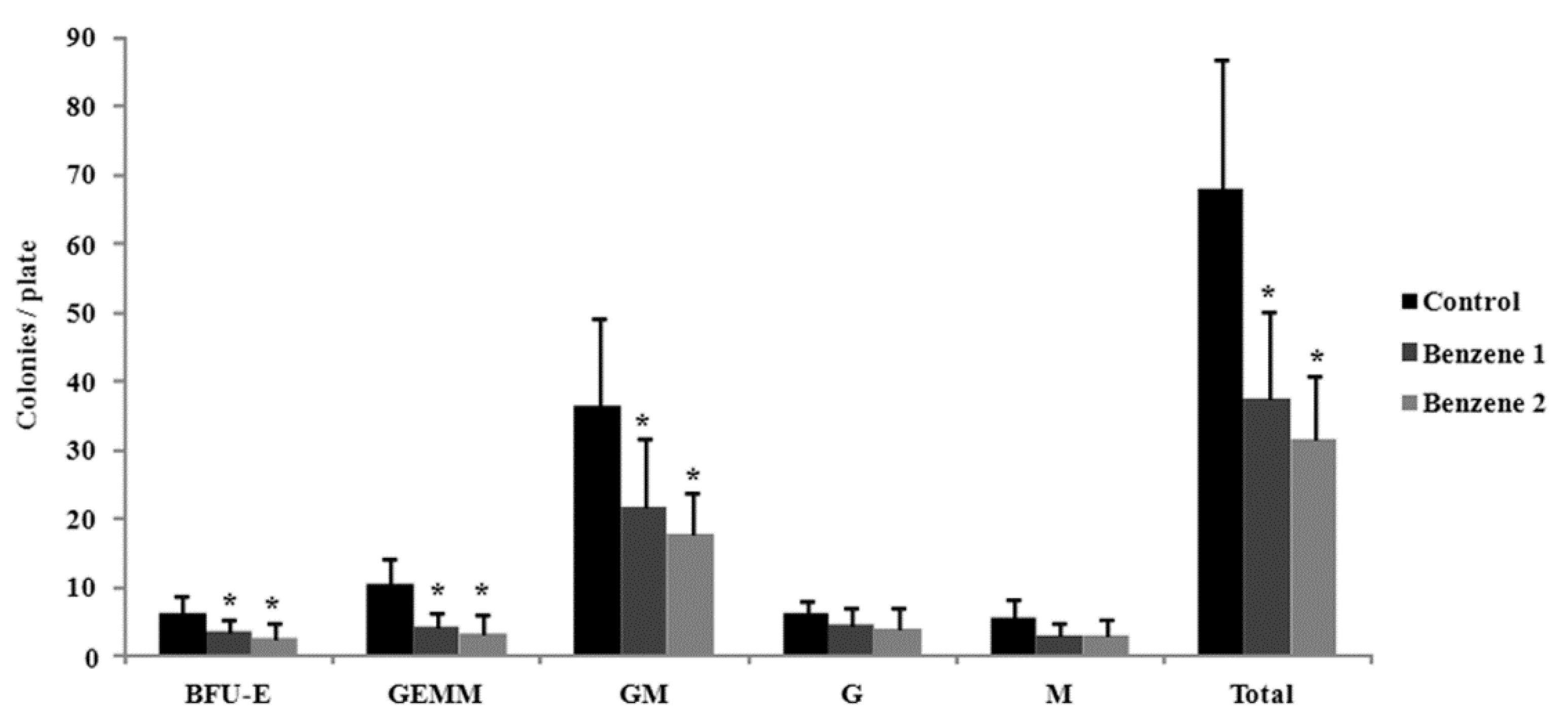
3.3. Frequency of LSK Cells and Number of Colony Forming
| Time Points | Blood Parameters (Mean ± SD) | ||||
|---|---|---|---|---|---|
| Group | WBC (109/L) | RBC (1012/L) | Hgb (g/L) | Pit (109/L) | |
| Three months after benzene exposure | Control | 5.05 ± 1.56 | 8.28 ± 0.32 | 138.42 ± 4.07 | 661.92 ± 87.01 |
| Benzene 1 | 1.60 ± 0.72* | 5.63 ± 1.15* | 114.00 ± 19.8* | 374.00 ± 144.18* | |
| Benzene 2 | 2.19 ± 1.16* | 5.84 ± 1.39* | 90.00 ± 37.98* | 318.67 ± 163.54* | |
| Ten months post-benzene exposure | Control | 7.43 ± 2.03 | 8.23 ± 1.22 | 130.27 ± 22.12 | 514.40 ± 85.60 |
| Benzene 1 | 5.81 ± 1.82 | 7.59 ± 1.07 | 117.75 ± 16.12 | 677.37 ± 155.22* | |
| Benzene 2 | 6.10 ± 2.49 | 6.75 ± 1.27*,# | 106.65 ± 13.10* | 701.73 ± 196.21* | |

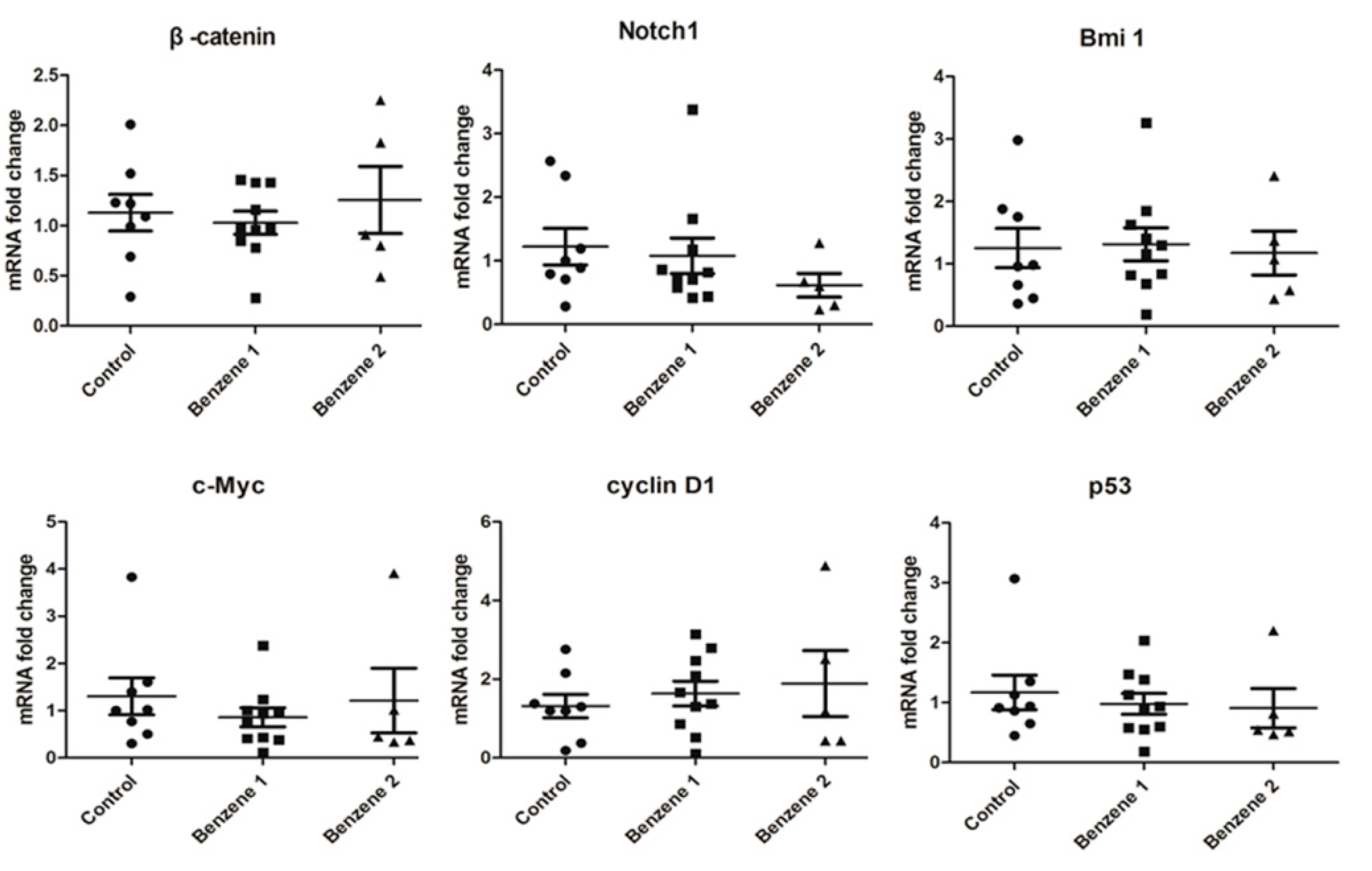
3.4. Expression of β-catenin, c-Myc, Notch1, Cyclin D1, Bmi 1 and P53

4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wilbur, S.; Wohlers, D.; Paikoff, S.; Keith, L.S.; Faroon, O. ATSDR evaluation of potential for human exposure to benzene. Toxicol. Ind. Health 2008, 24, 399–442. [Google Scholar] [CrossRef] [PubMed]
- IARC (International Agency for Research on Cancer). Monographs on the Evaluation of Carcinogenic Risks to Humans Overall Evaluations of Carcinogenicity. 2014. Available online: http://monographs.iarc.fr/ENG/Monographs/vol71/mono71.pdf (accessed on 3 March 2014).
- Khalade, A.; Jaakkola, M.S.; Pukkala, E.; Jaakkola, J.J. Exposure to benzene at work and the risk of leukemia: A systematic review and meta-analysis. Environ. Health 2010, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.T. Advances in understanding benzene health effects and susceptibility. Ann. Rev. Public Health 2010, 31, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Steinmaus, C.; Smith, A.H.; Jones, R.M.; Smith, M.T. Meta-analysis of benzene exposure and non-Hodgkin lymphoma: Biases could mask an important association. Occup. Environ. Med. 2008, 65, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Billet, S.; Paget, V.; Garcon, G.; Heutte, N.; Andre, V.; Shirali, P.; Sichel, F. Benzene-induced mutational pattern in the tumour suppressor gene Tp53 analysed by use of a functional assay, the functional analysis of separated alleles in yeast, in human lung cells. Arch. Toxicol. 