Synthesis of Alkyl-Glycerolipids Standards for Gas Chromatography Analysis: Application for Chimera and Shark Liver Oils

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
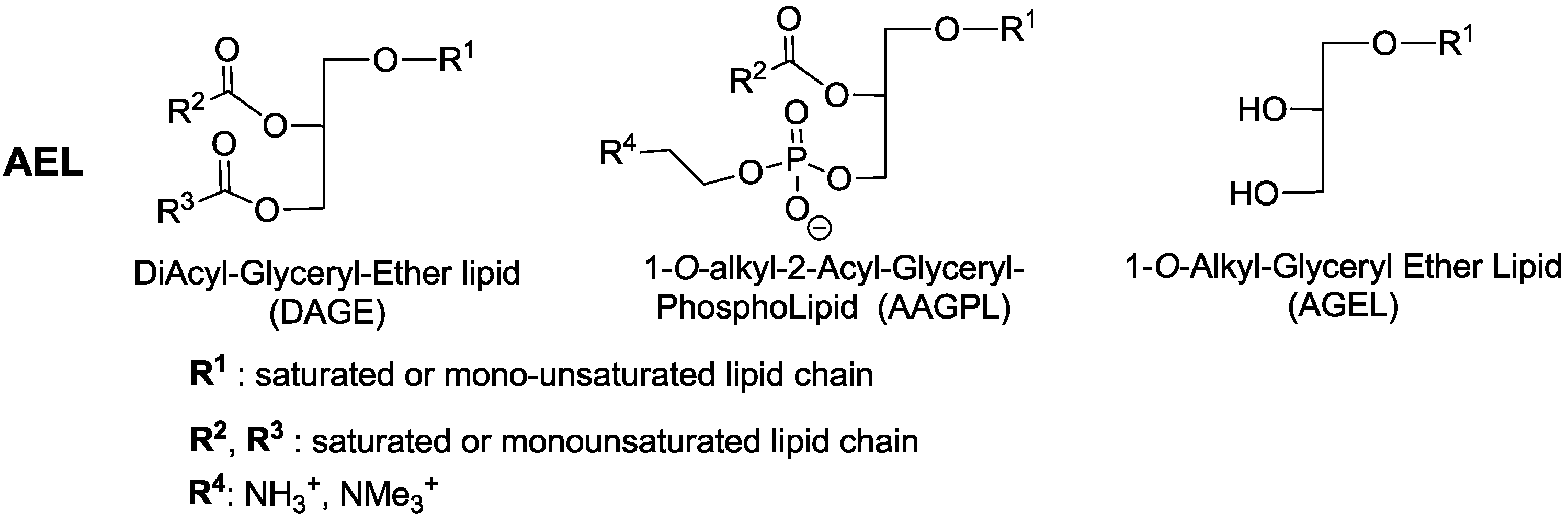
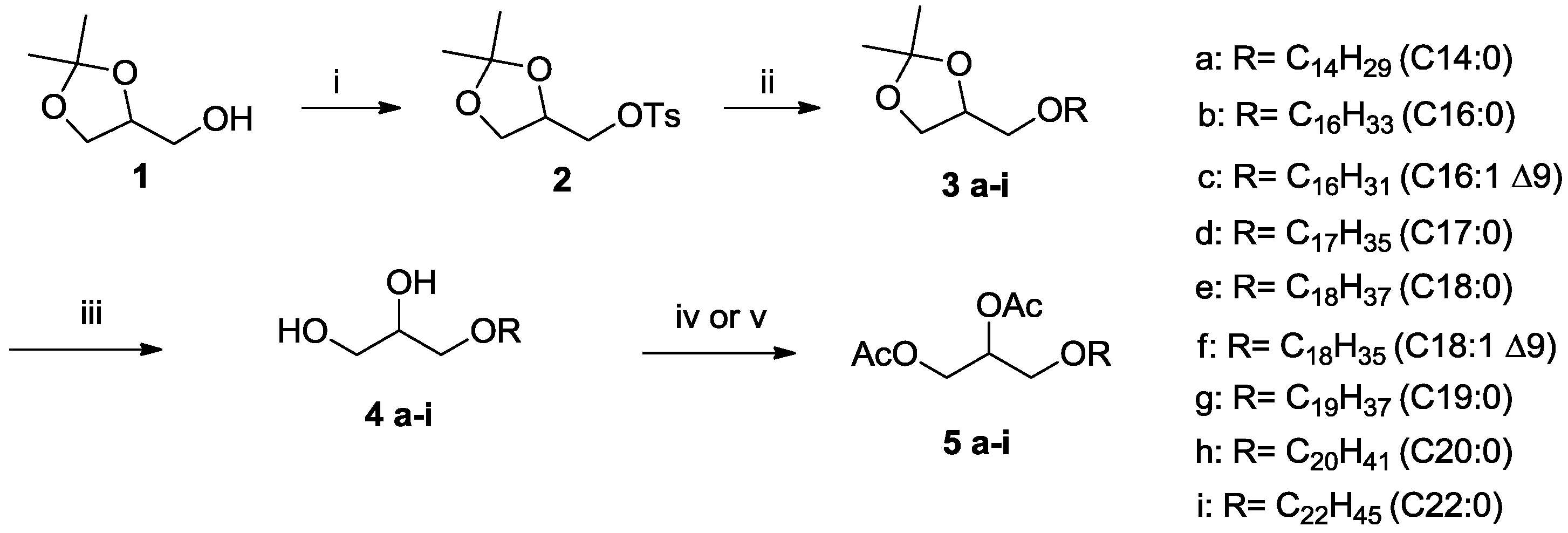
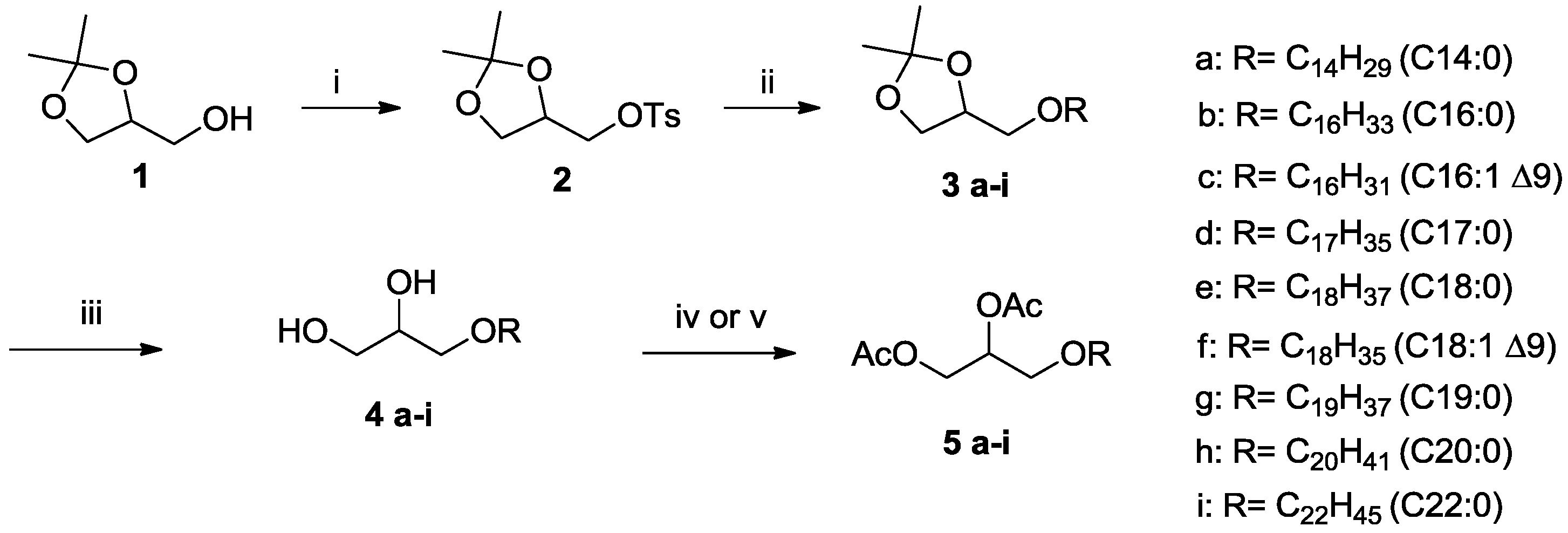
2.1. Alkyl-Glycerolipids Chemical Synthesis
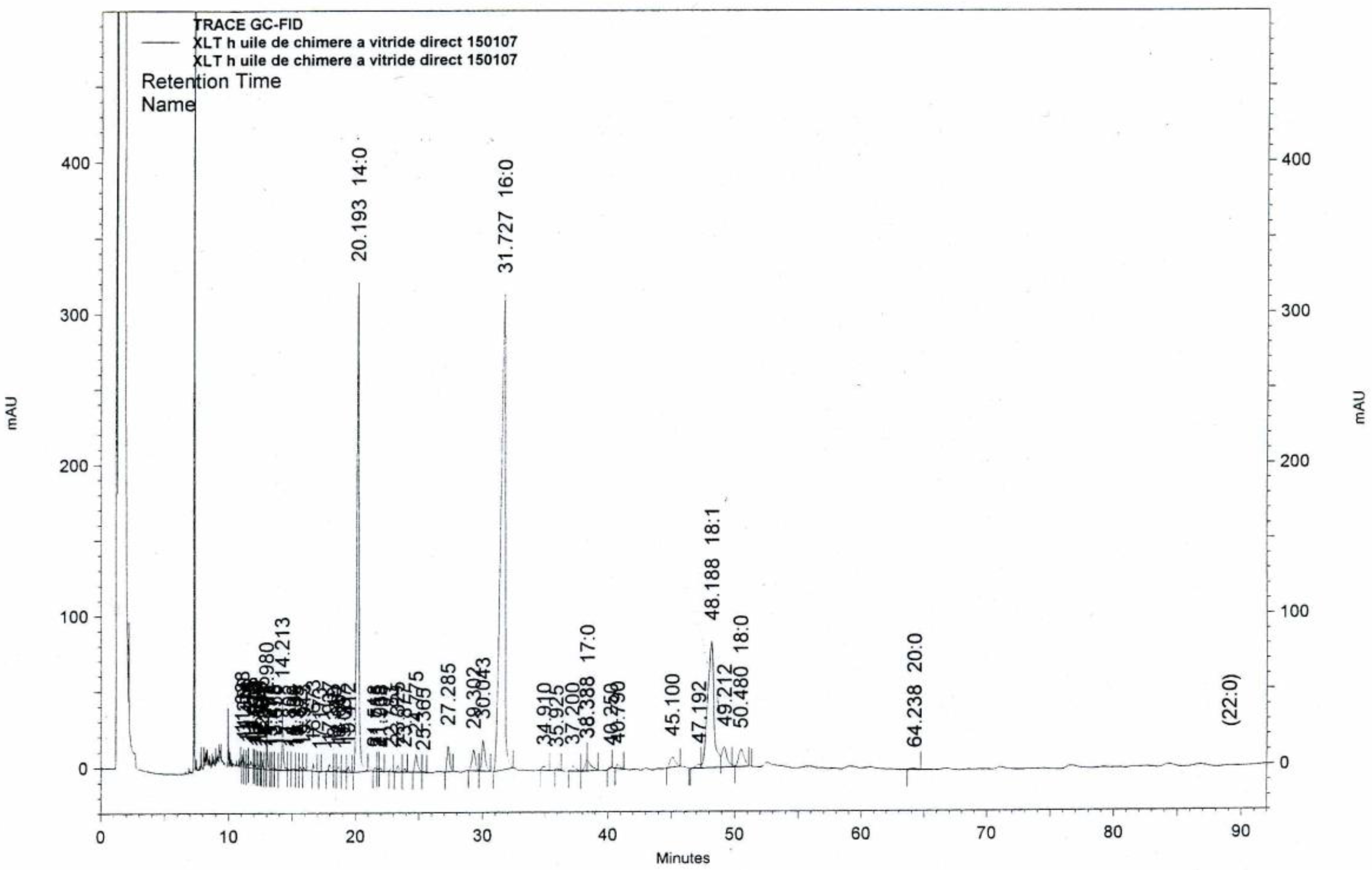
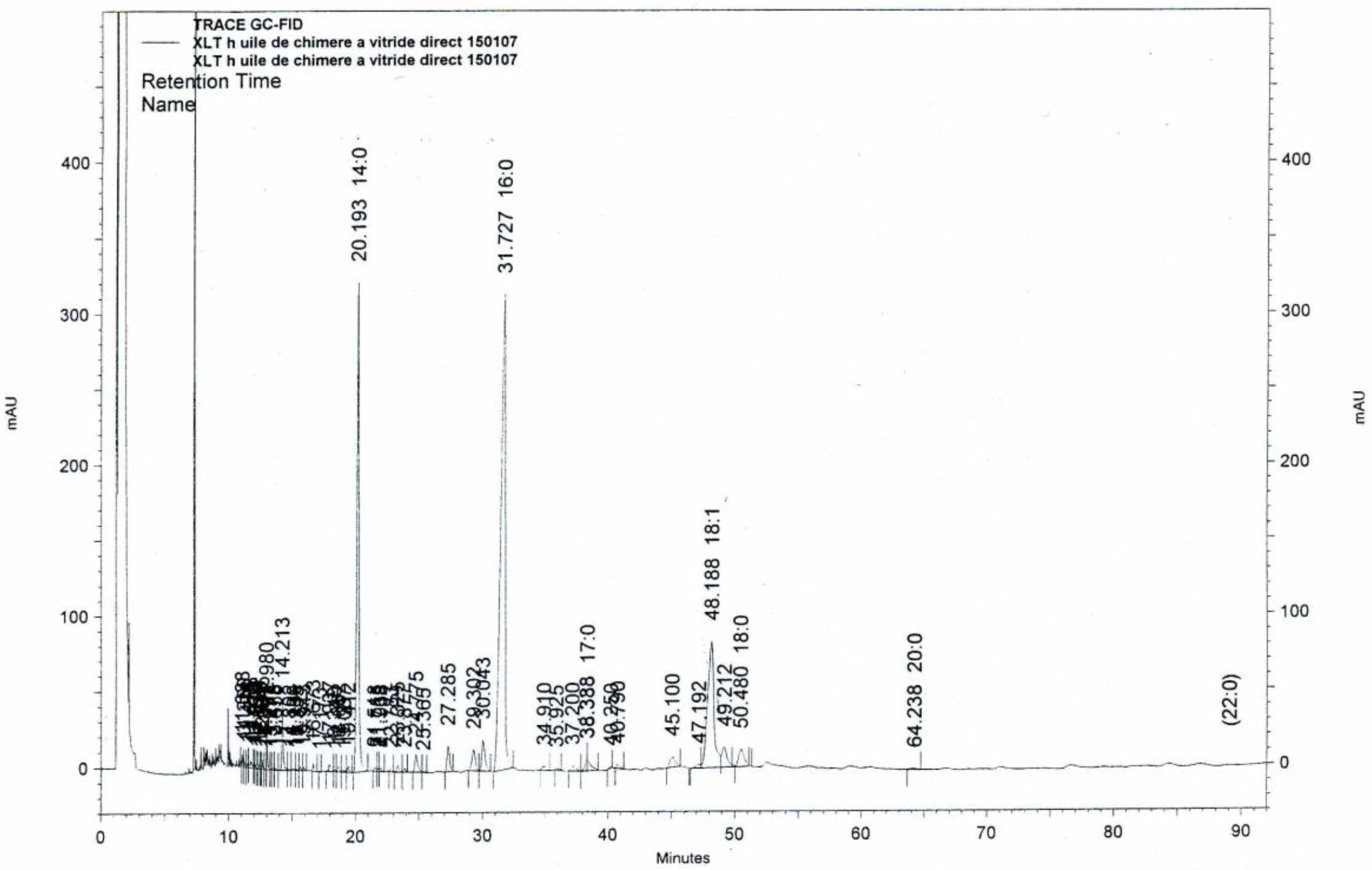
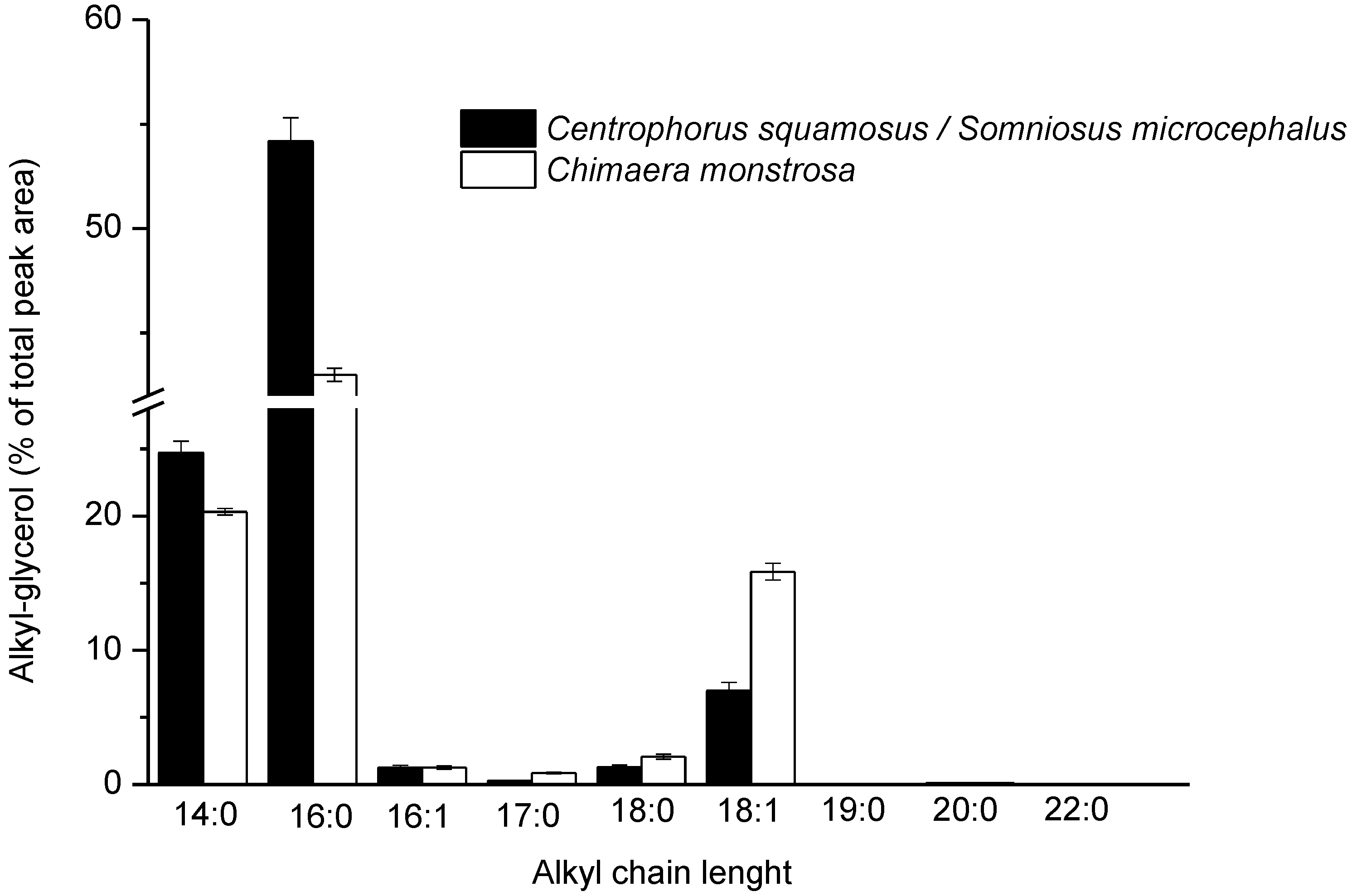
2.2. Liver Oils Alkyl-Glycerolipids Analysis
3. Materials and Methods
3.1. Chemical Synthesis
3.2. Analytical Chemistry
3.3. Mass Spectrometry
Optimization of the Separation Method
3.4. Experimentation Section
3.4.1. Preparation of the (2,2-Dimethyl-1,3-dioxolan-4-yl)methyl 4-methylbenzenesulfonate (2)
3.4.2. Preparation of 4-Alkoxymethyl-2,2-dimethyl-1,3-dioxolanes (3a–i)
3.4.3. Preparation of the Ether-Glycerol 4a-i by Hydrolysis of Acetal 3a–i
3.4.4. Derivation Procedure: Preparation of 3-(Alkoxy)propane-1,2-diacetate
3.5. Analytical Chemistry
Analytical Biochemistry
3.6. Validation of the Analytical Procedure
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Magnusson, C.D.; Haraldsson, G.G. Ether lipids. Chem. Phys. Lipids 2011, 164, 315–340. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.Y.; Jadhav, L.S. Effects of dietary alkylglycerols in lactating rats on immune responses in pups. Pediatr. Res. 1994, 36, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Linman, J.W.; Bethell, F.H.; Long, M.J. The erythropoietic simulatory activity of batyl alcohol. J. Lab. Clin. Med. 1958, 52, 596–604. [Google Scholar] [PubMed]
- Brohult, A.; Brohult, J.; Brohult, S. Regression of tumour growth after administration of alkoxyglycerols. Acta Obstet. Gynecol. Scand. 1978, 57, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Brohult, A.; Brohult, J.; Brohult, S.; Joelsson, I. Effect of alkoxyglycerols on the frequency of injuries following radiation therapy for carcinoma of the uterine cervix. Acta Obstet. Gynecol. Scand. 