Environmental Arsenic Exposure and Microbiota in Induced Sputum

Abstract
:1. Introduction
2. Experimental Section
2.1. Ethics Statement
2.2. Study Area and Selection of Subjects
2.3. Prior Sample Collection and DNA Extraction
2.4. PGM Sequencing
2.5. Data Analysis
3. Results and Discussion
3.1. Results

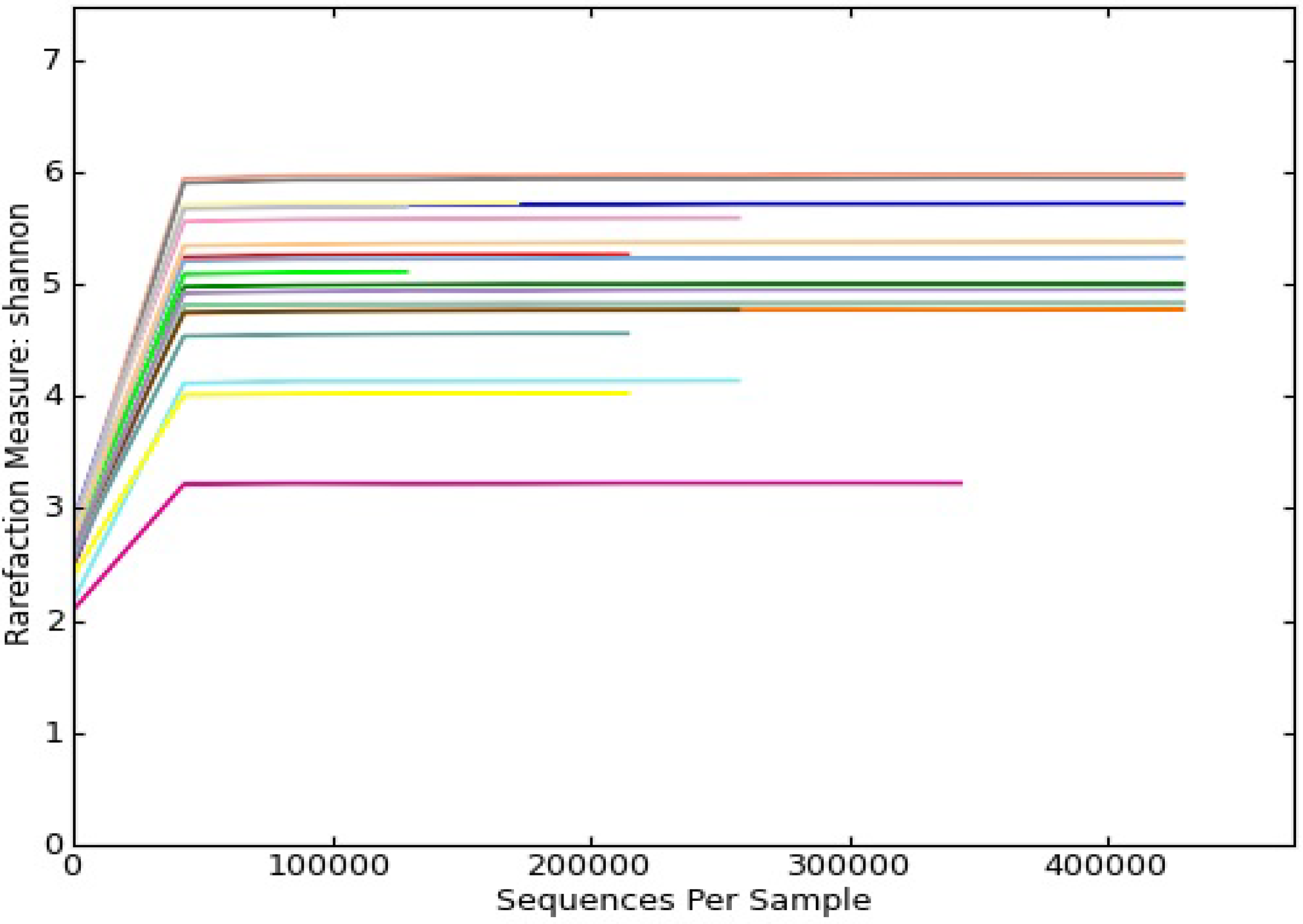{kind=link}
{kind=link}
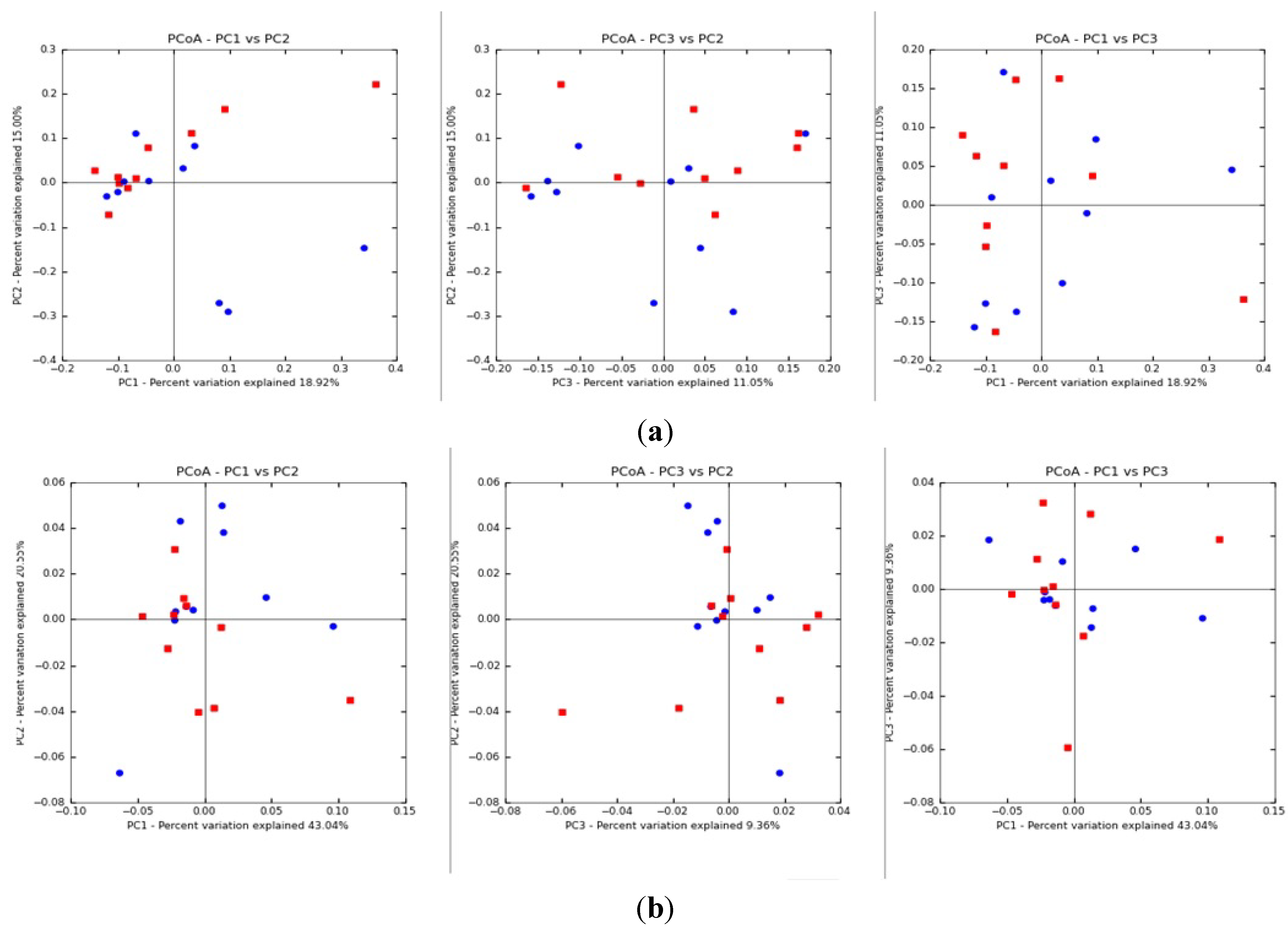{kind=link}
| Moderate Exposure | Low Exposure | p Value | |
|---|---|---|---|
| (n or mean ± SD) | (mean ± SD) | ||
| Number of Subjects | 10 | 10 | |
| Female | 8 | 9 | 0.500 |
| Age | 51.0 ± 21.3 | 41.5 ± 13.2 | 0.122 |
| Past smoking | 4 | 1 | 0.151 |
| Asthma | 0 | 2 | 0.237 |
| Chronic bronchitis or emphysema (COPD) | 0 | 2 | 0.237 |
| Diabetes | 2 | 0 | 0.237 |
| Cardiovascular disease | 4 | 2 | 0.314 |
| GI disease | 1 | 2 | 0.500 |
| Kidney disease | 1 | 2 | 0.500 |
| Liver disease | 0 | 1 | 0.500 |
| Neurological disease | 2 | 1 | 0.500 |
| As levels in water | 21.1 ± 6.4 | 2.4 ± 0.8 | <0.001 |
| (μg/L, range) a | (13.8–28.8) | (1.7–3.5) | |
