Quality not Quantity: The Role of Marine Natural Products in Drug Discovery and Reverse Chemical Proteomics


Department of Chemistry & Biomolecular Sciences, Macquarie University, Sydney, NSW 2109 Australia
*
Author to whom correspondence should be addressed.
Mar. Drugs 2005, 3(2), 36-63; https://doi.org/10.3390/md302036
Submission received: 15 April 2005
/
Accepted: 30 May 2005
/
Published: 1 June 2005
Abstract
:Reverse chemical proteomics combines affinity chromatography with phage display and promises to be a powerful new platform technology for the isolation of natural product receptors, facilitating the drug discovery process by rapidly linking biologically active small molecules to their cellular receptors and the receptors’ genes. In this paper we review chemical proteomics and reverse chemical proteomics and show how these techniques can add value to natural products research. We also report on techniques for the derivatisation of polystyrene microtitre plates with cleavable linkers and marine natural products that can be used in chemical proteomics or reverse chemical proteomics. Specifically, we have derivatised polystyrene with palau’amine and used reverse chemical proteomics to try and isolate the human receptors for this potent anticancer marine drug.
Introduction
As reactions in Nature are biased towards function, it follows that every natural product must have a biological receptor; only if one accepts this premise can the enormous biochemical expense of producing natural products be rationalised. Even though natural products may not have co-evolved with human proteins, they have emerged in nature to interact with biomolecules. As Jerrold Meinwald succinctly put it, “Natural products have evolved to interact with something, and that something may not be so different from human proteins” [1]. This assertion is supported by a recent survey [2], which found that 61% of the 877 new small molecule chemical entities introduced as drugs worldwide during 1981–2002 were either natural products, natural product derivatives or natural product mimics. The percentages were even higher when considering only the antibacterial (79%) and anticancer (74%) compounds. These figures are not surprising as natural products cover a far greater area of chemical space than synthetic compounds, and have property distributions that are similar to those of drugs currently in use [3]. For instance, when compared to synthetic compounds, natural products, on average, have higher molecular weights, incorporate fewer nitrogen, halogen or sulfur atoms, but more oxygen atoms, and are sterically more complex, with more bridgehead atoms, rings, and chiral centres.
Clearly, natural products are a rich source of drugs and drug leads. However, even when a natural product is found to exhibit biological activity, the cellular target and mode of action of the compound are rarely identified. This is also true of many natural products that are currently in clinical trials or have already been approved as pharmaceuticals. The absence of a definitive cellular target for a biologically active natural product hinders the rational design and development of more potent therapeutics. Therefore, there is a great need for new techniques to facilitate the rapid identification of cellular targets for biologically active natural products.
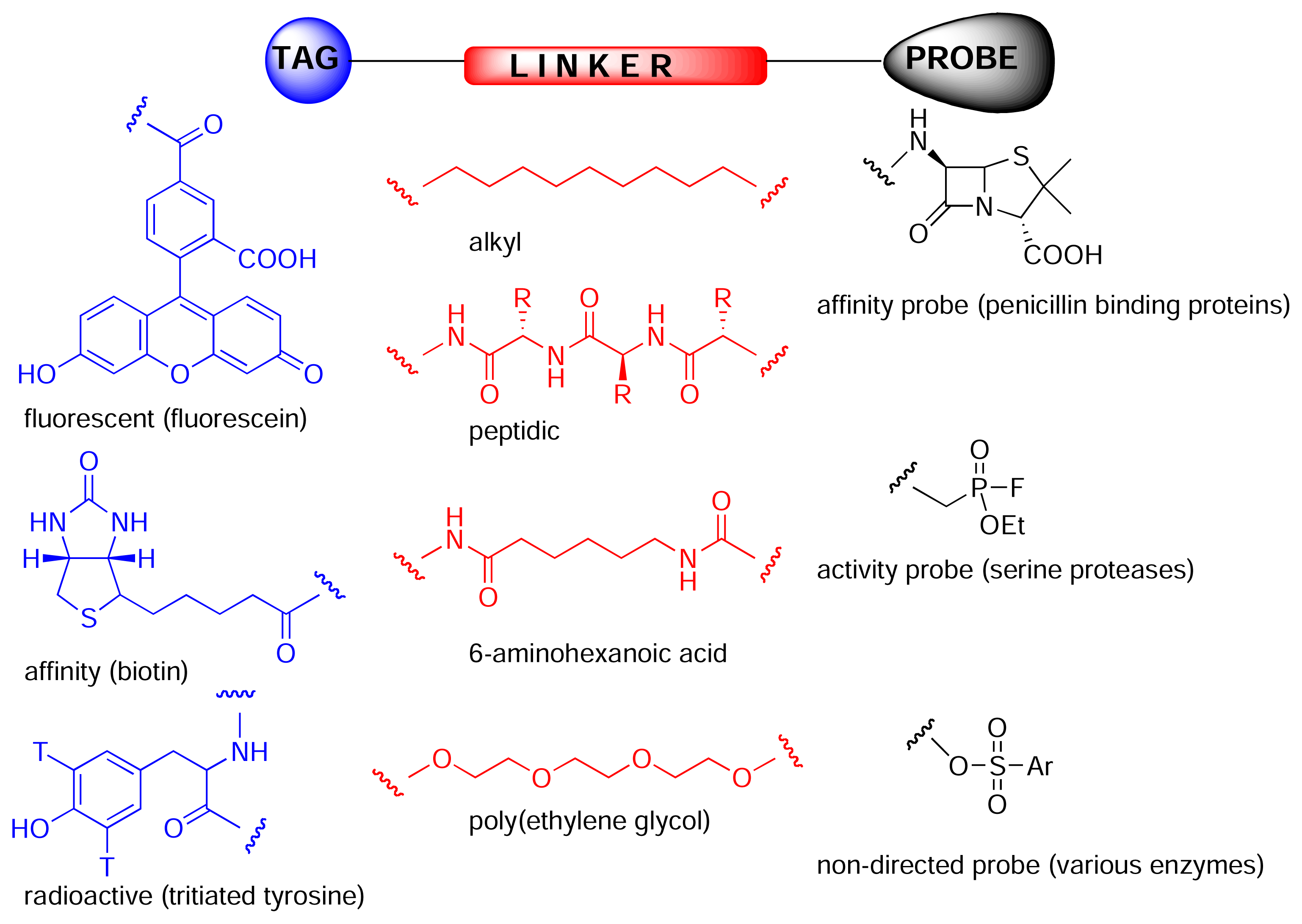
Chemical proteomics is a powerful tool for isolating and identifying cellular receptors for biologically active natural products, thereby facilitating subsequent rational drug design, and often providing valuable information regarding underlying biochemical and cellular processes. The key to chemical proteomics is the construction of an affinity probe. These probes are composed of three domains (Fig. 2). The tag can consist of either a radioactive/fluorescent label, to allow visualisation of bound proteins on an electrophoresis gel, or a solid-phase bead/surface, to allow affinity purification of proteins. Frequently, biotin is used as the tag as it allows both affinity purification, using a streptavidin resin, and in-gel visualisation, using streptavidin coupled to a reporter enzyme such as horseradish peroxidase. The probe can be any small molecule that reacts with a particular class of enzyme (activity probe) or binds to a particular protein or class of proteins (affinity probe). Affinity probes can be more or less specific for a particular protein or class of proteins or could be completely non-directed [4]. The probe and tag are usually separated from each other by a linker (Fig. 2), that can be simply an alkyl group, peptidic or polyethylene-glycol (PEG) based. The PEG based linkers are hydrophilic like the peptidic linkers, but without the disadvantage of possible cleavage in biological systems. The linker should also be as long as possible as the receptor for a particular small molecule may be deeply buried in the protein.
Activity based probes are designed to isolate specific classes of enzymes [5]. For example, Patricelli et al. fluorescently labelled a complex proteome with ethyl fluorophosphonate activity probes, thereby facilitating in-gel analysis of all putative serine proteases, without the need for further fractionation or purification [6]. Similarly, Speers and Cravatt used biotinylated and fluorescently labelled analogues of the epoxide-containing natural product, E64, to isolate putative members of the papain class of cysteine proteases [7]. Affinity based probes, on the other hand, do not rely on a protein’s enzymatic activity, but on the recognition of a particular motif. For instance, Zhao et al. used radioactively and fluorescently labelled penicillin analogues to detect penicillin-binding proteins in bacterial cell lysate [8].
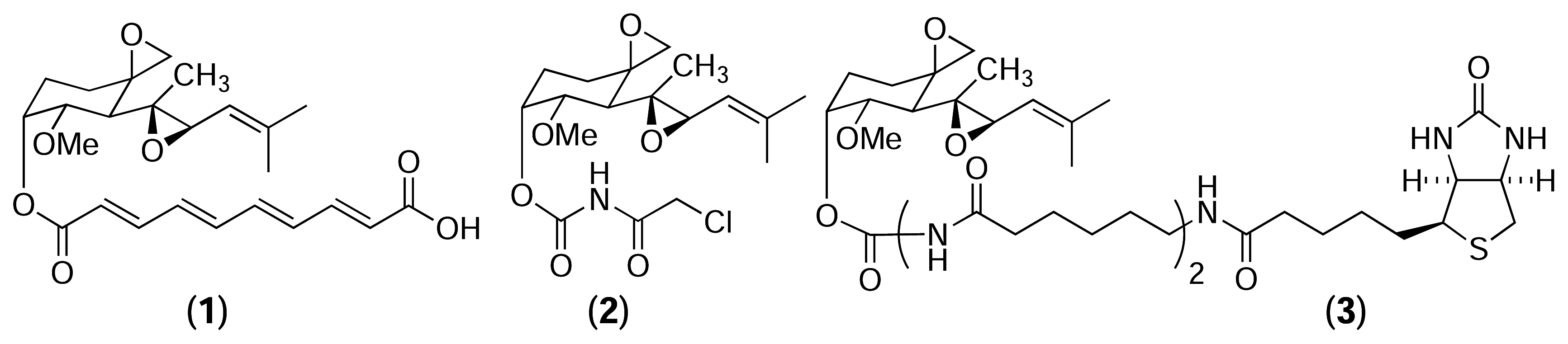
Chemical proteomics was first used by Taylor et al. in 1965 to identify tubulin as the cellular receptor for colchicine, a tropolone alkaloid known to disrupt mitosis [9–13]. Porcine brain extract was incubated with a tritiated analogue of colchicine and the resulting mixture was fractionated by ion exchange chromatography. Radioactive fractions were combined and further fractionated by gel filtration chromatography and analysed by gel electrophoresis that led eventually to the discovery of tubulin. Since then, the technique has been vastly improved and used to identify the cellular receptors for numerous other natural products [14]. In particular, the most impressive examples are from the Crews lab at Yale, who specialise in the isolation of protein receptors for epoxide containing natural products such as fumagillin [15], eponemycin and epoxomicin [16]. Fumagillin (1) and TNP-470 (2), a semisynthetic analogue of fumagillin, are potent inhibitor of angiogenesis, causing late G1 phase endothelial cell cycle arrest [17]. Despite the fact that TNP-470 had undergone numerous pharmacological studies and clinical trials, little was known about its cellular target or mode of action. Sin et al. used a biotinylated analogue of fumagillin (3) to identify its cellular target from human umbilical venous endothelial cells [15]. This led to the isolation of human methionyl aminopeptidase (MetAP-2) – a cobalt-dependent metalloprotease. Mass spectrometry and X-ray crystallography revealed the imidazole nitrogen of histidine-231 in the active site of MetAP-2 forms a covalent bond with the carbon of the spirocyclic epoxide of fumagillin [18, 19].
In marine natural products, ilimaquinone (4) is a sesquiterpene quinone first isolated in 1979 from the marine sponge Hippospongia metachromia [20]. In addition to possessing mild anti-bacterial, anti-viral, anti-fungal and anti-inflammatory activities, ilimaquinone has been found to break down the Golgi apparatus into small vesicles, thereby blocking cellular secretion [21, 22]. Snapper et al. used chemical proteomics to identify cellular receptors for ilimaquinone [23]. A tritiated azidobenzene moiety was attached to a synthetic chloroquinone analogue of ilimaquinone to form a photoaffinity reagent (5). Bovine liver extract was incubated with this reagent in the presence of ultraviolet light. The crude mixture was then separated by ion exchange chromatography and fractions containing high levels of radioactivity were further purified by size exclusion, yielding two major radioactive bands (48 and 55 kDa). Amino acid sequencing of the 48-kDa band revealed it to be the enzyme S-adenosylhomocysteinase (SAHase). SAHase plays a key role in cellular methylation chemistry by catalysing the breakdown of S-adenosylhomocysteine (SAH) to homocysteine and adenosine.

In a second experiment, the authors synthesised an ilimaquinone-agarose affinity resin (6), which was incubated with homogenised bovine liver and then washed extensively [24]. Proteins retained by the resin were separated by gel electrophoresis, yielding six main protein bands. Amino acid sequencing of these bands revealed three proteins involved in the activated methyl cycle – SAHase (as isolated previously), S-adenosylmethionine synthetase (SAM synthetase) and catechol-O-methyltransferase (COMT) – as well as three unrelated proteins. Subsequent enzymatic assays established that ilimaquinone is a competitive inhibitor of SAHase, but has little effect on the activity of SAM synthetase or COMT. The authors noted that a consequence of SAHase inhibition would be the intracellular accumulation of SAH, which is a potent feedback inhibitor of methyltransferases. These results support the assertion that methylation events play an important role in cellular secretory events and vesicle-mediated processes.
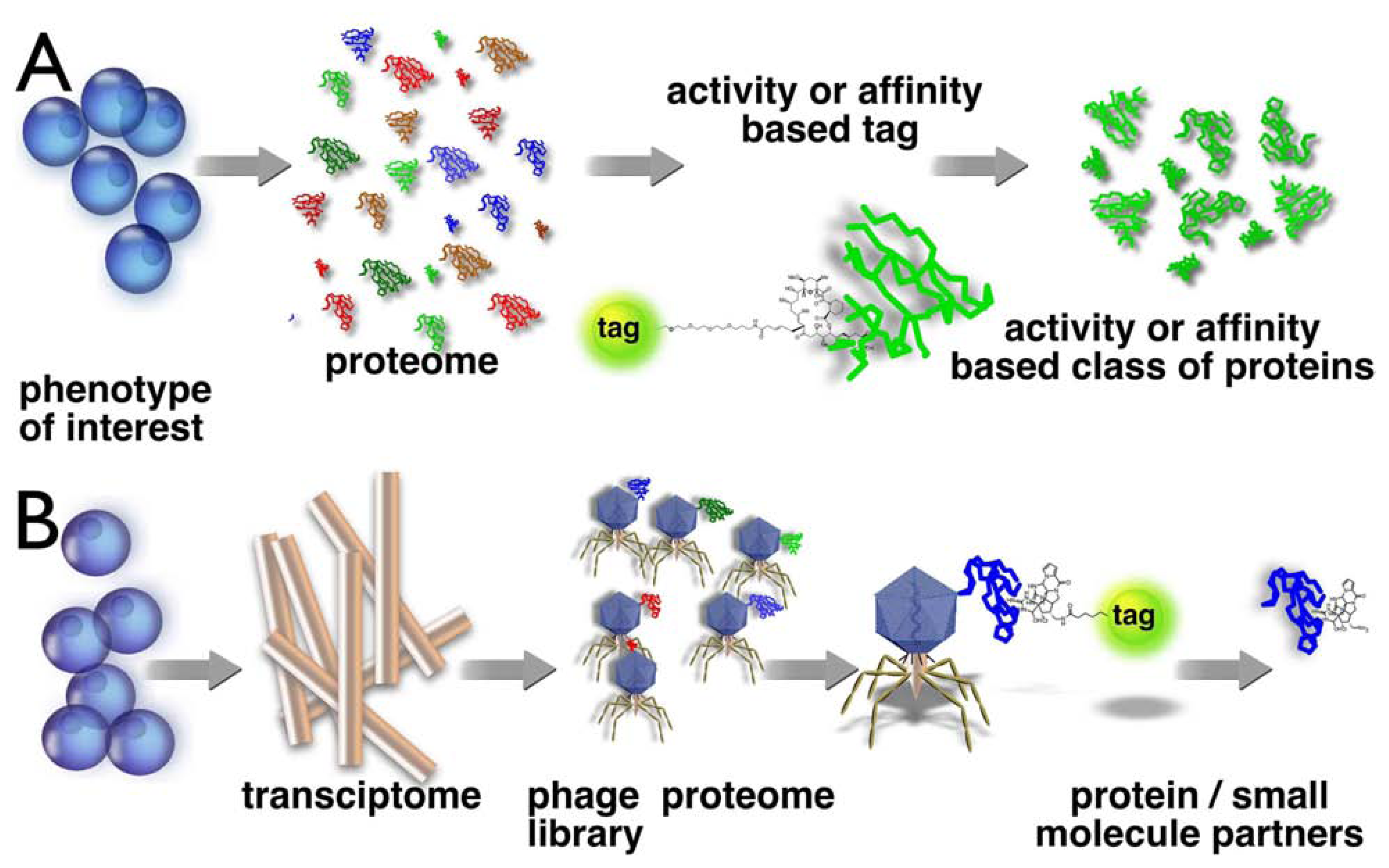
In reverse chemical proteomics (Fig 1B), the starting point is a transcriptome of a phenotype of interest. A cDNA library is cloned into an expression system and the resulting tagged proteome is screened against a small molecule. To date, only phage display has been used in a reverse chemical proteomics context, although other methods, such bacterial [25] and yeast [26] cell-surface display, are also possible, but need to be developed further. If an entire cDNA library is cloned into a phage display vector, each phage particle will contain a different gene insert and will express the protein encoded by that gene on its surface. The displayed proteins often behave as if they were free in solution, so phage displaying the target protein can be rescued from an entire library using an immobilised natural product, as per standard chemical proteomics experiments. However, the real power of phage display comes from the fact that rescued phages can be amplified by transfection into E. coli and then subjected to another round of affinity selection with the immobilised natural product. This cycle can be repeated numerous times, allowing the most avid binders to be identified, even if they are only present in very small amounts in the original cDNA library. This is a significant advantage over chemical proteomics where the most abundant binding protein is usually isolated at the expense of the most avid binders [27]. A significant disadvantage with phage display is that the proteins are not expressed in their native environments, so they will not contain any post-translational modifications and may not fold correctly. In addition, membrane-bound proteins and proteins comprised of multiple subunits are unlikely to be displayed correctly on the surface of a phage particle.
