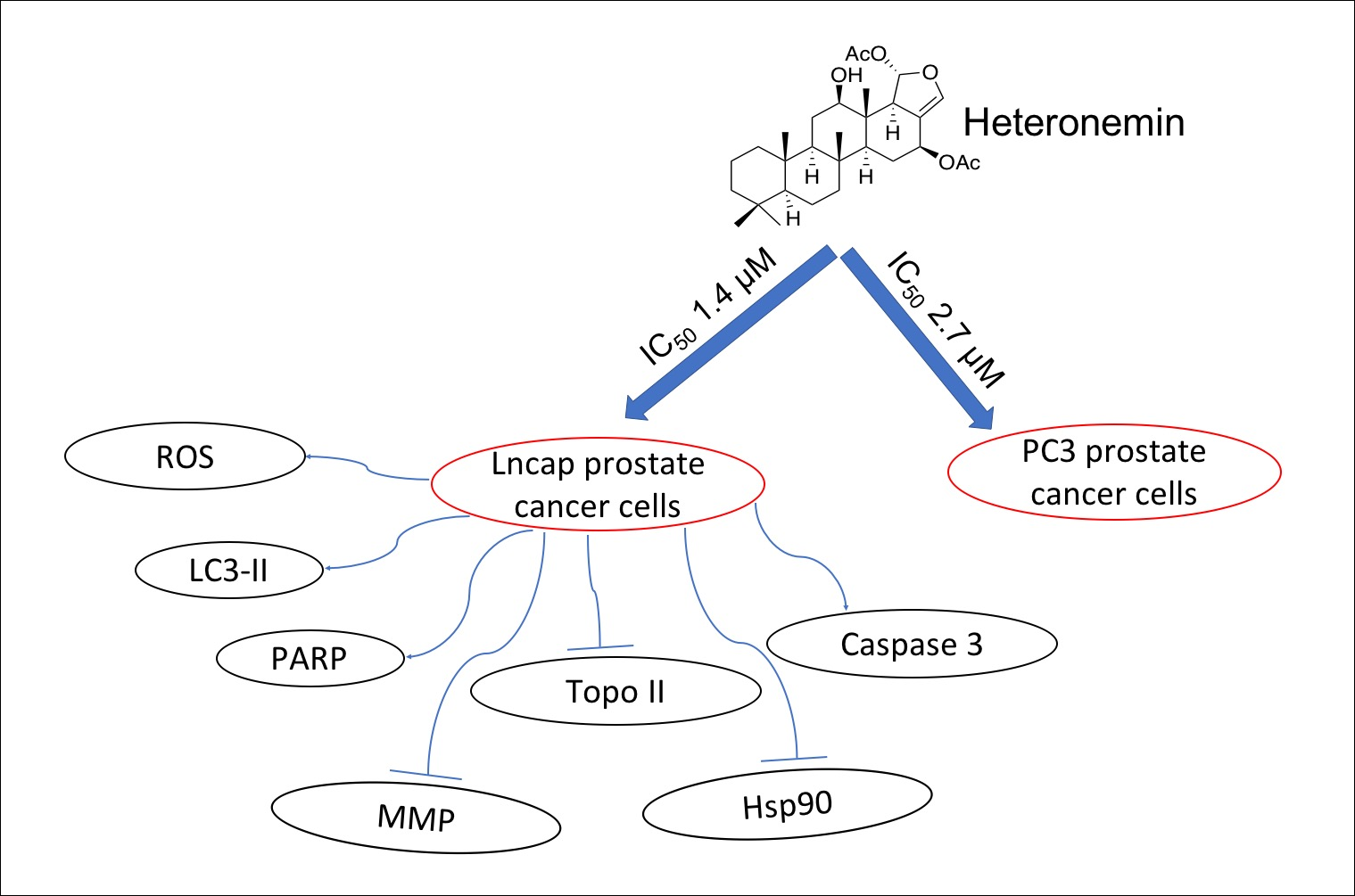
Heteronemin, a Marine Sesterterpenoid-Type Metabolite, Induces Apoptosis in Prostate LNcap Cells via Oxidative and ER Stress Combined with the Inhibition of Topoisomerase II and Hsp90

 , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
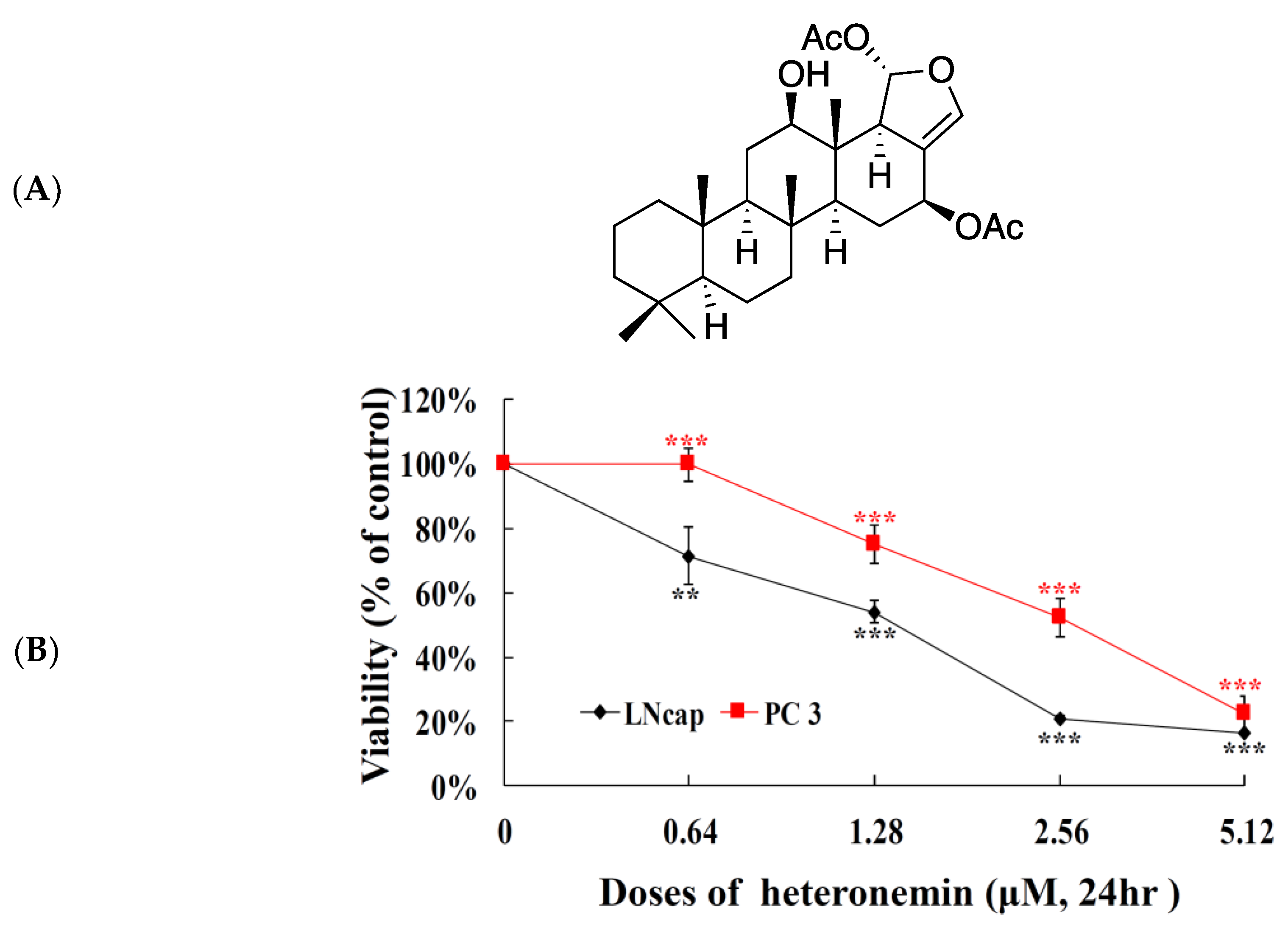
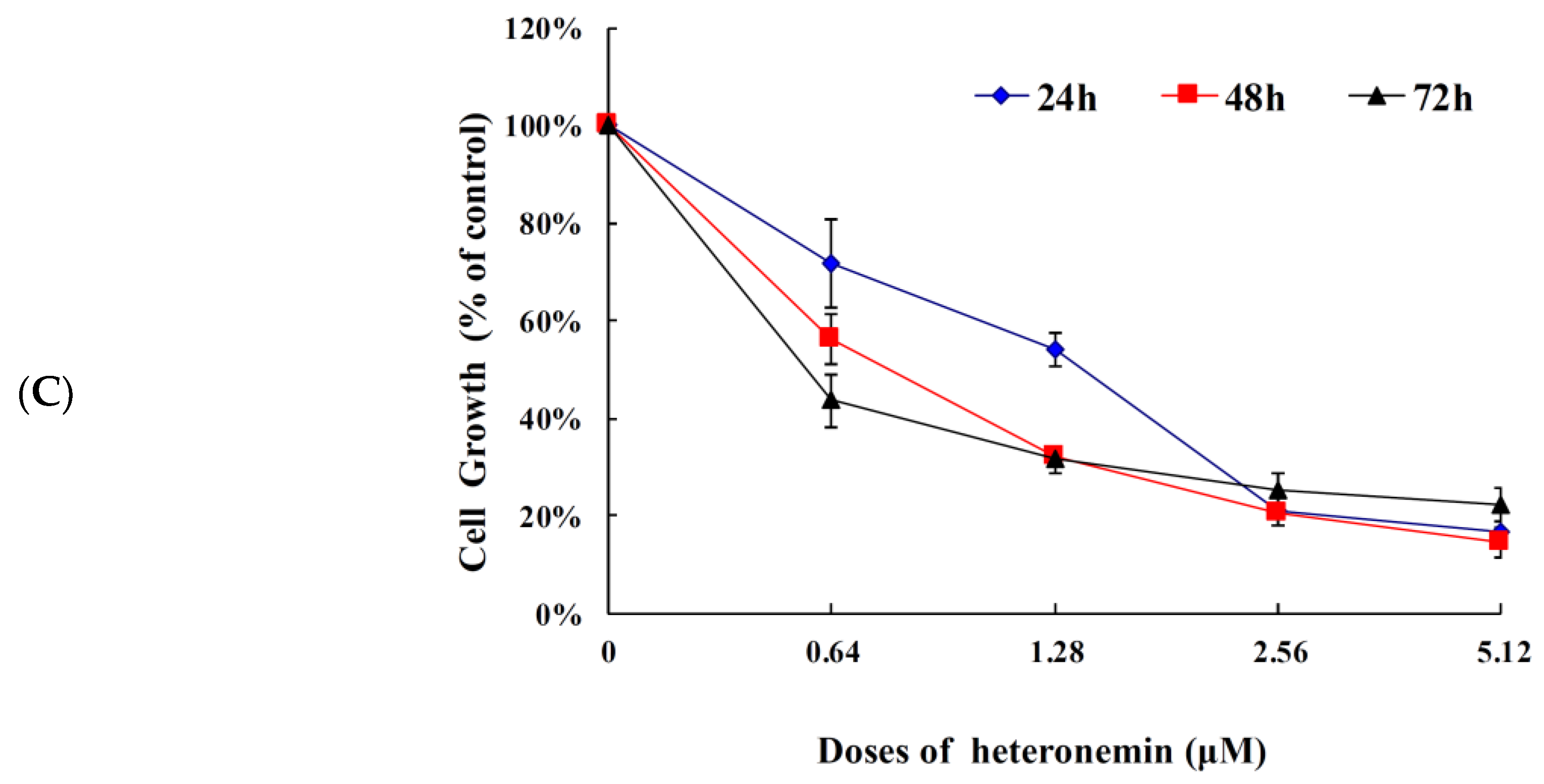
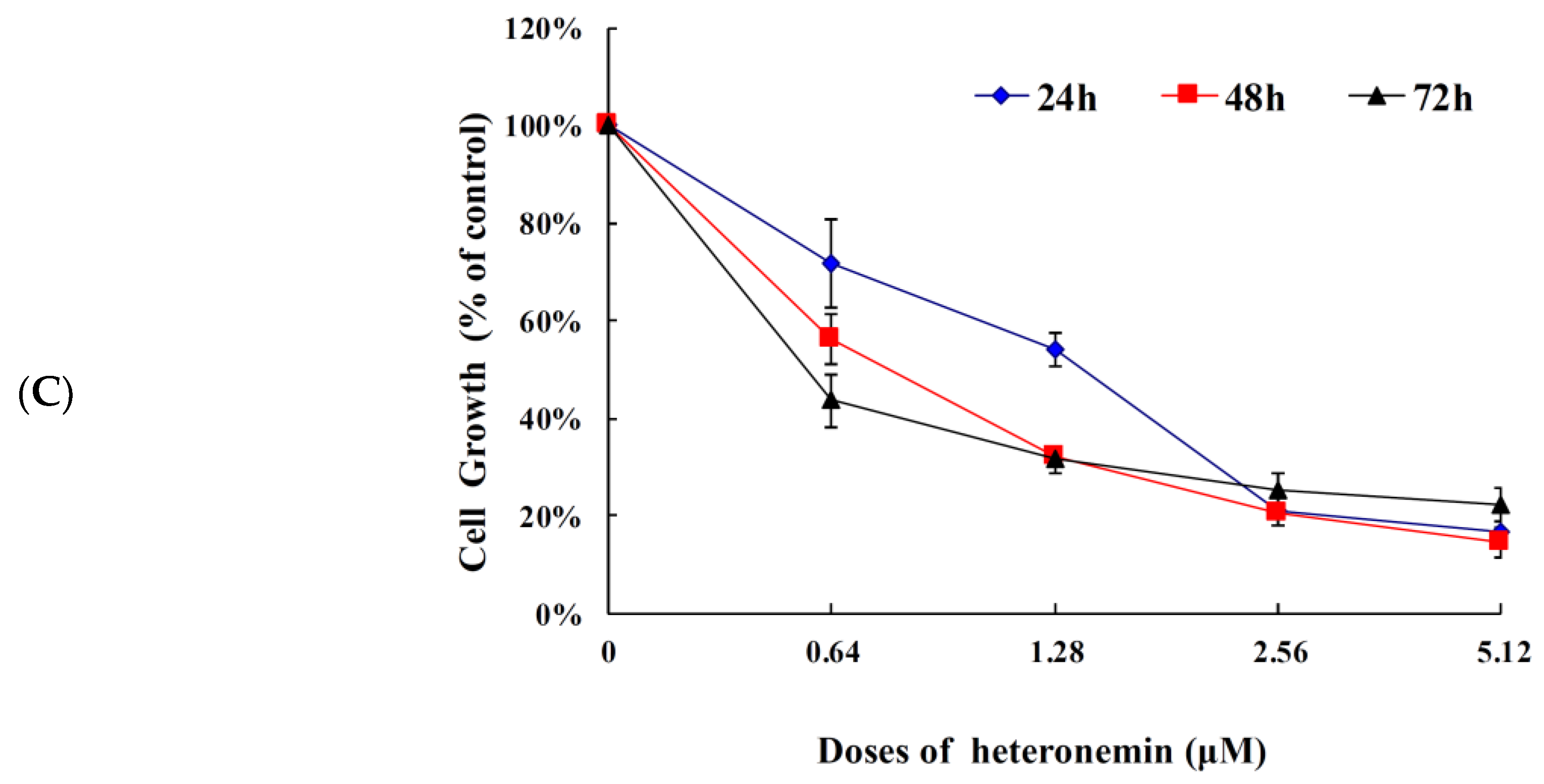
2.1. Effect of Heteronemin on Cellular Viability In Vitro Assay
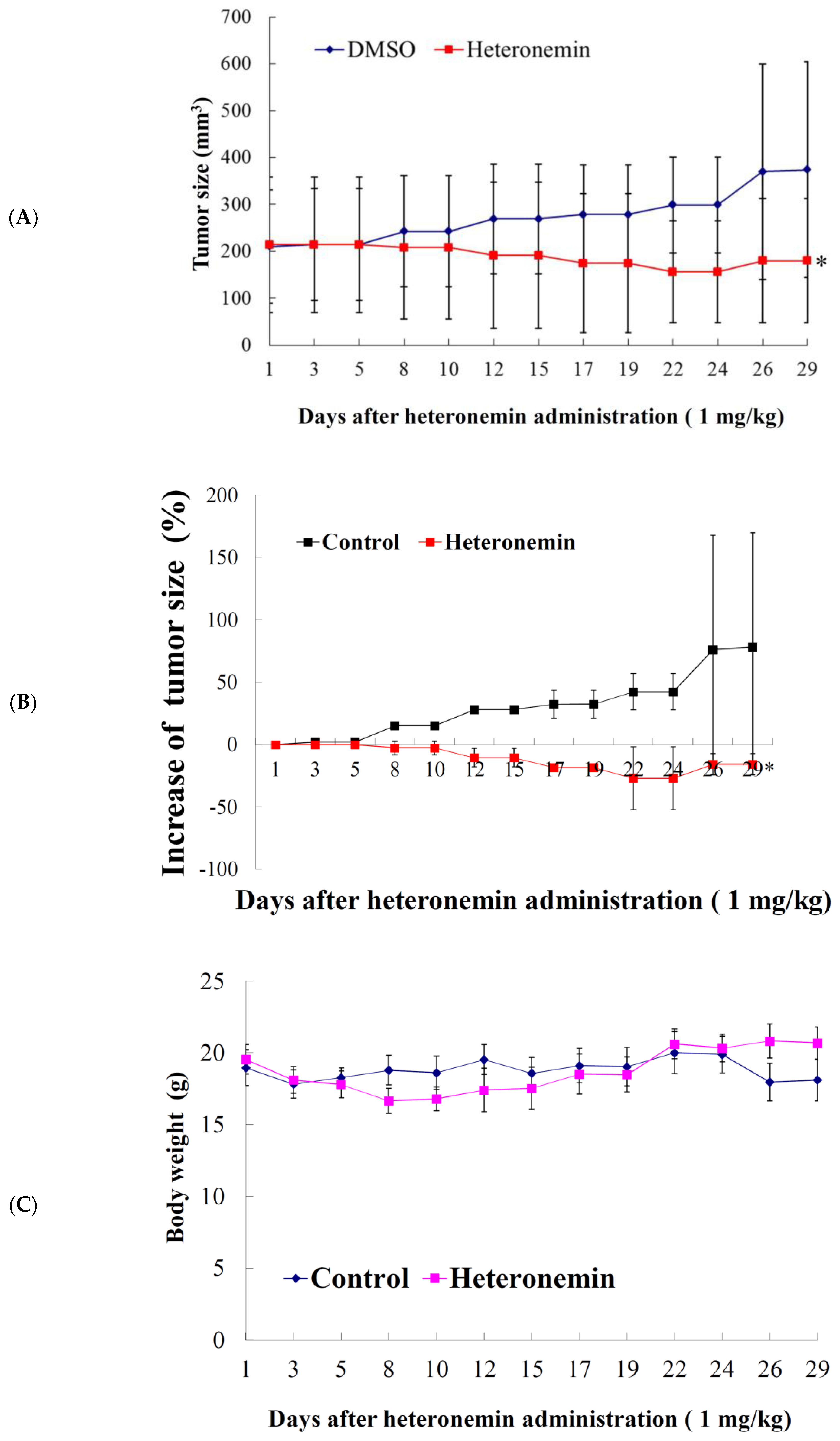
2.2. Effect of Heteronemin on the Growth of Human Prostate LNcap Tumor In Vivo Xenograft Animal Model
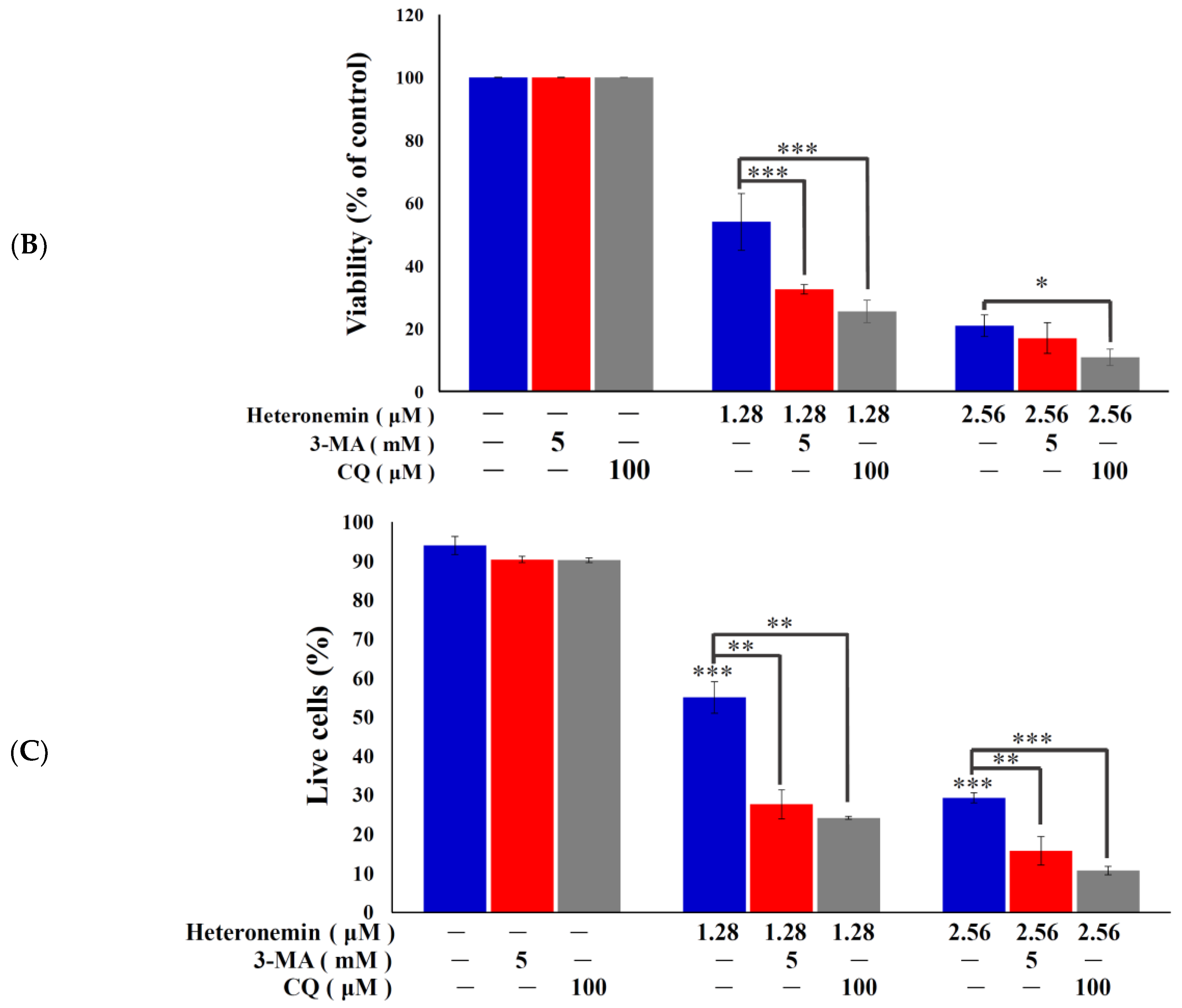
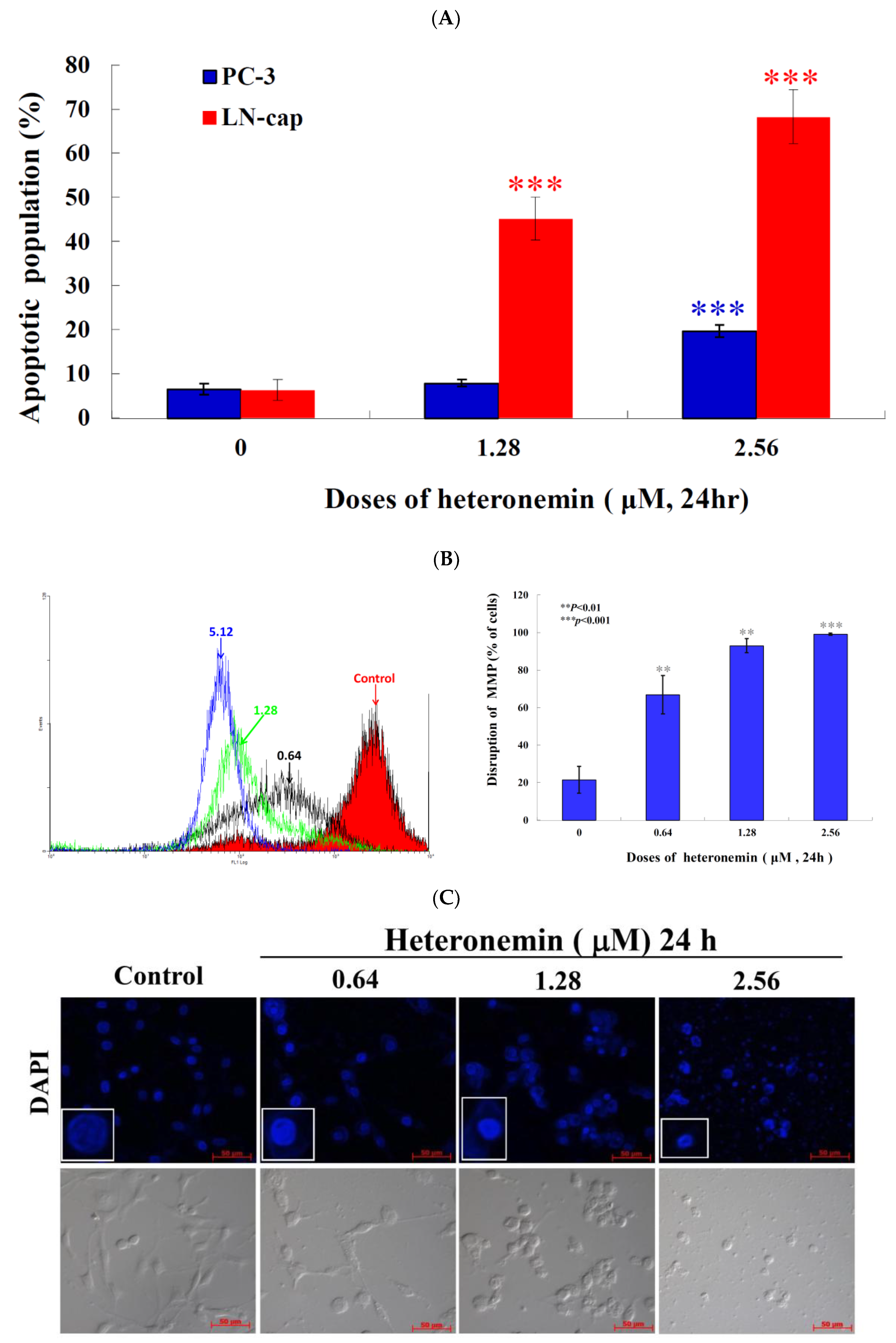
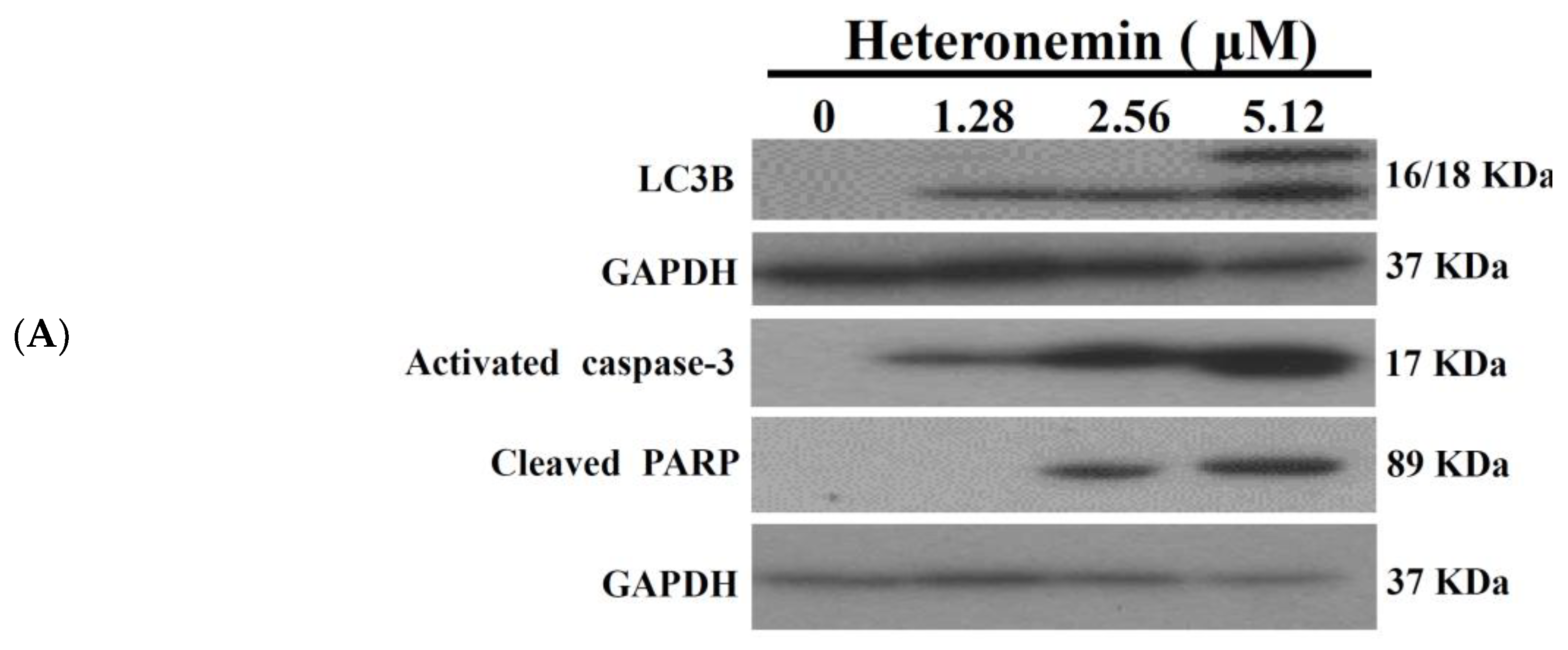
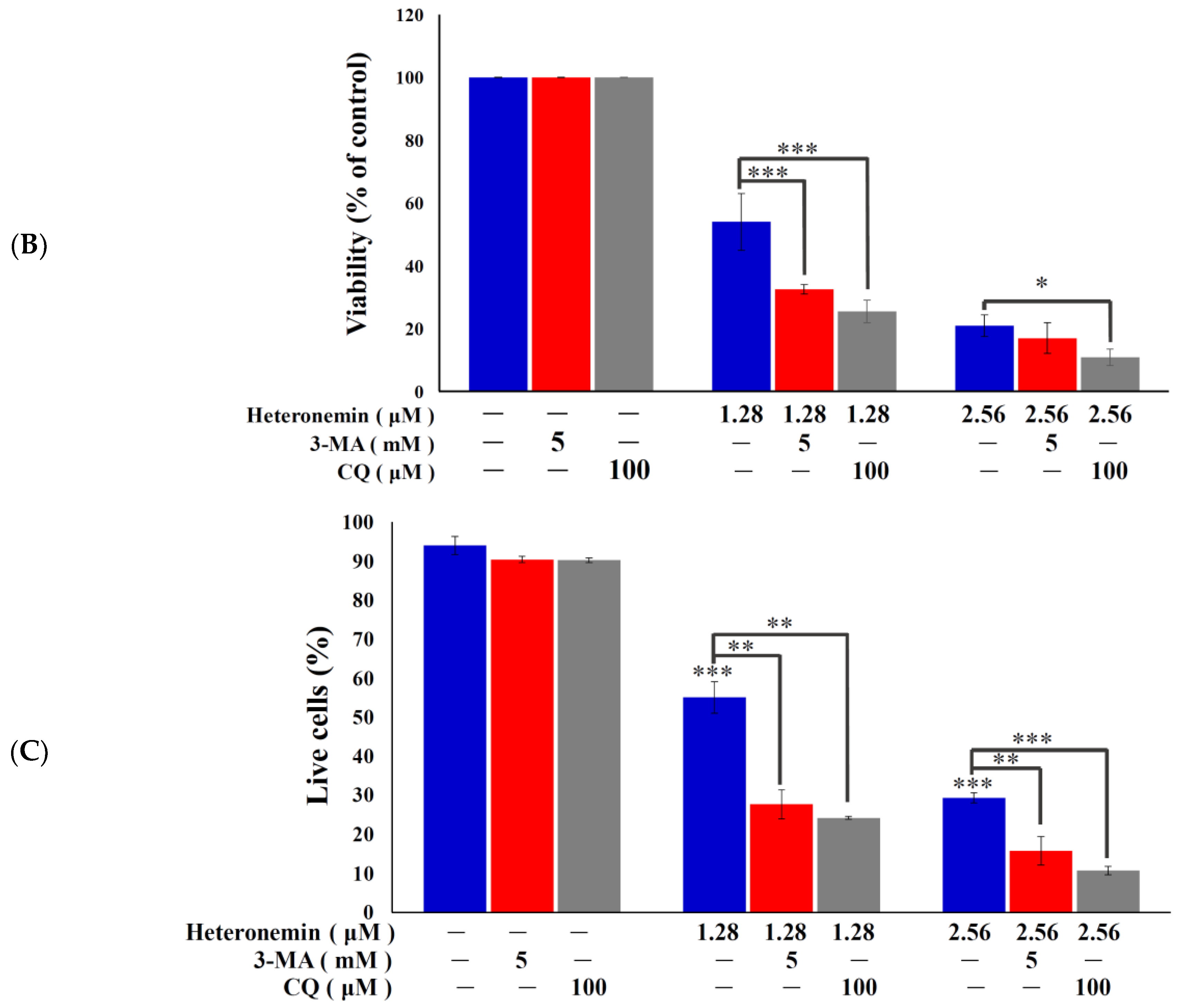
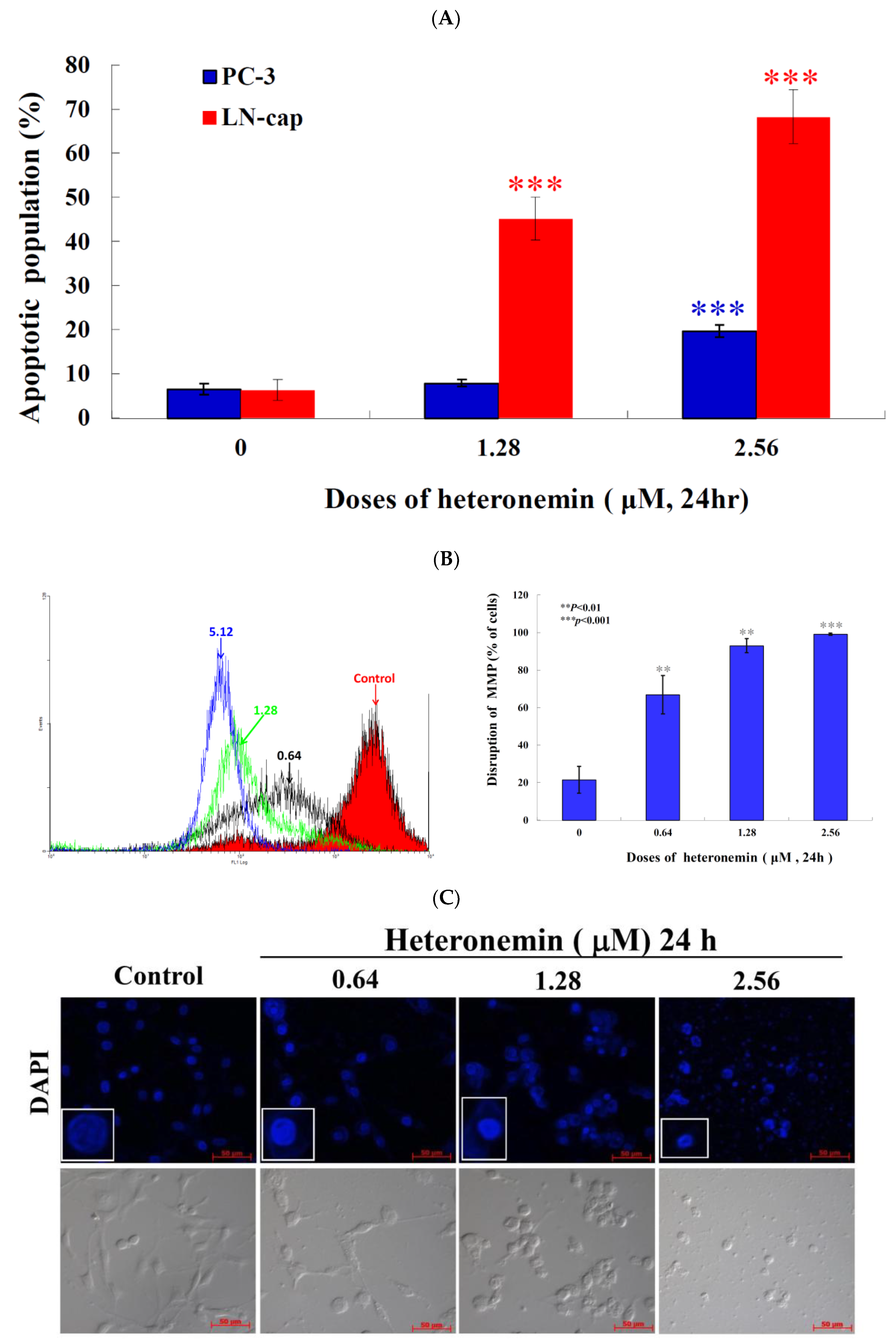
2.3. Effect of Heteronemin on Apoptotic and Autophagic Induction in LNcap Cells
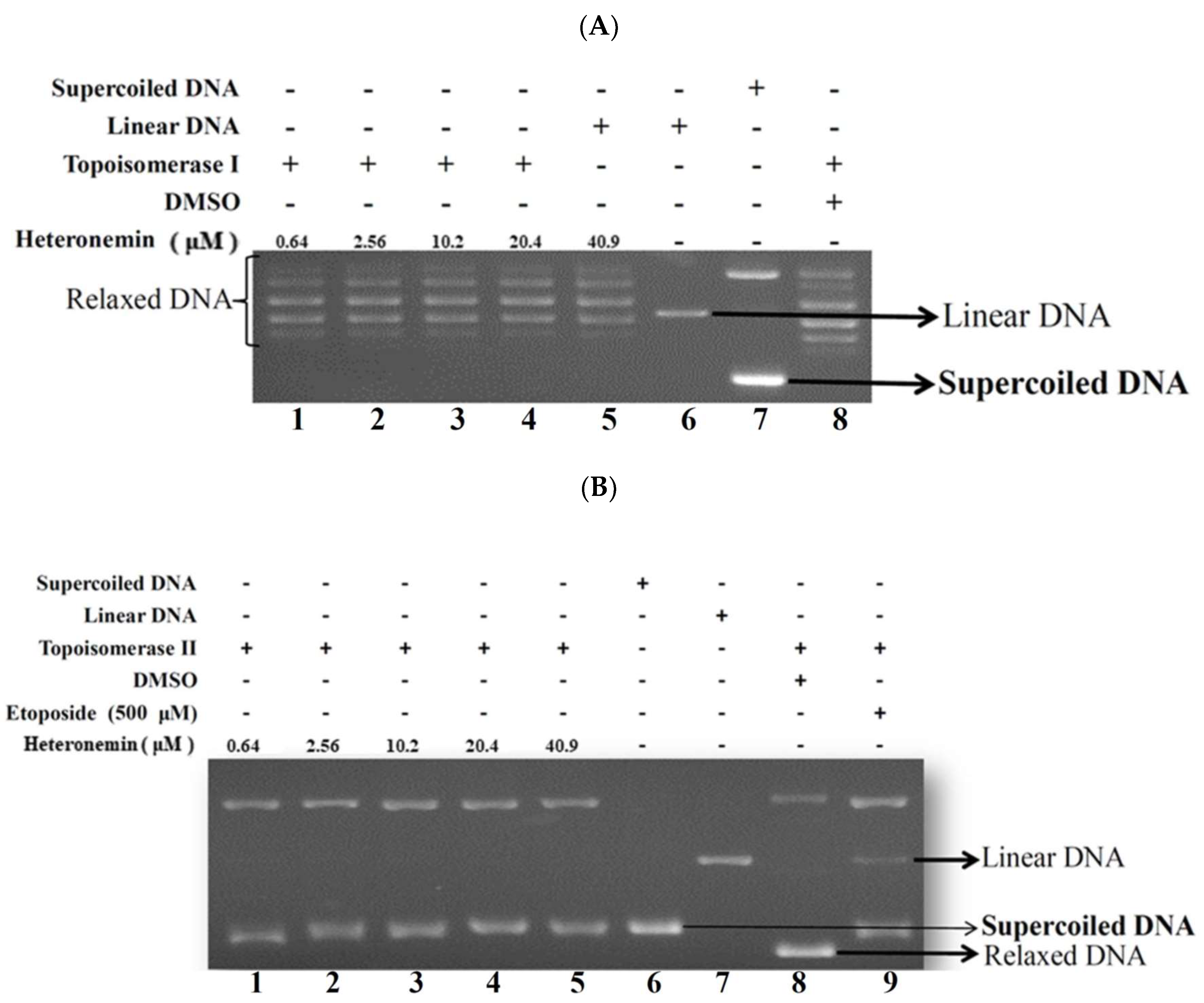
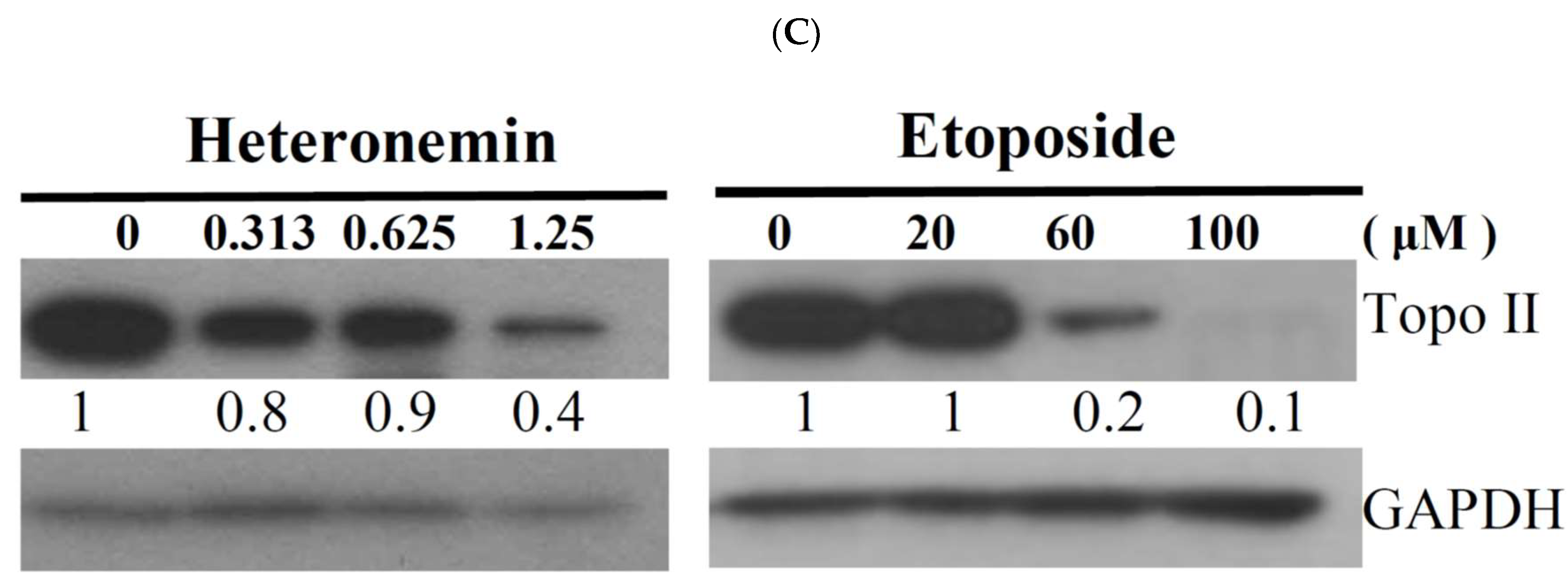
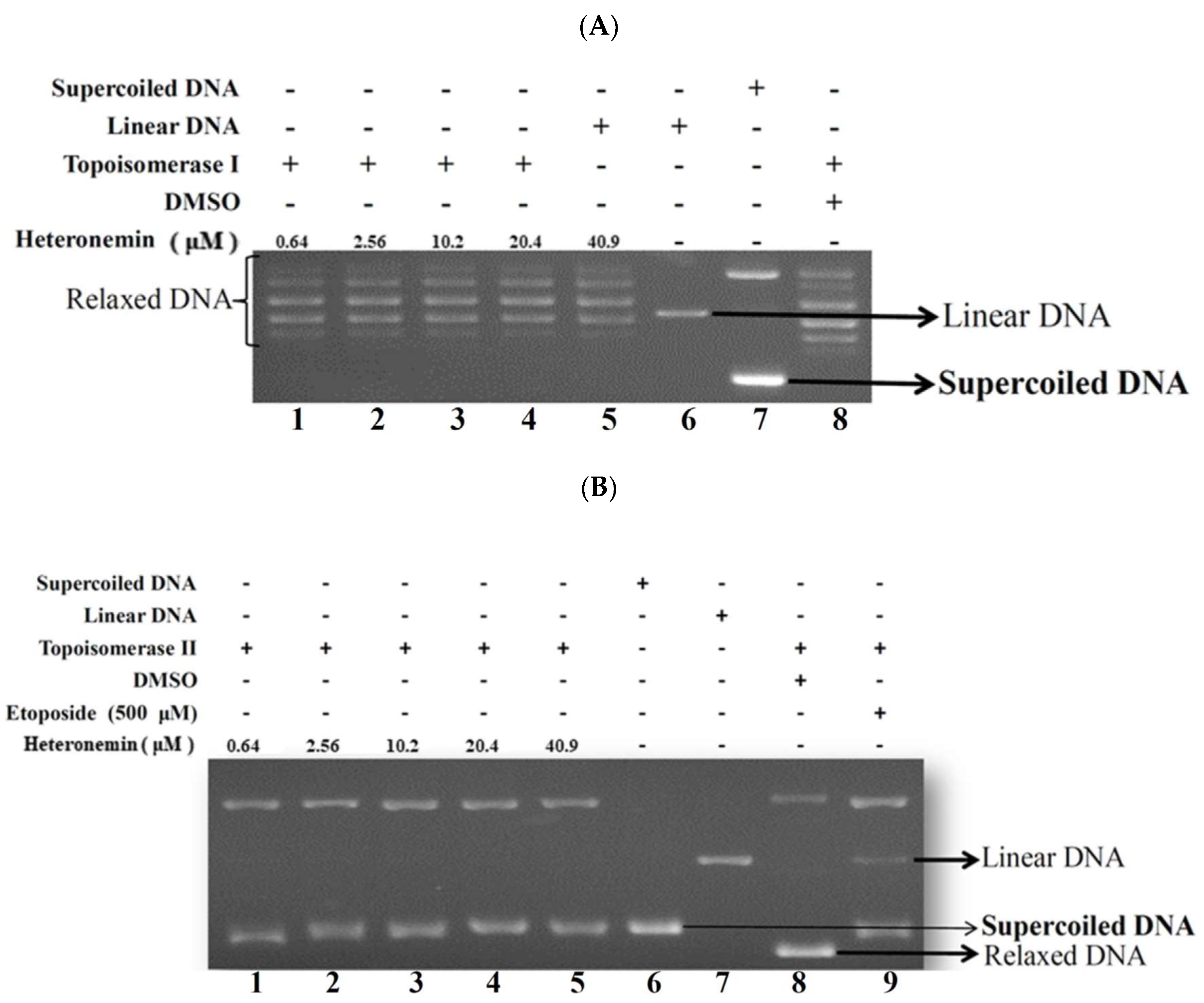
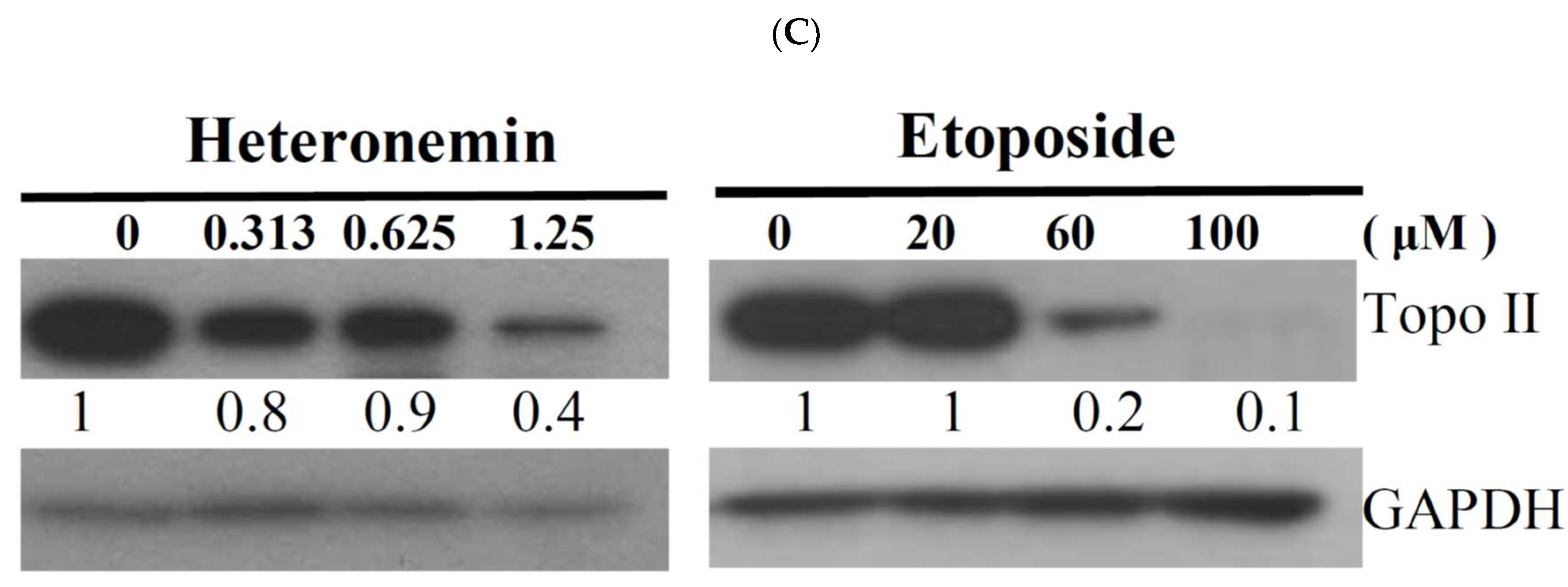
2.4. Effect of Heteronemin on Topoisomerase I and II Activity
2.5. Apoptotic Induction of Heteronemin Involves Disruption of Mitochondrial Membrane Potential (MMP) in LNcap Cells
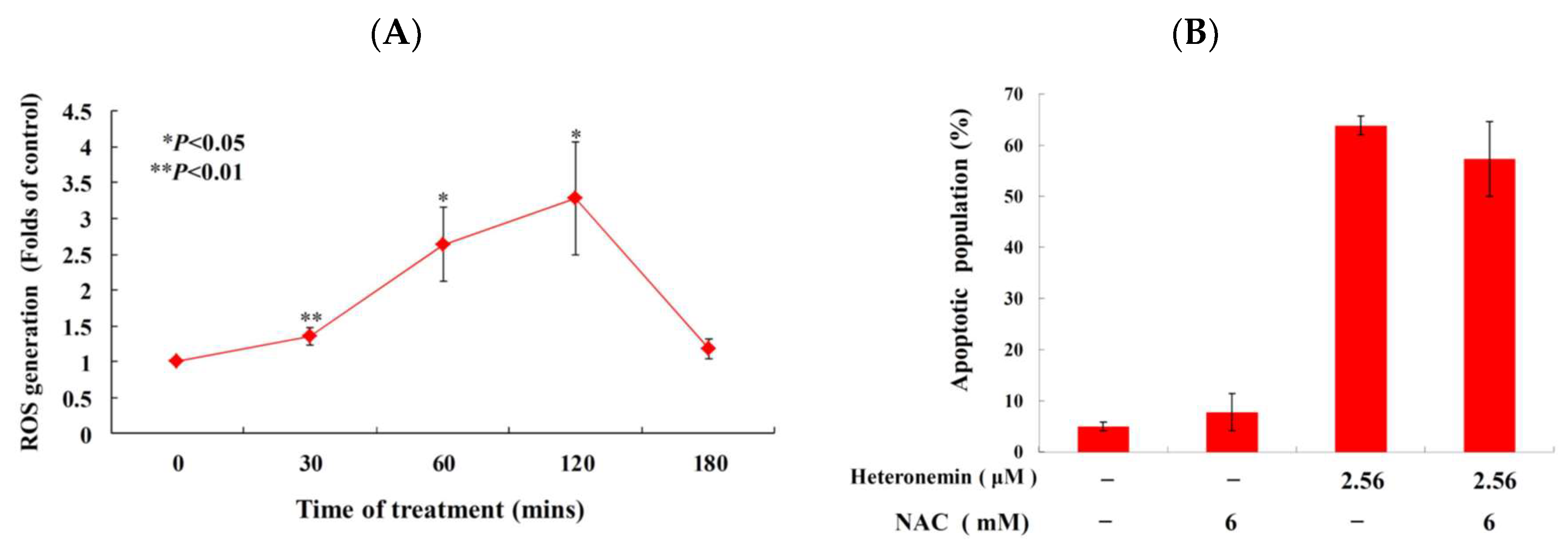
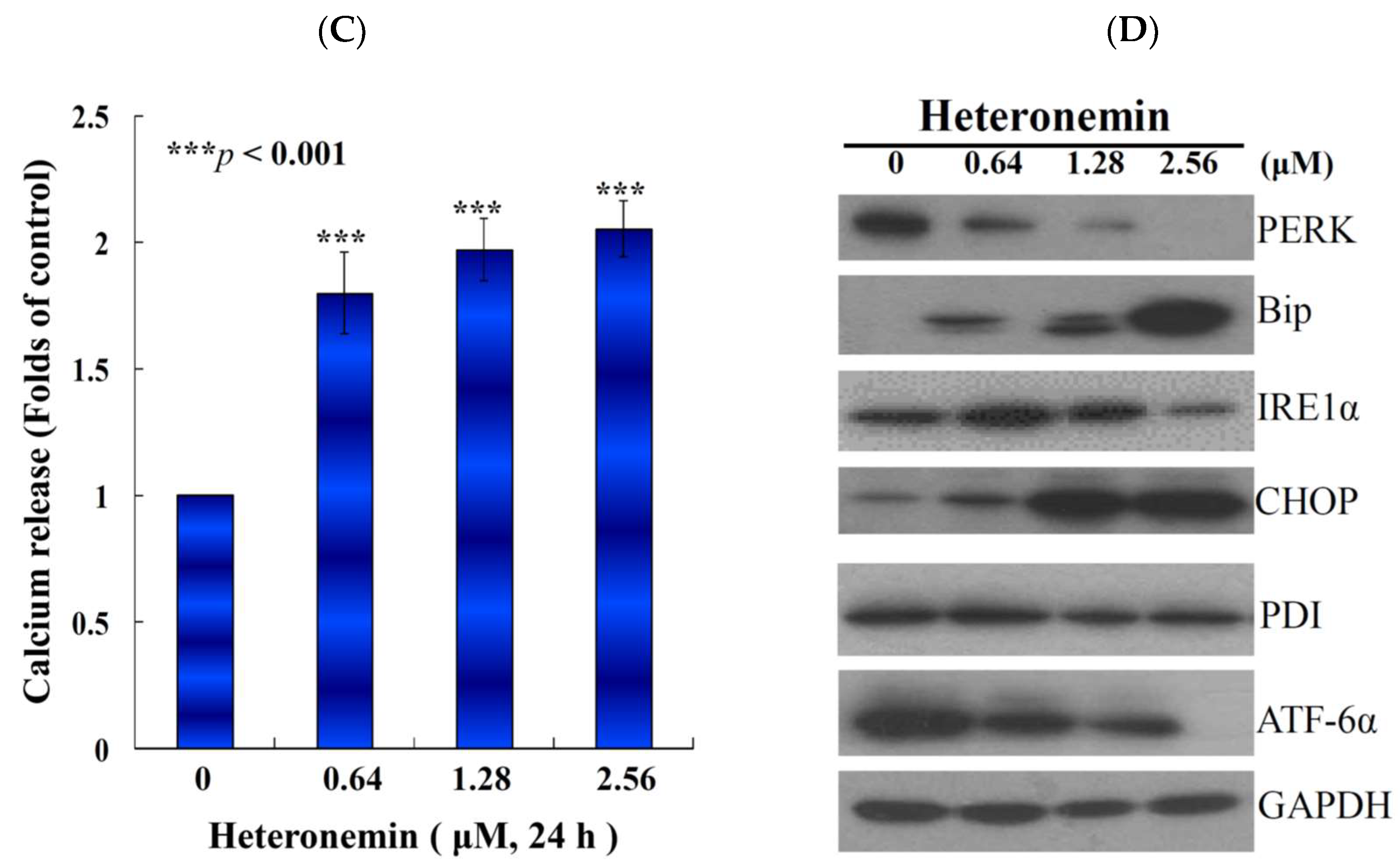
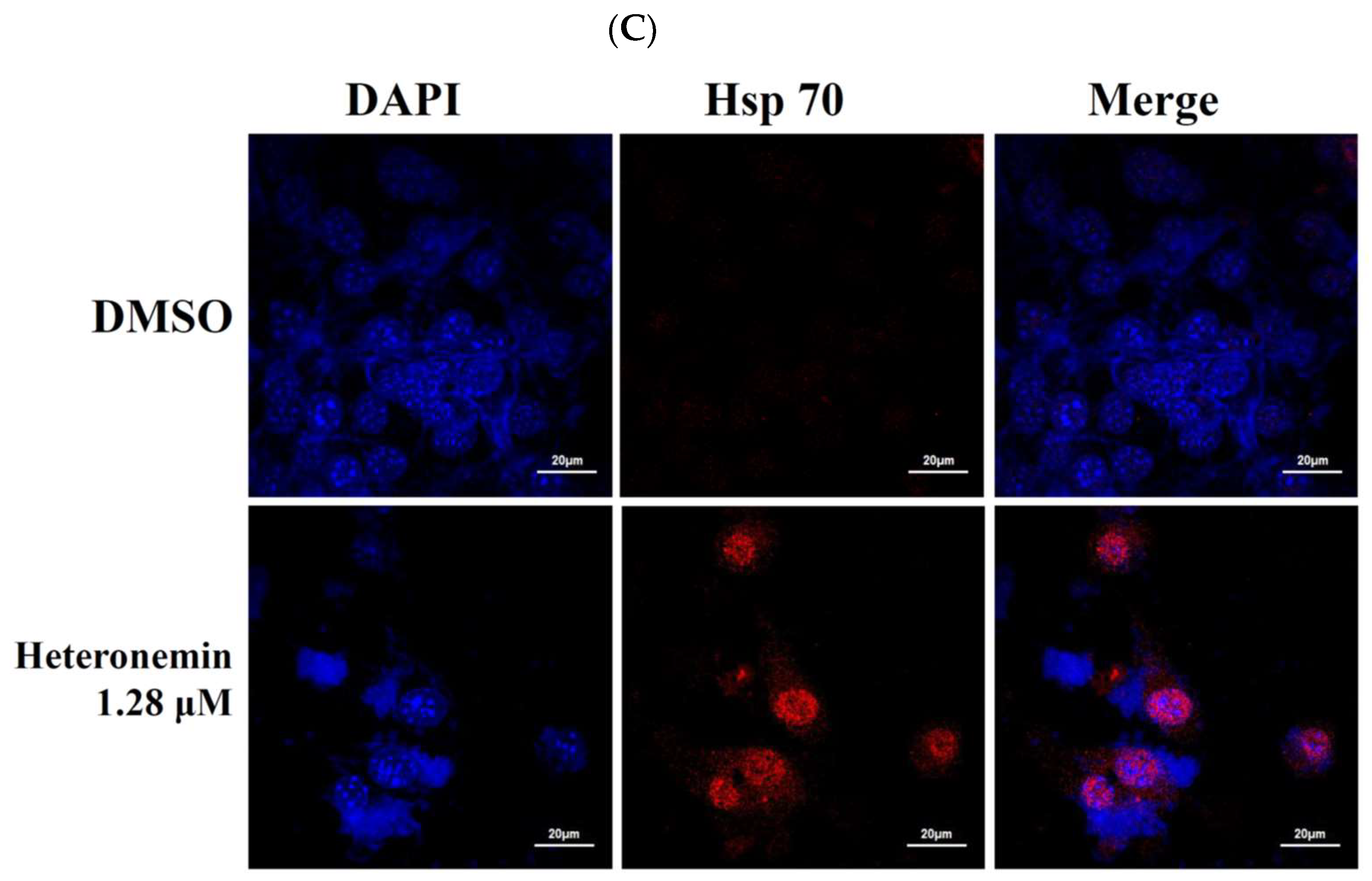
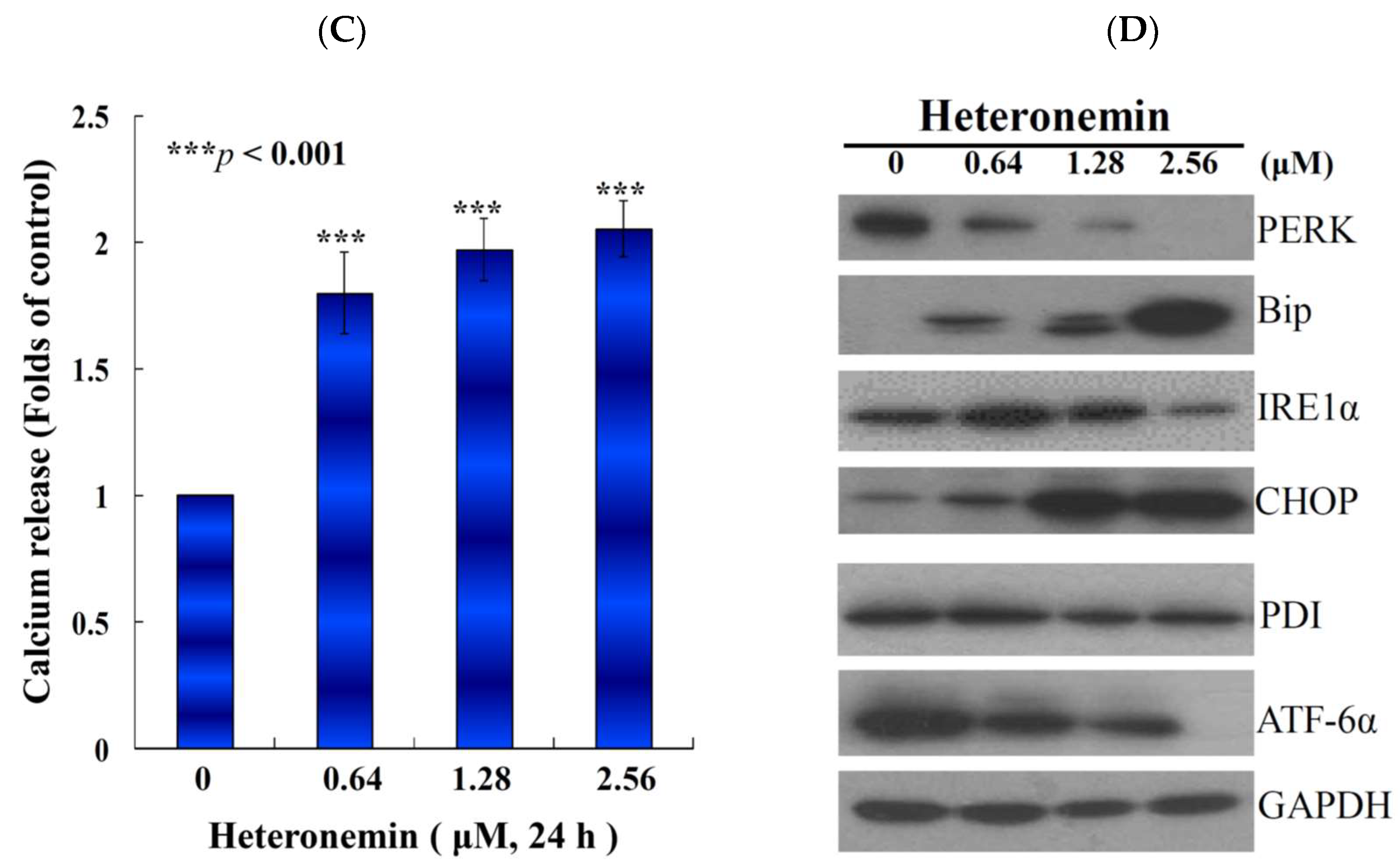
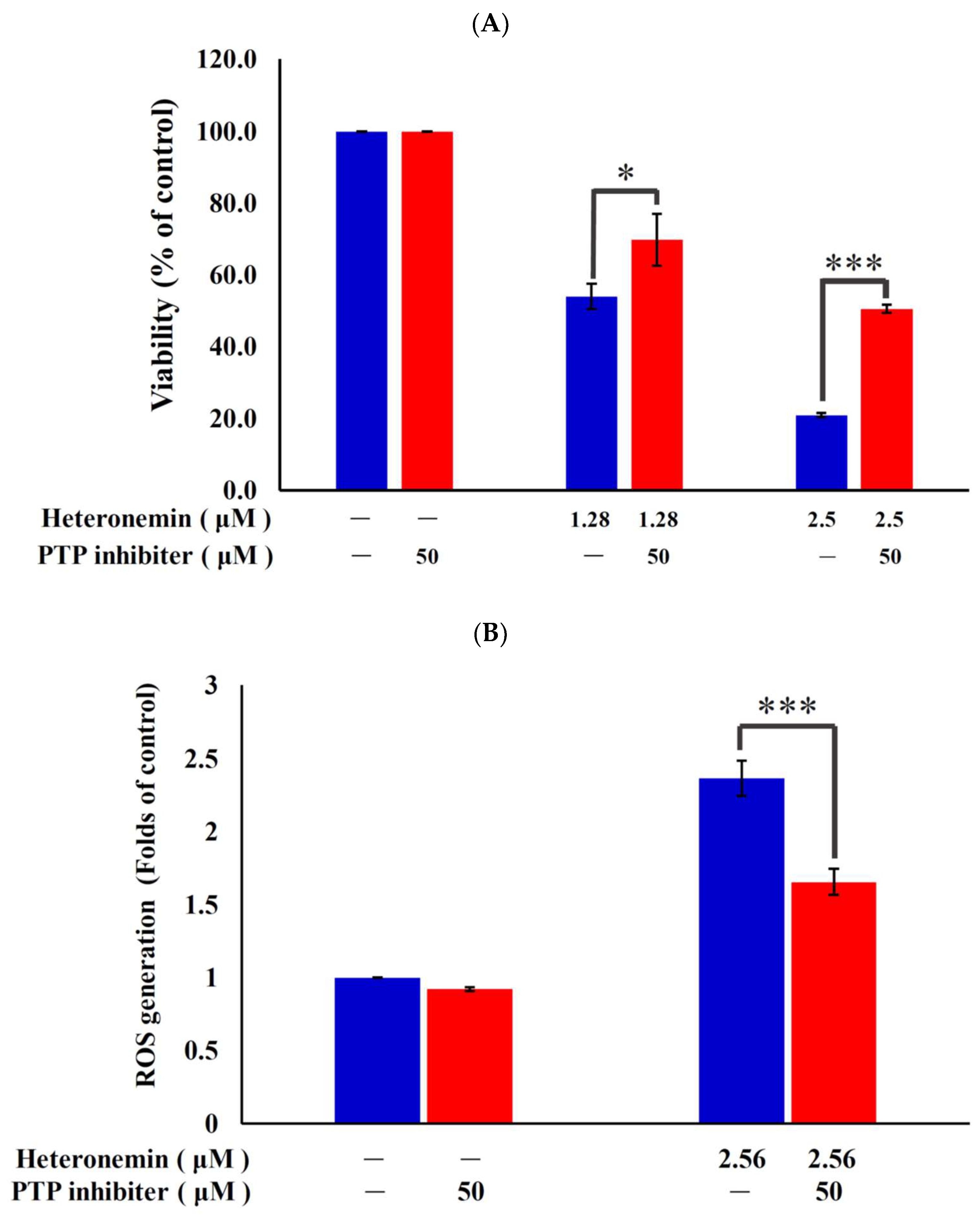
2.6. Effect of Heteronemin on Reactive Oxygen Species (ROS) Generation and ER Stress in LNcap Cells
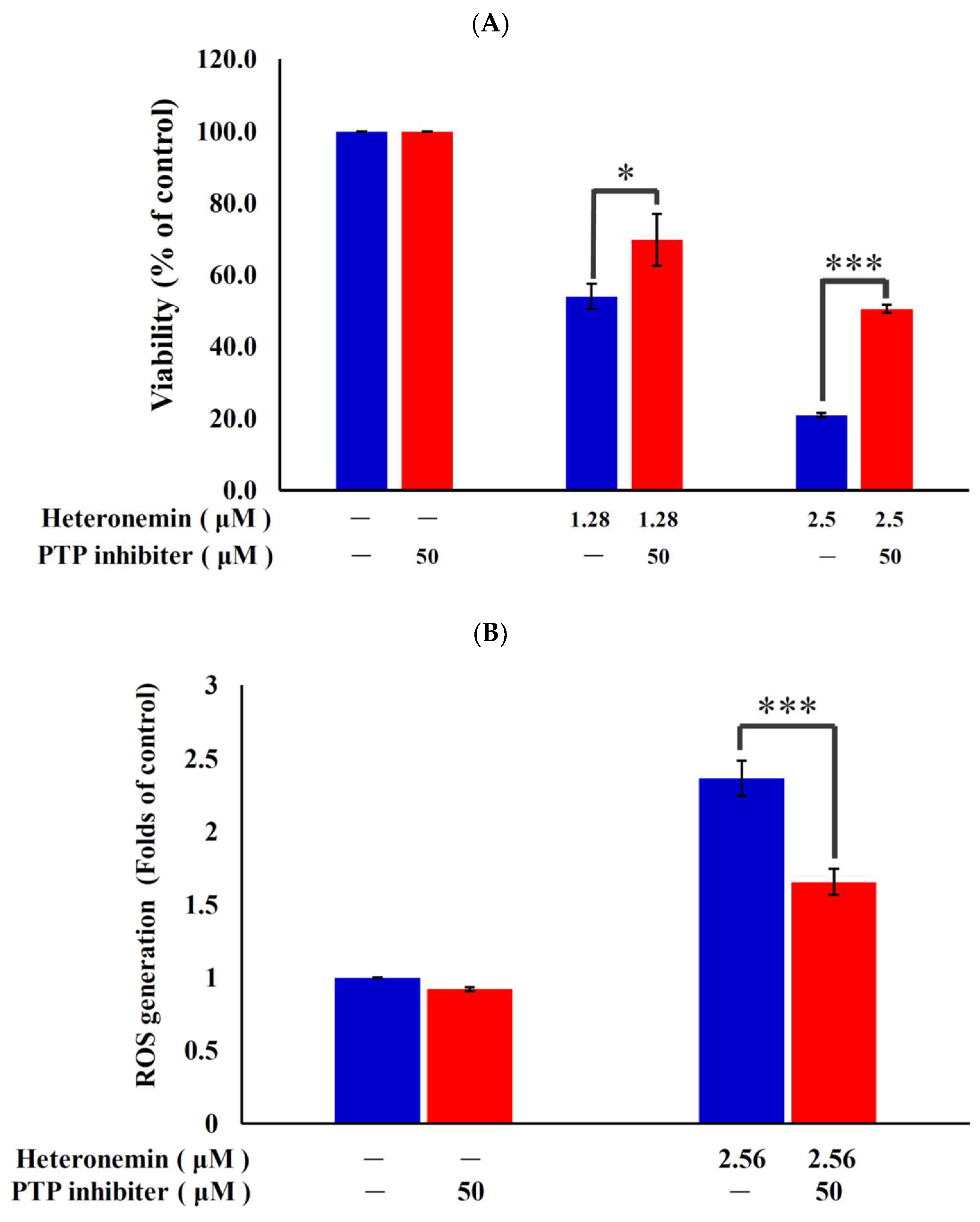
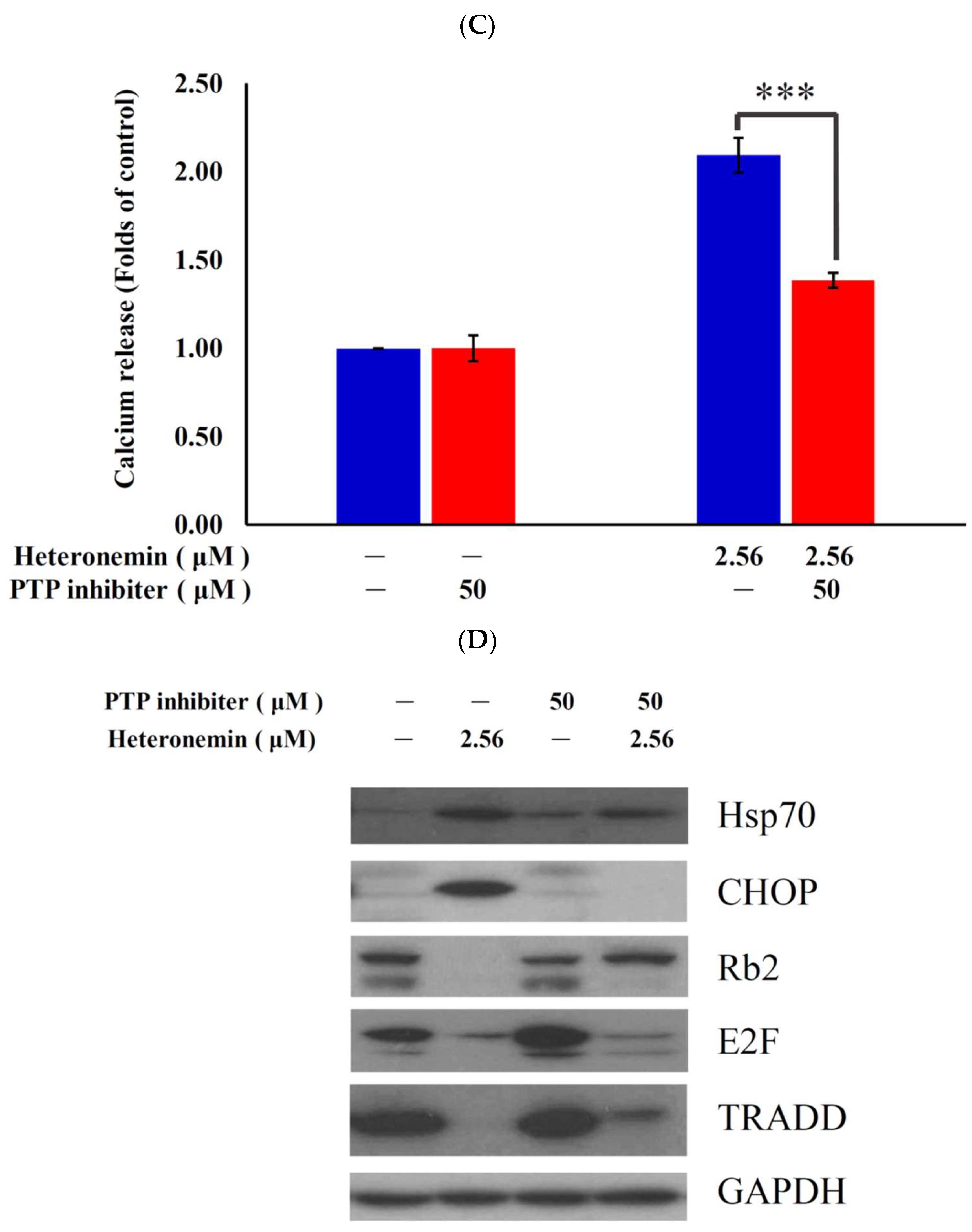
2.7. Antitumor Effect of Heteronemin Involves Protein Tyrosine Phosphatases PTP Activation
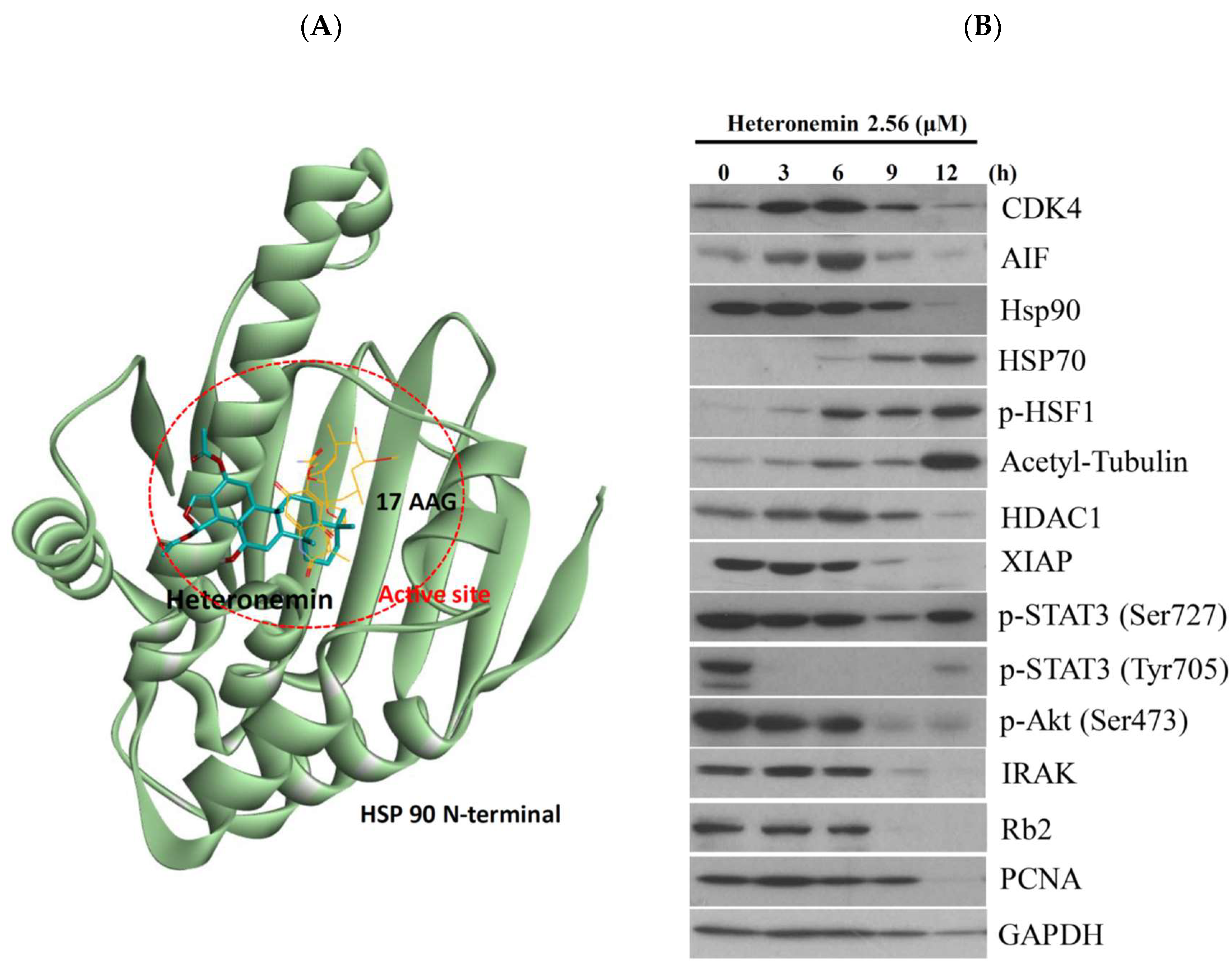
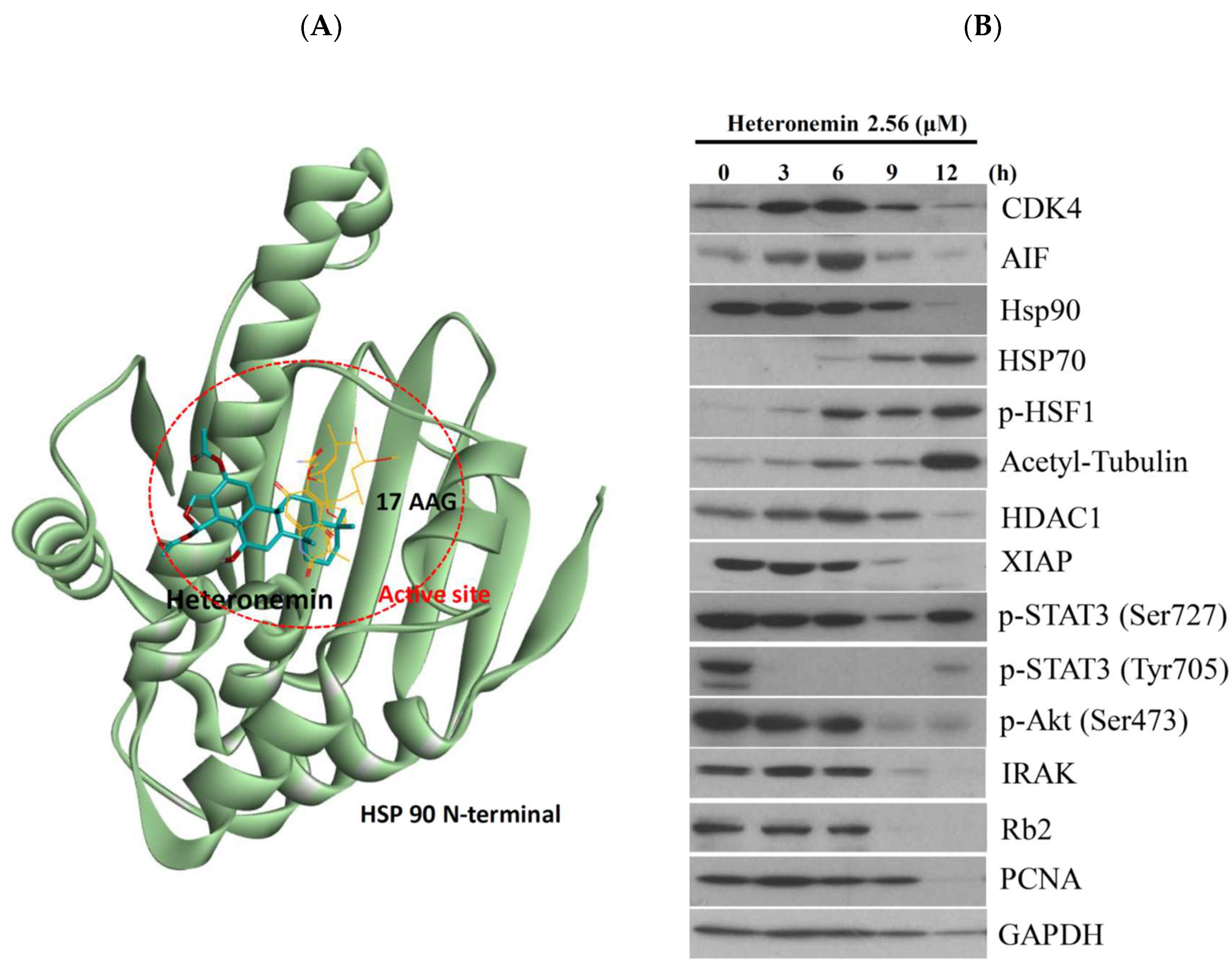
2.8. Heteronemin Acts as Potent Hsp90 Inhibitor
3. Discussion
4. Experimental Section
4.1. Bioassay Chemicals and Biological Materials
4.2. Stock Solution of Heteronemin
4.3. MTT Cell Proliferation Assay
4.4. Annexin V/PI Apoptotic Assay
4.5. Determination of ROS Generation, Calcium Accumulation, and MMP Disruption