1977, 56, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Pedrono, F.; Martin, B.; Leduc, C.; Le Lan, J.; Saiag, B.; Legrand, P.; Moulinoux, J.P.; Legrand, A.B. Natural alkylglycerols restrain growth and metastasis of grafted tumors in mice. Nutr. Cancer 2004, 48, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Mao, J.; Ai, J.; Deng, Y.; Roth, M.R.; Pound, C.; Henegar, J.; Welti, R.; Bigler, S.A. Identification of plasma lipid biomarkers for prostate cancer by lipidomics and bioinformatics. PLoS ONE 2012, 7, e48889. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, J. Platelet-activating factor, a new mediator of anaphylaxis and immune complex deposition from rabbit and human basophils. Nature 1974, 249, 581–582. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, J.; Vargaftig, B.B. Platelet-activating factor: An ether lipid with biological activity. In Ether Lipids: Biochemical and Biomedical Aspects; Mangold, H.K., Paltauf, F., Eds.; Academic Press: New York, NY, USA, 1983; pp. 355–376. [Google Scholar]
- Hichami, A.; Duroudier, V.; Leblais, V.; Vernhet, L.; Le Goffic, F.; Ninio, E.; Legrand, A. Modulation of platelet-activating-factor production by incorporation of naturally occurring 1-O-alkylglycerols in phospholipids of human leukemic monocyte-like THP-1 cells. Eur. J. Biochem. 1997, 250, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Clark, K.J.; Murray, A.W. Evidence that the bradykinin-induced activation of phospholipase D and of the mitogen-activated protein kinase cascade involve different protein kinase C isoforms. J. Biol. Chem. 1995, 270, 7097–7103. [Google Scholar] [CrossRef] [PubMed]
- Marigny, K.; Pedrono, F.; Martin-Chouly, C.A.; Youmine, H.; Saiag, B.; Legrand, A.B. Modulation of endothelial permeability by 1-O-alkylglycerols. Acta Physiol. Scand. 2002, 176, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Warne, T.R.; Buchanan, F.G.; Robinson, M. Growth-dependent accumulation of monoalkylglycerol in Madin-Darby canine kidney cells. Evidence for a role in the regulation of protein kinase C. J. Biol. Chem. 1995, 270, 11147–11154. [Google Scholar] [CrossRef] [PubMed]