Chemical proteomics and reverse chemical proteomics may seem like unusual tools for drug discovery as they require a known drug from the outset, and this is exactly what the pharma industry is interested in discovering. This is where natural products are useful. As reactions in Nature are heavily biased towards function, it follows that every natural product will have a biological receptor and that receptor will be a potentially drugable target. Chemical proteomics or reverse chemical proteomics can be used to identify this receptor and its corresponding gene, thereby completing the gene-protein-ligand trinity. This information can then be used to determine the function and role of the protein in the cell as well as to design new, more potent ligands. Therefore, all natural products should be considered as potential lead compounds in drug discovery.
Humans and natural products generally have not coevolved, so it is unlikely that the true molecular target of a human disease will have a natural product partner. Therefore, when natural products are used as starting points to target proteins other than those with which they coevolved, it can be argued that the uniqueness of these compounds is lost [1]. Genes do not appear randomly – they are conserved and the proteins they encode are also likely to be conserved in structure and function. Even though natural products may not have coevolved with human proteins, they have emerged in Nature to interact with something, and that something may not be so different from human proteins [1].
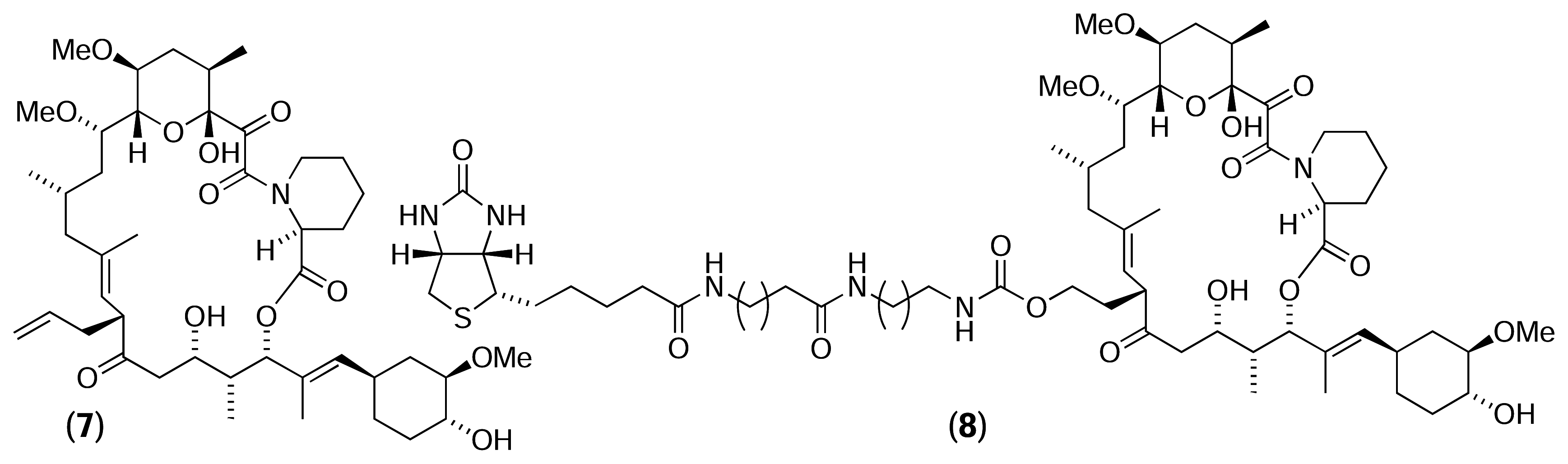
The potential of reverse chemical proteomics was first demonstrated by Dave Austin, who used the immunosuppressant drug FK506 (7) to isolate its known receptor, FKBP, from a T7 phage display library [28]. Initially, a biotinylated analogue of FK506 (8) was immobilised onto a monomeric avidin-agarose column. Lysate from T7 phage displaying a human brain cDNA library was passed through a column containing underivatised avidin-agarose to remove endogenous avidin binders, and then through a biotin-derivatised avidin-agarose column to remove endogenous biotin binders. Finally, the pre-treated lysate was passed through the FK506-derivatised affinity column, and the column was washed thoroughly with buffer. Phage particles retained by the column were eluted with free biotin and transfected into fresh E. coli. After lysis, the amplified phage were precipitated and washed to remove biotin from the previous elution step. Finally, the phage were resuspended in buffer and poured through a fresh FK506-derivatised affinity column for the next round of selection. Following each round of selection, five random phage plaques were picked and their DNA was amplified by PCR. After the second round of selection, all five clones gave rise to a DNA band of 450 bp, suggesting one clone had become the dominant member of the library. DNA sequencing of these bands revealed identical copies of a full-length, in-frame gene coding for human FKBP1a. Therefore, it appeared that after only two rounds of selection with the FK506 affinity resin, phage particles displaying FKBP on their surface had been amplified selectively to become the dominant members of the library. However, it was discovered subsequently that the FKBP clone isolated had actually originated from contamination by a positive control FKBP clone, rather than from the cDNA library [29].
Bacteriophage are small and hardy, and can travel long distances as aerosols, remaining infective for many years. Consequently, cross-contamination is a common problem associated with all phage display systems. However, the risk of contamination can be reduced by using disposable plasticware, minimising the formation of aerosols and disinfecting exposed surfaces with a dilute solution of bleach. The authors repeated the experiment, performing seven rounds of selection and taking particular care to avoid contamination [29]. Sixteen random clones were picked after the sixth round of selection and their DNA amplified by PCR. Five of these clones were found to be identical and contained the entire coding sequence of FKBP1a. This number increased to eleven out of sixteen clones in round seven. The results of this experiment show that it is possible to isolate the receptor for a natural product using a cDNA library displayed on T7 bacteriophage, thereby providing an important proof of concept. However, the FK506-FKBP interaction is particularly strong (Kd = 0.4 nM) and it is unclear whether experiments using natural products with more modest dissociation constants would be as successful. In such cases, it is likely that more rounds of selection would be required for the strongest binder to become the dominant clone in the library.
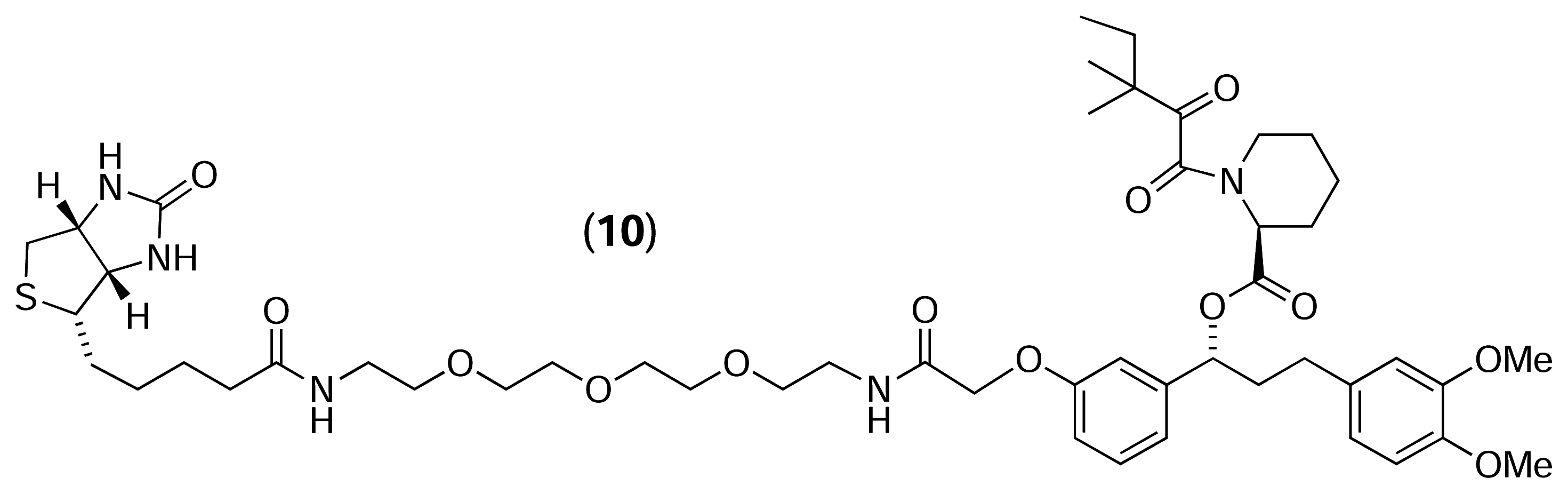
Austin et al. [30] also used reverse chemical proteomics to isolate cellular receptors for AP1497 – a synthetic analogue of FK506. Initially, biotinylated AP1497 (10) was immobilised on a streptavidin-coated polystyrene microtitre plate, and the resulting affinity support was used to probe a T7 phage-displayed human brain cDNA library. After three rounds of selection had been performed, 96 random plaques were selected and were tested individually for their ability to bind to an AP1497-derivatised plate. The clones displaying the highest level of affinity were submitted for DNA sequencing, revealing that 22 of the top 30 clones contained either an FKBP1a, FKBP1b or FKBP2 gene, all of which are known receptors for FK506. This experiment confirmed that the level of derivatisation attainable on the surface of a microtitre plate well is sufficient to isolate phages displaying the target protein from the plethora of other phages in a cDNA library. This was an important proof of concept as previous experiments had employed biotinylated FK506 immobilised on streptavidin-functionalised agarose resin [28, 29], which has a much higher loading capacity than the microtitre plate surface. However, like the FK506-FKBP interaction (Kd = 0.4 nM) [31, 32], the AP1497-FKBP interaction is particularly strong (Kd = 14 nM), so it still remains to be seen whether reverse chemical proteomics can be used to isolate receptors with more modest affinities for their ligands from the numerous non-specific binders present in a T7 phage-displayed cDNA library.
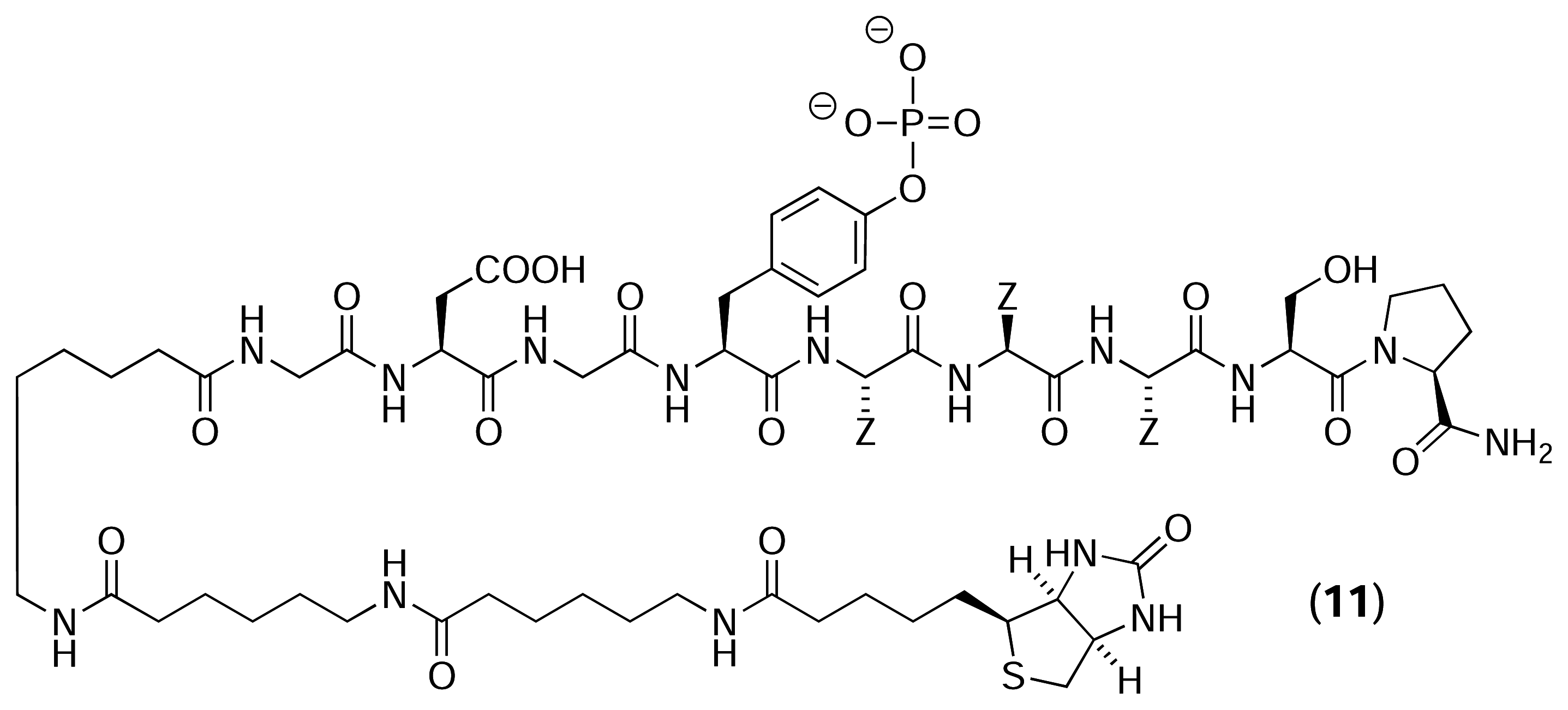
Austin et al. [33] also used a reverse chemical proteomics approach to isolate clones displaying proteins with phosphotyrosine binding (SH2) domains. A biotinylated phosphotyrosine diversity peptide, Biotin-YZZZ (11), was immobilised on a streptavidin-coated polystyrene microtitre plate, and the resulting affinity support was used to probe 11 different T7 phage-displayed human cDNA libraries. After three rounds of selection had been performed, 36 random plaques were selected from each library and were tested individually for their ability to bind to a Biotin-YZZZ-derivatised plate. The clones displaying the highest level of affinity (dissociation constant < 1 μM) were submitted for DNA sequencing, revealing that 40 of the top 50 clones displayed proteins containing SH2 domains. In a 2005 paper [34], the authors also isolated the entire N-terminal SH2 domain of the α subunit of phosphatidyl inositol-3 kinase from a T7 phage-displayed human liver cDNA library using a similar biotinylated phosphotyrosine peptide. The results of these experiments show that immobilised peptides can also be used as affinity probes to isolate phages displaying proteins of interest from a cDNA library.
Kwon and colleagues [35] used reverse chemical proteomics to identify the cellular receptor for HBC – a synthetic curcumin derivative that inhibits the proliferation of several tumour cell lines. A biotinylated analogue of HBC (12) was immobilised on a streptavidin-coated polystyrene microtitre plate and the resulting affinity support was used to probe a mixture of five different T7 phage-displayed human cDNA libraries. After four rounds of selection had been performed, 17 random plaques were selected and their DNA inserts were sequenced, revealing that 12 of the clones displayed the C-terminal (aa 86–149) of calmodulin. The authors used surface plasmon resonance to show that the HBC-calmodulin interaction exhibited a clear dose response, with the dissociation constant for the interaction calculated to be 8 μM. A flexible docking study also indicated that HBC is compatible with the binding cavity occupied by the known calmodulin inhibitor, W7. However, given that only a fragment of calmodulin was isolated, and that this fragment is rich in charged amino acid residues (38%), further biochemical studies are required to make this interaction convincing.

Clearly, reverse chemical proteomics is an exciting new technique that combines small-molecule affinity chromatography with phage display to identify the cellular receptors for biologically active natural products. While the potential of reverse chemical proteomics using T7 phage display has been demonstrated by Austin et al. on model systems, the technique has not been validated using a natural product with a completely unknown cellular receptor, and no similar technique has ever been applied to marine natural products. Our work aims to improve the technique by removing the need to biotinylate rare natural products by preparing a variety of solid phases capable of reacting irreversibly with specific functional groups in any small molecule, making these affinity supports available to chemists and non-chemists alike.