4.6. Immunofluorescence Analysis
4.7. Topoisomerase I and II Catalytic Inhibitors and Poisons
4.8. Xenograft Animal Model with Human Prostate LNcap Cells
4.9. Molecular Docking
4.10. Statistics
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Nagle, D.G.; Zhou, Y.D. Marine Natural Products as Inhibitors of Hypoxic Signaling in Tumors. Phytochem. Rev. 2009, 8, 415–429. [Google Scholar] [CrossRef] [PubMed]
- Villa, F.A.; Gerwick, L. Marine natural product drug discovery: Leads for treatment of inflammation, cancer, infections, and neurological disorders. Immunopharmacol. Immunotoxicol. 2010, 32, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, S.; Hodges, T.W.; Rajbhandari, I.; Gerwick, W.H.; Hamann, M.T.; Nagle, D.G. Marine natural products as novel antioxidant prototypes. J. Nat. Prod. 2003, 66, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Gademann, K.; Kobylinska, J. Antimalarial natural products of marine and freshwater origin. Chem. Rec. 2009, 9, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Salma, Y.; Lafont, E.; Therville, N.; Carpentier, S.; Bonnafe, M.J.; Levade, T.; Génisson, Y.; Andrieu-Abadie, N. The natural marine anhydrophytosphingosine, Jaspine, B.; induces apoptosis in melanoma cells by interfering with ceramide metabolism. Biochem. Pharmacol. 2009, 78, 477–485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, Z.; Wang, H. Cytotoxic Natural Products from Marine Sponge-Derived Microorganisms. Mar. Drugs 2017, 15, 68. [Google Scholar] [CrossRef] [PubMed]
- Kamel, H.N.; Kim, Y.B.; Rimoldi, J.M.; Fronczek, F.R.; Ferreira, D.; Ferreira, D.; Slattery, M. Scalarane sesterterpenoids: Semisynthesis and biological activity. J. Nat. Prod. 2009, 72, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.C.; Tseng, S.W.; Liu, L.L.; Chou, Y.; Ho, Y.S.; Lu, M.C.; Su, J.H. Cytotoxic sesterterpenoids from a sponge Hippospongia sp. Mar. Drugs 2012, 10, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Okamoto, T.; Hayashi, K.; Yokoyama, N.; Sasaki, T.; Kitagawa, I. Marine natural products. XXXII. Absolute configurations of C-4 of the manoalide family, biologically active sesterterpenes from the marine sponge Hyrtios erecta. Chem. Pharm. Bull. 1994, 42, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.A. Scalarane Sesterterpenoids. Curr. Bioact. Compd. 2010, 6, 178–206. [Google Scholar] [CrossRef]