| Sum of species (μg/L)b | 38.7 ± 18.0 | 19.5 ± 7.0 | <0.003 |
| Moderate Exposure | Low Exposure | p Value | |
|---|---|---|---|
| (mean ± SD) | (mean ± SD) | ||
| Sequence Reads a | 494,149.4 ± 112,755.1 | 442,070.4 ± 102,308.5 | 0.368 |
| OTUs | 2,440.6 ± 357.3 | 2,331.6 ± 270.1 | 0.405 |
| Chao1 | 3,491.1 ± 433.3 | 3,313.3 ± 342.5 | 0.376 |
| Shannon Index | 5.06 ± 0.25 | 5.00 ± 0.20 | 0.419 |


| Number of OTU | The Mean Proportion of OTUs in the Group (%) | ||||||
|---|---|---|---|---|---|---|---|
| Taxon | Moderate exposure | Low exposure | Moderate exposure a | Low exposure a | p-value a | Odds ratio b | 95% CI b |
| Actinobacteria /Actinomyces | 912 | 906 | 5.5% | 5.0% | 0.90 | 0.96 | 0.87–1.06 |
| Actinobacteria /Rothia | 326 | 415 | 1.6% | 2.4% | 0.15 | 0.75 | 0.64–0.87 |
| Bacteroidetes /Porphyromonas | 609 | 640 | 2.7% | 3.7% | 0.46 | 0.91 | 0.81–1.02 |
| Bacteroidetes /Prevotella | 2344 | 2497 | 14.4% | 12.7% | 0.80 | 0.89 | 0.83–0.94 |
| Bacteroidetse /Prevotellaceae | 202 | 484 | 0.9% | 2.4% | 0.19 | 0.39 | 0.33–0.47 |
| Firmicutes | 353 | 251 | 1.7% | 1.3% | 0.45 | 1.35 | 1.14–1.59 |