Since Merrifield’s pioneering work in 1963 [36], there has been an exponential increase in the use of polymeric supports for solid phase organic synthesis [37, 38], and a similar increase in range of cleavable linkers that are available to immobilise small molecules onto these solid supports [39, 40]. The incorporation of new photolabile [41], traceless [42] and safety catch [43] linkers allows the orthogonal cleavage of different reagents from the same solid phase, greatly increasing the versatility of the system. However, as these linkers were designed for solid phase synthesis in organic solvents, they are generally not water-soluble and are often quite short. Therefore, we synthesised a range of long, hydrophilic linkers, suitable for generating affinity supports for use in reverse chemical proteomics studies in polystyrene microtitre plates.
Polystyrene (PS) is cheap, rigid, transparent, shows very low water adsorption and can be extruded, injection moulded and foam moulded, so it is not surprising that the majority of microtitre plates currently used in the laboratory and for high throughput screening are made from this polymer. Clearly, it would be expedient to derivatise the surface of PS plate wells with small molecules for use in reverse chemical proteomics, as this would ensure widespread availability of the plates and would also maintain compatibility with robotic liquid handlers and other analytical instruments that are configured specifically for PS plates.
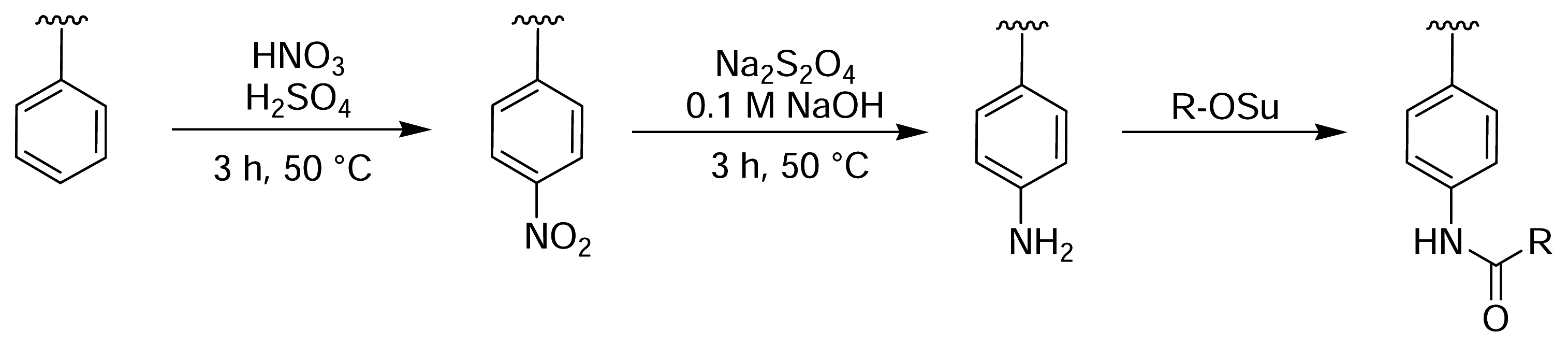
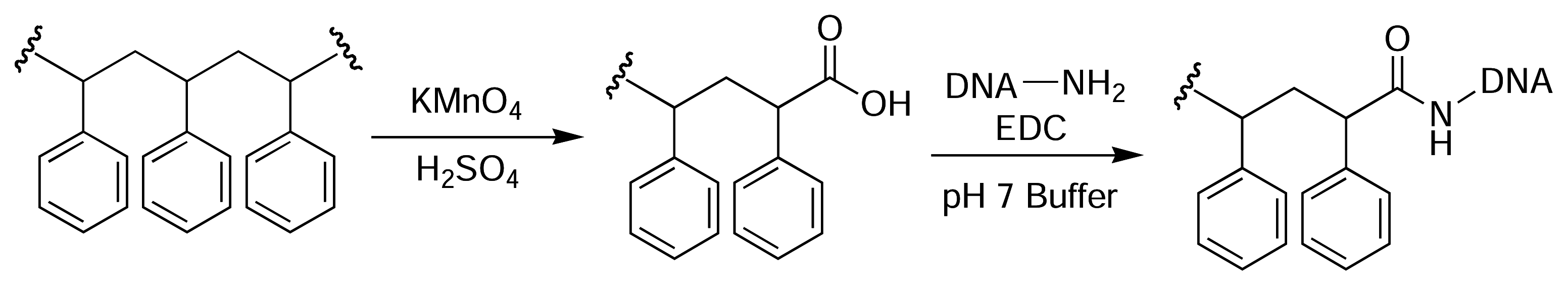
Rotmans and Delwel described a method of introducing amine groups onto the surface of PS plates for the covalent immobilisation of antigens [44]. The PS surface was first nitrated with fuming nitric acid and then reduced with sodium dithionite (Fig. 3). This yielded aromatic amines on the plate surface that could then be derivatised with an amine-reactive molecule of choice [45]. Zammatteo and colleagues oxidised the surface of PS microtitre plates with acidic potassium permanganate to generate free carboxyl groups on the polymer backbone, which were then activated with EDC and coupled with DNA probes for hybridisation assays [46] (Fig. 4).
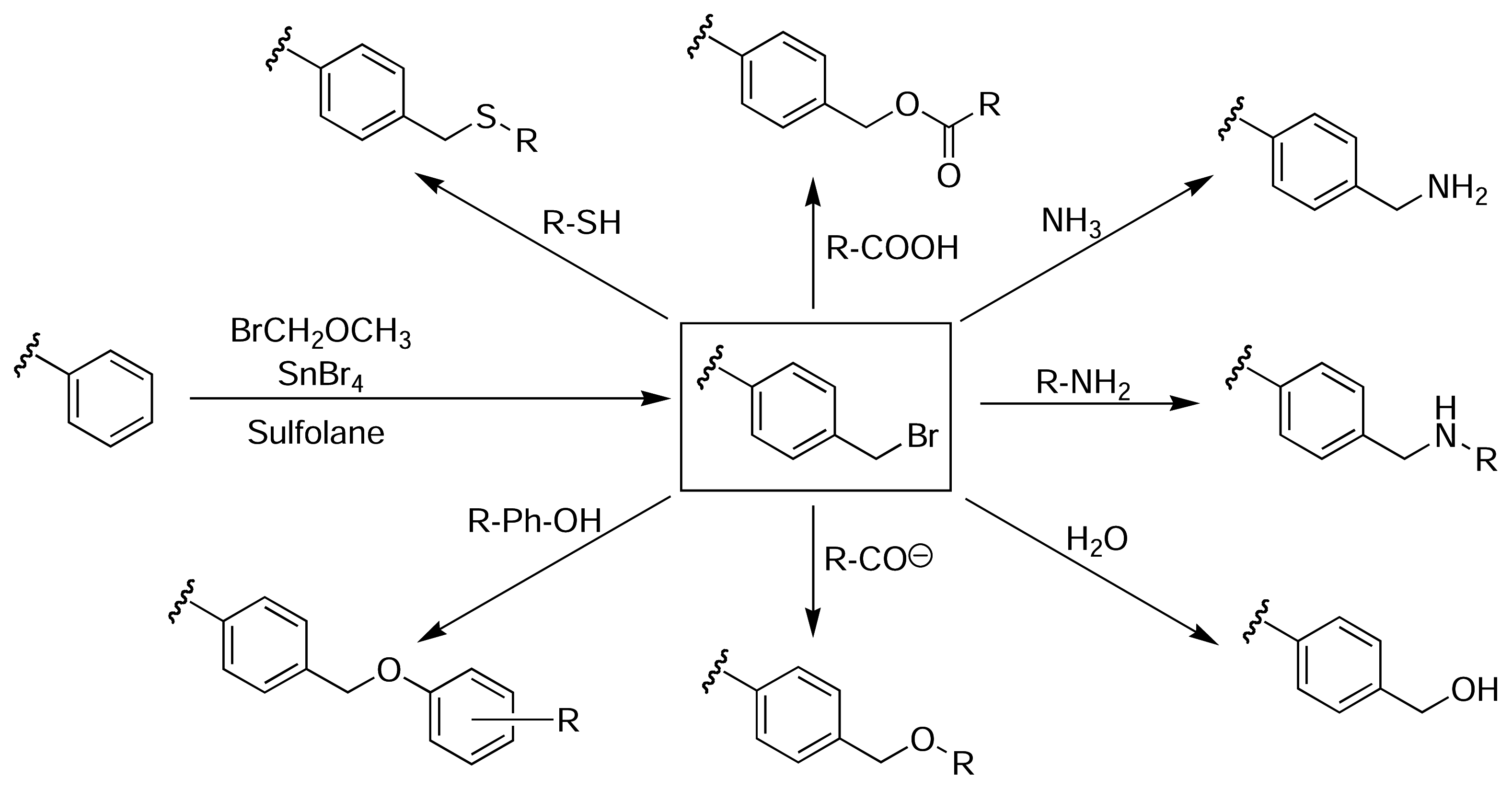
Although the nitration and oxidation methods were successful in attaching molecules to the surface of PS microtitre plates, they invariably caused some damage to the wells. Clark described a method of derivatising the surface of PS microtitre plates, whilst maintaining the structural integrity and optical clarity of the wells [47]. The method relies on an initial Friedel-Crafts alkylation of the plate with bromomethyl methyl ether and tin(IV) bromide in tetramethylene sulfone (sulfolane). The resulting bromomethylated PS, which is analogous to the chloromethylated Merrifield resin used for solid phase peptide synthesis [36], provides access to a wide range of functionality through subsequent nucleophilic displacement reactions (Fig. 5). Sulfolane is a polar aprotic solvent that is chemically and thermally stable, miscible with water and organic solvents, and does not damage PS. Therefore, it is an ideal solvent for performing many different reactions on the surface of PS plates, although its relatively high melting point (28 °C) introduces some difficulties in handling.
Nahar et al. used a photoactivated azide derivative to immobilise proteins [48] and small molecules [49] on the surface of PS microtitre plate wells. A methanolic solution of fluoronitroazidobenzene (FNAB) [50] was allowed to evaporate in the wells of a PS microtitre plate. The plate was then exposed to UV light (365 nm), converting the azide into a highly reactive nitrene, which is capable of grafting to the PS backbone. The resulting fluoronitrobenzene adduct could then undergo nucleophilic aromatic substitution with an amine-containing molecule of choice (Fig. 6). Similarly, Ito and co-workers photografted an azide onto the surface of PS plates [51]. Initially, the plates were treated with 4-azidobenzoic acid in the presence of UV light, and the resulting benzoic acid adduct was converted to the NHS ester for subsequent coupling with insulin.
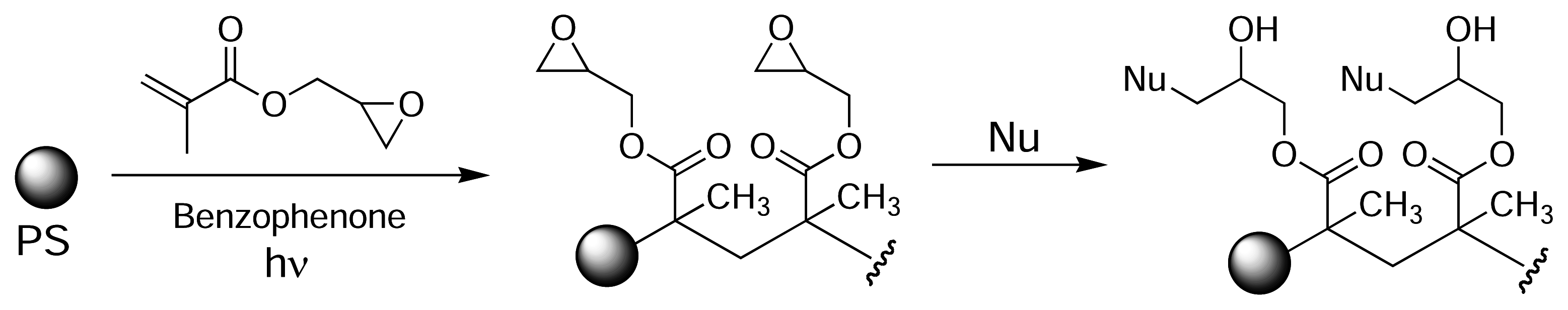
Eckert and colleagues used photografting to introduce epoxide groups onto the surface of PS [52]. Initially, a methanolic solution of glycidylmethacrylate was added to the wells of a PS microtitre plate and the plate was exposed to UV light, using benzophenone as a photoinitiator. This resulted in poly(glycidylmethacrylate) being grafted to the plate surface (Fig. 7). The immobilised epoxide groups could then react with a nucleophile of choice (amine, thiol, alcohol). Similarly, Larsson et al. introduced carboxyl groups on the surface of PS plates by photografting crotonic acid using radiation from a 60Co source [53, 54]. The carboxyl-modified polymer surface was used to immobilise various human antibodies via EDC-mediated amide bond formation.
Results and Discussion
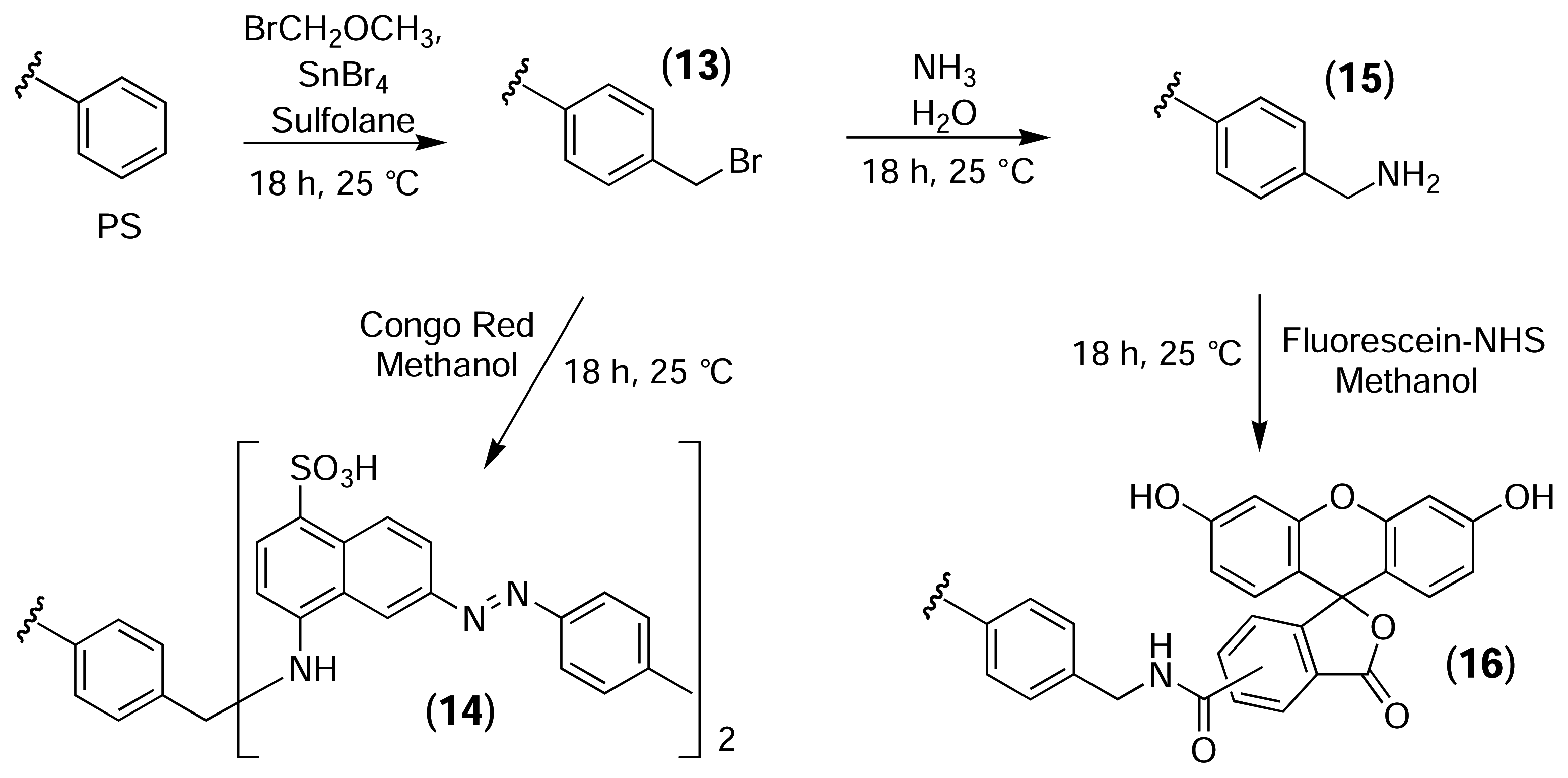
Cross-linked PS resin was used as a model system to evaluate the protocols described by Clark [47]. The high loading capacity of the resin (>1 mmol/g) allowed the use of fluorescent probes to follow the reactions visually (Fig. 8). Initially, PS resin was bromomethylated using the same conditions reported by Clark for PS microtitre plates. One portion of the activated resin (13) was treated with congo red (λem = 600 nm) to yield (14), while a second portion of activated resin was first converted to the aminomethylated derivative (15) and was then reacted with fluorescein-NHS (λem = 520 nm) to yield (16). Portions of the underivatised PS resin were also treated with the dyes as controls.