- O’Brian, C.; Vogel, V.G.; Singletary, S.E.; Ward, N.E. Elevated protein kinase C expression in human breast tumor biopsies relative to normal breast tissue. Cancer Res. 1989, 49, 3215–3217. [Google Scholar] [PubMed]
- Daniel, L.W.; Small, G.W.; Schmitt, J.D.; Marasco, C.J.; Ishaq, K.; Piantadosi, C. Alkyl-linked diglycerides inhibit protein kinase C activation by diacylglycerols. Biochem. Biophys. Res. Commun. 1988, 151, 291–297. [Google Scholar] [CrossRef]
- Berggren, M.I.; Gallegos, A.; Dressler, L.A.; Modest, E.J.; Powis, G. Inhibition of the signalling enzyme phosphatidylinositol-3-kinase by antitumor ether lipid analogues. Cancer Res. 1993, 53, 4297–4302. [Google Scholar] [PubMed]
- Zhou, X.; Lu, X.; Richard, C.; Xiong, W.; Litchfield, D.W.; Bittman, R.; Arthur, G. 1-O-octadecyl-2-O-methyl-glycerophosphocholine inhibits the transduction of growth signals via the MAPK cascade in cultured MCF-7 cells. J. Clin. Investig. 1996, 98, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Oishi, K.; Shoji, M.; Eibl, H.; Berdel, W.E.; Hajdu, J.; Vogler, W.R.; Kuo, J.F. Inhibition of protein kinase C, (sodium plus potassium)-activated adenosine triphosphatase, and sodium pump by synthetic phospholipid analogues. Cancer Res. 1990, 50, 3025–3031. [Google Scholar] [PubMed]
- Besson, P.; Gore, J.; Vincent, E.; Hoinard, C.; Bougnoux, P. Inhibition of Na+/H+ exchanger activity by an alkyl-lysophospholipid analogue in a human breast cancer cell line. Biochem. Pharmacol. 1996, 51, 1153–1158. [Google Scholar] [CrossRef]