| Firmicutes /Bacilli | 3910 | 3884 | 19.1% | 21.2% | 0.62 | 0.95 | 0.91–1.00 |
| Firmicutes /Gemella | 712 | 552 | 4.0% | 3.1% | 0.76 | 1.24 | 1.11–1.39 |
| Firmicutes /Lactobacillales | 347 | 267 | 1.8% | 1.4% | 0.63 | 1.25 | 1.06–1.47 |
| Firmicutes /Streptococcus | 865 | 478 | 6.0% | 2.3% | 0.11 | 1.76 | 1.57–1.97 |
| Firmicutes /Veillonella | 1269 | 1217 | 8.0% | 6.3% | 0.52 | 1.00 | 0.92–1.08 |
| Fusobacteria /Fusobacterium | 540 | 713 | 2.6% | 4.0% | 0.11 | 0.72 | 0.64–0.80 |
| Fusobacteria /Leptotrichia | 810 | 744 | 4.1% | 3.5% | 0.74 | 1.04 | 0.94–1.15 |
| Proteobacteria /Gammaproteobacteria | 964 | 969 | 4.6% | 5.3% | 0.64 | 0.95 | 0.86–1.04 |
| Proteobacteria /Neisseria | 493 | 317 | 2.2% | 1.8% | 0.65 | 1.50 | 1.29–1.73 |
| Proteobacteria /Neisseriaceae | 2930 | 3190 | 14.6% | 19.8% | 0.43 | 0.86 | 0.82–0.91 |
| Proteobacteria/Pasteurellaceae | 1311 | 542 | 6.2% | 3.5% | 0.34 | 2.39 | 2.15–2.64 |

3.2. Discussion
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Arsenic; Center for Disease Control: Atlanta, GA, USA, 2007.