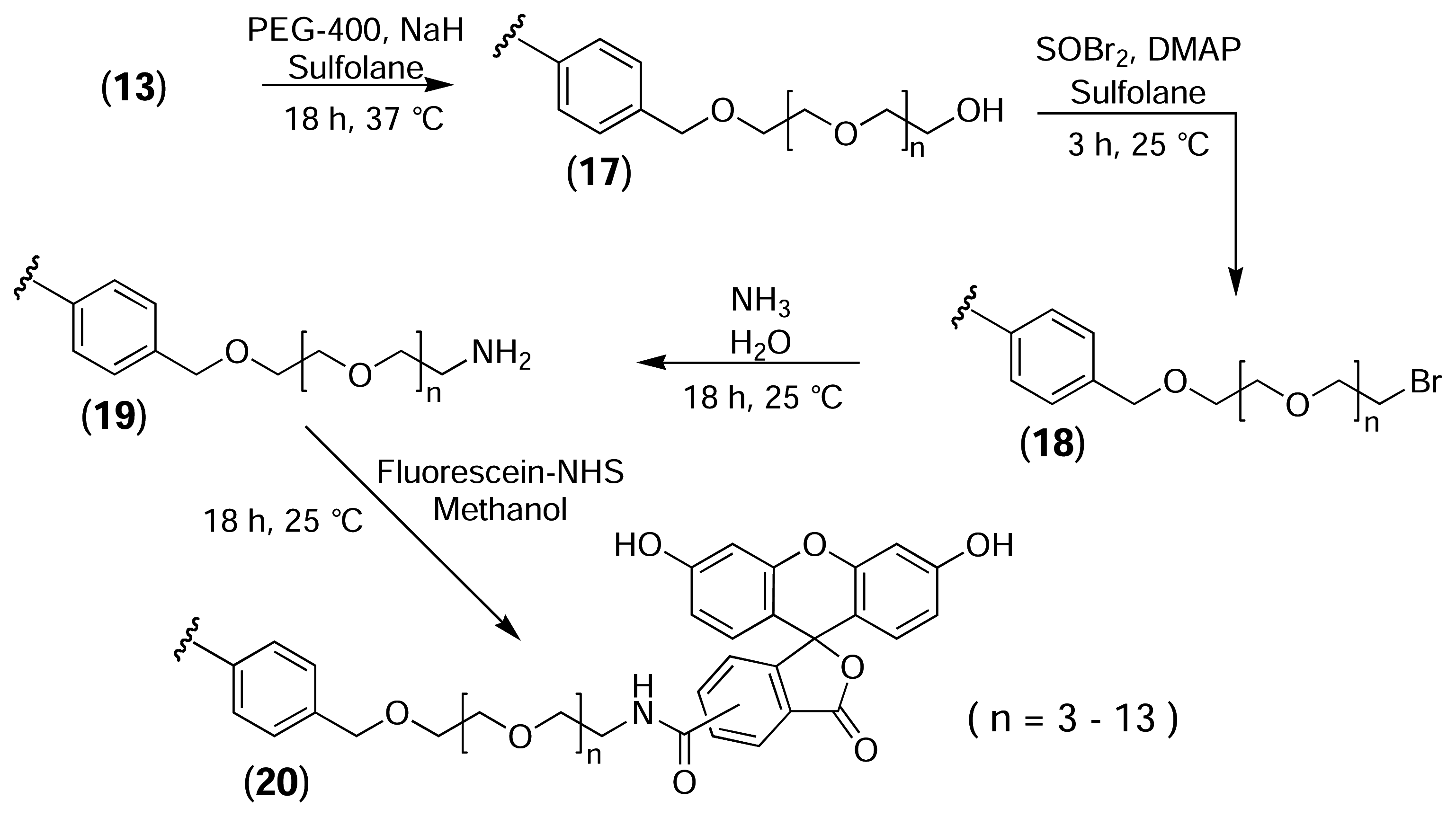
While the direct attachment of fluorescent probes to the surface of the PS resins is adequate for determining whether the desired reactions are occurring, and for optimising reaction conditions, a functional affinity support requires the incorporation of a long linker between the solid phase and the probe molecule to provide sufficient room for macromolecules to bind. Therefore, to mimic the construction of an affinity support, a poly(ethylene glycol) (PEG) linker with an average molecular weight of 400 was incorporated between the PS resin and the fluorescent probe (Fig. 10) PEG-400 was treated with sodium hydride to generate the alkoxide ion and was then added to bromomethylated PS resin, resulting in the immobilisation of PEG via a stable ether linkage (17). A large molar excess of PEG was used to minimise the formation of cross-links between adjacent activated sites on the resin. The terminal hydroxyl group was then brominated (18), converted to the amine (19), and derivatised with fluorescein-NHS (20).
Following derivatisation, the resins were washed extensively with methanol to remove any non-covalently adsorbed fluorophores. This rigorous washing step is particularly important for molecules containing aromatic rings (such as congo red and fluorescein), which have a high affinity for polystyrene due to π-π interactions. In fact, the non-covalent adsorption of proteins and other hydrophobic molecules onto the surface of PS microtitre plate wells is an immobilisation strategy commonly used in many biochemical assays [55]. After extensive washing, the congo red-derivatised resin appeared pale red to the naked eye, the two fluorescein-derivatised resins appeared pale yellow and the two control resins appeared light brown. A small quantity of each derivatised PS resin was suspended in water and examined under an epifluorescence microscope to determine whether the reactions had been successful. A micrograph of each sample was taken using identical exposure settings, thereby allowing a direct comparison of fluorescence intensity between the different resins.
The three derivatised resins (Fig. 11(c,e,f)) showed a high level of fluorescence, while neither of the control resins (Fig. 11(b,d)) showed any detectable fluorescence. With the Clark protocol validated, the derivatisation reactions were repeated on the surface of PS microtitre plate wells.
PS microtitre plates were next derivatised with palau’amine via a cleavable PEG linker (Fig. 12). Palau’amine is a bisguanidine alkaloid, first isolated from the marine sponge Stylotella agminata by Kinnel, Gehrken and Scheuer [56]. Although its acute toxicity is relatively low, palau’amine exhibits powerful antibacterial, antifungal, antitumour and immunosuppressive activities [57]. The complex hexacyclic ring system, dense stereochemistry, high degree of functionalisation and inherent instability of the molecule makes palau’amine a challenging synthetic target [58, 59, 60, 61], and a successful total synthesis has yet to be reported. As is frequently the case with biologically active natural products, the cellular receptors for palau’amine are not known, so palau’amine is an interesting candidate for investigation by reverse chemical proteomics.
Palau’amine was immobilised on a bromomethylated PS microtitre plate via a cleavable disulfide linker and the resulting affinity support was used to probe five different T7 phage-displayed human cDNA libraries. Following each round of selection, phages retained by the affinity support were released by reductive cleavage of the disulfide linker with DTT. After five rounds of selection had been performed, 16 individual plaques were picked from each sublibrary and their DNA inserts were amplified by PCR using generic T7 primers (Fig. 13). In addition, a subset of the amplified DNA inserts from each sublibrary was sequenced (Table 1).
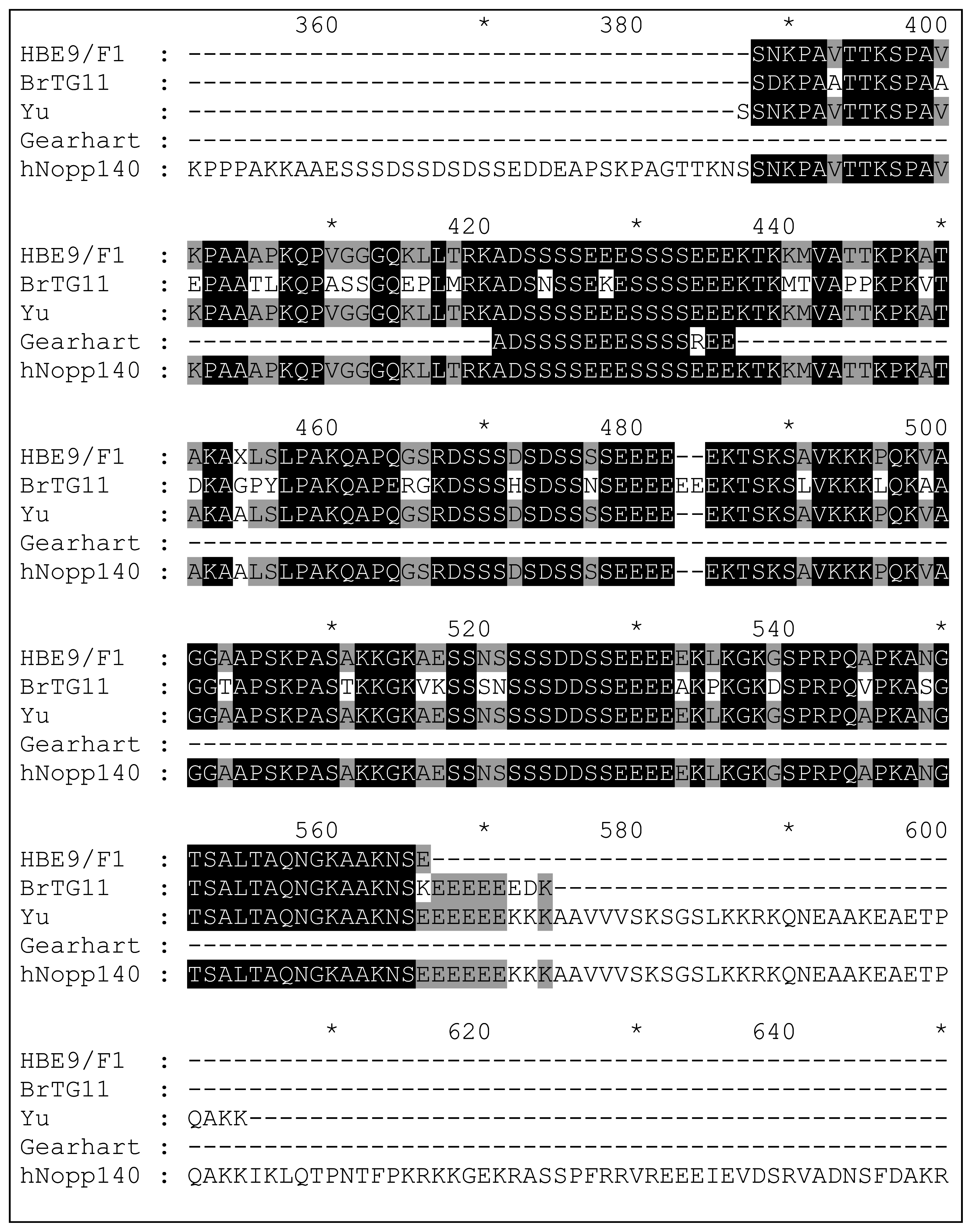
The DNA gels of the PCR products obtained from randomly selected plaques showed a range of different sized DNA inserts in all five libraries, indicating the selections had not reached a consensus after five rounds. Analysis of the DNA sequences obtained revealed the majority of clones contained DNA inserts that were either in backwards (25%), not in the correct reading frame (20%), not in the coding region of the encoded gene (10%) or expressed only short peptides (32%). However, two clones rescued from the T7Select10-3 human brain library (E9/F1) contained the C-terminal domain of nucleolar and coiled-body phosphoprotein 1 (hNopp140), and one clone rescued from the breast tumour library (G11) contained the C-terminal domain of the almost identical nucleolar phosphoprotein p130 (Fig. 14), both of which are involved in biogenesis of the nucleolus [62].
The primary sequence of hNopp140 consists of long runs of serine residues and a high proportion of charged amino acids, with 82 potential phosphorylation sites on the protein [65]. The level of phosphorylation changes throughout the cell cycle in vivo [66, 67], although no phosphorylation of T7 phage-displayed hNopp140 would occur as E. coli is not capable of performing post-translational modifications. The protein has also been shown to possess GTPase/ATPase activity and was able to form large complexes in the presence of fluoride and magnesium ions [68]. It is hypothesised that hNopp140 acts as a chaperone, shuttling proteins between the nucleolus and the cytoplasm [69].
Interestingly, hNopp140 has also been isolated by two other groups using T7 phage display (Fig. 14). Yu et al. isolated the same C-terminal domain of hNopp140 from a T7-displayed human liver cDNA library using a biotinylated doxorubicin affinity probe [63]. The authors over-expressed this protein fragment in E. coli and used fluorescence spectroscopy and surface plasmon resonance to show that doxorubicin only interacts with the C-terminal domain of hNopp140 when it is not phosphorylated. Gearhart and colleagues also rescued a small (16 aa) fragment of hNopp140, along with fragments of five other highly acidic proteins, using 2-methylnorharman adsorbed non-covalently on nitrocellulose discs [64]. Palau’amine, doxorubicin and 2-methylnorharman have markedly different biological properties and hence it is unlikely that hNopp140 is a receptor for all three natural products. Given its long runs of serine residues and high percentage of charged amino acids (Table 2), it is more likely that hNopp140 is a promiscuous non-specific binder, which can become the dominant member of a phage display library very quickly in the absence of a competing strong protein-ligand interaction. Therefore, the appearance of hNopp140 in a phage display experiment is probably indicative of an unsuccessful selection.
Two clones (Liver-F3 and Breast-G1) were found to contain an ATP synthase gene. These clones were isolated in a previous experiment [70] and are clearly the result of cross-contamination, highlighting one of the major pitfalls faced when performing reverse chemical proteomics.
There are many possible reasons why the derivatised PS plates failed to identify a cellular receptor for palau’amine. Firstly, the amino group through which the molecule was immobilised may be essential for biological activity or may have orientated the molecule in such a way that the cellular receptor could not bind. In addition, the cellular receptor for palau’amine may be a multimeric protein, a transmembrane protein or some other class of protein that is intractable to display cloning. Finally, palau’amine is relatively unstable, particularly above pH 6.5, so components of the phage lysate may have caused the compound to decompose during the selection procedure.
Experimental
General
Polystyrene 96-well microtitre plates were obtained from Disposable Plastics (Australia). Adhesive plate covers were obtained from Eppendorf (Germany). Teflon 96-well microtitre plate mats were obtained from Radleys (UK). DCC, Grubbs’ catalyst and 3,3′-dithiodipropionic acid was obtained from Fluka (Switzerland). Acetonitrile, chromium trioxide, glacial acetic acid, hydrochloric acid, potassium carbonate, potassium permanganate, sodium borate and sulfuric acid were obtained from Merck/BDH (Germany). Methanol was obtained from Fronine (Australia). Fluorescein-NHS was obtained from Pierce (USA). Congo red was obtained from George T. Gurr (London). FK506 was donated by Vertex Pharmaceuticals (USA). All other reagents were obtained from Aldrich (USA). Sulfolane and PEG-400 were dried over 4 Å molecular sieves. Methanol was distilled from anhydrous potassium carbonate before use. All other reagents were used without purification unless otherwise indicated.
Fluorescence was measured in black 96-well microtitre plates on a Fluostar Galaxy plate reader (BMG Labtechnologies, Germany) using a 485 nm excitation filter and a 520 nm emission filter. Epifluorescence micrographs were obtained on Kodak Elite Chrome 400 slide film using an Axioskop 2 epifluorescence microscope with a 100 W mercury vapour arc lamp, 450–490 nm bandpass excitation filter and 515 nm low pass emission filter (Carl Zeiss, Germany). Ultraviolet-visible spectra were recorded on a Cary 1-Bio spectrophotometer (Varian, USA) in 1 cm matched quartz cuvettes (Selbys Scientific, Australia). NMR spectra were recorded in 5 mm Pyrex tubes (Wilmad, USA) on a DPX-400 400 MHz spectrometer (Bruker, Germany). Scanning electron micrographs were obtained on a JSM840 scanning electron microscope (JEOL, Japan). Low magnification light microscopy was performed using a stereoscopic dissecting microscope (Carl Zeiss, Germany). Water was purified using a Milli-Q Ultrapure Water Purification System (Millipore, USA).