- Potier, M.; Chantome, A.; Joulin, V.; Girault, A.; Roger, S.; Besson, P.; Jourdan, M.L.; LeGuennec, J.Y.; Bougnoux, P.; Vandier, C. The SK3/K(Ca)2.3 potassium channel is a new cellular target for edelfosine. Br. J. Pharmacol. 2011, 162, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.R.; Thomas, V.L.; Snyder, F. Inhibition of cellular transport systems by alkyl phospholipid analogs in HL-60 human leukemia cells. Biochim. Biophys. Acta 1992, 1127, 74–80. [Google Scholar] [CrossRef]
- Arthur, G.; Bittman, R. Glycosylated antitumor ether lipids: Activity and mechanism of action. Anti-Cancer Agents Med. Chem. 2014, 14, 592–606. [Google Scholar] [CrossRef]
- Samadder, P.; Xu, Y.; Schweizer, F.; Arthur, G. Cytotoxic properties of d-gluco-, d-galacto- and d-manno-configured 2-amino-2-deoxy-glycerolipids against epithelial cancer cell lines and BT-474 breast cancer stem cells. Eur. J. Med. Chem. 2014, 78, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Sevrain, C.M.; Haelters, J.P.; Chantome, A.; Couthon-Gourves, H.; Gueguinou, M.; Potier-Cartereau, M.; Vandier, C.; Jaffres, P.A. DiGalactosyl-Glycero-Ether Lipid: Synthetic approaches and evaluation as SK3 channel inhibitor. Org. Biomol. Chem. 2013, 11, 4479–4487. [Google Scholar] [CrossRef] [PubMed]
- Girault, A.; Haelters, J.P.; Potier-Cartereau, M.; Chantome, A.; Jaffres, P.A.; Bougnoux, P.; Joulin, V.; Vandier, C. Targeting SKCa Channels in Cancer: Potential New Therapeutic Approaches. Curr. Med. Chem. 2012, 19, 697–713. [Google Scholar] [CrossRef] [PubMed]
- Sevrain, C.M.; Haelters, J.P.; Chantome, A.; Couthon-Gourves, H.; Girault, A.; Vandier, C.; Jaffres, P.A. Glyco-Phospho-Glycero Ether Lipid (GPGEL): Synthesis and evaluation as Small Conductance Ca2+-Activated K+ Channels (SK3) inhibitor. MedChemComm 2012, 3, 1471–1478. [Google Scholar] [CrossRef]
- Gueguinou, M.; Harnois, T.; Crottes, D.; Uguen, A.; Deliot, N.; Gambade, A.; Chantome, A.; Haelters, J.P.; Jaffres, P.A.; Jourdan, M.L.; et al. SK3/TRPC1/Orai1 complex regulates SOCE-dependent colon cancer cell migration: A novel opportunity to modulate anti-EGFR mAb action by the alkyl-lipid Ohmline. Oncotarget 2016, 7, 36168–36184. [Google Scholar] [CrossRef] [PubMed]