2010, 84, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Kolachana, P.; Subrahmanyam, V.V.; Meyer, K.B.; Zhang, L.; Smith, M.T. Benzene and its phenolic metabolites produce oxidative DNA damage in HL60 cells in vitro and in the bone marrow in vivo. Cancer. Res. 1993, 53, 1023–1026. [Google Scholar] [PubMed]
- McHale, C.M.; Zhang, L.; Lan, Q.; Vermeulen, R.; Li, G.; Hubbard, A.E.; Porter, K.E.; Thomas, R.; Portier, C.J.; Shen, M.; et al. Global gene expression profiling of a population exposed to a range of benzene levels. Environ. Health Perspect. 2011, 119, 628–634. [Google Scholar] [CrossRef] [PubMed]
- Schugar, R.C.; Robbins, P.D.; Deasy, B.M. Small molecules in stem cell self-renewal and differentiation. Gene Ther. 2008, 15, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Duncan, A.W.; Ailles, L.; Domen, J.; Scherer, D.C.; Willert, K.; Hintz, L.; Nusse, R.; Weissman, I.L. A role for Wnt signalling in self-renewal of haematopoietic stem cells. Nature 2003, 423, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Mikesch, J.H.; Steffen, B.; Berdel, W.E.; Serve, H.; Müller-Tidow, C. The emerging role of Wnt signaling in the pathogenesis of acute myeloid leukemia. Leukemia 2007, 21, 1638–1647. [Google Scholar] [CrossRef] [PubMed]
- Bafico, A.; Liu, G.; Goldin, L.; Harris, V.; Aaronson, S.A. An autocrine mechanism of constitutive Wnt pathway activation in human cancer cells. Cancer Cell 2004, 6, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; von Kries, J.P.; Kuhl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Pelengaris, S.; Khan, M.; Evan, G.I. c-MYC: More than just a matter of life and death. Nat. Rev. Cancer 2002, 2, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Bigas, A.; Robert-Moreno, A.; Espinosa, L. The Notch pathway in the developing hematopoietic system. Int. J. Dev. Biol. 2010, 54, 1175–1188. [Google Scholar] [CrossRef] [PubMed]
