The Tetrodotoxin Receptor of Voltage-Gated Sodium Channels—Perspectives from Interactions with μ-Conotoxins

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
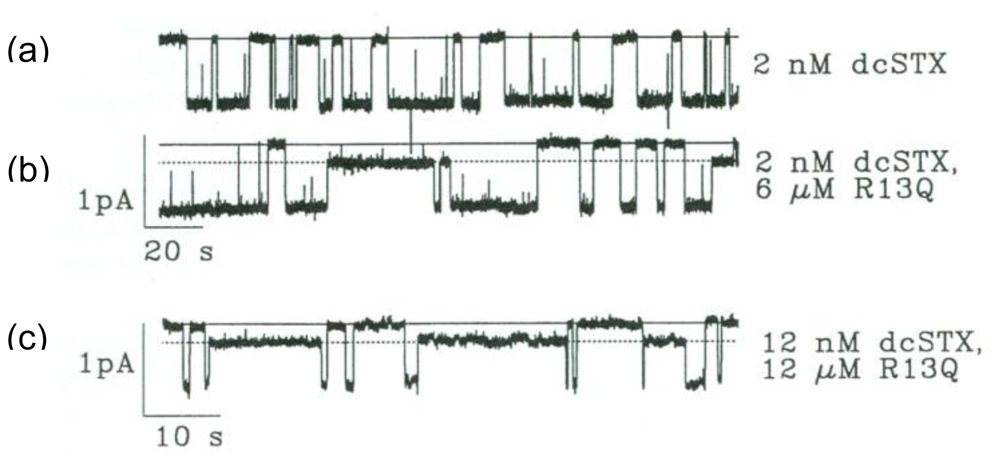
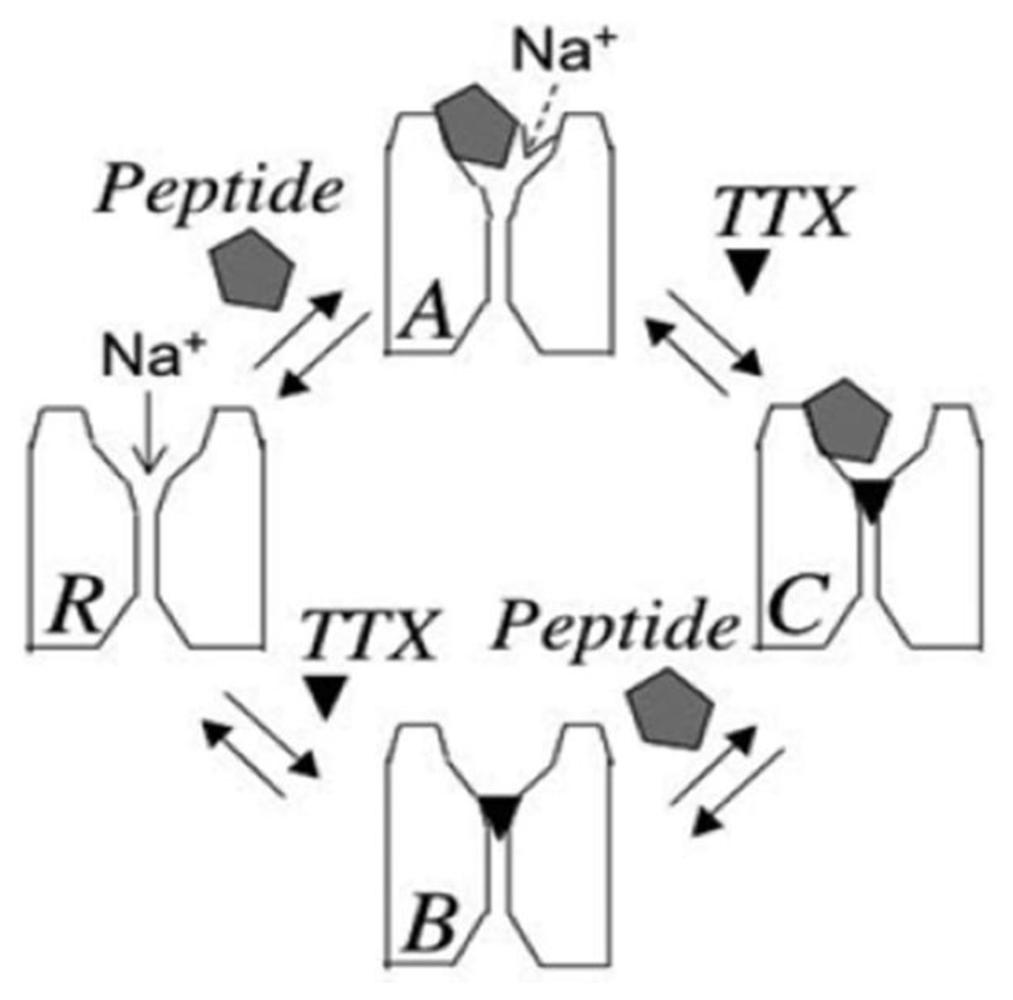
2. Newly Discovered Complexities of Interactions between TTX and μ-Conopeptides
