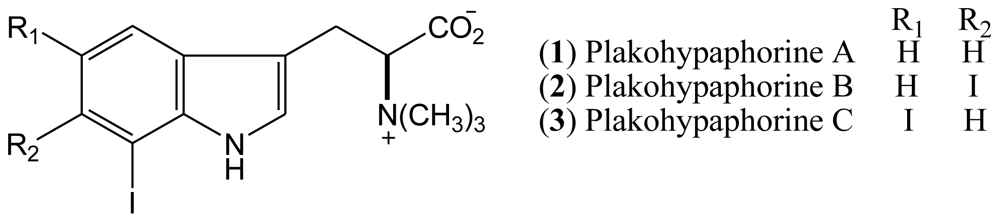
In this report we have focused on the halogenated indole alkaloids from marine invertebrates, particularly meridianins; their related compounds variolins, psammopemmins, and aplicyanins as well as the aplysinopsins and leptoclinidamines. Also summarized are the methods of structure determination, observed biological activities and a compilation of 13C-NMR spectral data is provided.
1.1. Biohalogenation
The halogenation of natural products is a frequent modification of secondary metabolism that allows for optimization of the bioactivity of small molecules, providing evolutionary advantage [
6].
Many biohalogenation enzymes have been isolated and characterized. Chloroperoxidase, bromoperoxidase, iodoperoxidase, and the enzymes involved in the biosynthesis of fluoroacetic acid (fluoroacetaldehyde dehydrogenase and 5′-fluorodeoxyadenosine synthase) are some examples [
8].
Halogenating enzymes have been discovered in a broad range of organisms and they can be grouped into two main classes: (i) highly substrate-specific halogenases requiring dioxygen for enzymatic activity and (ii) less specific haloperoxidases (HPO) utilizing hydrogen peroxide. In dioxygen-dependent halogenases, either flavin (FADH
2-dependent halogenases) or R-ketoglutarate (non-heme FeII/R-ketoglutarate/O
2- dependent halogenases) are found to function as co-substrates. Furthermore, methyltransferases are involved in the formation of the carbon halogen bonds of CH
3Cl, CH
3Br, and CH
3I, and other enzymes requiring
S-adenosyl-
l-methionine as catalyst have been identified to be involved in fluorination and chlorination [
13].
In the recent years, the understanding of biohalogenation processes has been extended extraordinarily. The cloning and sequencing of biosynthetic gene clusters have revealed new mechanisms leading to halogen incorporation and stimulated detailed mechanistic studies of these enzymes [
6,
8]. New groups of halogenating enzymes have been discovered and investigated at both biochemical and genetic levels. Each group of these enzymes performs halogenation reactions on chemically distinct substructures using a specific reaction mechanism. For instance, some FADH
2-dependent halogenases are directly involved in the halogenation of aromatic compounds, recognizing tryptophan or indole moieties, while other groups of FADH
2-dependent halogenases participate in the halogenation of aliphatic compounds [
13].
1.2. Meridianins
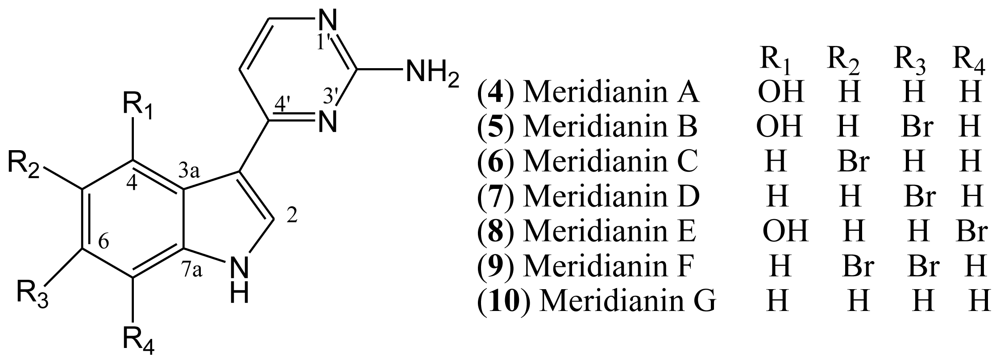
Meridianins are marine alkaloids which were first isolated from the Ascidian
Aplidium meridianum [
14]. Structurally, the meridianins comprise a brominated and/or hydroxylated indole nucleus substituted at C-3 by a 2-aminopyrimidine. Seven meridianins A–G (
4–
10) have been discovered so far. Bromine substitution occurs on position 5 for meridianin C (
6), on position 6 for B (
5) and D (
7), on position 7 for E (
8), and on positions 5 and 6 for F (
9) (
Figure 2).
Meridianins have been described as potent inhibitors of various protein kinases (
Table 1) and they display antitumor activity. Meridianins B (
5) and E (
8) are the most potent and, for this reason meridianin E was selected for further selectivity studies on 25 highly purified kinases [
15]. Essentially, all physiological processes and most human diseases involve protein phosphorylation. Phosphorylation of proteins on serine, threonine, and tyrosine residues by the 518 protein kinases encoded in the human genome constitutes one of the major mechanisms used by cells to regulate their metabolism and functions. The recent appreciation of the implication of abnormal protein phosphorylation in many human diseases has sparked considerable interest in the search for pharmacological inhibitors of kinases [
16–
18].
Protein phosphorylation regulates most aspects of cell life, whereas abnormal phosphorylation is a cause or consequence of diseases. For instance, among the 518 human kinases cyclin-dependent kinases (CDK) have attracted considerable interest given their involvement in many essential physiological pathways and numerous abnormalities in multiple human diseases, especially cancer and neurodegenerative diseases such as Alzheimer’s and Parkison’s diseases [
16,
18,
19].