- United States Environmental Protection Agency (EPA). National Primary Drinking Water Regulations; Office of Ground Water and Drinking Water, EPA: Washington, DC, USA, 2002.
- Tchounwou, P.B.; Patlolla, A.K.; Centeno, J.A. Carcinogenic and systemic health effects associated with arsenic exposure—A critical review. Toxicol. Pathol. 2003, 31, 575–588. [Google Scholar]
- International Agency for Research on Cancer (IARC). Some Drinking-water Disinfectants and Contaminants, Including Arsenic: IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; IARC Press: Lyon, France, 2004. [Google Scholar]
- Mazumder, D.N.; Steinmaus, C.; Bhattacharya, P.; von Ehrenstein, O.S.; Ghosh, N.; Gotway, M.; Sil, A.; Balmes, J.R.; Haque, R.; Hira-Smith, M.M.; et al. Bronchiectasis in persons with skin lesions resulting from arsenic in drinking water. Epidemiology 2005, 16, 760–765. [Google Scholar]
- Kozul, C.D.; Hampton, T.H.; Davey, J.C.; Gosse, J.A.; Nomikos, A.P.; Eisenhauer, P.L.; Weiss, D.J.; Thorpe, J.E.; Ihnat, M.A.; Hamilton, J.W. Chronic exposure to arsenic in the drinking water alters the expression of immune response genes in mouse lung. Environ. Health Persp. 2009, 117, 1108–1115. [Google Scholar] [CrossRef]
- Von Ehrenstein, O.S.; Mazumder, D.N.; Yuan, Y.; Samanta, S.; Balmes, J.; Sil, A.; Ghosh, N.; Hira-Smith, M.; Haque, R.; Purushothamam, R.; et al. Decrements in lung function related to arsenic in drinking water in West Bengal, India. Am. J. Epidemiol. 2005, 162, 533–541. [Google Scholar] [CrossRef]
- De, B.K.; Majumdar, D.; Sen, S.; Guru, S.; Kundu, S. Pulmonary involvement in chronic arsenic poisoning from drinking contaminated ground-water. J. Assoc. Physicians India 2004, 52, 395–400. [Google Scholar]
- Mazumder, D.N.; Haque, R.; Ghosh, N.; De, B.K.; Santra, A.; Chakraborti, D.; Smith, A.H. Arsenic in drinking water and the prevalence of respiratory effects in West Bengal, India. Int. J. Epidemiol. 2000, 29, 1047–1052. [Google Scholar] [CrossRef]
- Charlson, E.S.; Bittinger, K.; Haas, A.R.; Fitzgerald, A.S.; Frank, I.; Yadav, A.; Bushman, F.D.; Collman, R.G. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am. J. Respir. Crit. Care Med. 2011, 184, 957–963. [Google Scholar] [CrossRef]
- Erb-Downward, J.R.; Thompson, D.L.; Han, M.K.; Freeman, C.M.; McCloskey, L.; Schmidt, L.A.; Young, V.B.; Toews, G.B.; Curtis, J.L.; Sundaram, B.; et al. Analysis of the lung microbiome in the “healthy” smoker and in COPD. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Lipuma, J.J. The changing microbial epidemiology in cystic fibrosis. Clin. Microbiol. Rev. 2010, 23, 299–323. [Google Scholar] [CrossRef]
- Huang, Y.J.; Kim, E.; Cox, M.J.; Brodie, E.L.; Brown, R.; Wiener-Kronish, J.P.; Lynch, S.V. A persistent and diverse airway microbiota present during chronic obstructive pulmonary disease exacerbations. OMICS 2010, 14, 9–59. [Google Scholar] [CrossRef]