Sodium chloride, potassium dihydrogen phosphate, PBS, PWB (PBS + 0.05% Tween-20), IPTG, ethidium bromide and DNA molecular weight markers were obtained from Sigma-Aldrich (USA). Tryptone, yeast extract, agar and polystyrene Petri dishes were obtained from Bacto Laboratories (Australia). Glucose, agarose, carbenicillin sodium and Tris were obtained from AMRESCO (USA). Acetic acid, glycerol, ammonium chloride, disodium hydrogen phosphate and EDTA disodium salt were obtained from BDH (Germany). T7Select10-3 human disease cDNA libraries and E. coli strain BLT5615 were obtained from Novagen Inc. (USA). T7Select1-1b human brain cDNA library was a kind gift from Prof. David Austin (Department of Chemistry, Yale University). Nucleotides (dNTPs) were obtained from Bioline (UK). Oligonucleotides (primers) were obtained from Sigma-Genosys (Australia). Taq DNA polymerase and QIAquick PCR purification kits were obtained from QIAGEN (USA). HinfI restriction endonuclease and NEB buffer 2 were obtained from New England Biolabs Inc. (USA). Electrophoresis grade agarose was obtained from American Bioanalytical (USA). Nuclease-free water, 1 M magnesium chloride and 20% SDS were obtained from Ambion (USA). Reacti-bind HBC Neutravidin 8-well strip plates were obtained from Pierce (USA). Poly-Prep disposable plastic chromatography columns were obtained from Bio-Rad (USA). Disposable plastic syringes were obtained from Terumo (Japan). Polystyrene 96-well microtitre plates and flexible poly(vinyl chloride) 96-well assay plates were obtained from Corning (USA).
Bacterial cultures were incubated in a heated orbital shaker (Thermoline Scientific, Australia). Optical densities were recorded in 1 cm polystyrene semi-micro cuvettes (Sarstedt, Germany) using a CO65 analogue colorimeter containing a 600 nm filter (Walden Precision Apparatus, UK). Solutions were centrifuged with a 6K15 refrigerated centrifuge (Sigma, Germany). DNA was amplified with a GeneAmp PCR System 2400 Thermocycler (Perkin Elmer, USA). DNA sequencing was performed by the Macquarie University DNA Analysis Facility using an ABI Prism 377 Sequencer (Applied Biosystems, USA). Agarose gel electrophoresis was performed using a Mini-Sub Cell GT system (Bio-Rad, USA) and gels were visualised with a ChemiImager digital imaging system (Alpha Innotech, USA). Water was purified using a Milli-Q Ultrapure Water Purification System (Millipore, USA). Oligonucleotide primers were designed using the Primer3 software package. Linear and non-linear least squares regression analysis was performed using Origin 6.0 (Microcal, USA).
Derivatisation of Polystyrene (PS) Resins
PS-Bromomethyled Resin (13)
Adapting the procedure by Clark [47], cross linked (2%) PS resin (5.0 g) was added to a solution of tin(IV) bromide (0.60 mL, 4.6 mmol) and bromomethyl methyl ether (0.40 mL, 4.9 mmol) in warm (37 °C) anhydrous sulfolane (20 mL) under argon and the resulting suspension was stirred vigorously for 18 h at room temperature under argon. The resin was then collected at the pump, washed with anhydrous acetonitrile (5 × 10 mL), dried under high vacuum and stored in a desiccator until required.
PS-Congo Red Resin (14)
Bromomethylated PS resin (13) (10 mg) was added to a solution of congo red (0.7 mg, 0.1 μmol) in methanol (1 mL) and the resulting suspension was stirred vigorously for 18 h at room temperature. The resin was then collected at the pump, washed extensively with methanol (20 × 1 mL, allowing the resin to soak for 1 min between washes), dried under high vacuum and stored in a desiccator until required.
PS-NH2 Resin (15)
Bromomethylated PS resin (13) (200 mg) was added to ammonium hydroxide (28%; 1 mL) and the resulting suspension was stirred vigorously for 18 h at room temperature. The resin was then collected at the pump, washed with water (3 × 2 mL) and methanol (3 × 2 mL), dried under high vacuum and stored in a desiccator until required.
PS-Fluorescein Resin (16)
PS-NH2 resin (15) (10 mg) was added to a solution of fluorescein-NHS (0.5 mg, 0.1 μmol) in anhydrous methanol (1 mL) and the resulting suspension was stirred vigorously for 18 h at room temperature. The resin was then collected at the pump, washed extensively with methanol (20 × 1 mL, allowing the resin to soak for 1 min between washes), dried under high vacuum and stored in a desiccator until required.
PS-PEG-OH Resin (17)
Sodium hydride 50% dispersion in mineral oil (120 mg, 2.5 mmol) was washed with light petroleum (3 × 5 mL) and added to a solution of PEG-400 (1.0 g, 2.5 mmol) in warm (37 °C) anhydrous sulfolane (5 mL) to generate an excess of the monoanion. An atmosphere of argon was maintained over the solution until the evolution of gas ceased. Bromomethylated PS resin (13) (1.0 g) was then added and the resulting suspension was stirred vigorously for 18 h at 37 °C. Finally, the resin was collected at the pump, washed with methanol (5 × 5 mL), dried under high vacuum and stored in a desiccator until required.
PS-PEG-Br Resin (18)
PS-PEG-OH resin (17) (200 mg) was added to a solution of thionyl bromide (100 mg, 0.50 mmol) and DMAP (1.0 mg, cat.) in warm (37 °C) anhydrous sulfolane (1 mL) and the resulting suspension was stirred vigorously for 3 h at room temperature under argon. The resin was then collected at the pump, washed with anhydrous acetonitrile (5 × 2 mL), dried under high vacuum and stored in a desiccator until required.
PS-PEG-NH2 Resin (19)
PS-PEG-Br resin (18) (200 mg) was derivatised with ammonium hydroxide as described for compound 64.
PS-Bromomethylated Plate (13)
PS plates were bromomethylated using a modified version of the protocol described by Clark [47]. Briefly, a solution of tin(IV) bromide (6 mL, 46 mmol) and bromomethyl methyl ether (4 mL, 49 mmol) in warm (37 °C) anhydrous sulfolane (40 mL) was prepared and an aliquot (200 μL) of the resulting straw-coloured solution was added to each well of a 96-well PS microtitre plate under argon. The plate was sealed with an adhesive cover and allowed to react for 18 h at room temperature inside a desiccator filled with argon. The reaction mixture was then aspirated from the wells and each well was rinsed with anhydrous acetonitrile (3 × 200 μL). Finally, the plate was dried under high vacuum and stored in a desiccator until required.
PS-PEG-OH Plate (17)
Sodium hydride 50% dispersion in mineral oil (48 mg, 1.0 mmol) was washed with light petroleum (3 × 1 mL) and added to a solution of PEG-400 (400 mg, 1 mmol) in warm (37 °C) anhydrous sulfolane (50 mL). An atmosphere of argon was maintained over the solution until the evolution of gas ceased. An aliquot of the resulting solution (200 μL) was added to each well of a bromomethylated PS microtitre plate (13) under argon. The plate was sealed with an adhesive cover and left to react for 18 h at 37 °C. The reaction mixture was then aspirated from the wells and each well was rinsed with methanol (3 × 200 μL). Finally, the plate was dried under high vacuum and stored in a desiccator until required.
PS-PEG-Br Plate (18)
Thionyl bromide (210 mg, 1.0 mmol) and DMAP (1.0 mg, cat.) were dissolved in warm (37 °C) anhydrous sulfolane (50 mL) and an aliquot (200 μL) of the resulting solution was added to each well of a PS-PEG-OH microtitre plate (17) under argon. The plate was sealed with an adhesive cover and left to react for 3 h at room temperature. The reaction mixture was then aspirated from the wells and each well was rinsed with anhydrous acetonitrile (3 × 200 μL). Finally, the plate was dried under high vacuum and stored in a desiccator until required.
PS-NH2 Plate (15)
Ammonium hydroxide (28%; 200 μL) was added to each well of a bromomethylated PS microtitre plate (13). The plate was sealed with polyethylene “cling wrap” and left to react for 18 h at room temperature. The ammonia solution was then aspirated from the wells and each well was rinsed with water (3 × 200 μL). Finally, the plate was dried under high vacuum and stored in a desiccator until required.
PS-PEG-NH2 Plate (19)
A PS-PEG-Br microtitre plate (18) was derivatised with ammonium hydroxide as described for 15.
PS-PEG-S-S-OSu Plate (21)
Lomant’s reagent (3-mercaptopropionic acid disulfide) (400 mg, 1.0 mmol) and DMAP (1.0 mg, cat.) were dissolved in warm (37 °C) anhydrous sulfolane (50 mL) and an aliquot (200 μL) of the resulting solution was added to each well of a PS-NH2 microtitre plate (19) under argon. The plate was sealed with an adhesive cover and left to react for 18 h at 30 °C. The reaction mixture was then aspirated from the wells and each well was rinsed with anhydrous acetonitrile (3 × 200 μL). Finally, the plate was dried under high vacuum and stored in a desiccator until required.
PS-PEG-S-S-Palau’amine Plate (22)
Palau’amine (1.0 mg, 2.4 μmol) was dissolved in anhydrous methanol (50 mL) and an aliquot (200 μL) of the resulting solution was added to each well of a PS-PEG-OSu microtitre plate (21) under argon. The plate was sealed with an adhesive cover and allowed to react for 18 h at room temperature. The reaction mixture was then aspirated from the wells and each well was rinsed with methanol (3 × 200 μL). Finally, the plate was dried under high vacuum and desiccated at 4 °C until required.
Phage Display
The display cloning protocols presented below were adapted from those described in the Novagen T7Select System Handbook [71] as well as from those used by Prof. David Austin (Department of Chemistry, Yale University, http://ursula.chem.yale.edu/~austinlab/display_cloning.html).
Preparation of Bacterial Cultures
A stock of E. coli strain BLT5615 was stored at −80 °C in 10% glycerol. An initial culture was prepared by streaking a small quantity of this frozen stock onto an LB agar plate and incubating the plate at 33 °C for 16 h. The plate was stored at 4 °C and used to inoculate subsequent cultures for up to one month.
A saturated overnight culture of BLT5615 was prepared by inoculating M9TB (20 mL) with a single bacterial colony from an LB agar plate and then incubating at 33 °C for 16 h with gentle swirling. A fresh culture of BLT5615 ready for infection by T7 bacteriophage was prepared by inoculating M9TB (100 mL) with saturated overnight culture (5 mL) and incubating at 37 °C with vigorous shaking until an OD600 of 0.4 was reached (2–3 h). IPTG (24%; 100 μL) was added and incubation continued for a further 30 min. The culture was then stored on slushy ice (for up to 24 h) until required.
Growth of T7 Lysates
IPTG-treated BLT5615 cells (100 mL) were infected with a T7Select cDNA library (1 μL) and incubated at 37 °C with vigorous shaking until lysis had occurred (1–2 h), as indicated by a marked decrease in OD600. Immediately following lysis, the lysate was centrifuged at 4700 rpm for 10 min at 4 °C to precipitate cellular debris and the supernatant was decanted into a clean tube containing Tween-20 (1%; 1 mL). The clarified lysate containing 0.01% Tween-20 was stored on slushy ice until required.
Affinity Selections
All microtitre plate wells were pre-incubated with PBS (100 μL) for 2 h at room temperature before use. Clarified T7 phage lysate (100 μL) was added to one well of a bromomethylated PS plate that had been derivatised with a control compound, and was left to incubate for 4 h at room temperature. The lysate was then transferred to a second well of the plate that had been derivatised with the target molecule via a cleavable disulfide linker, and was left to incubate overnight at 4 °C. The next morning, the well was washed with ice-cold PWB (3 × 250 μL) and then incubated with DTT (1 mM; 100 μL) for 2 h at 37 °C. Finally, an aliquot (10 μL) of the DTT eluate was added to fresh IPTG-treated E. coli BLT5615 cells (10 mL) for the next round of selection. This procedure was repeated until five rounds of selection had been completed. The stringency of the washing step was increased with each successive round of selection, from 3 × 250 μL PWB over 1 min in Round 1 to 20 × 250 μL PWB over 5 min in Round 5.
Picking Plaques
Amplified phage lysate from the final round of selection was serially diluted with 2xYT from 10−1 to 10−7 in a flexible 96-well assay plate. A top agarose plate containing an aliquot of the 10−7 phage dilution (50 μL) was incubated at 37 °C until plaques were clearly visible against the lawn of bacteria (3–4 h). Individual plaques (96) were collected by stabbing the centre of each plaque with a 10 μL micropipette tip and transferring the tip to IPTG-treated BLT5615 cells (100 μL per well) in a 96-well microtitre plate. After 96 plaques had been picked, the microtitre plate was incubated at 37 °C until complete lysis of the bacterial cells in each well was observed (1–2 h). The plate was then centrifuged at 4300 rpm for 10 min at 4 °C, and the supernatant from each well (40 μL) was transferred into a clean 96-well microtitre plate containing 80% glycerol (10 μL per well) and stored at −80 °C until required.
Amplification, Sequencing and Fingerprinting of cDNA Inserts
Phage lysate (1 μL) was added to PCR master mix (49 μL) and the resulting solution was subjected to 35 rounds of thermocycling using the protocol shown in Table 3. An aliquot of the amplified DNA solution (20 μL) was then incubated with DNA fingerprinting mix (30 μL) at 37 °C for 2 h.
Gel Electrophoresis
Electrophoresis-grade agarose (0.6 g) was suspended in 1× TAE (40 mL) and the suspension was boiled in a microwave oven until the agarose had dissolved completely. The 1.5% agarose solution was cooled to 50 °C and ethidium bromide (5 mg/mL; 4 μL) was added. The solution was mixed thoroughly, poured into a casting tray (10 × 7 cm) containing two 15 well combs, and allowed to set for 1 h at room temperature. The solidified gel was then transferred to a gel tank, flooded with 1× TAE, and the combs removed. Each amplified cDNA insert or digested fingerprinting sample (5 μL) was diluted with nuclease free water (5 μL), mixed with 6x DNA loading buffer (2 μL) and an aliquot (5 μL) loaded onto the gel with a micropipette. After all samples had been loaded, the gel was run at 80 V until the bromophenol blue dye had migrated approximately half way down each half of the gel (25–35 min). The gel was then removed from the tank and visualised using a UV transilluminator. A photograph of the gel was taken with a CCD video camera attached to the transilluminator.
DNA Sequencing
An aliquot of PCR-amplified DNA (15 μL) was purified using a QIAquick PCR purification kit following the manufacturer’s instructions. The purified DNA was then centrifuged at 10,000 rpm for 10 min at 4 °C to precipitate any resin that had carried through from the QIAquick column. Finally, an aliquot of the purified DNA (8 μL) was combined with one PCR primer (1 μM; 4 μL, 4 pmol) and the resulting solution was submitted for DNA sequencing.
Figure 1.
Schematic representation of chemical proteomics and reverse chemical proteomics. In chemical proteomics (A), one uses a small molecule (e.g. marine natural product) to construct an affinity or activity probe to isolate specific proteins or a family of proteins. In reverse chemical proteomics (B), we start with the transcriptome, which is cloned into an amplifiable vector (e.g. a virus) that expresses a single protein from the proteome on its surface. A tagged natural product can be used to probe the tagged proteome in an iterative manner.
Figure 1.
Schematic representation of chemical proteomics and reverse chemical proteomics. In chemical proteomics (A), one uses a small molecule (e.g. marine natural product) to construct an affinity or activity probe to isolate specific proteins or a family of proteins. In reverse chemical proteomics (B), we start with the transcriptome, which is cloned into an amplifiable vector (e.g. a virus) that expresses a single protein from the proteome on its surface. A tagged natural product can be used to probe the tagged proteome in an iterative manner.
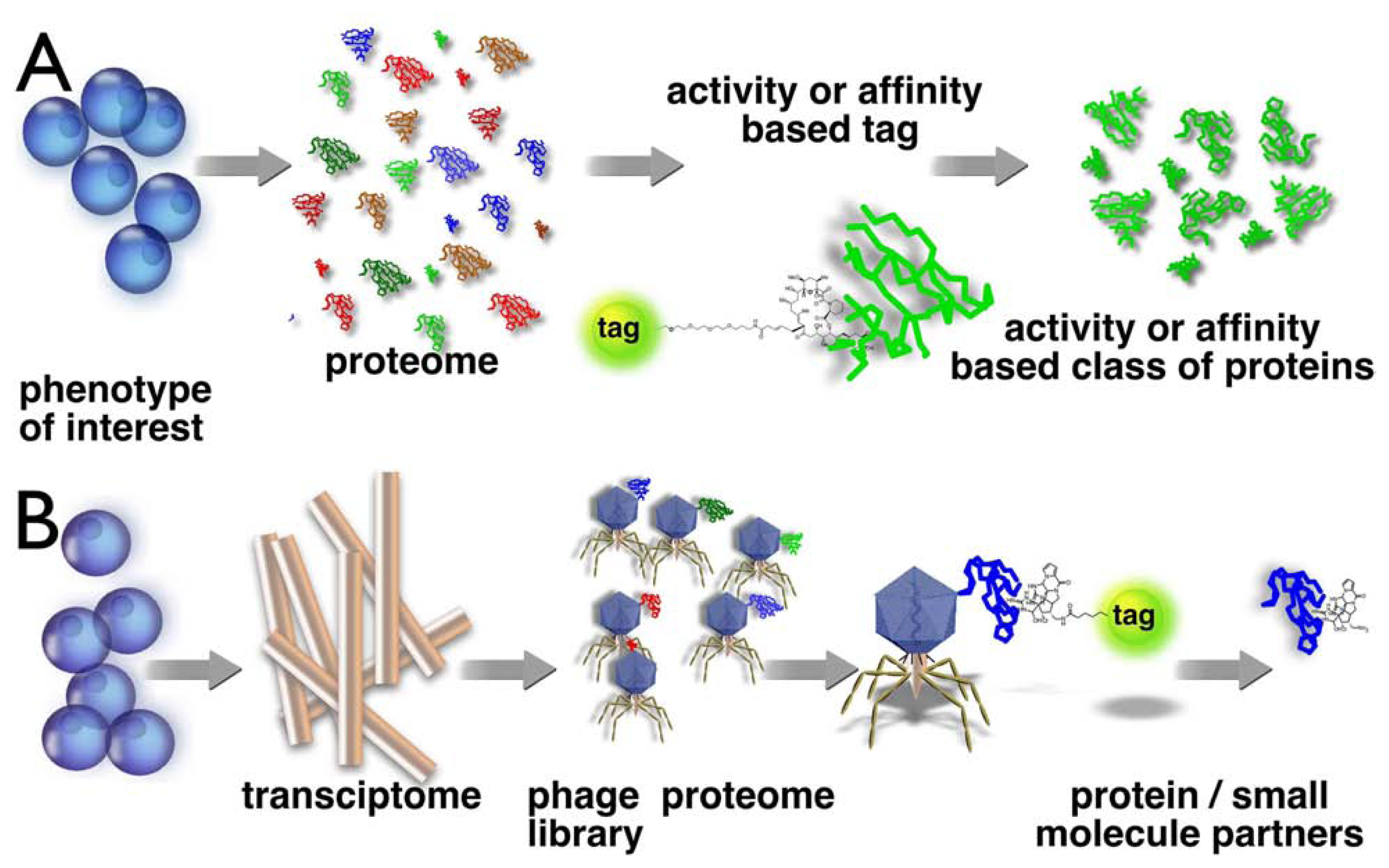
Figure 2.