Investigations of structure-activity relationships of meridianins have revealed that the substitution at C-5 and the methylation of the indole nitrogen are important for either kinase inhibitory activity or
in vitro antiproliferative activities. Related to CDK1 and CDK5, the bromine substitution on position 7 and the hydroxyl on position 4 provide the best inhibitory activity. A single bromine substitution on position 5 or 6 of the indole ring results in considerable improvement in potency. On the other hand, two bromide substitutions slightly reduce the inhibitory potency [
20,
21].
Meridianins B, C, D, and E (
5–
8) display cytotoxicity toward LMM3 (murine mammalian adenocarcinoma cell line) with IC
50 values of 11.4 μM, 9.3 μM, 33.9 μM, and 11.1 μM, respectively [
14]. Certainly, meridianins constitute a new scaffold exhibiting micromolar inhibition of protein kinases from which more potent and selective inhibitors can be designed [
15].
Meridianins are closely related to the variolins, a class of marine alkaloids from the Antarctic sponge
Kirkpatrickia varialosa [
22,
23].
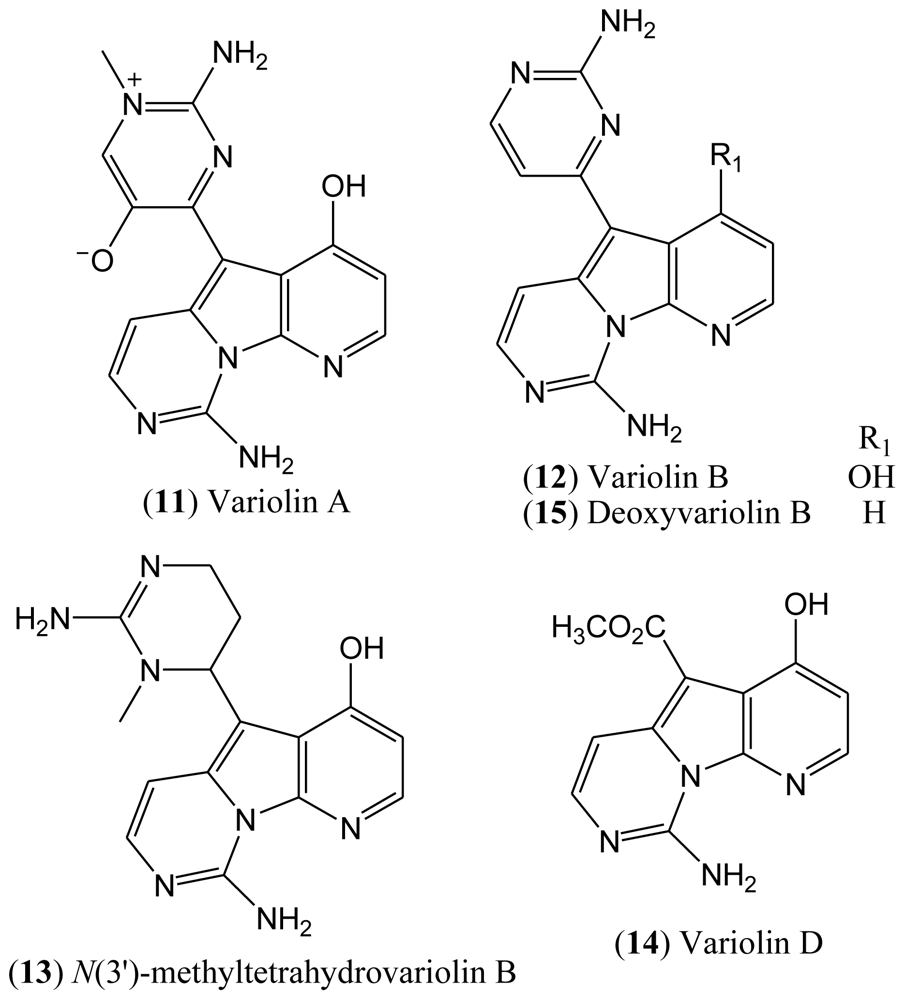
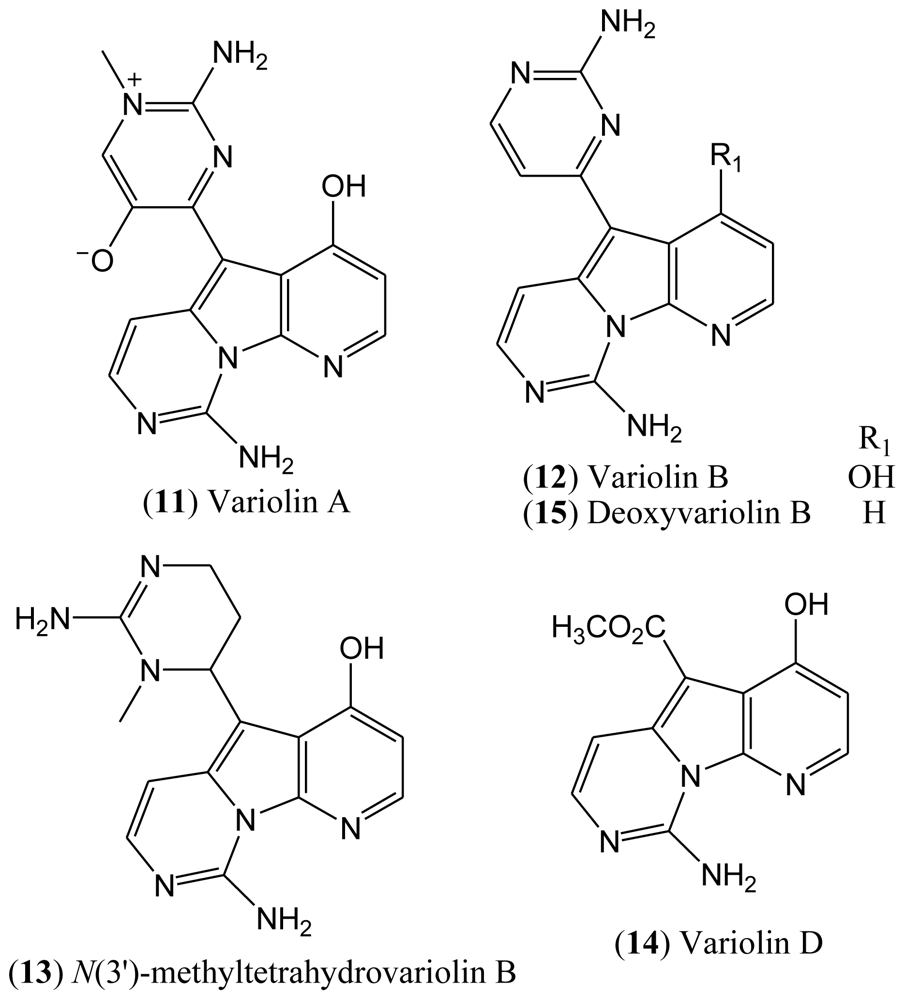
1.3. Variolins
In 1994, the Blunt, Munro and Faulkner laboratories reported the isolation and structural elucidation of the variolins from the rare Antarctic sponge
Kirkpatrickia varialosa [
22,
23]. Variolins are the first examples of either terrestrial or marine natural products with a pyrido[3′,2′:4,5]pyrrolo[1,2-c]pyrimidine system. This rare pyridopyrrolopyrimidine skeleton has made the variolins an interesting class of alkaloids from both structural and biogenetic viewpoints. Variolins can also be considered as guanidine-based alkaloids in which the guanidine moiety is found in the guise of a 2-aminopyrimidine ring [
