Increased NQO1 but Not c-MET and Survivin Expression in Non-Small Cell Lung Carcinoma with KRAS Mutations

Abstract
:1. Introduction
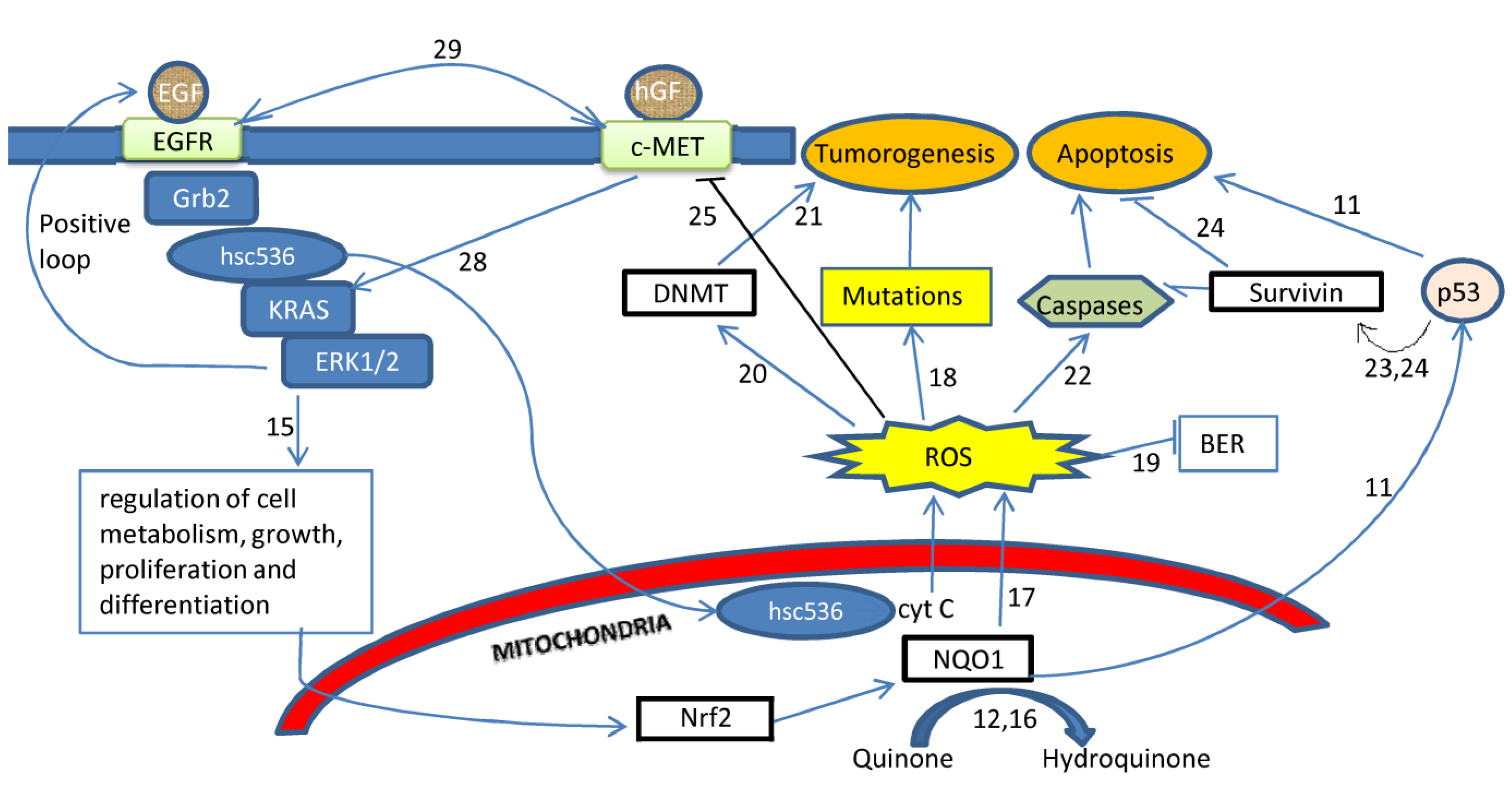
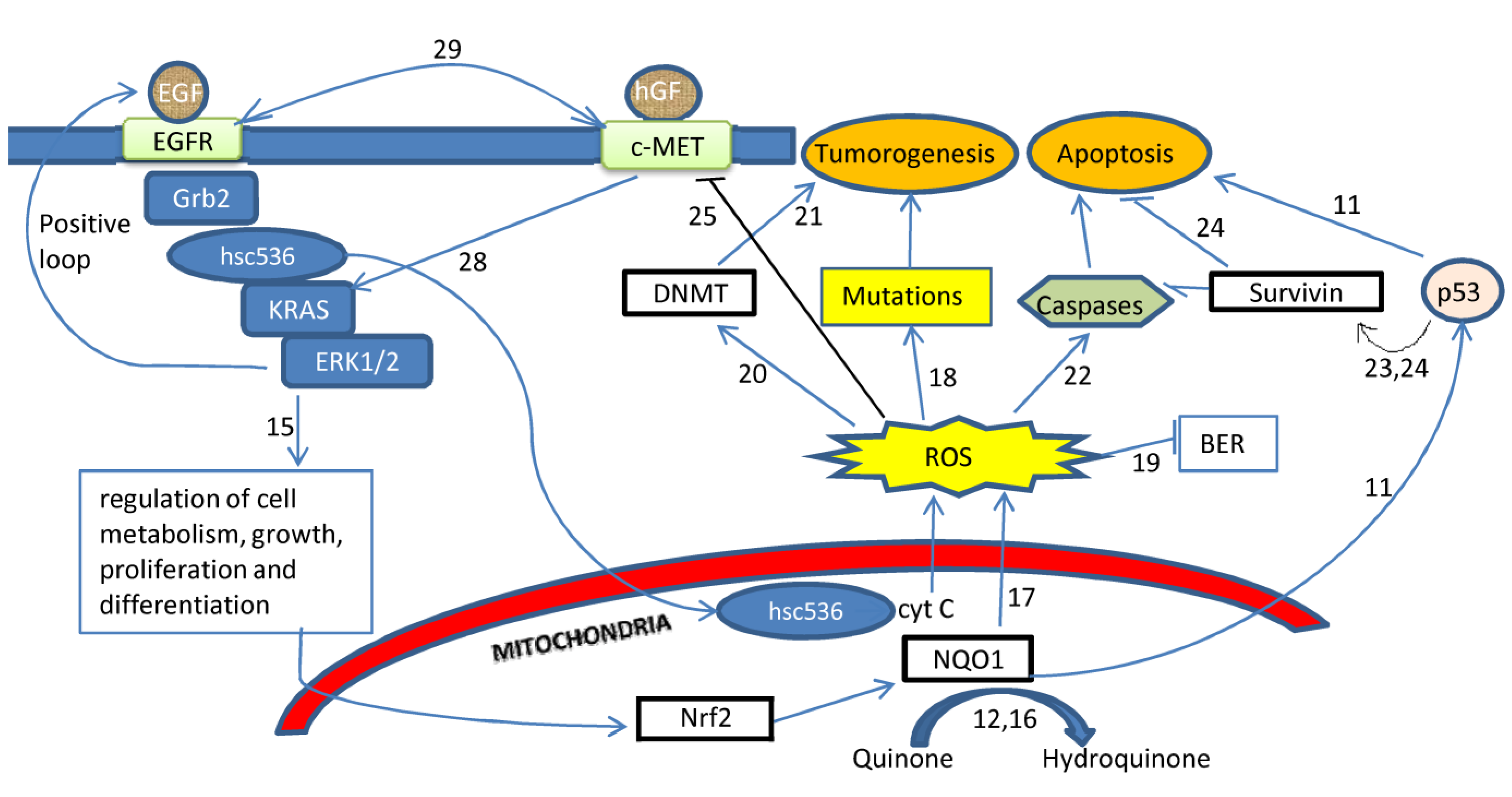
2. Results

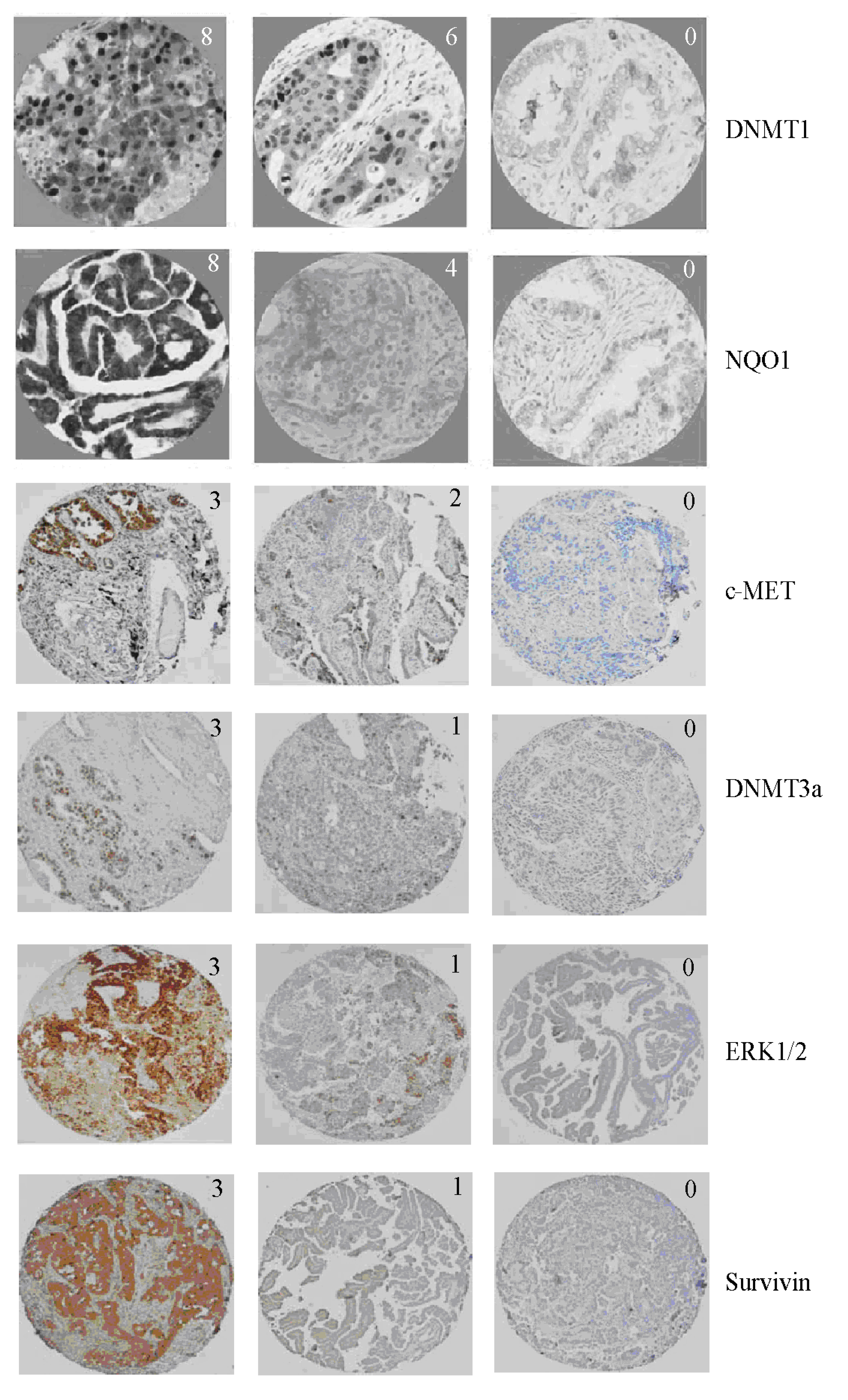
{kind=link}
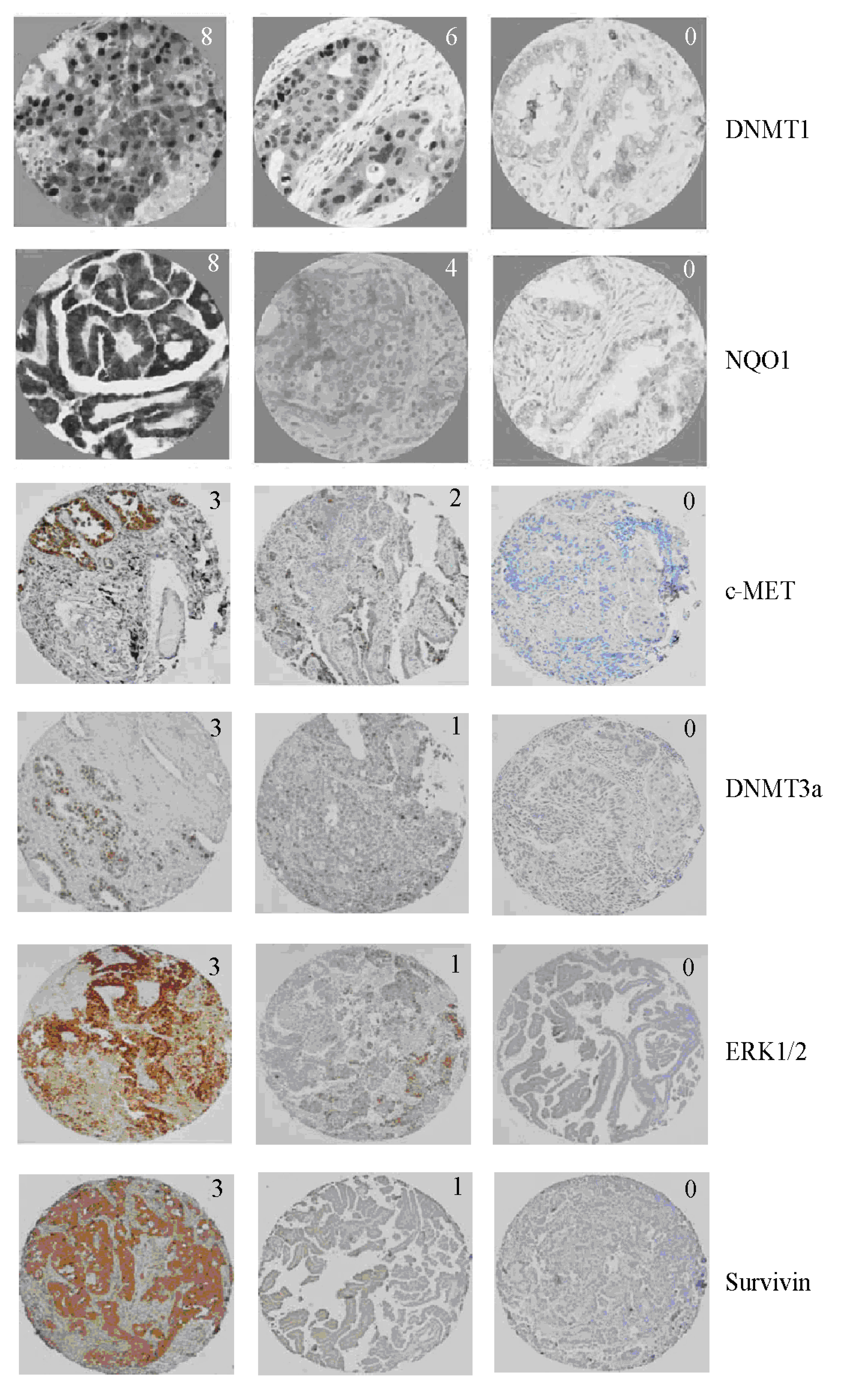{kind=link}
| Protein | IHC a | KRAS Wild Type b | KRAS Mutated b | Total | P c |
|---|---|---|---|---|---|
| NQO1 | Negative | 17 | 2 | 19 | <0.001 * |
| Positive | 16 | 24 | 40 | ||
| DNMT1 | Negative | 14 | 1 | 15 | <0.001 * |
| Positive | 19 | 25 | 44 | ||
| DNMT3a | Negative | 24 | 10 | 34 | 0.01 * |
| Positive | 10 | 16 | 26 | ||
| ERK1/2 | Negative | 17 | 11 | 28 | 0.002 * |