Chemical proteomics probes [5] are composed of a tag (or anchor such as polystyrene), a linker (spacer) and probe or reactive group. The probe can be a drug, natural product, peptide, reactive group or anything and can react covalently or non-covalently with its biological receptor, which can have more or less specificity for the probe or react with a specific type of enzyme.
Figure 2.
Chemical proteomics probes [5] are composed of a tag (or anchor such as polystyrene), a linker (spacer) and probe or reactive group. The probe can be a drug, natural product, peptide, reactive group or anything and can react covalently or non-covalently with its biological receptor, which can have more or less specificity for the probe or react with a specific type of enzyme.
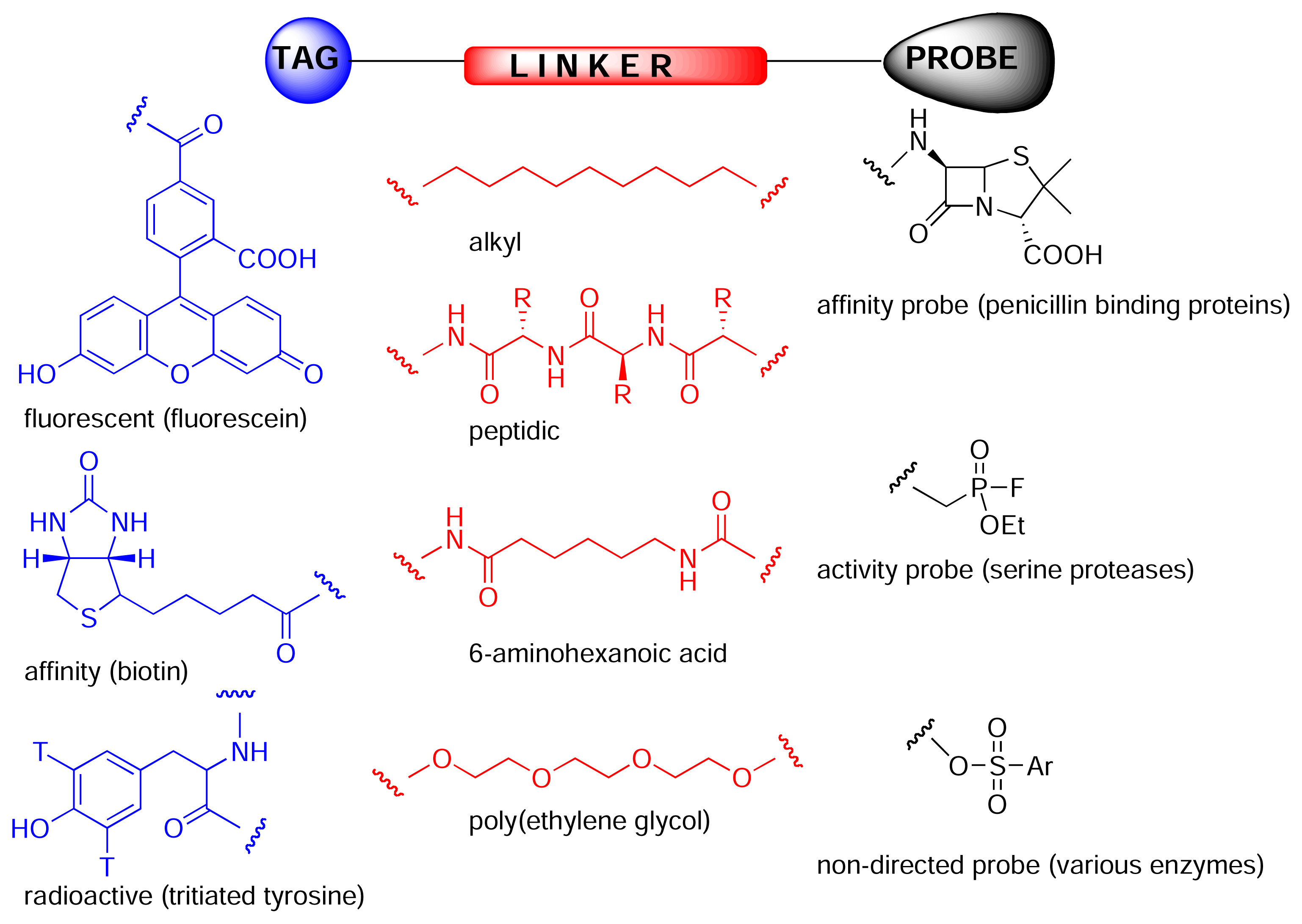
Figure 3.
Nitration of PS microtitre plate wells with fuming nitric acid, followed by reduction with sodium dithionite, yields aromatic amine groups on the polymer surface [44].
Figure 3.
Nitration of PS microtitre plate wells with fuming nitric acid, followed by reduction with sodium dithionite, yields aromatic amine groups on the polymer surface [44].

Figure 4.
Oxidation of the backbone of PS yields carboxyl groups on the polymer surface [46].
Figure 4.
Oxidation of the backbone of PS yields carboxyl groups on the polymer surface [46].
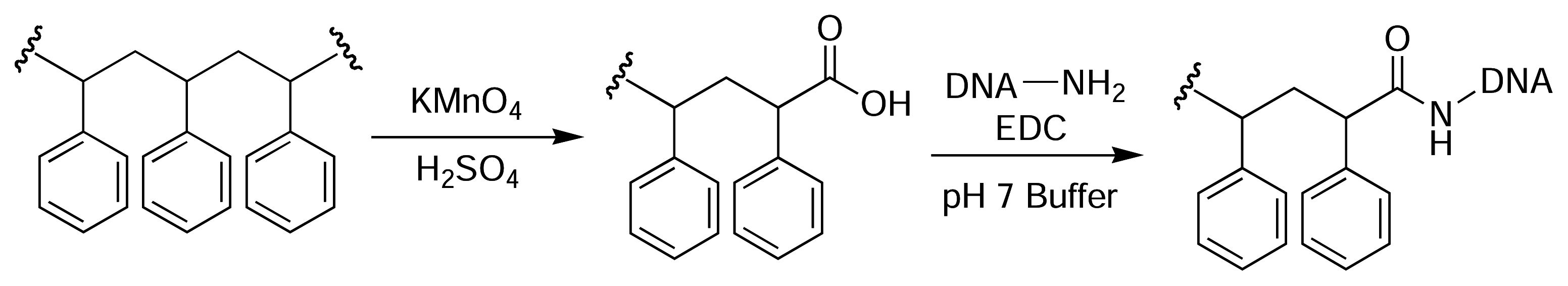
Figure 5.
Bromomethylation of PS microtitre plates via a Friedel-Crafts alkylation with bromomethyl methyl ether and tin(IV) bromide in sulfolane provides access to a wide range of different functionalisation, whilst maintaining the structural integrity and optical clarity of the plate [47].
Figure 5.
Bromomethylation of PS microtitre plates via a Friedel-Crafts alkylation with bromomethyl methyl ether and tin(IV) bromide in sulfolane provides access to a wide range of different functionalisation, whilst maintaining the structural integrity and optical clarity of the plate [47].
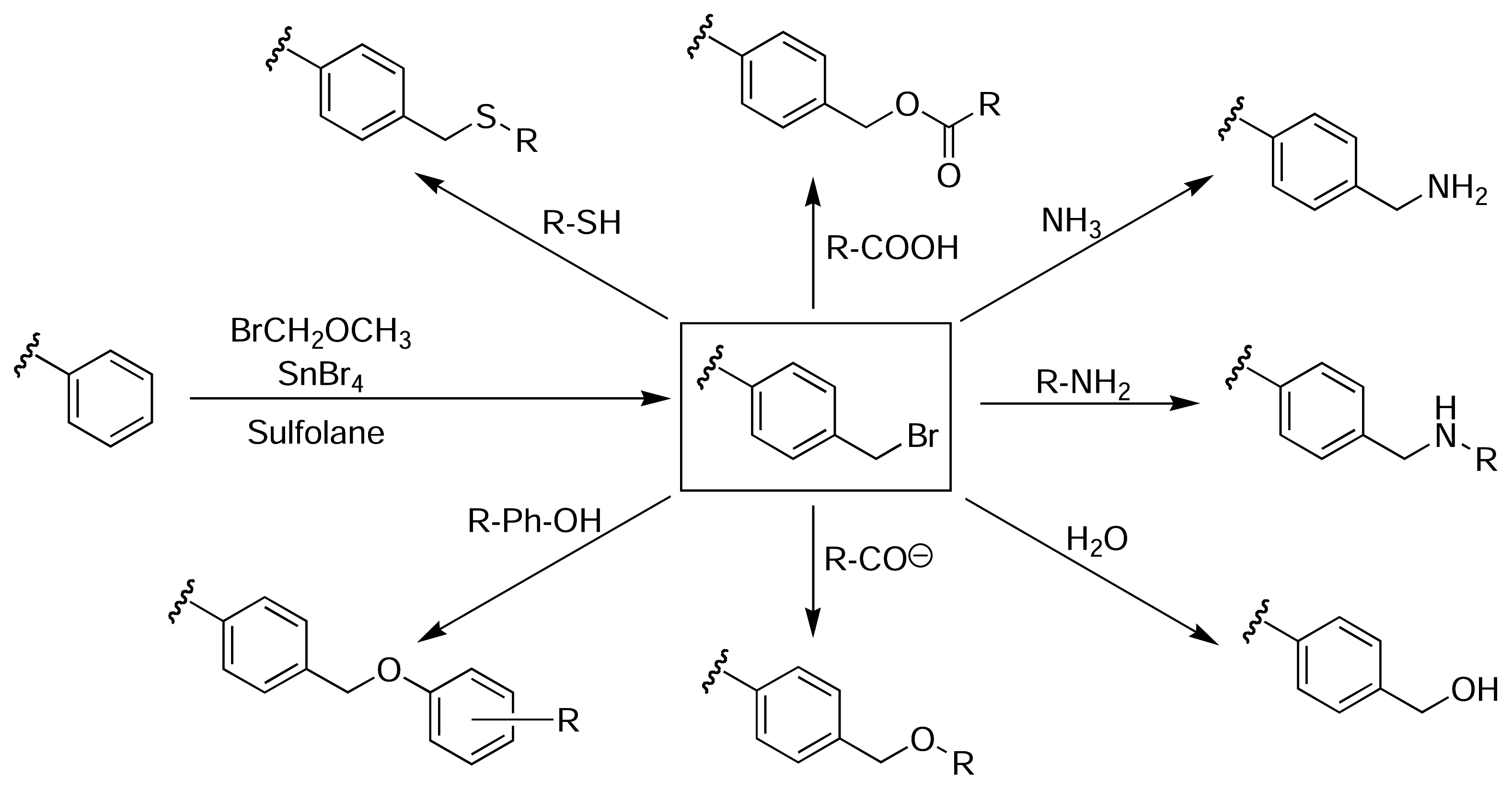
Figure 6.
Derivatisation of PS using UV-photoactivated FNAB for the immobilisation of amine-containing molecules [48].
Figure 6.
Derivatisation of PS using UV-photoactivated FNAB for the immobilisation of amine-containing molecules [48].

Figure 7.
Photografting of glycidylmethacrylate onto PS microtitre plates using UV light in the presence of a benzophenone photoinitiator [52].
Figure 7.
Photografting of glycidylmethacrylate onto PS microtitre plates using UV light in the presence of a benzophenone photoinitiator [52].

Figure 8.
Bromomethylation of PS and subsequent derivatisation with fluorescent probes.
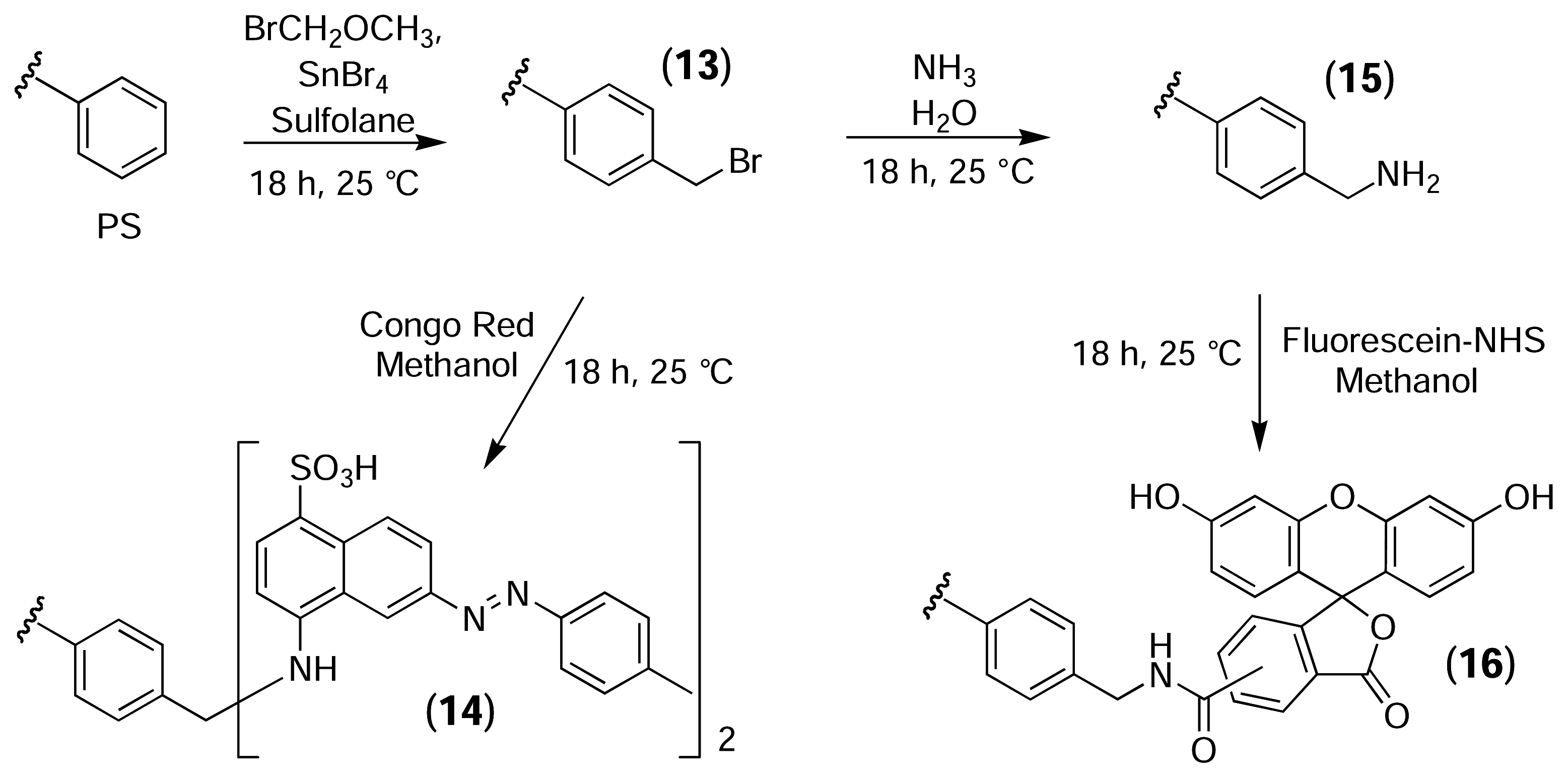
Figure 10.
Derivatisation of bromomethylated PS with fluorescein via a non-cleavable PEG linker.