24–
26].
The isolated compounds included variolin A (
11), variolin B (
12),
N(3′)-methyl tetrahydrovariolin B (
13), and variolin D (
14), the latter of which was reported to be an artifact of the extraction process produced by aerial oxidation of the variolins (
Figure 3). This type of compounds exhibit a potent cytotoxic activity against P388 murine leukemia cell line, also being effective against Herpes simplex type I. Variolin B (
12) is the most active of this family of natural products [
26].
There has been considerable interest in the synthesis of variolins due to the novelty of their structures, not to mention their biological properties and low natural occurrence [
25]. To date, four total syntheses of variolin B have been reported in the literature [
21,
27–
33], and the preparation of the synthetic deoxyvariolin B (
15) has also been described [
34,
35]. The synthesis of new derivatives of variolin B with different substituents at positions C-5 and C-7 has also been reported [
26].
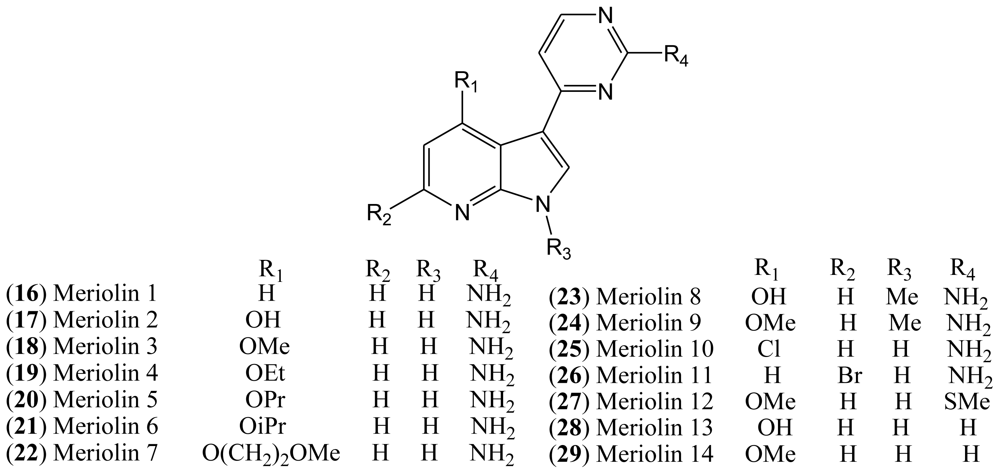
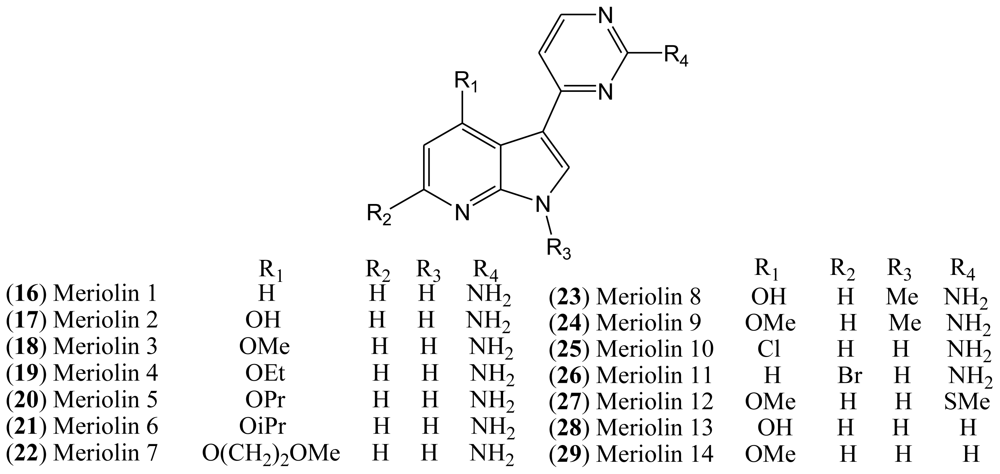
Although the natural variolins isolated are not halogenated, this type of skeleton along with the structure of meridianins have been an inspiration for the synthesis of the hybrid meriolins 1–14 (
16–
29,
Figure 4), including the halogenated meriolins 10 (
25) and 11 (
26) [
18].