- Suresh, S.; Irvine, A.E. The NOTCH signaling pathway in normal and malignant blood cell production. J. Cell. Commun. Signal. 2015, 9, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Rattis, F.M.; Dimascio, L.N.; Congdon, K.L.; Pazianos, G.; Zhao, C.; Yoon, K.; Cook, J.M.; Willert, K.; Gaiano, N.; et al. Integration of Notch and Wnt signaling in haematopoietic stem cell maintenance. Nat. Immunol. 2005, 6, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Lobry, C.; Ntziachristos, P.; Ndiaye-Lobry, D.; Oh, P.; Cimmino, L.; Zhu, N.; Araldi, E.; Hu, W.; Freund, J.; Abdel-Wahab, O.; et al. Notch pathway activation targets AML-initiating cell homeostasis and differentiation. J. Exp. Med. 2013, 210, 301–319. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.; Kohlmann, A.; Dugas, M.; Kern, W.; Hiddemann, W.; Schnittger, S.; Haferlach, T. Genomic gains and losses influence expression levels of genes located within the affected regions: A study on acute myeloidleukemi as with trisomy 8, 11, or 13, Monosomy7, ordeletion5q. Leukemia 2005, 19, 1224–1228. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Zeng, W.; Miyazato, A.; Billings, E.; Maciejewski, J.P.; Kajigaya, S.; Sloand, E.M.; Young, N.S. Distinctive gene expression profiles of CD34 Cells from patients with myelodys plastic syndrome characterized by specific chromosomal abnormalities. Blood 2004, 104, 4210–4218. [Google Scholar] [CrossRef] [PubMed]
- Towbridge, J.J.; Scott, M.P.; Bhatia, M. Hedgehog modulates cell cycle regulators in stem cells to control hematopoietic regeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 14134–14139. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, R.U.; Memarzadeh, S.; Wu, H.; Witte, O.N. Bmi-1 is a crucial regulator of prostate stem cell self-renewal and malignant transformation. Cell Stem Cell 2010, 7, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Beigi, R.; Guo, G.R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansionof Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Elf, S.E.; Miyata, Y.; Sashida, G.; Liu, Y.; Huang, G.; di Giandomenico, S.; Lee, J.M.; Deblasio, A.; Menendez, S.; et al. p53 Regulates hematopoietic stem cell quiescence. Cell Stem Cell 2009, 4, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Park, I.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 is required for maintenance of adult self-renewing hematopoietic stem cells. Nature 2003, 423, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, Y.; Yoon, B.I.; Li, G.X.; Fujii-Kuriyama, Y.; Kaneko, T.; Kanno, J.; Inoue, T. Benzene-induced hematopoietic toxicity transmitted by AhR in wild-type mouse and nullified by repopulation with AhR-deficient bone marrow cells: Time after benzene treatment and recovery. Chemosphere 2008, 73, S290–S294. [Google Scholar] [CrossRef] [PubMed]