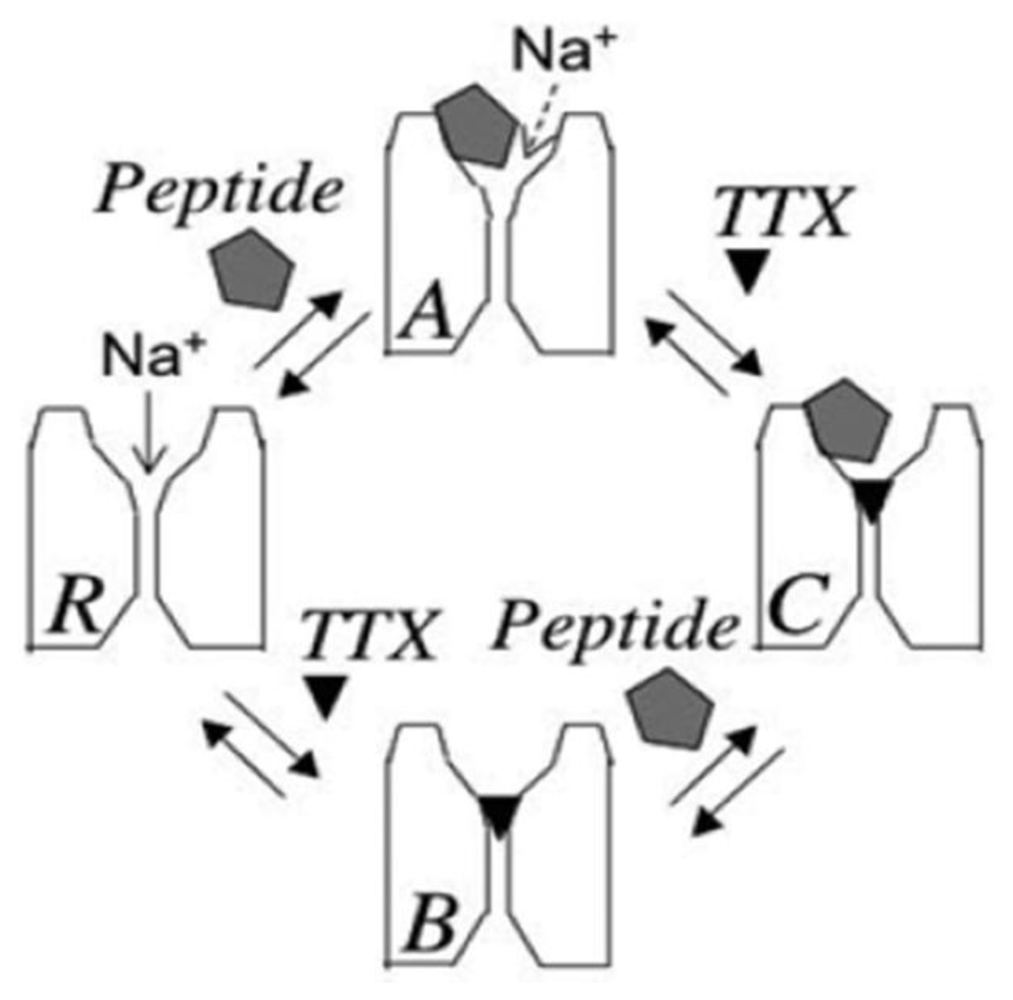
3. Potential Applications of Double Occupancy of Site 1
3.1. Syntoxins
3.2. Contratoxins