1.4. Meriolins
Variolins with a pyridopyrrolopyrimidine system and meridianins possessing a pyrimidyl-substituted indole skeleton bear some structural similarities. Through a combination of the common features of these natural products, a new class of 7-azaindole-containing analogues (
16–
29) known as meriolins has been designed by Meijer and co-workers [
21].
Meriolins [3-(pyrimidin-4-yl)-7-azaindoles], a chemical hybrid of the variolins and meridianins, display potent inhibitory activity toward CDKs (especially CDK2 and CDK9). This class of compounds also exhibit better antiproliferative and proapoptotic properties in cell cultures compared with their “inspirational parent” molecules [
18,
19].
The resemblance between the chemical structures of the two natural products meridianins and variolin B has inspired the synthesis of a hybrid structure referred to as meriolins, which display better antiproliferative and proapoptotic properties in human tumor cell cultures than their parent molecules. A selectivity study performed on 32 kinases has shown that, compared with variolin B, meriolins exhibit enhanced specificity toward CDKs, with marked potency on CDK2 and CDK9 [
19].
The structures of pCDK2/cyclin A/meriolin 3, pCDK2/cyclin A/meriolin 5, and pCDK2/cyclin A/variolin B complexes have been determined by X-ray crystallography, which revealed that these inhibitors bind within the ATP binding site of the kinase, but in different orientations [
18,
19,
21].
All synthesized meriolins 1–14, along with variolin B as a reference, were tested on seven purified protein kinases, namely CDK1/cyclin B, CDK2/cyclin A, CDK5/p25, CDK9/cyclin T, GSK-3 δ/β, CK1δ/ɛ, and DYRK1A (
Table 2). Structure-Activity studies complemented with the crystal structure have provided some clarification on the action mechanisms of these molecules on their CDK target [
18].
In the case of meriolin 11 (
26), addition of a bromide atom at C-5 leads to a drop in inhibitory activity for almost all tested protein kinases, but this effect is particularly pronounced against CDK9 and GSK-3. CDK1, CDK2, and CDK5 are less affected by the bromide addition. Moreover, addition of a chloride atom at C-4 in meriolin 10 (
25) results in decreased potency compared to the non-halogenated meriolin 1 (
16). Taken together, these observations suggest that meriolins constitute a new CDK inhibitory scaffold with promising antitumor activity, and they can be derived from molecules initially isolated from marine organisms [
19].
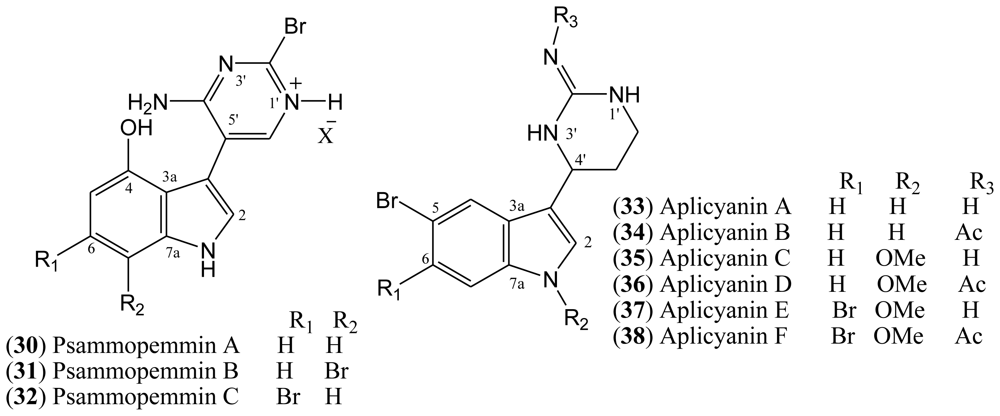
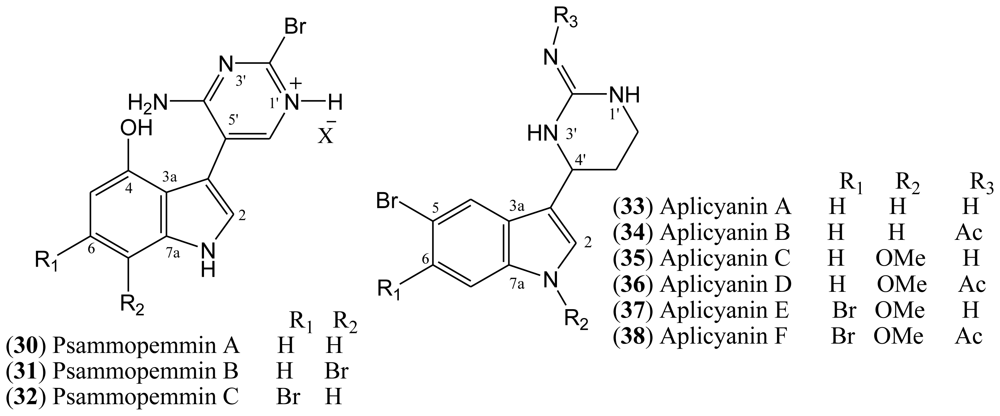
1.6. Aplicyanins
A new family of indole alkaloids was recently isolated from the Antarctic tunicate
Aplidium cyaneum by Reyes and co-workers [
37]. The aplicyanins A–F (
33–
38,
Figure 5) contain a bromoindole nucleus and a 6-tetrahydropyrimidine substituent at C-3. The main structural variations present in aplicyanins include additional bromination of indole ring and the presence of
N-methoxy group as shown in aplicyanins C–F (
35–
38). The aplicyanins share a common 3-(pyrimid-4-yl)indole structure with meridianins A–G (
4–
10), the psammopemmins A–C (
30–
32) and variolins A–D (
11–
14). The tetrahydropyrimidine system of the aplicyanins has a stereocenter at C 4′, in contrast with the planar pyrimidine ring of the meridianins [
21].