- Wu, S.Y.; Sung, P.J.; Chang, Y.L.; Pan, S.L.; Teng, C.M. Heteronemin, a Spongean Sesterterpene, Induces Cell Apoptosis and Autophagy in Human Renal Carcinoma Cells. Biomed. Res. Int. 2015, 2015, 738241. [Google Scholar] [CrossRef] [PubMed]
- Wonganuchitmeta, S.N.; Yuenyongsawad, S.; Keawpradub, N.; Plubrukarn, A. Antitubercular sesterterpenes from the Thai sponge Brachiaster sp. J. Nat. Prod. 2004, 67, 1767–1770. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, M.; Cerella, C.; Eifes, S.; Chateauvieux, S.; Morceau, F.; aspars, M.; Dicato, M.; Diederich, M. Heteronemin, a spongean sesterterpene, inhibits TNF alpha-induced NF-kappa B activation through proteasome inhibition and induces apoptotic cell death. Biochem. Pharmacol. 2010, 79, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Folmer, F.; Jaspars, M.; Schumacher, M.; Dicato, M.; Diederich, M. Marine natural products targeting phospholipases A2. Biochem. Pharmacol. 2010, 80, 1793–1800. [Google Scholar] [CrossRef] [PubMed]
- Kamal, A.; Nayak, V.L.; Bagul, C.; Vishnuvardhan, M.V.; Mallareddy, A. Investigation of the mechanism and apoptotic pathway induced by 4beta cinnamido linked podophyllotoxins against human lung cancer cells A549. Apoptosis 2015, 20, 1518–1529. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C.; Wang, C.T.; Hung, H.C.; Wu, W.J.; Wu, D.C.; Chang, M.C.; Sung, P.J.; Chou, Y.W.; Wen, Z.H.; Tai, M.H. Heteronemin Is a Novel c-Met/STAT3 Inhibitor Against Advanced Prostate Cancer Cells. Prostate 2016, 76, 1469–1483. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Her, C. Inhibition of Topoisomerase (DNA) I (TOP1): DNA Damage Repair and Anticancer Therapy. Biomolecules 2015, 5, 1652–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitiss, J.L. DNA topoisomerase, I.I. and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Nitiss, J.L. Targeting DNA