| Positive | 4 | 20 | 24 | ||
| c-MET | Negative | 5 | 9 | 14 | 0.43 |
| Positive | 15 | 16 | 31 | ||
| Survivin | Negative | 3 | 10 | 13 | 0.19 |
| Positive | 19 | 20 | 39 |
3. Discussion
4. Experimental Section
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Ezzati, M.; Henley, S.J.; Lopez, A.D.; Thun, M.D. Role of smoking in global and regional cancer epidemiology: Current patterns and data needs. Int. J. Cancer 2005, 116, 963–971. [Google Scholar]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar]
- Ferlay, J.; Shin, H.R.; Bray, F.; Forman, D.; Mathers, C.; Parkin, D.M. Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int. J. Cancer 2010, 127, 2893–2917. [Google Scholar]
- Campos-Parra, A.D.; Zuloaga, C.; Manríquez, M.E.; Avilés, A.; Borbolla-Escoboza, J.; Cardona, A.; Meneses, A.; Arrieta, O. KRAS mutation as the biomarker of response to chemotherapy and EGFR-TKIs in patients with advanced non-small cell lung cancer: Clues for its potential use in second-line therapy decision making. Am. J. Clin. Oncol. 2013, in press. [Google Scholar]
- Karachaliou, N.; Mayo, C.; Costa, C.; Magrí, I.; Gimenez-Capitan, A.; Molina-Vila, M.A.; Rosell, R. KRAS mutations in lung cancer. Clin. Lung Cancer 2013, 14, 205–214. [Google Scholar] [CrossRef]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 2007, 14, 3291–3310. [Google Scholar] [CrossRef]