Aplycianins are cytotoxic to the human tumor cell lines MDA-MB-231 (breast adenocarcinoma), A549 (lung carcinoma), and HT-29 (colorectal carcinoma). They also exhibit antimitotic activity [
38]. Lastly, given the high cytotoxicity typical of bromoindole derivatives, the presence of a bromoindole moiety in some aplicyanins warrants their investigation as anticancer drugs. Recently, the first total synthesis of (±)-aplicyanins A, B, and E and 17 analogues has been reported [
38].
Regarding the aplicyanin family of indole alkaloids, the six variants of aplicyanins isolated were evaluated for cytotoxicity against a panel of three human tumor cell lines, colon (HT-29), lung (A-549), and breast (MDA-MB-231). The antimitotic activity of these variants has also been assessed. Cytotoxic activity in the submicromolar range as well as antimitotic properties have been found for aplicyanin B (
34), D (
36), and F (
38), with IC
50 values in the low to sub-μM range. On the other hand, aplicyanin A (
33) and C (
35) proved to be inactive at the highest concentrations tested, whereas aplicyanin E (
37) displayed weak cytotoxic properties (
Table 3). These results indicate a key role for the presence of the acetyl group in the biological activity of the aplicyanin family [
37].
In order to establish the structure-activity relationships of the aplicyanins, the total synthesis of (±)-aplicyanins A, B, and E, plus 17 analogues was carried out by Sísa and co-workers in 2009 [
38]. The compounds were again screened for cytotoxicity against the same three human tumor cell lines used for the natural compounds. Racemic (±)-aplicyanin A exhibited activity in the submicromolar range, despite the inactivity of the corresponding natural product. Racemic (±)-aplicyanin B was as active as its corresponding natural product in all three tested cellular lines, whereas aplicyanin E maintained the activity only towards the MDA-MB-231cell line (
Table 3). The decreased cytotoxicity observed for racemic aplicyanin E compared to the natural product, indicates that one enantiomer is more active than the other [
38].
Fourteen of the synthesized compounds also exhibited considerable cytotoxic activity, and these results suggest that the bromine at position 5 of the indole nucleus strongly favors antiproliferative activity, and the acetyl group at the imine nitrogen also acts in some compounds. These results demonstrate the potential of aplycianins structure as a scaffold for anticancer drug discovery [