- Berthe, W.; Sevrain, C.M.; Chantome, A.; Bouchet, A.M.; Gueguinou, M.; Fourbon, Y.; Potier-Cartereau, M.; Haelters, J.P.; Couthon-Gourves, H.; Vandier, C.; et al. New Disaccharide-Based Ether Lipids as SK3 Ion Channel Inhibitors. ChemMedChem 2016, 11, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Chantome, A.; Potier-Cartereau, M.; Clarysse, L.; Fromont, G.; Marionneau-Lambot, S.; Gueguinou, M.; Pages, J.C.; Collin, C.; Oullier, T.; Girault, A.; et al. Pivotal role of the lipid Raft SK3-Orai1 complex in human cancer cell migration and bone metastases. Cancer Res. 2013, 73, 4852–4861. [Google Scholar] [CrossRef] [PubMed]
- Girault, A.; Haelters, J.P.; Potier-Cartereau, M.; Chantome, A.; Pinault, M.; Marionneau-Lambot, S.; Oullier, T.; Simon, G.; Couthon-Gourves, H.; Jaffres, P.A.; et al. New alkyl-lipid blockers of SK3 channels reduce cancer cell migration and occurrence of metastasis. Curr. Cancer Drug Targets 2011, 11, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Jaffres, P.A.; Gajate, C.; Bouchet, A.M.; Couthon-Gourves, H.; Chantome, A.; Potier-Cartereau, M.; Besson, P.; Bougnoux, P.; Mollinedo, F.; Vandier, C. Alkyl ether lipids, ion channels and lipid raft reorganization in cancer therapy. Pharmacol. Ther. 2016, 165, 114–131. [Google Scholar] [CrossRef] [PubMed]
- Herrera, F.E.; Sevrain, C.M.; Jaffrès, P.A.; Couthon, H.; Grélard, A.; Dufourc, E.J.; Chantôme, A.; Potier-Cartereau, M.; Vandier, C.; Bouchet, A.M. Singular Interaction between an Antimetastatic Agent and the Lipid Bilayer: The Ohmline Case. ACS Omega 2017, 2, 6361–6370. [Google Scholar] [CrossRef]
- Hallgren, B.; Larsson, S. The glycerol ethers in man and cow. J. Lipid Res. 1962, 3, 39–43. [Google Scholar]
- Hallgren, B.; Larsson, S. The glyceryl ethers in the liver oils of elasmobranch fish. J. Lipid Res. 1962, 3, 31–38. [Google Scholar]