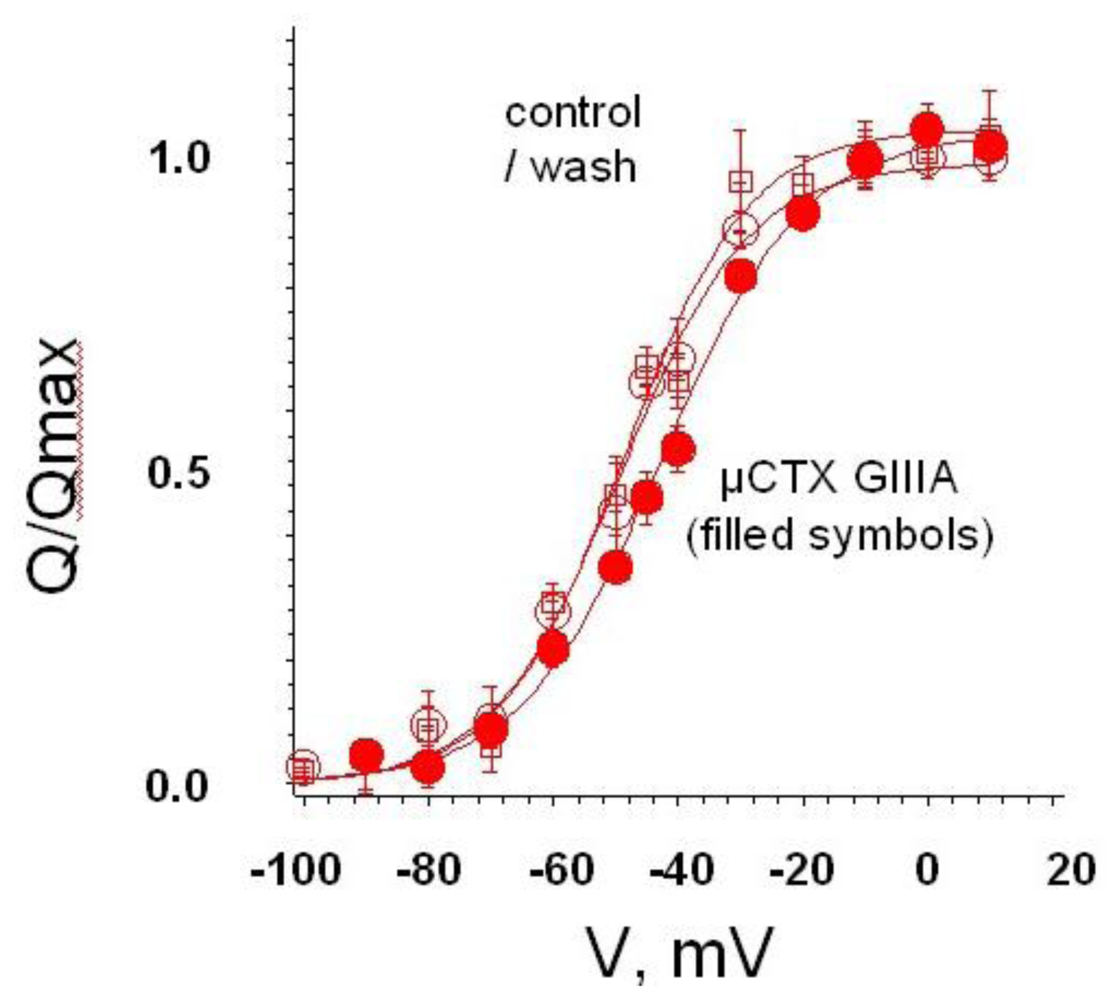
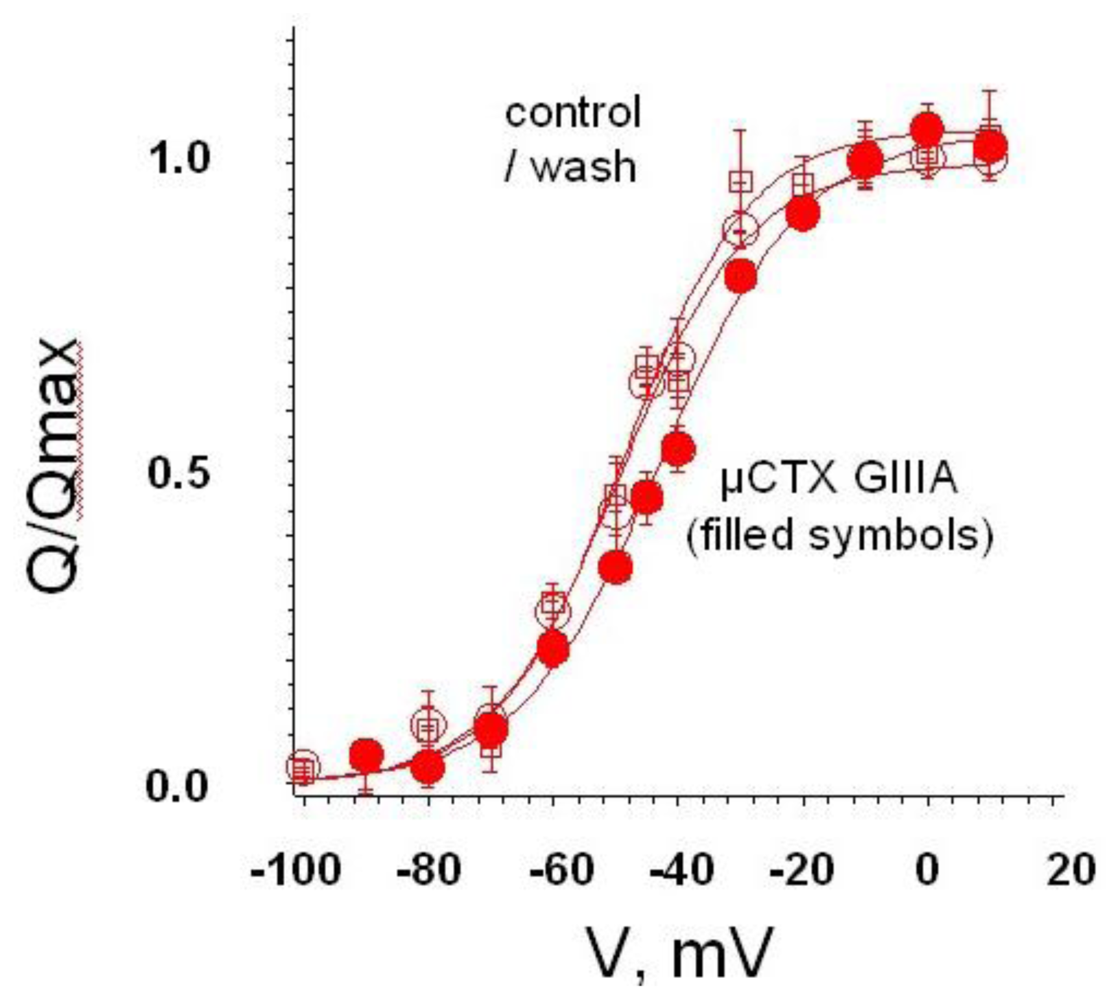
4. Recent Results
Acknowledgements
- Samples Availability: Available from the authors.