38].
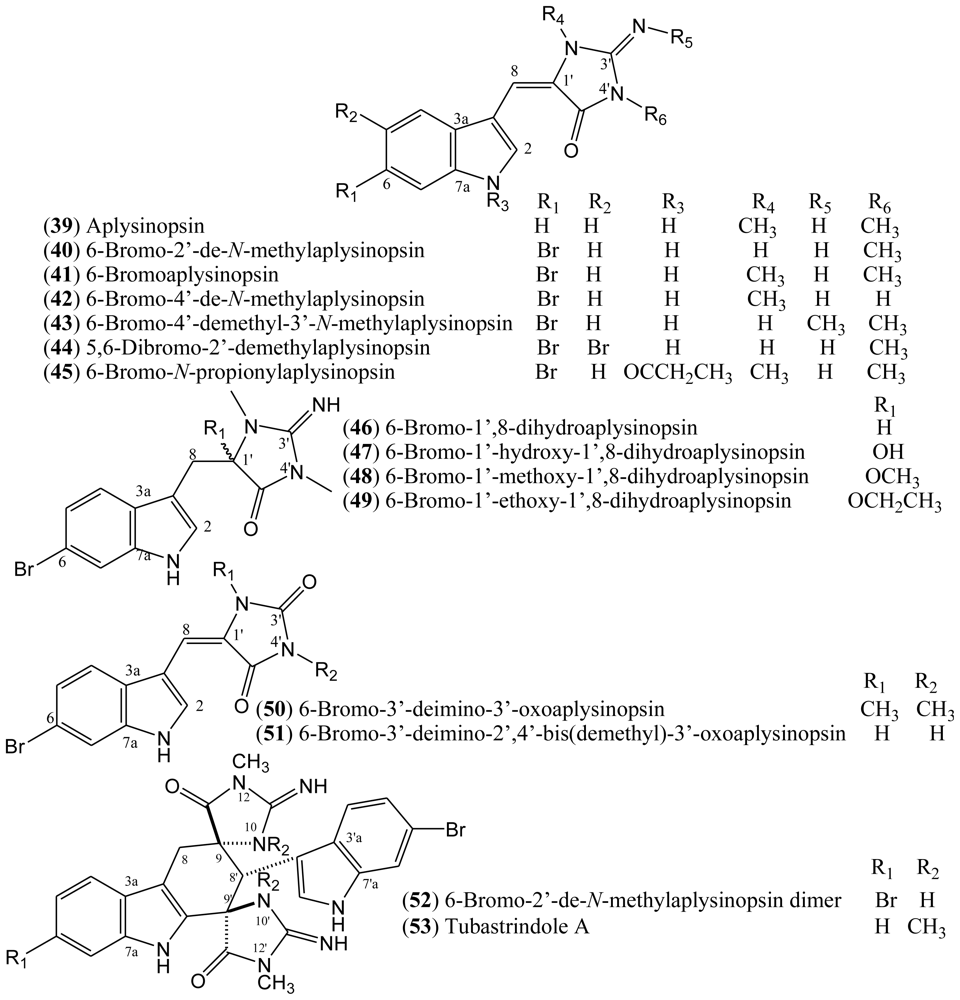
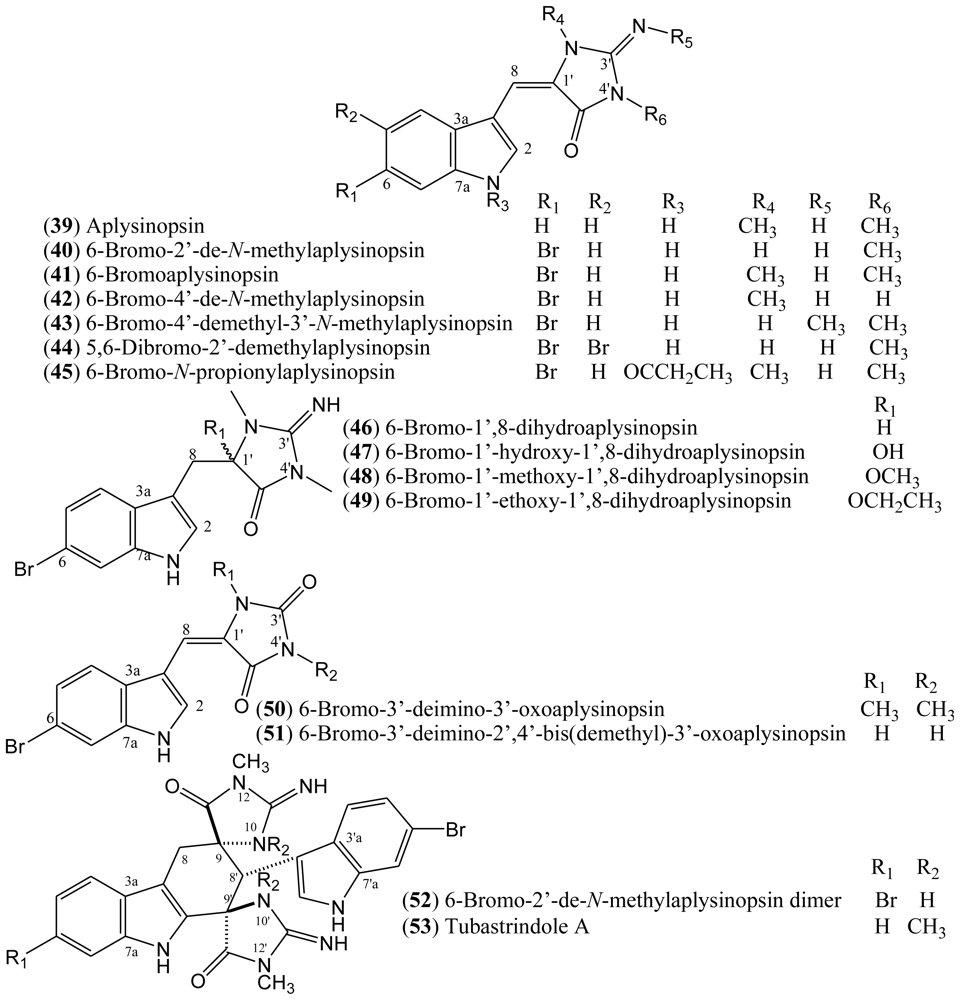
1.7. Aplysinopsins
In 1977, Kazlauskas, Rymantas, and co-workers reported the isolation of aplysinopsin (
39) from the dictoyoceratid sponge
Aplysinopsis [
39,
40]. Aplysinopsin derivatives belong to a class of indole alkaloids and they have also been found in other dictyoceratid and astrophorid sponges as well as in dendrophylliid scleractinian corals [
41]. Additionally, aplysinopsins have been described in anemone, in a symbiotic association, and in a mollusk that feeds on the coral
Tubastrea coccinea [
39].
The halogenated aplysinopsins natural derivatives (
Figure 6) contain a 6-bromoindole moiety, and an iminoimidazolidinone or imidazolidinedione system, both varying in terms of the number and position of
N-methylation. The iminoimidazolidinone portion of compounds
39–
45 are shown as the exocyclic imino tautomer. Only compound
44 contains an additional bromine at the C-5 of the indole core. The aplysinopsins derivatives also differ in terms of the presence and absence of the C-8-C-1′ double bond. Thus, aplysinopsins with C-8-C-1′ double bonds, the most abundant type, can occur as two geometrical isomers (
E/
Z). Also, it has been observed that (
Z)-aplysinopsins are generally less abundant than the (
E)-isomers [
41,
42]. Aplysinopsins substituted at the nitrogen atom of the indole ring and dimers have also been isolated or identified, although compound
45 could be an artifact [
43–
45].
Aplysinopsins exhibit cytoxicity towards tumour cells, as well as some antimalarial and antimicrobial activities. However, properties related to neurotransmission modulation seem to be the most significant pharmacological feature of these compounds. Aplysinopsins have the potential to influence monoaminooxidase (MAO) and nitric oxide synthase (NOS) activities. They have also been found to modulate serotonin receptors [
39].
Aplysinopsin-type compounds have been reported from multiple sources, with brominated aplysinopsins being described from sponges [
46–
49], corals [
41–
45], anemone, and mollusks [
50,
51]. Natural aplysinopsins differ in the bromination pattern of the indole ring. Almost all natural occurring aplysinopsins display halogenations at the position 6 of the indole ring. The only exception is the compound 5,6-dibromo-2′-demethylaplysinopsin (
44), which an additional bromine atom at C-5 [
39].