- Hallgren, B.; Niklasson, A.; Stallberg, G.; Thorin, H. On the occurrence of 1-O-alkylgly-cerols and 1-O-(2-methoxyalkyl)glycerols in human colostrum, human milk, cow’s milk, sheep’s milk, human red bone marrow, red cells, blood plasma and a uterine carcinoma. Acta Chem. Scand. B 1974, 28, 1029–1034. [Google Scholar] [CrossRef] [PubMed]
- Snyder, F.; Wood, R. Alkyl and alk-1-enyl ethers of glycerol in lipids from normal and neoplastic human tissues. Cancer Res. 1969, 29, 251–257. [Google Scholar] [PubMed]
- Prinz, H.; Six, L.; Ruess, K.P.; Liefländer, M.L. Stereoselektive Synthese von langkettigen l-O-(β-D-Maltosyl)- 3-O-alkyl-sn-glycerinen (Alkylglycerylether-Lysoglycolipide). Liebigs Ann. Chem. 1985, 217–225. [Google Scholar]
- Aranda, G.; Lallemand, J.Y. Proposal for Modulable Photoaffinity Labelling. Synth. Commun. 1990, 20, 85–96. [Google Scholar] [CrossRef]
- Huang, Z.; Szoka, F.C. Sterol-Modified Phospholipids: Cholesterol and Phospholipid Chimeras with Improved Biomembrane Properties. J. Am. Chem. Soc. 2008, 130, 15702–15712. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.K.; Hajdu, J. Stereospecific Synthesis of 2-Substituted Ether Phospholipids. Synthesis 1989, 1, 16–20. [Google Scholar] [CrossRef]
- Nelson, W.L.; Wennerstrom, J.E.; Sankar, S.R. Absolute configuration of glycerol derivatives. 3. Synthesis and Cupra A circular dichroism spectra of some chiral 3-aryloxy-1,2-propanediols and 3-aryloxy-1-amino-2-propanols. Circ. Res. 1977, 42, 1006–1012. [Google Scholar]
- Cymerman, C.; Hamon, D.P.G. Studies Directed toward the Synthesis of Plasmalogens. II. (±)-cis- and -trans-3-(n-Hexadec-1’-enyloxy)-1,2-propanediol1. J. Org. Chem. 1965, 30, 4168–4175. [Google Scholar]
- Bonner, T.G.; McNamara, P. The Pyridine-catalysed Acetylation of Phenolsand Alcohols. J. Med. Soc. 1968, 0, 795–797. [Google Scholar]
- George, B.; Jamieson, R.; Reid, E.H. The analysis of oils and fats by gas chromatography : III. Separation factors of acetates, alcohols and hydrocarbons. J. Chromatogr. A 1967, 26, 8–16. [Google Scholar]