- Hilty, M.; Burke, C.; Pedro, H.; Cardenas, P.; Bush, A.; Bossley, C.; Davies, J.; Ervine, A.; Poulter, L.; Pachter, L.; et al. Disordered microbial communities in asthmatic airways. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Sze, M.A.; Dimitriu, P.A.; Hayashi, S.; Elliott, W.M.; McDonough, J.E.; Gosselink, J.V.; Cooper, J.; Sin, D.D.; Mohn, W.W.; Hogg, J.C. The lung tissue microbiome in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2012, 185, 1073–1080. [Google Scholar] [CrossRef]
- Charlson, E.S.; Chen, J.; Custers-Allen, R.; Bittinger, K.; Li, H.; Sinha, R.; Hwang, J.; Bushman, F.D.; Collman, R.G. Disordered microbial communities in the upper respiratory tract of cigarette smokers. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Bisgaard, H.; Hermansen, M.N.; Buchvald, F.; Loland, L.; Halkjaer, L.B.; Bonnelykke, K.; Brasholt, M.; Heltberg, A.; Vissing, N.H.; Thorsen, S.V.; et al. Childhood asthma after bacterial colonization of the airway in neonates. New Engl. J. Med. 2007, 357, 1487–1495. [Google Scholar] [CrossRef]
- Josyula, A.B.; Poplin, G.S.; Kurzius-Spencer, M.; McClellen, H.E.; Kopplin, M.J.; Sturup, S.; Clark Lantz, R.; Burgess, J.L. Environmental arsenic exposure and sputum metalloproteinase concentrations. Environ. Res. 2006, 102, 283–290. [Google Scholar] [CrossRef]
- Lantz, R.C.; Lynch, B.J.; Boitano, S.; Poplin, G.S.; Littau, S.; Tsaprailis, G.; Burgess, J.L. Pulmonary biomarkers based on alterations in protein expression after exposure to arsenic. Environ. Health Persp. 2007, 115, 586–591. [Google Scholar] [CrossRef]
- Olsen, C.E.; Liguori, A.E.; Zong, Y.; Lantz, R.C.; Burgess, J.L.; Boitano, S. Arsenic upregulates MMP-9 and inhibits wound repair in human airway epithelial cells. Am. J. Physiol. Lung C. 2008, 295, L293–L302. [Google Scholar] [CrossRef]
- Roberge, J.; O’Rourke, M.K.; Meza-Montenegro, M.M.; Gutierrez-Millan, L.E.; Burgess, J.L.; Harris, R.B. Binational arsenic exposure survey: Methodology and estimated arsenic intake from drinking water and urinary arsenic concentrations. Int. J. Environ. Res. Public Health 2012, 9, 1051–1067. [Google Scholar] [CrossRef]
- Meza, M.M.; Kopplin, M.J.; Burgess, J.L.; Gandolfi, A.J. Arsenic drinking water exposure and urinary excretion among adults in the Yaqui Valley, Sonora, Mexico. Environ. Res. 2004, 96, 119–126. [Google Scholar] [CrossRef]
- Burgess, J.L.; Nanson, C.J.; Hysong, T.A.; Gerkin, R.; Witten, M.L.; Lantz, R.C. Rapid decline in sputum IL-10 concentration following occupational smoke exposure. Inhal. Toxicol. 2002, 14, 133–140. [Google Scholar]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef]
- Query Input and Database Selection. Available online: http://www.ncbi.nlm.nih.gov/BLAST/blastcgihelp.shtml (accessed on 13 December 2012).