- Campbell, S.L.; Khosravi-Far, R.; Rossman, K.L.; Clark, G.J.; Der, C.J. Increasing complexity of Ras signaling. Oncogene 1998, 17, 1395–1413. [Google Scholar]
- Noda, N.; Matsuzoe, D.; Konno, T.; Kawahara, K.; Yamashita, Y.; Shirakusa, T. K-Ras gene mutations in non-small cell lung cancer in Japanese. Oncol. Rep. 2001, 8, 889–892. [Google Scholar]
- Ronai, Z.A.; Gradia, S.; Peterson, L.A.; Hecht, S.S. G to A transitions and G to T transversions in codon 12 of the Ki-Ras oncogene isolated from mouse lung tumors induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and related DNA methylating and pyridyloxobutylating agents. Carcinogenesis 1993, 14, 2419–2422. [Google Scholar]
- Cheng, K.C.; Cahill, D.S.; Kasai, H.; Nishimura, S.; Loeb, L.A. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G-T and A-C substitutions. J. Biol. Chem. 1992, 267, 166–172. [Google Scholar]
- Asher, G.; Lotem, J.; Kama, R.; Sachs, L.; Shaul, L. NQO1 stabilizes p53 through a distinct pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 3099–3104. [Google Scholar] [CrossRef]
- Beall, H.D.; Winski, S.I. Mechanisms of action of quinone-containing alkylating agents. I: NQO1-directed drug development. Front. Biosci. 2000, 5, D639–D648. [Google Scholar] [CrossRef]
- PubMed Home Page. Available online: http://www.pubmed.gov (accessed on 27 May 2014).