The compounds 6-bromo-2′-de-
N-methylaplysinopsin (
40) and 6-bromoaplysinopsin (
41) isolated from the Jamaican sponge
Smenospongia aurea displayed high-affinity [
3H]antagonist binding from cloned human serotonin 5-HT
2C receptors expressed in a mammalian cell line (
Ki = 2.3 μM and
Ki = 0.33 μM, respectively). Compound
41 also displayed high-affinity [
3H]antagonist binding from the 5-HT
2A receptor subtype (
Ki = 2.0 μM) compared with serotonin affinity values
Ki = 0.32 μM at the 5-HT
2A receptor and
Ki = 0.13 μM at the 5-HT
2C receptor [
46].
The structure-activity relationship data reveal a role for the R
1, R
2, and R
3 functional groups at positions 6, 2′, and 3′, respectively, in the binding to human serotonin 5-HT
2 receptors. The length of the alkyl chain at the R
3 position as well as the bromination at position R
1 seems to be important for activity. In addition, bromination at the R
1 position is also relevant for the binding affinity of aplysinopsins and for their selective binding to the 5-HT
2C receptor subtype, since both compounds
40 and
41 are brominated and both selectively bind the 5-HT
2C receptor subtype over the 5-HT
2A receptor subtype. Methylation at the R
2 position facilitates binding to the 5-HT
2A receptor subtype. A larger number of analogues will be required to confirm this proposed structure-activity relationship [
46]. Pharmacological and genetic studies have revealed that these receptors influence feeding, glucose homeostasis, and the energy efficiency of physical activity, sleep, sensory processing and learning, affective functioning, and the pathophysiology of several neuropsychiatric disorders [
52,
53].
6-Bromo-2′-de-
N-methylaplysinopsin (
40) and 6-bromoaplysinopsin (
41) have also been tested
in vitro against a D6 clone of
Plasmodium falciparum for their
in vitro antimalarial activity. 6-Bromoaplysinopsin (
41) exhibited activity at 0.34 μg/mL with selective index 14 (S.I. = [IC
50 (Vero cells)/IC
50 (
P. falciparum)], while 6-bromo-2′-de-
N-methylaplysinopsin (
40) showed moderate activity at 1.1 μg/mL with low selectivity. Moreover, compound
40 inhibited the antimalarial target plasmepsin II enzyme with IC
50 53 μM (FRET) and 66 μM (FP) [
46].
Additionally, 6-bromoaplysinopsin (
41) has been reported to be involved in the symbiotic association between
Radianthus kuekentbali (sea anemone) and
Amphiprum perideraion (anemone fish) [
50].
A number of aplysinopsin alkaloids have also been evaluated for their neuromodulatory activity in two types of nitric oxide synthase (NOS) isozymes. Nitric oxide (NO) is known to be an important second messenger having numerous functions which regulate many physiological processes; e.g., inflammation, blood pressure regulation, platelet adhesion, neurotransmission, and defense mechanisms. The biosynthesis of NO is catalyzed by nitric oxide synthase (NOS), which is classified into three isoforms: inducible NOS (iNOS), endothelial NOS (eNOS), and neuronal NOS (nNOS). Therefore, a selective inhibitor of NOS isozymes would be expected to have significant therapeutic potential as a neuromodulator [
47]. 6-Bromo-2′-de-
N-methylaplysinopsin (
40) and the isomers 5,6-dibromo-2′-demethylaplysinopsin
Z-
44 and
E-
44 isolated from the sponge
Hyrtios erecta display selective inhibitory activity against nNOS, with 100% inhibition of nNOS at 125 μg/mL [
47]. Compounds
Z-
44 and
E-
44 showed no inhibitory activity against iNOS. In turn, aplysinopsin
40 inhibited only 7.5% of iNOS activity at a concentration of 125 μg/mL [
47].
Regarding the antimicrobial potential of halogenated aplysinopsins, Koh and Sweatman [
54] have reported the screening of the Australian coral
Tubastraea faulkneri extract for antimicrobial assay against seven species of microbes (
Vibrio alginolyticus,
V. harveyi,
V. parahaemolyticus,
Photobacterium damsela,
Alteromonas rubra,
Staphylococcus aureus, and
Synechococcus sp). Aplysinopsin (
39), 6-bromoaplysinopsin (
41), 6-bromo-2′-de-
N-methylaplysinopsin (
40), and its dimer
52 were the compounds isolated accounting for 72% of the activity of the
T. faulkneri methanol extract. This study also suggested that these aplysinopsins are toxic to the larvae of other coral species that are potential competitors and could act as allelochemicals [
54]. The aplysinopsins
46–
49 isolated from the sponge
Thorectandra sp were evaluated for antimicrobial activity against
Staphylococcus epidermidi. All of the compounds were found to have either weak or moderate minimum inhibitory concentrations (MIC) ranging from 6.25 to 100 μg/mL as compared to the standard vancomycin (0.625 μg/mL) [
48].
1.10. Structural Elucidation
This section reports a compilation of the
13C chemical shifts of the halogenated marine indole alkaloids derivatives, meridianins (
5–
9), psammopemmins (
30 and
32), aplicyanins (
33–
38), aplysinopsins (
40–
41,
43–
44,
46–
51,
53), and leptoclinidamines (
56), which have in common the presence of a 3-substituted indole nucleus. Additionally, the
13C data of
10 and
39 are presented for comparison of the
13C chemical shifts with halogenated examples. The literature data are listed in
Tables 5,
6,
7 and
8. The solvent (A = DMSO-
d6, B = CD
3OD, and C = CDCl
3) and references are shown in the first line of the tables.
Inspection of the
13C-NMR data of compounds
5,
8,
30 and
32 as compared with
10 (
Table 5) reveals that introduction of a hydroxyl group in the C-4 indole moiety results in downfield signals at the α carbon. Additionally, comparison of the
13C data of meridianin G (
10), which bears only a 3-substituted indole core, with the other bromine indole derivatives shows that introduction of a bromine in the indole skeleton results in upfield signals at the α carbon.