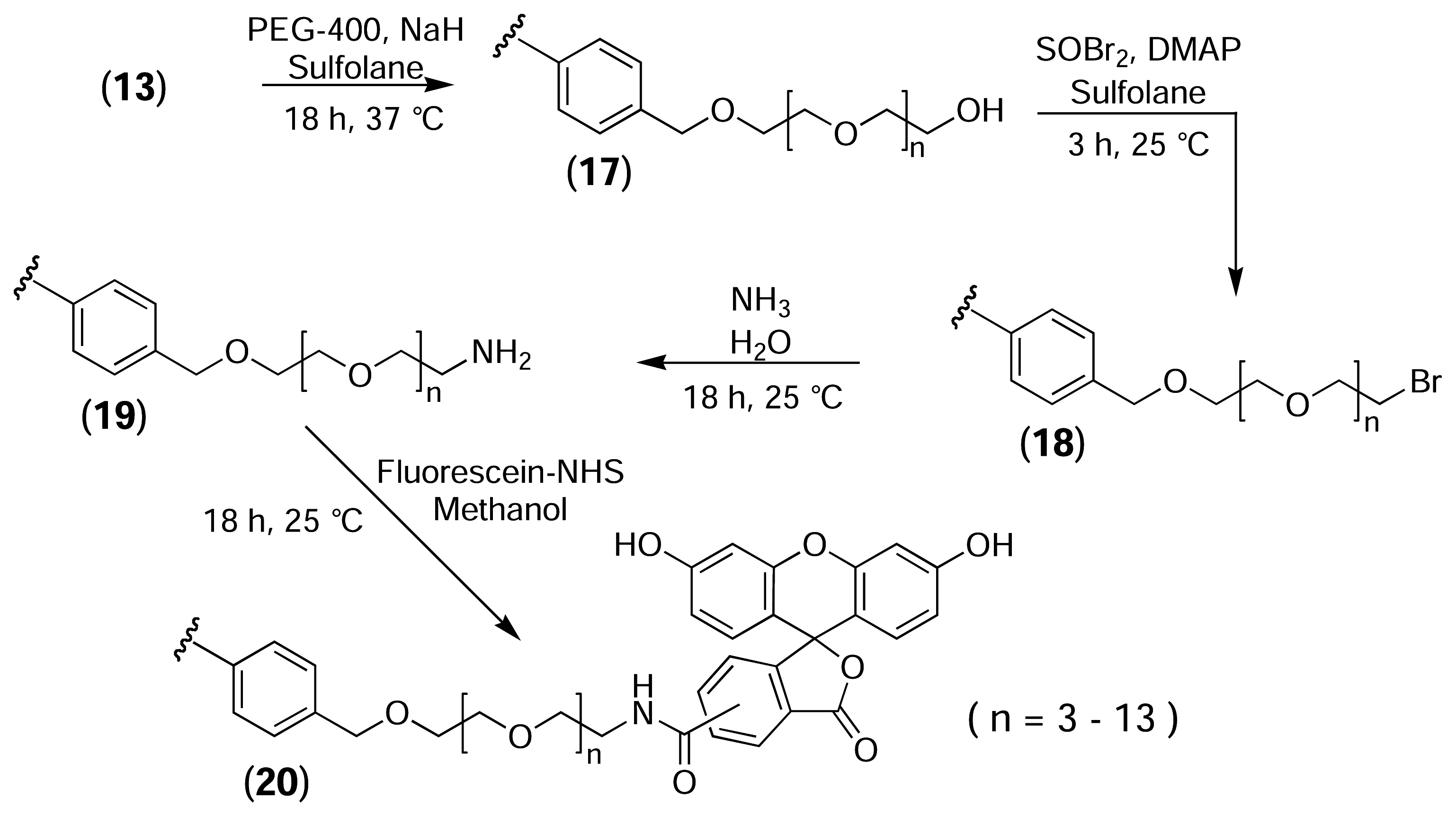
Figure 11.
Epifluorescence micrographs of PS resins (100× magnification, 450–490 nm bandpass excitation filter, 520 nm low pass emission filter). (a) Underivatised PS resin as viewed under phase contrast illumination. (b) Underivatised PS resin treated with congo red (control). (c) Bromomethylated PS resin treated with congo red (14). (d) Underivatised PS resin treated with fluorescein-NHS (control). (e) PS-NH2 resin treated with fluorescein-NHS (16). (f) PS-PEG-NH2 resin treated with fluorescein-NHS (20).
Figure 11.
Epifluorescence micrographs of PS resins (100× magnification, 450–490 nm bandpass excitation filter, 520 nm low pass emission filter). (a) Underivatised PS resin as viewed under phase contrast illumination. (b) Underivatised PS resin treated with congo red (control). (c) Bromomethylated PS resin treated with congo red (14). (d) Underivatised PS resin treated with fluorescein-NHS (control). (e) PS-NH2 resin treated with fluorescein-NHS (16). (f) PS-PEG-NH2 resin treated with fluorescein-NHS (20).

Figure 12.
Derivatisation of PS microtitre plates with palau’amine via a cleavable (disulfide-containing) PEG linker.
Figure 12.
Derivatisation of PS microtitre plates with palau’amine via a cleavable (disulfide-containing) PEG linker.

Figure 13.
Agarose gel electrophoresis of PCR products obtained from various phage-displayed human cDNA libraries after five rounds of selection with palau’amine immobilised on a PS plate via a cleavable disulfide-containing linker. The DNA inserts were amplified using generic T7 primers.
Figure 13.
Agarose gel electrophoresis of PCR products obtained from various phage-displayed human cDNA libraries after five rounds of selection with palau’amine immobilised on a PS plate via a cleavable disulfide-containing linker. The DNA inserts were amplified using generic T7 primers.
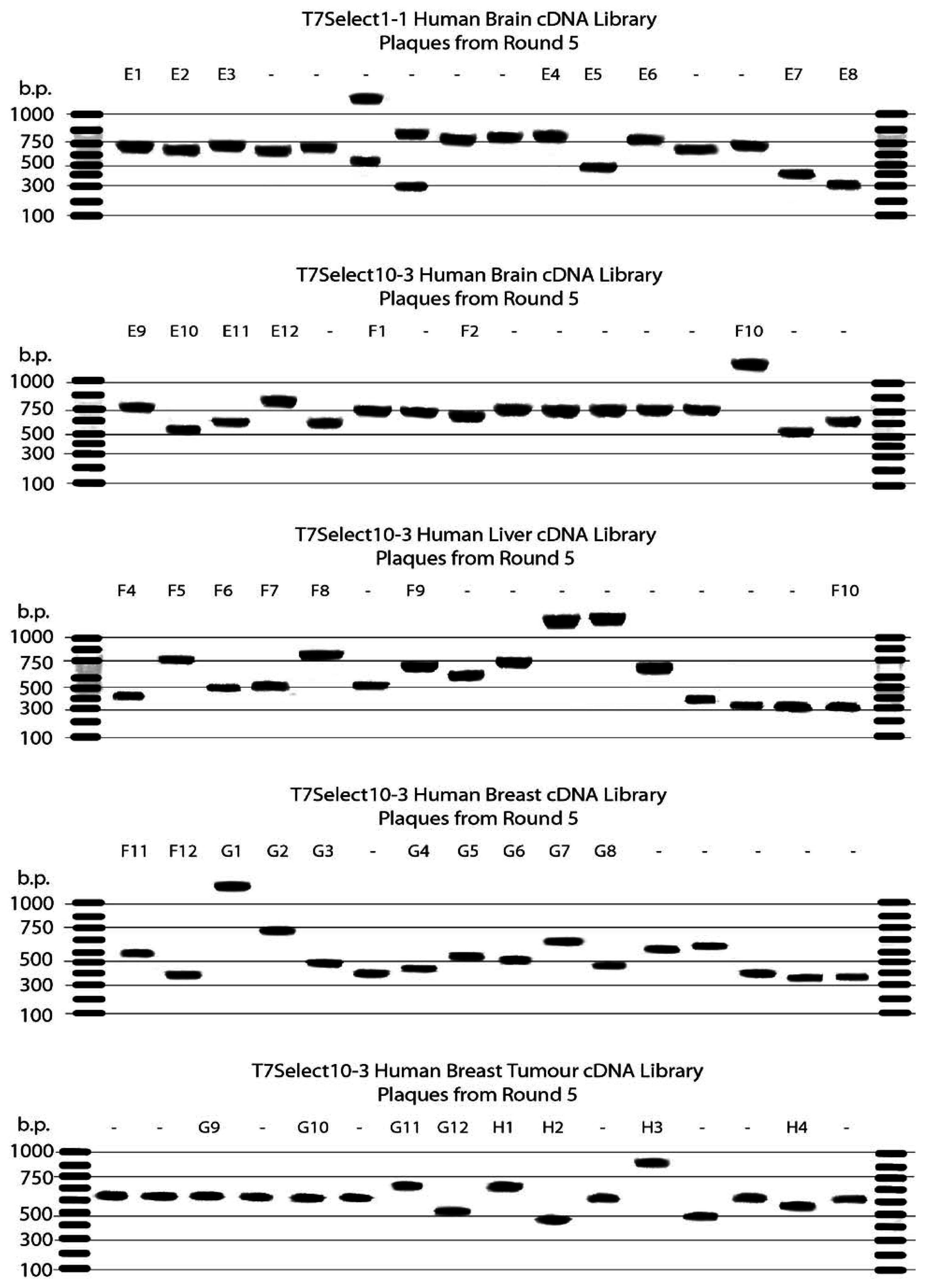
Figure 14.
Alignment of the C-terminal domain (aa 350–650 of 699) of hNopp140 (Swiss-Prot: Q14978) with clones rescued from human brain (HB) and human breast tumour (BrT) cDNA libraries using palau’amine immobilised on PS microtitre plate wells. Similar clones isolated from T7 phage-displayed libraries by Yu et al. [63] (using biotinylated doxorubicin immobilised on a streptavidin-coated plate) and Gearhart et al. [64] (using 2-methylnorharman adsorbed to nitrocellulose discs) are also shown. Black shading = 80% homology; Grey shading = 60% homology.
Figure 14.
Alignment of the C-terminal domain (aa 350–650 of 699) of hNopp140 (Swiss-Prot: Q14978) with clones rescued from human brain (HB) and human breast tumour (BrT) cDNA libraries using palau’amine immobilised on PS microtitre plate wells. Similar clones isolated from T7 phage-displayed libraries by Yu et al. [63] (using biotinylated doxorubicin immobilised on a streptavidin-coated plate) and Gearhart et al. [64] (using 2-methylnorharman adsorbed to nitrocellulose discs) are also shown. Black shading = 80% homology; Grey shading = 60% homology.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
DNA sequencing of PCR products obtained from 40 individual plaques after five rounds of selection with palau’amine immobilised on a PS plate via a cleavable disulfide-containing linker.
| Library | Plaques | Gene Identified from DNA Sequence | Notes |
|---|---|---|---|
| 1-1 Brain | E1, E3, E6 | Complete mitochondrial genome, haplotype B4a2, individual Ya015 | Fragment In Backwards |
| 1-1 Brain | E2 | Prader-Willi/Angelman syndrome region on chromosome 15 | Fragment |
| 1-1 Brain | E4 | Homo sapiens Chromosome 16 BAC clone CIT987SK-44M2 | Fragment |
| 1-1 Brain | E5 | Human DNA sequence from clone RP11-364G4 on chromosome 13 | Fragment |
| 1-1 Brain | E7 | Homo sapiens cDNA FLJ42917 fis, clone BRHIP3026335. | Fragment |
| 1-1 Brain | E8 | Human DNA sequence from clone RP11-4F1 on chromosome 10 | Fragment In Backwards |
| 10-3 Brain | E9, F1 | Homo sapiens nucleolar and coiled-body phosphoprotein 1 (hNopp140) | Fragment In Frame |
| 10-3 Brain | E10 | Homo sapiens BAC clone RP11-696N14 from 4 | Fragment In Backwards |
| 10-3 Brain | E11 | Complete mitochondrial genome, haplotype B4a1a individual Am145. | Fragment In Backwards |
| 10-3 Brain | E12 | Leucine zipper, putative tumour suppressor 1 (LZTS1) | Fragment Not in CDS |
| 10-3 Brain | F2 | Homo sapiens phosphatase and actin regulator 3 (PHACTR3) | Fragment In Backwards |
| Liver | F3 | ATP synthase, H+transporting | Contamination |
| Liver | F4 | Diazepam binding inhibitor (GABA receptor modulator) | Fragment Not in CDS |
| Liver | F5 | Homo sapiens chromosome clone RP11-323I15 | Fragment In Backwards |
| Liver | F6 | Homo sapiens complement component 1, s subcomponent | Fragment Not in CDS |
| Liver | F7 | Human serum albumin (ALB) gene | Fragment |
| Liver | F8 | Homo sapiens 12 BAC RP11-218M22 | Fragment |
| Liver | F9 | Homo sapiens cDNA FLJ11946 fis, clone HEMBB1000709 | Fragment In Backwards |
| Liver | F10 | Homo sapiens ferritin light polypeptide | Fragment Frame 3 |
| Breast | F11 | Homo sapiens splicing factor 3b, subunit 2 (SF3B2) | Fragment Frame 2 |
| Breast | F12 | Homo sapiens chromosome 5 clone RP11-394O4 | Fragment |
| Breast | G1 | ATP synthase, H+transporting | Contamination |
| Breast | G2 | Homo sapiens snRNA activating protein complex (SNAP190) | Fragment Frame 3 |
| Breast | G3 | Homo sapiens ribosomal protein L10 (RPL10) | Fragment Frame 2 |
| Breast | G4 | Homo sapiens metastasis associated lung adenocarcinoma | Fragment |
| Breast | G5 | Lectin, galactoside-binding, soluble, 1 (galectin 1) | Fragment Frame 3 |
| Breast | G6 | Homo sapiens isolate H5-08 mitochondrion | Fragment |
| Breast | G7 | Human DNA sequence from clone RP11-337N19 on chromosome 10 | Fragment |
| Breast | G8 | Human mRNA for erythrocyte adducin alpha subunit | Fragment Not in CDS |
| Breast Tumour | G9, G10 | Heterogeneous nuclear ribonucleoprotein H3 (2H9)(HNRPH3) | Fragment Frame 3 |
| Breast Tumour | G11 | Homo sapiens nucleolar phosphoprotein p130 (hNopp140) | Fragment In Frame |
| Breast Tumour | G12 | Homo sapiens SMT3 suppressor of mif two 3 homolog 2 | Fragment Frame 2 |
| Breast Tumour | H1 | Homo sapiens BAC clone RP11-572N21 from 2 | Fragment |
| Breast Tumour | H2 | Homo sapiens cDNA FLJ26671 fis, clone MPG03325. | Fragment In Backwards |
| Breast Tumour | H3 | Chromosome 14 clone RP11-45G3 containing neurexin III gene | Fragment |
| Breast Tumour | H4 | Human DNA sequence from clone RP11-84A14 on chromosome 1 | Fragment |
Table 2.
Amino acid composition of the C-terminal domain of hNopp140 (aa 400–699), as isolated from human brain and breast tumour libraries using palau’amine immobilised on a PS plate. Note the high percentage of lysine, glutamic acid and serine residues.
| Charged | Polar | Intermediate | Hydrophobic | ||||
|---|---|---|---|---|---|---|---|
| Arg | 5.0% | Asn | 3.0% | Ala | 12.0% | Ile | 1.3% |
| Asp | 3.7% | Gln | 4.0% | Cys | 0.0% | Leu | 3.0% |
| Glu | 11.4% | Ser | 15.4% | Gly | 6.7% | Phe | 2.0% |
| His | 0.3% | Thr | 4.3% | Met | 0.3% | Trp | 0.3% |
| Lys | 17.1% | Pro | 5.4% | Tyr | 0.3% | ||
| Val | 4.3% | ||||||
| TOTAL | 37.5% | TOTAL | 26.7% | TOTAL | 24.4% | TOTAL | 11.2% |
| CYCLES | TEMPERATURE | TIME |
|---|---|---|
| 1 | 94 °C | 2 min 30 s |
| 94 °C | 45 s | |
| 35 | 55 °C | 60 s |
| 72 °C | 30 s | |
| 1 | 72 °C | 10 min |
| 1 | 4 °C | ∞ |
Acknowledgements
We would like to thank Prof. Dave Austin (Yale University) for introducing us to phage display and for hosting a sabbatical visit by PK in 2000/2001. We would also like to thank Prof. Paul Scheuer (University of Hawai’i) for the generous gift of palau’amine.
- Sample availability: Not available.