topoisomerase, I.I. in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Shih, H.C.; El-Shazly, M.; Juan, Y.S.; Chang, C.Y.; Su, J.H.; Chen, Y.C.; Shih, S.P.; Chen, H.M.; Wu, Y.C.; Lu, M.C. Cracking the cytotoxicity code: Apoptotic induction of 10-acetylirciformonin B is mediated through ROS generation and mitochondrial dysfunction. Mar. Drugs 2014, 12, 3072–3090. [Google Scholar] [CrossRef] [PubMed]
- Doi, Y.; Shigemori, H.; Ishibashi, M.; Mizobe, F.; Kawashima, A.; Nakaike, S.; Kobayashi, J.I. New sesterterpenes with nerve growth factor synthesis-stimulating activity from the Okinawan marine sponge Hyrtios sp. Chem. Pharm. Bull. 1993, 41, 2190–2191. [Google Scholar] [CrossRef] [PubMed]
- Shih, S.P.; Lee, M.G.; El-Shazly, M.; Juan, Y.S.; Wen, Z.H.; Du, Y.C.; Su, J.H.; Sung, P.J.; Chen, Y.C.; Yang, J.C.; et al. Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities. Mar. Drugs 2015, 13, 3132–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kania, E.; Pajak, B.; Orzechowski, A. Calcium homeostasis and, E.R. stress in control of autophagy in cancer cells. Biomed. Res. Int. 2015, 2015, 352794. [Google Scholar] [CrossRef] [PubMed]
- Ichas, F.; Jouaville, L.S.; Mazat, J.P. Mitochondria are excitable organelles capable of generating and conveying electrical and calcium signals. Cell 1997, 89, 1145–1153. [Google Scholar] [CrossRef]
- Kato, H.; Nishitoh, H. Stress responses from the endoplasmic reticulum in cancer. Front. Oncol. 2015, 5, 93. [Google Scholar] [CrossRef] [PubMed]
- Thastrup, O.; Cullen, P.J.; Drobak, B.K.; Hanley, M.R.; Dawson, A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. USA 1990, 87, 2466–2470. [Google Scholar] [CrossRef] [PubMed]
- Morales, L.D.; Pena, K.; Kim, D.J.; Lieman, J.H. SHP-2 and PTP-pest induction during Rb-E2F associated apoptosis. Cell. Mol. Biol. Lett. 2012, 17, 422–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Zhang, Y.; Ge, L.; Lin, Y.; Kwok, H.F. The Roles of Protein Tyrosine Phosphatases in Hepatocellular Carcinoma. Cancers 2018, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Hellmuth, K.; Grosskopf, S.; Lum, C.T.; Wurtele, M.; Roder, N.; von Kries, J.P.; Rosario, M.; Rademann, J.; Birchmeier, W. Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. Proc. Natl. Acad. Sci. USA 2008, 105, 7275–7280. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Su, Z.; Xu, H.; Niu, M.; Chen, X.; Min, H.; Zhang, B.; Sun, G.; Xie, S.; Wang, H.; et al. Luteolin selectively kills STAT3 highly activated gastric cancer cells through enhancing the binding of STAT3 to SHP-1. Cell Death Dis. 2017, 8, e2612. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.H.; Liu, Y.C.; Su, J.H.; El-Shazly, M.; Wu, C.F.; Du, Y.C.; Hsu, Y.M.; Yang, J.C.; Weng, M.K.; Chou, C.H.; et al. Antileukemic Scalarane Sesterterpenoids and Meroditerpenoid from Carteriospongia (Phyllospongia) sp., Induce Apoptosis via Dual Inhibitory Effects on Topoisomerase, I.I. and Hsp90. Sci. Rep. 2016, 6, 36170. [Google Scholar] [CrossRef] [PubMed]
- Schramm, C.; Edwards, M.A.; Krenz, M. New approaches to prevent LEOPARD syndrome-associated cardiac hypertrophy by specifically targeting Shp2-dependent signaling. J. Biol. Chem. 2013, 288, 18335–18344. [Google Scholar] [CrossRef] [PubMed]
- Irandoust, M.; van den Berg, T.K.; Kaspers, G.J.; Cloos, J. Role of tyrosine phosphatase inhibitors in cancer treatment with emphasis on SH2 domain-containing tyrosine phosphatases (SHPs). Anticancer Agents Med. Chem. 2009, 9, 212–220. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Yu, Z.H.; Zhang, R.Y.; Wu, L.; Gunawan, A.M.; Lane, B.S.; Shim, J.S.; Zeng, L.F.; He, Y.; Chen, L.; et al. Exploring the Existing Drug Space for Novel pTyr Mimetic and SHP2 Inhibitors. ACS Med. Chem. Lett. 2015, 6, 782–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chun, J.; Li, R.J.; Cheng, M.S.; Kim, Y.S. Alantolactone selectively suppresses STAT3 activation and exhibits potent anticancer activity in MDA-MB-231 cells. Cancer Lett. 2015, 357, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.M.; Lawrence, H.R.; Sebti, S.M.; Lawrence, N.J.; Wu, J. Targeting protein tyrosine phosphatases for anticancer drug discovery. Curr. Pharm. Des. 2010, 16, 1843–1862. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Comoglio, P.M.; Trusolino, L. Beta4 integrin activates a Shp2-Src signaling pathway that sustains HGF-induced anchorage-independent growth. J. Cell Biol. 2006, 175, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Ostman, A.; Hellberg, C.; Bohmer, F.D. Protein-tyrosine phosphatases and cancer. Nat. Rev. Cancer 2006, 6, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Andersen, J.N.; Jansen, P.G.; Echwald, S.M.; Mortensen, O.H.; Fukada, T.; Del Vecchio, R.; Tonks, N.K.; Møller, N.P.H. A genomic perspective on protein tyrosine phosphatases: Gene structure, pseudogenes, and genetic disease linkage. FASEB J. 2004, 18, 8–30. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Xu, F.; Luo, R.; Zheng, Q.; Lu, J.; Yang, Y.; Qin, T.; Yuan, Z.; Shi, Y.; Jiang, W.; et al. The prognostic significance of topoisomerase, I.I. alpha protein in early stage luminal breast cancer. BMC Cancer 2018, 18, 331. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Sun, Y.; Ji, P.; Kopetz, S.; Zhang, W. Topoisomerase IIalpha in chromosome instability and personalized cancer therapy. Oncogene 2015, 34, 4019–4031. [Google Scholar] [CrossRef] [PubMed]
- Wall, M.E.; Wani, M.C. Camptothecin and taxol: Discovery to clinic--thirteenth Bruce F. Cain Memorial Award Lecture. Cancer Res. 1995, 55, 753–760. [Google Scholar] [PubMed]
- Pommier, Y. DNA topoisomerase I inhibitors: Chemistry, biology, and interfacial inhibition. Chem. Rev. 2009, 109, 2894–2902. [Google Scholar] [CrossRef] [PubMed]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Tu, J.; Dou, C.; Zhang, J.; Yang, L.; Liu, X.; Lei, K.; Liu, Z.; Wang, Y.; Li, L.; et al. HSP90 promotes cell glycolysis, proliferation and inhibits apoptosis by regulating PKM2 abundance via Thr-328 phosphorylation in hepatocellular carcinoma. Mol. Cancer 2017, 16, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saini, J.; Sharma, P.K. Clinical, Prognostic and Therapeutic Significance of Heat Shock Proteins in Cancer. Curr. Drug Targets 2017. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Liu, T.; Rios, Z.; Mei, Q.; Lin, X.; Cao, S. Heat Shock Proteins and Cancer. Trends Pharmacol. Sci. 2017, 38, 226–256. [Google Scholar] [CrossRef] [PubMed]