- Cronkite, E.P.; Bullis, J.; Inoue, T.; Drew, R.T. Benzene inhalation produces leukemia in mice. Toxicol. Appl. Pharmacol. 1984, 75, 358–361. [Google Scholar] [CrossRef]
- Sun, R.; Zhang, J.; Xiong, M.; Chen, Y.; Yin, L.; Pu, Y. Metabonomics biomarkers for subacute toxicity screening for benzene exposure in mice. J. Toxicol. Environ. Health A 2012, 75, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Zhang, J.; Yin, L.; Pu, Y. Investigation into Variation of Endogenous Metabolites in Bone Marrow Cells and Plasma in C3H/He Mice Exposed to Benzene. Int. Mol. Sci. 2014, 15, 4994–5010. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, A. The role of DNA repair in benzene-induced carcinogenesis. Chem. Biol. Interact. 2010, 184, 269–272. [Google Scholar] [CrossRef] [PubMed]
- Vigneron, A.; Vousden, K.H. P53, ROS and senescence in the control of aging. Aging 2010, 2, 471–474. [Google Scholar] [PubMed]
- Robert, S. Leukemia and benzene. Int. J. Environ. Res. Public Health 2012, 9, 2875–2893. [Google Scholar]
- McHale, C.M.; Zhang, L.; Smith, M.T. Current understanding of the mechanism of benzene-induced leukemia in humans: implications for risk assessment. Carcinogenesis 2012, 33, 240–252. [Google Scholar] [CrossRef] [PubMed]
- Bogadi-Sare, A.; Turk, R.; Zavalic, M. Medical surveillance studies of workers exposed to low level benzene. Arh. Hig. Rada. Toksikol. 1995, 46, 391–398. [Google Scholar] [PubMed]
- Robert Schnatter, A.; Kerzic, P.J.; Zhou, Y.; Chen, M.; Nicolich, M.J.; Lavelle, K.; Armstrong, T.W.; Bird, M.G.; Lin, L.; Fu, H.; et al. Peripheral blood effects in benzene-exposed workers. Chem. Biol. Interact. 2010, 19, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Zhang, L.; Li, G.; Vermeulen, R.; Weinberg, R.S.; Dosemeci, M.; Rappaport, S.M.; Shen, M.; Alter, B.P.; Wu, Y.; et al. Hematotoxicity in workers exposed to low levels of benzene. Science 2004, 306, 1774–1776. [Google Scholar] [CrossRef] [PubMed]