Reference and Notes
- Rouhi, A. M. Rediscovering Natural Products. Chem. Eng. News 2003, 81, 77–91. [Google Scholar]
- Newman, D. J.; Cragg, G. M.; Snader, K. M. Natural products as sources of new drugs over the period 1981–2002. J. Nat. Prod 2003, 66, 1022–1037. [Google Scholar]
- Feher, M.; Schmidt, J. M. Property distributions: Differences between drugs, Natural Products, and molecules from combinatorial chemistry. J. Chem. Inf. Comput. Sci 2003, 43, 218–227. [Google Scholar]
- Adam, G. C.; Sorensen, E. J.; Cravatt, B. F. Proteomic profiling of mechanistically distinct enzyme classes using a common chemotype. Nat. Biotechnol 2002, 20, 805–809. [Google Scholar]
- Jeffery, D. A.; Bogyo, M. Chemical proteomics and its application to drug discovery. Curr. Opin. Biotechnol 2003, 14, 87–95. [Google Scholar]
- Patricelli, M. P.; Giang, D. K.; Stamp, L. M.; Burbaum, J. J. Direct visualization of serine hydrolase activities in complex proteomes using fluorescent active site-directed probes. Proteomics 2001, 1, 1067–1071. [Google Scholar]
- Speers, A. E.; Cravatt, B. F. Chemical strategies for activity-based proteomics. ChemBioChem 2004, 5, 41–47. [Google Scholar]
- Zhao, G.; Meier, T. I.; Kahl, S. D.; Gee, K. R.; Blaszczak, L. C. BOCILLIN FL, a sensitive and commercially available reagent for detection of penicillin-binding proteins. Antimicrob. Agents Chemother 1999, 43, 1124–1128. [Google Scholar]
- Taylor, E. W. The mechanism of colchicine inhibition of mitosis. I. Kinetics of inhibition and the binding of colchicine-3H. J. Cell Biol 1965, 25, 145–160. [Google Scholar]
- Borisy, G. G.; Taylor, E. W. The mechanism of action of colchicine. Binding of colchicine-3H to cellular protein. J. Cell Biol 1967, 34, 525–533. [Google Scholar]
- Borisy, G. G.; Taylor, E. W. The mechanism of action of colchicine. Colchicine binding to sea urchin eggs and the mitotic apparatus. J. Cell Biol 1967, 34, 535–548. [Google Scholar]
- Shelanski, M. L.; Taylor, E. W. Isolation of a protein subunit from microtubules. J. Cell Biol 1967, 34, 549–554. [Google Scholar]
- Weisenberg, R. C.; Broisy, G. G.; Taylor, E. W. Colchicine-binding protein of mammalian brain and its relation to microtubules. Biochemistry 1968, 7, 4466–4479. [Google Scholar]
- Piggott, A. M.; Karuso, P. Quality, not quantity: The role of natural products and chemical proteomics in modern drug discovery. Comb. Chem. High Throughput Screen 2004, 7, 607–630. [Google Scholar]
- Sin, N.; Meng, L.; Wang, M. Q. W.; Wen, J. J.; Bornmann, W. G.; Crews, C. M. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc. Nat. Acad. Sci. USA 1997, 94, 6099–6103. [Google Scholar]
- Sin, N.; Meng, L.; Auth, H.; Crews, C. M. Eponemycin analogues: syntheses and use as probes of angiogenesis. Bioorg. Med. Chem 1998, 6, 1209–1217. [Google Scholar]
- Abe, J.; Zhou, W.; Takuwa, N.; Taguchi, J.; Kurokawa, K.; Kumada, M.; Takuwa, Y. A fumagillin derivative angiogenesis inhibitor, AGM-1470, inhibits activation of cyclin-dependent kinases and phosphorylation of retinoblastoma gene product but not protein tyrosyl phosphorylation or protooncogene expression in vascular endothelial cells. Cancer Res 1994, 54, 3407–3412. [Google Scholar]
- Lowther, W. T.; McMillen, D. A.; Orville, A. M.; Matthews, B. W. The anti-angiogenic agent fumagillin covalently modifies a conserved active-site histidine in the Escherichia coli methionine aminopeptidase. Proc. Nat. Acad. Sci. USA 1998, 95, 12153–12157. [Google Scholar]
- Liu, S.; Widom, J.; Kemp, C. W.; Crews, C. M.; Clardy, J. Structure of human methionine aminopeptidase-2 complexed with fumagillin. Science 1998, 282, 1324–1327. [Google Scholar]
- Luibrand, R. T.; Erdman, T. R.; Vollmer, J. J.; Scheuer, P. J.; Finer, J.; Clardy, J. Ilimaquinone, a sesquiterpenoid quinone from a marine sponge. Tetrahedron 1979, 35, 609–612. [Google Scholar]
- Takizawa, P. A.; Yucel, J. K.; Veit, B.; Faulkner, D. J.; Deerinck, T.; Soto, G.; Ellisman, M.; Malhotra, V. Complete vesiculation of Golgi membranes and inhibition of protein transport by a novel sea sponge metabolite, ilimaquinone. Cell 1993, 73, 1079–1090. [Google Scholar]
- Veit, B.; Yucel, J. K.; Malhotra, V. Microtubule independent vesiculation of Golgi membranes and the reassembly of vesicles into Golgi stacks. J. Cell Biol 1993, 122, 1197–1206. [Google Scholar]
- Radeke, H. S.; Snapper, M. L. Photoaffinity study of the cellular interactions of ilimaquinone. Bioorg. Med. Chem 1998, 6, 1227–1232. [Google Scholar]
- Radeke, H. S.; Digits, C. A.; Casaubon, R. L.; Snapper, M. L. Interactions of (−)-ilimaquinone with methylation enzymes: implications for vesicular-mediated secretion. Chem. Biol 1999, 6, 639–647. [Google Scholar]
- Lee, S. Y.; Choi, J. H.; Xu, Z. Microbial cell-surface display. Trends Biotech 2003, 21, 45–52. [Google Scholar]
- Kondo, A.; Ueda, M. Yeast cell-surface display - applications of molecular display. Appl. Microbiol. Biotechnol 2004, 64, 28–40. [Google Scholar]
- Crews, C. M.; Collins, J. L.; Lane, W. S.; Snapper, M. L.; Schreiber, S. L. GTP-dependent binding of the antiproliferative agent didemnin to elongation factor 1α. J. Biol. Chem 1994, 269, 15411–15414. [Google Scholar]
- Sche, P. P.; McKenzie, K. M.; White, J. D.; Austin, D. J. Display cloning: functional identification of natural product receptors using cDNA-phage display. Chem. Biol 1999, 6, 707–716. [Google Scholar]
- Sche, P. P.; McKenzie, K. M.; White, J. D.; Austin, D. J. Display cloning: Functional identification of natural product receptors using cDNA-phage display. [Erratum to document cited in CA132:45554]. Chem. Biol 2001, 8, 399–400. [Google Scholar]
- McKenzie, K. M.; Videlock, E. J.; Splittgerber, U.; Austin, D. J. Simultaneous identification of multiple protein targets by using complementary-DNA phage display and a Natural-Product-mimetic probe. Angew. Chem. Int. Ed 2004, 43, 4052–4055. [Google Scholar]
- Siekierka, J. J.; Hung, S. H. Y.; Poe, M.; Lin, C. S.; Sigal, N. H. A cytosolic binding protein for the immunosuppressant FK506 has peptidyl-prolyl isomerase activity but is distinct from cyclophilin. Nature 1989, 341, 755–757. [Google Scholar]
- Harding, M. W.; Galat, A.; Uehling, D. E.; Schreiber, S. L. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature 1989, 341, 758–760. [Google Scholar]
- Videlock, E. J.; Chung, V. K.; Mohan, M. A.; Strok, T. M.; Austin, D. J. Two-dimensional diversity: Screening human cDNA phage display libraries with a random diversity probe for the display cloning of phosphotyrosine binding domains. J. Am. Chem. Soc 2004, 126, 3730–3731. [Google Scholar]
- Videlock, E. J.; Chung, V. K.; Hall, J. M.; Hines, J.; Agapakis, C. M.; Austin, D. J. Identification of a molecular recognition role for the activation loop phosphotyrosine of the Src tyrosine kinase. J. Am. Chem. Soc. 2005, 127. [Google Scholar]
- Shim, J. S.; Lee, J.; Park, H-J.; Park, S-J.; Kwon, H. J. A new curcumin derivative, HBC, interferes with the cell cycle progression of colon Cancer cells via antagonization of the Ca2+/calmodulin function. Chem. Biol. 2004, 11, 1455–1463. [Google Scholar]
- Merrifield, R. B. Solid phase peptide synthesis. I. The synthesis of a tetrapeptide. J. Am. Chem. Soc 1963, 85, 2149–2154. [Google Scholar]
- Delgado, M.; Janda, K. D. Polymeric supports for solid phase organic synthesis. Curr. Org. Chem 2002, 6, 1031–1043. [Google Scholar]
- Blaney, P.; Grigg, R.; Sridharan, V. Traceless solid-phase organic synthesis. Chem. Rev 2002, 102, 2607–2624. [Google Scholar]
- Guillier, F.; Orain, D.; Bradley, M. Linkers and cleavage strategies in solid phase organic synthesis and combinatorial chemistry. Chem. Rev 2000, 100, 2091–2157. [Google Scholar]
- James, I. W. Linkers for solid phase organic synthesis. Tetrahedron 1999, 55, 4855–4946. [Google Scholar]
- Bochet, C. G. Photolabile protecting groups and linkers. J. Chem. Soc. Perkin Trans. 1 2002, 2, 125–142. [Google Scholar]
- Plunkett, M. J.; Ellman, J. A. A Silicon-Based Linker for Traceless Solid-Phase Synthesis. J. Org. Chem 1995, 60, 6006–6007. [Google Scholar]
- Patek, M.; Lebl, M. Safety-catch and multiply cleavable linkers in solid-phase synthesis. Biopolymers 1998, 47, 353–363. [Google Scholar]
- Rotmans, J. P.; Delwel, H. R. Cross-linking of Schistosoma mansoni antigens and their covalent binding on the surface of polystyrene microtitration trays for use in the ELISA. J. Immunol. Methods 1983, 57, 87–98. [Google Scholar]
- Aleixo, J. A. G.; Swaminathan, B.; Minnich, S. A.; Wallshein, V. A. Enzyme immunoassay: binding of Salmonella antigens to activated microtiter plates. J. Immunoass 1985, 6, 391–407. [Google Scholar]
- Zammatteo, N.; Girardeaux, C.; Delforge, D.; Pireaux, J-J.; Remacle, J. Amination of polystyrene microwells: application to the covalent grafting of DNA probes for hybridization assays. Anal. Biochem. 1996, 236, 85–94. [Google Scholar]
- Clark, B. R. Derivatized polystyrene and other polymer supports for spectroscopic studies; Aventis Pharmaceuticals Holdings Inc.: USA, 2003; p. 7. [Google Scholar]
- Bora, U.; Chugh, L.; Nahar, P. Covalent immobilization of proteins onto photoactivated polystyrene microtiter plates for enzyme-linked immunosorbent assay procedures. J. Immunol. Methods 2002, 268, 171–177. [Google Scholar]
- Nahar, P.; Wali, N. M.; Gandhi, R. P. Light-induced activation of an inert surface for covalent immobilization of a protein ligand. Anal. Biochem 2001, 294, 148–153. [Google Scholar]
- Fleet, G. W. J.; Porter, R. R.; Knowles, J. R. Affinity labeling of antibodies with aryl nitrene as reactive group. Nature 1969, 224, 511–512. [Google Scholar]
- Ito, Y.; Chen, G.; Imanishi, Y. Photoimmobilization of insulin onto polystyrene dishes for protein-free cell culture. Biotechnol. Prog 1996, 12, 700–702. [Google Scholar]
- Eckert, A. W.; Gröbe, D.; Rothe, U. Surface-modification of polystyrene-microtitre plates via grafting of glycidylmethacrylate and coating of poly-glycidylmethacrylate. Biomaterials 2000, 21, 441–447. [Google Scholar]
- Larsson, P. H.; Eklund, A.; Johansson, S. G. O.; Larsson, K. Covalent binding of proteins to grafted plastic surfaces suitable for immunoassays. II. Picograms of IgE detected in BAL fluid in sarcoidosis. J. Immunol. Methods 1997, 210, 41–49. [Google Scholar]
- Larsson, P. H.; Johansson, S. G. O.; Hult, A.; Göthe, S. Covalent binding of proteins to grafted plastic surfaces suitable for immunoassays. J. Immunol. Methods 1987, 98, 129–135. [Google Scholar]
- Butler, J. E. Solid supports in enzyme-linked immunosorbent assay and other solid-phase immunoassays. Methods Mol. Med 2004, 94, 333–372. [Google Scholar]
- Kinnel, R. B.; Gehrken, H-P.; Scheuer, P. J. Palau’amine: a cytotoxic and immunosuppressive hexacyclic bisguanidine antibiotic from the sponge Stylotella agminata. J. Am. Chem. Soc. 1993, 115, 3376–3377. [Google Scholar]
- Kinnel, R. B.; Gehrken, H-P.; Swali, R.; Skoropowski, G.; Scheuer, P. J. Palau’amine and its congeners: A family of bioactive bisguanidines from the marine sponge Stylotella aurantium. J. Org. Chem. 1998, 63, 3281–3286. [Google Scholar]
- Garrido-Hernandez, H.; Nakadai, M.; Vimolratana, M.; Li, Q.; Doundoulakis, T.; Harran, P. G. Spirocycloisomerization of tethered alkylidene glycocyamidines: Synthesis of a base template common to the palau’amine family of alkaloids. Angew. Chem. Int. Ed 2005, 44, 765–769. [Google Scholar]
- Katz, J. D.; Overman, L. E. Studies towards the total synthesis of palau’amine. Formation of 4,5-dihydropyrrole-2-carboxylate intermediates by alkene-enamide ring-closing metathesis. Tetrahedron 2004, 60, 9559–9568. [Google Scholar]
- Dilley, A. S.; Romo, D. Enantioselective Strategy to the Spirocyclic Core of Palau’amine and Related Bisguanidine Marine Alkaloids. Org. Lett 2001, 3, 1535–1538. [Google Scholar]
- Overman, L. E.; Rogers, B. N.; Tellew, J. E.; Trenkle, W. C. Stereocontrolled synthesis of the tetracyclic core of the bisguanidine alkaloids Palau’amine and Styloguanidine. J. Am. Chem. Soc 1997, 119, 7159–7160. [Google Scholar]
- Meier, U. T.; Blobel, G. A nuclear localization signal binding protein in the nucleolus. J. Cell Biol 1990, 111, 2235–2245. [Google Scholar]
- Jin, Y.; Yu, J.; Yu, Y. G. Identifcation of hNopp140 as a binding partner for doxorubicin with phage display cloning method. Chem. Biol 2002, 9, 157–162. [Google Scholar]
- Gearhart, D. A. T.; Patricia, F.; Warren Beach, J. Identification of brain proteins that interact with 2-methylnorharman. An analog of the parkinsonian-inducing toxin MPP+. Neurosci. Res. 2002, 44, 255–265. [Google Scholar]
- Kim, Y-K.; Jin, Y.; Vukoti, K. M.; Park, J. K.; Kim, E. E.; Lee, K-J.; Yu, Y. G. Purification and characterization of human nucleolar phosphoprotein 140 expressed in Escherichia coli. Prot. Expr. Purif. 2003, 31, 260–264. [Google Scholar]
- Pai, C-Y.; Chen, H-K.; Sheu, H-L.; Yeh, N-H. Cell cycle-dependent alterations of a highly phosphorylated nucleolar protein p130 are associated with nucleologenesis. J. Cell Sci. 1995, 108, 1911–1920. [Google Scholar]
- Pai, C-Y.; Yeh, N-H. Cell proliferation-dependent expression of two isoforms of the nucleolar phosphoprotein p130. Biochem. Biophys. Res. Commun. 1996, 221, 581–587. [Google Scholar]
- Chen, H-K.; Yeh, N-H. The nucleolar phosphoprotein P130 is a GTPase/ATPase with intrinsic property to form large complexes triggered by F-and Mg2+. Biochem. Biophys. Res. Commun. 1997, 230, 370–375. [Google Scholar]
- Meier, U. T.; Blobel, G. Nopp140 shuttles on tracks between nucleolus and cytoplasm. Cell 1992, 70, 127–138. [Google Scholar]
- Savinov, S. N.; Austin, D. J. The cloning of human genes using cDNA phage display and small-molecule chemical probes. Comb. Chem. High Throughput Screen 2001, 4, 593–597. [Google Scholar]
- Novagen, T7Select System Manual. USA. 2000, 1–21, Sample availability: Not available..
© 2005 by MDPI Reproduction is permitted for noncommercial purposes.
Share and Cite
MDPI and ACS Style
Piggott, A.M.; Karuso, P. Quality not Quantity: The Role of Marine Natural Products in Drug Discovery and Reverse Chemical Proteomics. Mar. Drugs 2005, 3, 36-63. https://doi.org/10.3390/md302036
AMA Style
Piggott AM, Karuso P. Quality not Quantity: The Role of Marine Natural Products in Drug Discovery and Reverse Chemical Proteomics. Marine Drugs. 2005; 3(2):36-63. https://doi.org/10.3390/md302036
Chicago/Turabian StylePiggott, Andrew M., and Peter Karuso. 2005. "Quality not Quantity: The Role of Marine Natural Products in Drug Discovery and Reverse Chemical Proteomics" Marine Drugs 3, no. 2: 36-63. https://doi.org/10.3390/md302036