- Roe, S.M.; Prodromou, C.; O’Brien, R.; Ladbury, J.E.; Piper, P.W.; Pearl, L.H. Structural basis for inhibition of the Hsp90 molecular chaperone by the antitumor antibiotics radicicol and geldanamycin. J. Med. Chem. 1999, 42, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Taipale, M.; Jarosz, D.F.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Usmani, S.Z.; Bona, R.; Li, Z. 17 AAG for HSP90 inhibition in cancer—From bench to bedside. Curr. Mol. Med. 2009, 9, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.A.; Hostein, I.; Banerji, U.; Stefano, F.D.; Maloney, A.; Walton, M.; Judson, I.; Workman, P. Gene expression profiling of human colon cancer cells following inhibition of signal transduction by 17-allylamino-17-demethoxygeldanamycin, an inhibitor of the hsp90 molecular chaperone. Oncogene 2000, 19, 4125–4133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacey, S.; Gore, M.; Chao, D.; Banerji, U.; Larkin, J.; Sarker, S.; Owen, K.; Asad, Y.; Raynaud, F.; Walton, M.; et al. A Phase, I.I. trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Investig. New Drugs 2012, 30, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Kopf, S.; Viola, K.; Atanasov, A.G.; Jarukamjorn, K.; Rarova, L.; Kretschy, N.; Teichmann, M.; Vonach, C.; Saiko, P.; Giessrigl, B.; et al. In vitro characterisation of the anti-intravasative properties of the marine product heteronemin. Arch. Toxicol. 2013, 87, 1851–1861. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.H.; Kuo, S.M.; Wu, Y.J.; Su, J.H. Improvement and enhancement of antibladder carcinoma cell effects of heteronemin by the nanosized hyaluronan aggregation. Int. J. Nanomed. 2016, 11, 1237–1251. [Google Scholar] [CrossRef] [PubMed]