- Bordier, C.G.; Sellier, N.; Foucault, A.P.; Le Goffic, F. Purification and characterization of deep sea shark Centrophorus squamosus liver oil 1-O-alkylglycerol ether lipids. Lipids 1996, 31, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Spener, F.; Mangold, H.K. The alkyl moieties in wax esters and alkyl diacyl glycerols of sharks. J. Lipid Res. 1971, 12, 12–16. [Google Scholar] [PubMed]
- Deniau, A.L.; Mosset, P.; Pedrono, F.; Mitre, R.; Le Bot, D.; Legrand, A.B. Multiple beneficial health effects of natural alkylglycerols from shark liver oil. Mar. Drugs 2010, 8, 2175–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Kuksis, A. Fatty Acids and Glycerides. Handb. Lipid Res. 2012, 3, 126. [Google Scholar]
- Rapport, M.M. The discovery of plasmalogen structure. J. Lipid Res. 1984, 25, 1522–1527. [Google Scholar] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alkyl Chain R = | Retention Time (min) | Molecular Weight (g. × mol−1) | Most Characteristic Peaks (m/z, (%)) |
|---|---|---|---|
| C14H29 (C14:0) | 13.6 | 372.54 | 43 (base peak), 57 (60), 71 (35), 83 (30), 97 (26), 111 (13), 117 (19), 159 (119), 223 (4), 269 (6), 313 (35) |
| C16H31 (C16:1) | 15.0 | 398.58 | 43 (base peak), 57 (63), 69 (44), 81 (75), 95(39), 117 (31), 159 (63), 222 (15), 281 (12), 339 (25) |
| C16H33 (C16:0) | 15.7 | 400.59 | 43 (base peak), 57 (62), 71 (38), 83 (28), 97 (27), 111 (14), 159 (7), 219 (<5), 251 (5), 255 (<5), 297 (6), 341 (67) |
| C17H35 (C17:0) | 16.8 | 414.62 | 43 (base peak), 57 (62), 83 (28), 97 (27), 111 (15), 117 (22) 159 (25), 209 (<5), 267 (8), 311 (12), 355 (54) |
| C18H35 (C18:1) | 17.5 | 426.63 | 43 (base peak), 55 (52), 67 (48), 81(72), 95 (44), 117 (18), 159 (75), 191 (7), 219 (<5), 281 (12), 341 (7), 367 (25) |
| C18H37 (C18:0) | 17.8 | 428.65 | 43 (base peak), 57 (70), 71(45), 85 (34), 97 (33), 111 (18), 117 (27), 159 (27), 191 (<5), 281 (10), 325 (9), 369 (37) |