- Badham, H.J.; Winn, L.M. In Utero Exposure to Benzene Disrupts Fetal Hematopoietic Progenitor Cell Growth via Reactive Oxygen Species. Toxicol. Sci. 2010, 113, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, C.H.; Ailles, L.E.; Dylla, S.J.; Muijtjens, M.; Jones, C.; Zehnder, J.L.; Gotlib, J.; Li, K.; Manz, M.G.; Keating, A.; et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N. Engl. J. Med. 2004, 351, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.; Grandage, V.L.; Linch, D.C.; Khwaja, A. Constitutive activation of the Wnt/b-catenin signalling pathway in acute myeloid leukaemia. Oncogene 2005, 24, 2410–2420. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Krivtsov, A.V.; Sinha, A.U.; North, T.E.; Goessling, W.; Feng, Z.; Zon, L.I.; Armstrong, S.A. TheWnt/β-Catenin Pathway Is Required for the Development of Leukemia Stem Cells in AML. Science 2010, 327, 1650–1653. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Murphy, M.J.; Oskarsson, T.; Kaloulis, K.; Bettess, M.D.; Oser, G.M.; Pasche, A.C.; Knabenhans, C.; MacDonald, H.R.; Trumpp, A. c-Myc controls the balance between hematopoietic stem cell self-renewa land differentiation. Genes Dev. 2004, 18, 2747–2763. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Q.; Cui, Y.; Liu, Z.Y.; Zhao, W.; Wang, C.L.; Dong, Y.; Hou, L.; Hu, G.; Luo, C.; et al. Knockdown of cyclin D1 inhibits proliferation, induces apoptosis, and attenuates the invasive capacity of human glioblastoma cells. J. Neurooncol. 2012, 106, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Wang, C.; Li, Z.; Sakamaki, T.; Pestell, R.G. Mini review: Cyclin D1: Normal and abnormal functions. Endocrinology 2004, 145, 5439–5447. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Mukhopadhyay, M.K.; Ghosh, P.D.; Nath, D. Effect of Methanolic Leaf Extract of Ocimum basilicum L. on benzene-induced hematotoxicity in Mice. Evid. Based. Complement. Alternat. Med. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.M.; Yen, C.C.; Wang, W.S.; Lin, Y.J.; Chu, C.J.; Chiou, T.J.; Liu, J.H.; Yang, M.H. Down-regulation of Notch-1 expression decreases PU. 1-mediatedmyeloid differentiation signaling in acute myeloid leukemia. Int. J. Oncol. 2008, 32, 1335–1341. [Google Scholar] [PubMed]