- Su, J.H.; Chen, Y.C.; El-Shazly, M.; Du, Y.C.; Su, C.W.; Tsao, C.W.; Liu, L.L.; Chou, Y.; Chang, W.B.; Su, Y.D.; et al. Towards the small and the beautiful: A small dibromotyrosine derivative from Pseudoceratina sp. sponge exhibits potent apoptotic effect through targeting IKK/NFkappaB signaling pathway. Mar. Drugs 2013, 11, 3168–3185. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Xu, M.L.; Lee, C.S.; Woo, M.H.; Chang, H.W.; Son, J.K. Cytotoxicity and DNA topoisomerases inhibitory activity of constituents from the sclerotium of Poria cocos. Arch. Pharm. Res. 2004, 27, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stebbins, C.E.; Russo, A.A.; Schneider, C.; Rosen, N.; Hartl, F.U.; Pavletich, N.P. Crystal structure of an Hsp90-geldanamycin complex: Targeting of a protein chaperone by an antitumor agent. Cell 1997, 89, 239–250. [Google Scholar] [CrossRef]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph Model 1999, 17, 57–61. [Google Scholar] [PubMed]
- Hubert, J.G.; Furkert, D.P.; Brimble, M.A. Preparation of cis-gamma-hydroxycarvone derivatives for synthesis of sesterterpenoid natural products: Total synthesis of phorbin A. J. Org. Chem. 2015, 80, 2231–2239. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.-G.; Liu, Y.-C.; Lee, Y.-L.; El-Shazly, M.; Lai, K.-H.; Shih, S.-P.; Ke, S.-C.; Hong, M.-C.; Du, Y.-C.; Yang, J.-C.; et al. Heteronemin, a Marine Sesterterpenoid-Type Metabolite, Induces Apoptosis in Prostate LNcap Cells via Oxidative and ER Stress Combined with the Inhibition of Topoisomerase II and Hsp90. Mar. Drugs 2018, 16, 204. https://doi.org/10.3390/md16060204
Lee M-G, Liu Y-C, Lee Y-L, El-Shazly M, Lai K-H, Shih S-P, Ke S-C, Hong M-C, Du Y-C, Yang J-C, et al. Heteronemin, a Marine Sesterterpenoid-Type Metabolite, Induces Apoptosis in Prostate LNcap Cells via Oxidative and ER Stress Combined with the Inhibition of Topoisomerase II and Hsp90. Marine Drugs. 2018; 16(6):204. https://doi.org/10.3390/md16060204
Chicago/Turabian StyleLee, Man-Gang, Yi-Chang Liu, Yi-Lun Lee, Mohamed El-Shazly, Kuei-Hung Lai, Shou-Ping Shih, Seng-Chung Ke, Ming-Chang Hong, Ying-Chi Du, Juan-Cheng Yang, and et al. 2018. "Heteronemin, a Marine Sesterterpenoid-Type Metabolite, Induces Apoptosis in Prostate LNcap Cells via Oxidative and ER Stress Combined with the Inhibition of Topoisomerase II and Hsp90" Marine Drugs 16, no. 6: 204. https://doi.org/10.3390/md16060204