References
- Narahashi, T. Tetrodotoxin: a brief history. Proc. Jpn. Acad., Ser. B Phys. Biol. Sci 2008, 84, 147–154. [Google Scholar]
- Catterall, WA. Neurotoxins that act on voltage-sensitive sodium channels in excitable membranes. Ann. Rev. Pharmacol. Toxicol 1980, 20, 15–43. [Google Scholar]
- Catterall, WA; Cestele, S; Yarov-Yarovoy, V; Yu, FH; Konoki, K; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [Green Version]
- Cestele, S; Catterall, WA. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar]
- Hall, S; Strichartz, G; Moczydlowski, E; Ravindran, A; Reichardt, P. The saxitoxins. Sources, chemistry, and pharmacology. In Marine Toxins: Origin, Structure and Molecular Pharmacology; American Chemical Society: Washington, DC, USA, 1990; pp. 29–65. [Google Scholar]
- Terlau, H; Olivera, BM. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiol. Rev 2004, 84, 41–68. [Google Scholar]
- Cruz, LJ; Gray, WR; Olivera, BM; Zeikus, RD; Kerr, L; Yoshikami, D; Moczydlowski, E. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. J. Biol. Chem 1985, 260, 9280–9288. [Google Scholar]
- Moczydlowski, E; Olivera, BM; Gray, WR; Strichartz, GR. Discrimination of muscle and neuronal Na-channel subtypes by binding competition between [3H]saxitoxin and μ-conotoxins. Proc. Natl. Acad. Sci. USA 1986, 83, 5321–5325. [Google Scholar]
- Fiedler, B; Zhang, MM; Buczek, O; Azam, L; Bulaj, G; Norton, RS; Olivera, BM; Yoshikami, D. Specificity, affinity and efficacy of iota-conotoxin RXIA, an agonist of voltagegated sodium channels Na(V)1.2, 1.6 and 1.7. Biochem. Pharmacol 2008, 75, 2334–2344. [Google Scholar]
- Al-Sabi, A; McArthur, J; Ostroumov, V; French, RJ. Marine toxins that target voltage-gated sodium channels. Mar. Drugs 2006, 4, 157–192. [Google Scholar]
- Becker, S; Prusak-Sochaczewski, E; Zamponi, G; Beck-Sickinger, AG; Gordon, RD; French, RJ. Action of derivatives of μ-conotoxin GIIIA on sodium channels. Single amino acid substitutions in the toxin separately affect association and dissociation rates. Biochemistry 1992, 31, 8229–8238. [Google Scholar]
- Chang, NS; French, RJ; Lipkind, GM; Fozzard, HA; Dudley, S, Jr. Predominant interactions between μ-conotoxin Arg-13 and the skeletal muscle Na+ channel localized by mutant cycle analysis. Biochemistry 1998, 37, 4407–4419. [Google Scholar]
- Dudley, SC, Jr; Chang, N; Hall, J; Lipkind, G; Fozzard, HA; French, RJ. μ-Conotoxin interactions with the voltage-gated Na+ channel predict a clockwise arrangement of domains. J. Gen. Physiol 2000, 116, 679–689. [Google Scholar]
- Choudhary, G; Aliste, MP; Tieleman, DP; French, RJ; Dudley, SC, Jr. Docking orientation of μ-conotoxin GIIIA in the sodium channel outer vestibule. Channels 2007, 1, 344–352. [Google Scholar]
- Li, RA; Hui, K; French, RJ; Sato, K; Henrikson, CA; Tomaselli, GF; Marbán, E. Dependence of μ-conotoxin block of sodium channels on ionic strength but not the permeating [Na+]: Implications for the distinctive mechanistic interactions between Na+ and K+ channel poreblocking toxins and their molecular targets. J. Biol. Chem 2003, 278, 30912–30919. [Google Scholar]
- Hui, K; Lipkind, G; Fozzard, HA; French, RJ. Electrostatic and steric contributions to block of the skeletal muscle sodium channel by μ-conotoxin. J. Gen. Physiol 2002, 119, 45–54. [Google Scholar]
- Hui, K; McIntyre, D; French, RJ. Conotoxins as sensors of local pH and electrostatic potential in the outer vestibule of the sodium channel. J. Gen. Physiol 2003, 122, 63–79. [Google Scholar]
- French, RJ; Prusak-Sochaczewski, E; Zamponi, GW; Becker, S; Kularatna, AS; Horn, R. Interactions between a pore-blocking peptide and the voltage sensor of the sodium channel: an electrostatic approach to channel geometry. Neuron 1996, 16, 407–413. [Google Scholar]
- Ma, Q; Pavlov, E; Britvina, T; Zamponi, GW; French, RJ. Trans-channel interactions in batrachotoxin-modified rat skeletal muscle sodium channels. Kinetic analysis of mutual inhibition between μ-conotoxin GIIIA derivatives and amine blockers. Biophys. J 2008, 95, 4266–4276. [Google Scholar]
- Pavlov, E; Britvina, T; Ma, Q; Sierralta, IC; Zamponi, GW; French, RJ. Trans-channel interactions in batrachotoxin-modified skeletal muscle sodium channels. Voltage dependent block by cytoplasmic amines, and the influence of μ-conotoxin GIIIA derivatives and permeant ions. Biophys. J 2008, 95, 4277–4288. [Google Scholar]
- Sheets, MF; French, RJ. Effect of μ-conotoxin GIIIA on gating charge movement in voltagegated sodium channels 1998. Unpublished work.