- Bhattacharyya, J.; Mihara, K.; Yasunaga, S.; Tanaka, H.; Hoshi, M.; Takihara, Y.; Kimura, A. BMI-1 expression is enhanced through transcriptional and posttranscriptional regulation during the progression of chronic myeloid leukemia. Ann. Hematol. 2009, 88, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Saudy, N.S.; Fawzy, I.M.; Azmy, E.; Goda, E.F.; Eneen, A.; Abdul Salam, E.M. BMI1 gene expression in myeloid leukemias and its impact on prognosis. Blood. Cells. Mol. Dis. 2014, 53, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Sablina, A.A.; Budanov, A.V.; Ilyinskaya, G.V.; Agapova, L.S.; Kravchenko, J.E.; Chumakov, P.M. The antioxidant function of the p53 tumor suppressor. Nat. Med. 2005, 11, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Oike, Y.; Onoyama, I.; Iwama, A.; Arai, F.; Takubo, K.; Mashimo, Y.; Oguro, H.; Nitta, E.; Ito, K.; et al. Fbxw7 acts as acritical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev. 2008, 22, 986–991. [Google Scholar] [CrossRef] [PubMed]
- Pant, V.; Quintás-Cardama, A.; Lozano, G. The p53 pathway in hematopoiesis: lessons from mouse models, implications for humans. Blood 2012, 120, 5118–5127. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, R.; Zhang, J.; Xiong, M.; Wei, H.; Tan, K.; Yin, L.; Pu, Y. Altered Expression of Genes in Signaling Pathways Regulating Proliferation of Hematopoietic Stem and Progenitor Cells in Mice with Subchronic Benzene Exposure. Int. J. Environ. Res. Public Health 2015, 12, 9298-9313. https://doi.org/10.3390/ijerph120809298
Sun R, Zhang J, Xiong M, Wei H, Tan K, Yin L, Pu Y. Altered Expression of Genes in Signaling Pathways Regulating Proliferation of Hematopoietic Stem and Progenitor Cells in Mice with Subchronic Benzene Exposure. International Journal of Environmental Research and Public Health. 2015; 12(8):9298-9313. https://doi.org/10.3390/ijerph120809298
Chicago/Turabian StyleSun, Rongli, Juan Zhang, Mengzhen Xiong, Haiyan Wei, Kehong Tan, Lihong Yin, and Yuepu Pu. 2015. "Altered Expression of Genes in Signaling Pathways Regulating Proliferation of Hematopoietic Stem and Progenitor Cells in Mice with Subchronic Benzene Exposure" International Journal of Environmental Research and Public Health 12, no. 8: 9298-9313. https://doi.org/10.3390/ijerph120809298