| C19H39 (C19:0) | 18.7 | 442.67 | 43 (base peak), 57 (65), 83 (38), 71 (38), 97 (36), 111 (20), 117(32), 159 (30), 233 (<5), 281 (7), 339 (6), 383 (47) |
| C20H41 (C20:0) | 19.7 | 456.70 | 43 (base peak), 57 (70), 71 (42), 83 (36), 97 (36), 111 (18), 117 (32),159 (27), 125 (10), 207 (23), 295 (12), 355 (32), 398(40) |
| C22H45 (C22:0) | 21.4 | 484.75 | 43 (base peak), 57 (70), 71 (38), 83 (28), 97 (38), 111 (22), 117 (12), 125 (12) 159 (30), 207 (22), 281 (8), 355 (10), 381 (6), 423 (27) |
| Parameters Studied | Results |
|---|---|
| Selectivity | Correct |
| Fidelity | |
| Repeatability | Correct (CVr = 3.73%) |
| Reproducibility | Correct (CVR = 3.75%) |
| Linearity | Correct (no deviation from linearity, R2 = 0.992) |
| Detection limit LOD | 2.9 pg (on-column injector, FID) |
| Quantification limit LOQ | 17.5 pg (on-column injector, FID) |
| Stability | 1 year at −80 °C |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinault, M.; Guimaraes, C.; Couthon, H.; Thibonnet, J.; Fontaine, D.; Chantôme, A.; Chevalier, S.; Besson, P.; Jaffrès, P.-A.; Vandier, C. Synthesis of Alkyl-Glycerolipids Standards for Gas Chromatography Analysis: Application for Chimera and Shark Liver Oils. Mar. Drugs 2018, 16, 101. https://doi.org/10.3390/md16040101
Pinault M, Guimaraes C, Couthon H, Thibonnet J, Fontaine D, Chantôme A, Chevalier S, Besson P, Jaffrès P-A, Vandier C. Synthesis of Alkyl-Glycerolipids Standards for Gas Chromatography Analysis: Application for Chimera and Shark Liver Oils. Marine Drugs. 2018; 16(4):101. https://doi.org/10.3390/md16040101
Chicago/Turabian StylePinault, Michelle, Cyrille Guimaraes, Hélène Couthon, Jérôme Thibonnet, Delphine Fontaine, Aurélie Chantôme, Stephan Chevalier, Pierre Besson, Paul-Alain Jaffrès, and Christophe Vandier. 2018. "Synthesis of Alkyl-Glycerolipids Standards for Gas Chromatography Analysis: Application for Chimera and Shark Liver Oils" Marine Drugs 16, no. 4: 101. https://doi.org/10.3390/md16040101