- Andrade, F.G.; Furtado-Silva, J.M.; Gonçalves, B.A.; Thuler, L.C.S.; Barbosa, T.C.; Emerenciano, M.; Siqueira, A.; Pombo-de-Oliveira, M.S.; Brazilian collaborative study group of infant acute leukaemia. RAS mutations in early age leukaemia modulated by NQO1 rs1800566 (C609T) are associated with second-hand smoking exposures. BMC Cancer 2014, 14. [Google Scholar] [CrossRef]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.S.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef]
- Ralph, S.J.; Rodríguez-Enríquez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sánchez, R. The causes of cancer revisited: “mitochondrial malignancy” and ROS-induced oncogenic transformation—Why mitochondria are targets for cancer therapy. Mol. Aspects Med. 2010, 31, 145–170. [Google Scholar] [CrossRef]
- Pelicano, H.; Feng, L.; Zhou, Y.; Carew, J.S.; Hileman, E.O.; Plunkett, W.; Plunkett, M.J.; Huang, J. Inhibition of mitochondrial respiration: A novel strategy to enhance drug-induced apoptosis in human leukemia cells by a reactive oxygen species-mediated mechanism. J. Biol. Chem. 2003, 278, 37832–37839. [Google Scholar] [CrossRef]
- Hakim, I.A.; Harris, R.; Garland, L.; Cordova, C.A.; Mikhael, D.A.; Chow, H-H.S. Gender difference in systemic oxidative stress and antioxidant capacity in current and former heavy smokers. Cancer Epidemiol. Biomark. Prev. 2012, 21, 2193–2200. [Google Scholar] [CrossRef]
- Chen, B.; Zhong, Y.; Peng, W.; Sun, Y.; Hu, Y.-J.; Yang, Y.; Kong, W.-J. Increased mitochondrial DNA damage and decreased base excision repair in the auditory cortex of D-galactose-induced aging rats. Mol. Biol. Rep. 2011, 38, 3635–3642. [Google Scholar] [CrossRef]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef]
- Belinsky, S.A.; Nikula, K.J.; Baylin, S.B.; Issa, J.P. Increased cytosine DNA—Methyltransferase activity is target-cell-specific and an early event in lung cancer. Proc. Natl. Acad. Sci. USA 1996, 93, 4045–4050. [Google Scholar] [CrossRef]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Wang, Y.A.; Kamarova, Y.; Shen, K.C.; Jiang, Z.L.; Hahn, M.-J.; Wang, Y.L.; Brooks, S.C. DNA methyltransferase-3a interacts with p53 and represses p53-mediated gene expression. Cancer Biol. Ther. 2005, 4, 1138–1143. [Google Scholar] [CrossRef]
- Hervouet, E.; Vallette, F.M.; Cartron, P.F. Impact of the DNA methyltransferases expression on the methylation status of apoptosis-associated genes in glioblastoma multiforme. Cell. Death Dis. 2010, 1. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Y. Suppression of HGF receptor gene expression by oxidative stress is mediated through the interplay between Sp1 and Egr-1. Am. J. Physiol. Renal Physiol. 2003, 284, F1216–F1225. [Google Scholar]
- Korobko, I.V.; Zinov’eva, M.V.; Allakhverdiev, A.K.; Zborovskaia, I.B.; Svwrdlov, E.D. c-Met and HGF expression in non-small-cell lung carcinomas. Mol. Gen. Mikrobiol. Virusol. 2007, 2, 18–21. [Google Scholar]
- Prat, M.; Narsimhan, R.P.; Crepaldi, T.; Nicotra, M.R.; Natali, P.G.; Comoglio, P.M. The receptor encoded by the human c-MET oncogene is expressed in hepatocytes, epithelial cells and solid tumors. Int. J. Cancer. 1991, 49, 323–328. [Google Scholar] [CrossRef]