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef]
- Kuczynski, J.; Stombaugh, J.; Walters, W.A.; Gonzalez, A.; Caporaso, J.G.; Knight, R. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Curr. Protoc. Bioinform. 2011, 36. [Google Scholar] [CrossRef]
- Chao, A. Nonparametric-estimation of the number of classes in a population. Scand. J. Stat. 1984, 11, 265–270. [Google Scholar]
- Shannon, C.E. A mathematical theory of communication. ATT Tech. J. 1948, 27, 623–656. [Google Scholar]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 2009, 5. [Google Scholar] [CrossRef]
- A Language and Environment for Statistical Computing. Available online: http://www.lsw.uni-heidelberg.de/users/christlieb/teaching/UKStaSS10/R-refman.pdf (accessed on 8 June 2013).
- American Conference of Government Industrial Hygienists (ACGIH). Documentation of Biological Exposure Indices; ACGIH Worldwide: Cincinnati, OH, USA, 2004. [Google Scholar]
- Beck, J.M.; Young, V.B.; Huffnagle, G.B. The microbiome of the lung. Transl. Res. 2012, 160, 258–266. [Google Scholar] [CrossRef]
- Dewhirst, F.E.; Klein, E.A.; Thompson, E.C.; Blanton, J.M.; Chen, T.; Milella, L.; Buckley, C.M.; Davis, I.J.; Bennett, M.L.; Marshall-Jones, Z.V. The canine oral microbiome. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Sheik, C.S.; Mitchell, T.W.; Rizvi, F.Z.; Rehman, Y.; Faisal, M.; Hasnain, S.; McInerney, M.J.; Krumholz, L.R. Exposure of soil microbial communities to chromium and arsenic alters their diversity and structure. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Davies, M.R.; Shera, J.; van Domselaar, G.H.; Sriprakash, K.S.; McMillan, D.J. A novel integrative conjugative element mediates genetic transfer from group G Streptococcus to other β-hemolytic Streptococci. J. Bacteriol. 2009, 191, 2257–2265. [Google Scholar] [CrossRef] [Green Version]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–169. [Google Scholar]
- Mardis, E.R. The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24, 133–141. [Google Scholar] [CrossRef]
- Robison, K. Semiconductors charge into sequencing. Nat. Biotechnol. 2011, 29, 805–807. [Google Scholar] [CrossRef]
- Whiteley, A.S.; Jenkins, S.; Waite, I.; Kresoje, N.; Payne, H.; Mullan, B.; Allcock, R.; O’Donnell, A. Microbial 16S rRNA Ion Tag and community metagenome sequencing using the Ion Torrent (PGM) Platform. J. Microbiol. Methods 2012, 91, 80–88. [Google Scholar] [CrossRef]
- Junemann, S.; Prior, K.; Szczepanowski, R.; Harks, I.; Ehmke, B.; Goesmann, A.; Stoye, J.; Harmsen, D. Bacterial community shift in treated periodontitis patients revealed by ion torrent 16S rRNA gene amplicon sequencing. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Liu, Z.; Lozupone, C.; Hamady, M.; Bushman, F.D.; Knight, R. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res. 2007, 35. [Google Scholar] [CrossRef]
- Flores, G.E.; Henley, J.B.; Fierer, N. A direct PCR approach to accelerate analyses of human-associated microbial communities. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Bangladesh National Drinking Water Quality Survey of 2009. Available online: http://www.unicef.org/bangladesh/knowledgecentre_6868.htm (accessed on 8 June 2013).