The meridianin family skeleton can be recognized by the typical
1H-NMR signals, as for example, in the case of compound
10: a pair of doublet for the pyrimidine protons (δ 7.02 and 8.05,
J = 5.5 Hz), together with a singlet for H-2, the typical pattern of a 3-substituted indole nucleus. The
13C-NMR downfield signals at δ C-2′, C-4′, and CH-6′ corroborate the presence of 2-aminopyrimidine at C-3 in compounds
5–
10 [
60].
The basic difference between the psammopemmins and the meridianins is the presence of a 5′-substituted 4′-amino-2′-bromopyrimidine at C-3 of the indole nucleus. The distinguishing
1H-NMR signals of the heterocyclic ring of the psammopemmins class can be recognized by the signals at δ 7.12 (d,
J = 5.4 Hz) and 8.12 (br d,
J = 5.4 Hz), attributed to the pyrimidine proton H-6′ and to NH at position 1′, as in the case of compound
30. The
13C-NMR downfield signals at δ C-2′, C-4′, and C-5′confirm the presence of 5′-substituted 4′-amino-2′-bromopyrimidine [
28].
The aplicianins’
13C-NMR spectra differ from those of the meridianins and psammopemmins because of the presence of the signals due to a guanidine group at low field (C-2′) and three chemicals shifts at upfield, ascribed to C-4′ (CH), C-5′ (CH
2), and C-6′ (CH
2). Additionally, the
1H-NMR coupling constants of the 6-tetrahydropyrimidine protons are important to establish the difference between aplicyanins, meridianins, and psammopemmins [
37].
Aplysinopsins (
39–
41,
43–
44), with the iminoimidazolidinone substituted at the C-3 of the indole core, normally show a
1H-NMR spectrum with signals due to
N-methyl groups in the range of δ 3.0 to 3.5 (s, 3H), as well as a singlet characteristic of an olefinic proton in the δ 6.38–6.46 range. The
13C NMR spectrum reveals the signals for two olefinic carbons C-8 (CH) and C-1′ (C), methyl, guanidine, and amide carbonyl, as well as those of the indole ring, as already mentioned [
43,
47,
48]. Analysis of the
13C data of
39, which bears a 3-substituted indole core, and comparison with data of the other bromine indole derivatives show that the presence of bromine in the indole moiety results in upfield signals at the carbon α.
The spectra of aplysinopsins (
50–
51) differ in terms of the signals at C-8, C-3′, and C-5′, if compared with data for 3-iminoimidazolidinone, where C=NH (C-3′) is replaced by C=O (C-3′) [
41].
The
E or
Z-configuration of the double bond at C-8 could be assigned on the basis of a
1H,
13C heteronuclear coupling constant. The coupling constant value obtained for the
E isomer was larger than in the
Z [
41,
42]. The geometry of the C-8-C-1′ olefin could be determined by comparison of the chemical shift of the H-2 proton and C-8 carbon. In the
Z isomer, the δ values of C-8 and H-2 were upfield compared to the values obtained for the
E isomer [
41,
42,
47]. Aplysinopsin type compounds without substituents at N-2′ are predominantly of
Z configuration, whereas the converse is true for compounds bearing a methyl group at N-2′. Although it is important to note that,
Z and
E aplysinopsin alkaloids undergo rapid isomerization [
41,
42,
61].
Comparison of the
13C-NMR data of
46–
49 with previous aplysinopsins reveals that the C-8 and C-1′ signals are shifted upfield according to R
1 at C-1′, thereby confirming that the double bond at C-8-C-1′ is absent. Segraves and Crews considered that
48 and
49 are artifacts formed from
47 during the extraction process [
48].
The
13C-NMR data of compound
53 indicates the presence of two indoles and two iminoimidazolidinones. Biogenetically, this compound could be formed from an enzymatic Diels–Alder cycloaddition of two molecules of aplysinopsin, which were probably derived from tryptophan and guanidine, followed by some modifications [
44].
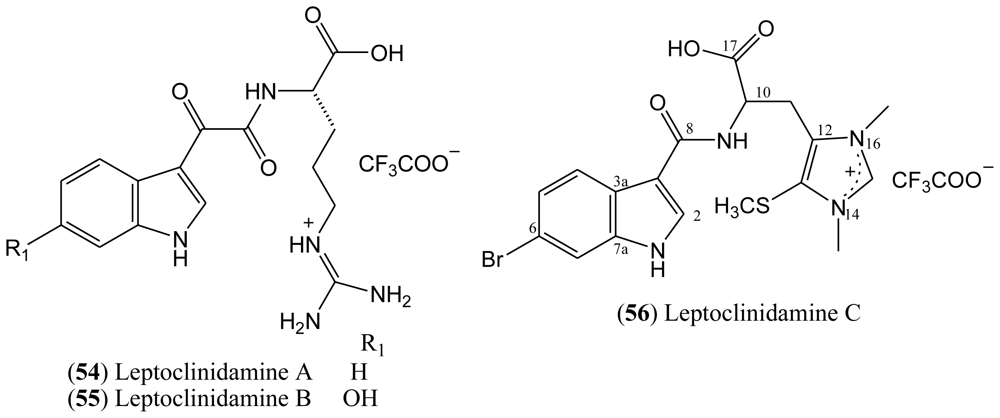
The structure of leptoclinidamine C (
56) has been established as a 3,6-disubstituted indole and a β-substituted alanine by 1D and 2D NMR data. The
13C data indicate the presence of two
N-methyl groups at C-14 and C-16; a third methyl group at C-13 is attributable to an S-methyl. As mentioned, the chemical shift of the quaternary carbon C-6 (δ 114.6) indicates that the bromine was substituted at this position [
55].


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}