- Yang, Y.; Wislez, M.; Fujimoto, N.; Prudkin, L.; Izzo, J.G.; Uno, F.; Ji, L.; Hanna, A.E.; Langley, R.R.; Liu, D. A selective small molecule inhibitor of c-Met, PHA-665752, reverses lung premalignancy induced by mutant K-ras. Mol. Cancer Ther. 2008, 7, 952–960. [Google Scholar] [CrossRef]
- Matsubara, D.; Ishikawa, S.; Sachiko, O.; Aburatani, H.; Fukayama, M.; Niki, T. Co-activation of epidermal growth factor receptor and c-MET defines a distinct subset of lung adenocarcinomas. Am. J. Pathol. 2010, 177, 2191–2204. [Google Scholar] [CrossRef]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.-Q.; Murray, T.; Thun, M.J. Cancer statistics, 2008. CA Cancer J. Clin. 2008, 58, 71–96. [Google Scholar] [CrossRef]
- Kamangar, F.; Dores, G.M.; Anderson, W.F. Patterns of cancer incidence, mortality, and prevalence across five continents: Defining priorities to reduce cancer disparities in different geographic regions of the world. J. Clin. Oncol. 2006, 24, 2137–2150. [Google Scholar] [CrossRef]
- Bonomi, P.; Kim, K.; Fairclough, D.; Cella, D.; Kugler, J.; Rowinsky, E.; Jiroutek, M.; Johnson, D. Comparison of survival and quality of life in advanced non-small-cell lung cancer patients treated with two dose levels of paclitaxel combined with cisplatin versus etoposide with cisplatin: Results of an Eastern Cooperative Oncology Group trial. J. Clin. Oncol. 2000, 18, 623–631. [Google Scholar]
- Eberhard, D.A.; Johnson, B.E.; Amler, L.C.; Goddard, A.D.; Heldens, S.L.; Herbst, R.S.; Ince, W.L.; Jänne, P.A.; Januario, T.; Johnson, D.H. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J. Clin. Oncol. 2005, 23, 5900–5909. [Google Scholar] [CrossRef]
- Aviel-Ronen, S.; Blackhall, F.H.; Shepherd, F.A.; Tsao, M.S. K-ras mutations in non-small-cell lung carcinoma: A review. Clin. Lung Cancer. 2006, 8, 30–38. [Google Scholar] [CrossRef]
- Sunaga, N.; Shames, D.S.; Girard, L.; Peyton, M.; Larsen, J.E.; Imai, H.; Soh, J.; Sato, M.; Yanagitani, N.; Kaira, K.; et al. Knockdown of oncogenic KRAS in non-small cell lung cancers suppresses tumor growth and sensitizes tumor cells to targeted therapy. Mol. Cancer Ther. 2011, 10, 336–346. [Google Scholar] [CrossRef]
- Keller, J.W.; Franklin, J.L.; Graves-Deal, R.; Friedman, D.B.; Whitwell, C.W.; Coffey, R.J. Oncogenic KRAS provides a uniquely powerful and variable oncogenic contribution among RAS family members in the colonic epithelium. J. Cell. Physiol. 2007, 210, 740–749. [Google Scholar] [CrossRef]
- Minamoto, T.; Mai, M.; Ronai, Z. K-ras mutation: Early detection in molecular diagnosis and risk assessment of colorectal, pancreas, and lung cancers—A review. Cancer Detect. Prev. 2000, 24, 1–12. [Google Scholar]
- Siegel, D.; Kepa, J.K.; Ross, D. NAD(P)H:quinone oxidoreductase 1 (NQO1) localizes to the mitotic spindle in human cells. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Song, S.Y.; Jeong, S.Y.; Park, H.J.; Park, S.; Kim, D.K.; Kim, Y.H.; Shin, S.S.; Lee, S.-W.; Ahn, S.D.; Kim, J.H.; et al. Clinical significance of NQO1 C609T polymorphisms after postoperative radiation therapy in completely resected non-small cell lung cancer. Lung Cancer 2010, 68, 278–282. [Google Scholar] [CrossRef]
- Yang, S.Y.; Yang, T.Y.; Li, Y.J.; Chen, K.C.; Liao, K.M.; Hsu, K.H.; Tsai, C.R.; Chen, C.H.; Hsu, C.P.; Hsia, J.Y.; et al. EGFR exon 19 in-frame deletion and polymorphisms of DNA repair genes in never-smoking female lung adenocarcinoma patients. Int. J. Cancer 2013, 132, 449–458. [Google Scholar] [CrossRef]