- Andrew, A.S.; Bernardo, V.; Warnke, L.A.; Davey, J.C.; Hampton, T.; Mason, R.A.; Thorpe, J.E.; Ihnat, M.A.; Hamilton, J.W. Exposure to arsenic at levels found in U.S. drinking water modifies expression in the mouse lung. Toxicol. Sci. 2007, 100, 75–87. [Google Scholar] [CrossRef]
- Parvez, F.; Chen, Y.; Brandt-Rauf, P.W.; Slavkovich, V.; Islam, T.; Ahmed, A.; Argos, M.; Hassan, R.; Yunus, M.; Haque, S.E.; et al. A prospective study of respiratory symptoms associated with chronic arsenic exposure in Bangladesh: Findings from the Health Effects of Arsenic Longitudinal Study (HEALS). Thorax 2010, 65, 528–533. [Google Scholar] [CrossRef]
- Islam, L.N.; Nabi, A.H.; Rahman, M.M.; Zahid, M.S. Association of respiratory complications and elevated serum immunoglobulins with drinking water arsenic toxicity in human. J. Environ. Sci. Health 2007, 42, 1807–1814. [Google Scholar] [CrossRef]
- Chen, C.J.; Hsu, L.I.; Wang, C.H.; Shih, W.L.; Hsu, Y.H.; Tseng, M.P.; Lin, Y.C.; Chou, W.L.; Chen, C.Y.; Lee, C.Y.; et al. Biomarkers of exposure, effect, and susceptibility of arsenic-induced health hazards in Taiwan. Toxicol. Appl. Pharmacol. 2005, 206, 198–206. [Google Scholar] [CrossRef]
- Lindberg, A.L.; Rahman, M.; Persson, L.A.; Vahter, M. The risk of arsenic induced skin lesions in Bangladeshi men and women is affected by arsenic metabolism and the age at first exposure. Toxicol. Appl. Pharm. 2008, 230, 9–16. [Google Scholar] [CrossRef]
- Rahman, M.; Vahter, M.; Sohel, N.; Yunus, M.; Wahed, M.A.; Streatfield, P.K.; Ekstrom, E.C.; Persson, L.A. Arsenic exposure and age and sex-specific risk for skin lesions: a population-based case-referent study in Bangladesh. Environ. Health Persp. 2006, 114, 1847–1852. [Google Scholar]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar]
- Lin, A.; Bik, E.M.; Costello, E.K.; Dethlefsen, L.; Haque, R.; Relman, D.A.; Singh, U. Distinct distal gut microbiome diversity and composition in healthy children from Bangladesh and the United States. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Structure, Function and Diversity of the Healthy Human Microbiome. Available online: http://www.nature.com/nature/journal/v486/n7402/abs/nature11234.html#corres-auth (accessed on 14 June 2012).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
White, A.G.; Watts, G.S.; Lu, Z.; Meza-Montenegro, M.M.; Lutz, E.A.; Harber, P.; Burgess, J.L. Environmental Arsenic Exposure and Microbiota in Induced Sputum. Int. J. Environ. Res. Public Health 2014, 11, 2299-2313. https://doi.org/10.3390/ijerph110202299
White AG, Watts GS, Lu Z, Meza-Montenegro MM, Lutz EA, Harber P, Burgess JL. Environmental Arsenic Exposure and Microbiota in Induced Sputum. International Journal of Environmental Research and Public Health. 2014; 11(2):2299-2313. https://doi.org/10.3390/ijerph110202299
Chicago/Turabian StyleWhite, Allison G., George S. Watts, Zhenqiang Lu, Maria M. Meza-Montenegro, Eric A. Lutz, Philip Harber, and Jefferey L. Burgess. 2014. "Environmental Arsenic Exposure and Microbiota in Induced Sputum" International Journal of Environmental Research and Public Health 11, no. 2: 2299-2313. https://doi.org/10.3390/ijerph110202299