- Sheets, MF; Kyle, JW; Krueger, S; Hanck, DA. Optimization of a mammalian expression system for the measurement of sodium channel gating currents. Am. J. Physiol 1996, 271, C1001–C1006. [Google Scholar]
- Martin-Moutot, N; Mansuelle, P; Alcaraz, G; Dos Santos, RG; Cordeiro, MN; De Lima, ME; Seagar, M; Van, RC. Phoneutria nigriventer toxin 1: a novel, state-dependent inhibitor of neuronal sodium channels that interacts with μ-conotoxin binding sites. Mol. Pharmacol 2006, 69, 1931–1937. [Google Scholar]
- Zhang, MM; Green, BR; Catlin, P; Fiedler, B; Azam, L; Chadwick, A; Terlau, H; McArthur, JR; French, RJ; Gulyas, J; Rivier, JE; Smith, BJ; Norton, RS; Olivera, BM; Yoshikami, D; Bulaj, G. Structure/function characterization of μ-conotoxin KIIIA, an analgesic, nearly irreversible blocker of neuronal mammalian sodium channels. J. Biol. Chem 2007, 282, 30699–30706. [Google Scholar]
- Zhang, MM; McArthur, JR; Azam, L; Bulaj, G; Olivera, BM; French, RJ; Yoshikami, D. Synergistic and antagonistic interactions between tetrodotoxin and μ-conotoxin in blocking voltage-gated sodium channels. Channels (Austin) 2009, 3, 32–38. [Google Scholar]
- Zhang, MM; Gruszczynski, P; Walewska, A; Bulaj, G; Olivera, BM; Yoshikami, D. Cooccupancy of the Outer Vestibule of Voltage-gated Sodium Channels by {micro}-Conotoxin KIIIA and Saxitoxin or Tetrodotoxin. J. Neurophysiol 2010. [Google Scholar] [CrossRef]
- Zhang, MM; Han, TS; Olivera, BM; Bulaj, G; Yoshikami, D. μ-Conotoxin KIIIA Derivatives with Divergent Affinities versus Efficacies in Blocking Voltage-Gated Sodium Channels. Biochemistry 2010, 49, 4804–4812. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
French, R.J.; Yoshikami, D.; Sheets, M.F.; Olivera, B.M. The Tetrodotoxin Receptor of Voltage-Gated Sodium Channels—Perspectives from Interactions with μ-Conotoxins. Mar. Drugs 2010, 8, 2153-2161. https://doi.org/10.3390/md8072153
French RJ, Yoshikami D, Sheets MF, Olivera BM. The Tetrodotoxin Receptor of Voltage-Gated Sodium Channels—Perspectives from Interactions with μ-Conotoxins. Marine Drugs. 2010; 8(7):2153-2161. https://doi.org/10.3390/md8072153
Chicago/Turabian StyleFrench, Robert J., Doju Yoshikami, Michael F. Sheets, and Baldomero M. Olivera. 2010. "The Tetrodotoxin Receptor of Voltage-Gated Sodium Channels—Perspectives from Interactions with μ-Conotoxins" Marine Drugs 8, no. 7: 2153-2161. https://doi.org/10.3390/md8072153