- Iskander, K.; Barrios, R.J.; Jaiswal, A.K. Disruption of NAD(P)H:quinone oxidoreductase 1 gene in mice leads to radiation-induced myeloproliferative disease. Cancer Res. 2008, 68, 7915–7922. [Google Scholar] [CrossRef]
- An, H.J.; Lee, H.; Paik, S.G. Silencing of BNIP3 results from promoter methylation by DNA methyltransferase 1 induced by the mitogen-activated protein kinase pathway. Mol. Cells 2011, 31, 579–583. [Google Scholar] [CrossRef]
- Tamm, I.; Wang, Y.; Sausville, E.; Scudiero, D.A.; Vigna, N.; Oltersdorf, T.; Reed, J.C. IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs. Cancer Res. 1998, 58, 5315–5320. [Google Scholar]
- Chen, Y.; Wang, X.; Li, W.; Zhang, H.; Zhao, C.; Li, Y.; Wang, Z.; Chen, C. Sp1 upregulates survivin expression in adenocarcinoma of lung cell line A549. Anat. Rec. (Hoboken) 2011, 294, 774–780. [Google Scholar] [CrossRef]
- Gaikwad, A.; Long, D.J., 2nd; Stringer, J.L.; Jaiswal, A.K. In vivo role of NAD(P)H: quinone oxidoreductase 1 (NQO1) in the regulation of intracellular redox state and accumulation of abdominal adipose tissue. J. Biol. Chem. 2001, 276, 22559–22564. [Google Scholar]
- Chu, J.; Wu, S.; Xing, D. Survivin mediates self-protection through ROS/cdc25c/CDK1 signaling pathway during tumor cell apoptosis induced by high fluence low-power laser irradiation. Cancer Lett. 2010, 297, 207–219. [Google Scholar] [CrossRef]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef]
- Altieri, D.C. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer. 2003, 3, 46–54. [Google Scholar] [CrossRef]
- Ryan, B.M.; O’Donovan, N.; Duffy, M.J. Survivin: A new target for anti-cancer therapy. Cancer Treat. Rev. 2009, 35, 553–562. [Google Scholar] [CrossRef]
- Han, P.H.; Li, X.J.; Qin, H.; Yao, J.; Du, N.; Ren, H. Upregulation of survivin in non-small cell lung cancer and its clinicopathological correlation with p53 and Bcl-2 (In Chinese). Xi Bao Yu Fen Zi Mian Yi Xue. Za. Zhi. 2009, 25, 710–713. [Google Scholar]
- Allred, D.C.; Harvey, J.M.; Berardo, M.; Clark, G.M. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod. Pathol. 1998, 11, 155–168. [Google Scholar]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yilmaz, A.; Mohamed, N.; Patterson, K.A.; Tang, Y.; Shilo, K.; Villalona-Calero, M.A.; Davis, M.E.; Zhou, X.; Frankel, W.; Otterson, G.A.; et al. Increased NQO1 but Not c-MET and Survivin Expression in Non-Small Cell Lung Carcinoma with KRAS Mutations. Int. J. Environ. Res. Public Health 2014, 11, 9491-9502. https://doi.org/10.3390/ijerph110909491
Yilmaz A, Mohamed N, Patterson KA, Tang Y, Shilo K, Villalona-Calero MA, Davis ME, Zhou X, Frankel W, Otterson GA, et al. Increased NQO1 but Not c-MET and Survivin Expression in Non-Small Cell Lung Carcinoma with KRAS Mutations. International Journal of Environmental Research and Public Health. 2014; 11(9):9491-9502. https://doi.org/10.3390/ijerph110909491
Chicago/Turabian StyleYilmaz, Ahmet, Nehad Mohamed, Kara A. Patterson, Yan Tang, Konstantin Shilo, Miguel A. Villalona-Calero, Michael E. Davis, Xiaoping Zhou, Wendy Frankel, Gregory A. Otterson, and et al. 2014. "Increased NQO1 but Not c-MET and Survivin Expression in Non-Small Cell Lung Carcinoma with KRAS Mutations" International Journal of Environmental Research and Public Health 11, no. 9: 9491-9502. https://doi.org/10.3390/ijerph110909491