Synthesis and Biological Evaluation of a New Structural Simplified Analogue of cADPR, a Calcium-Mobilizing Secondary Messenger Firstly Isolated from Sea Urchin Eggs

 , ,
, ,  , ,
, , 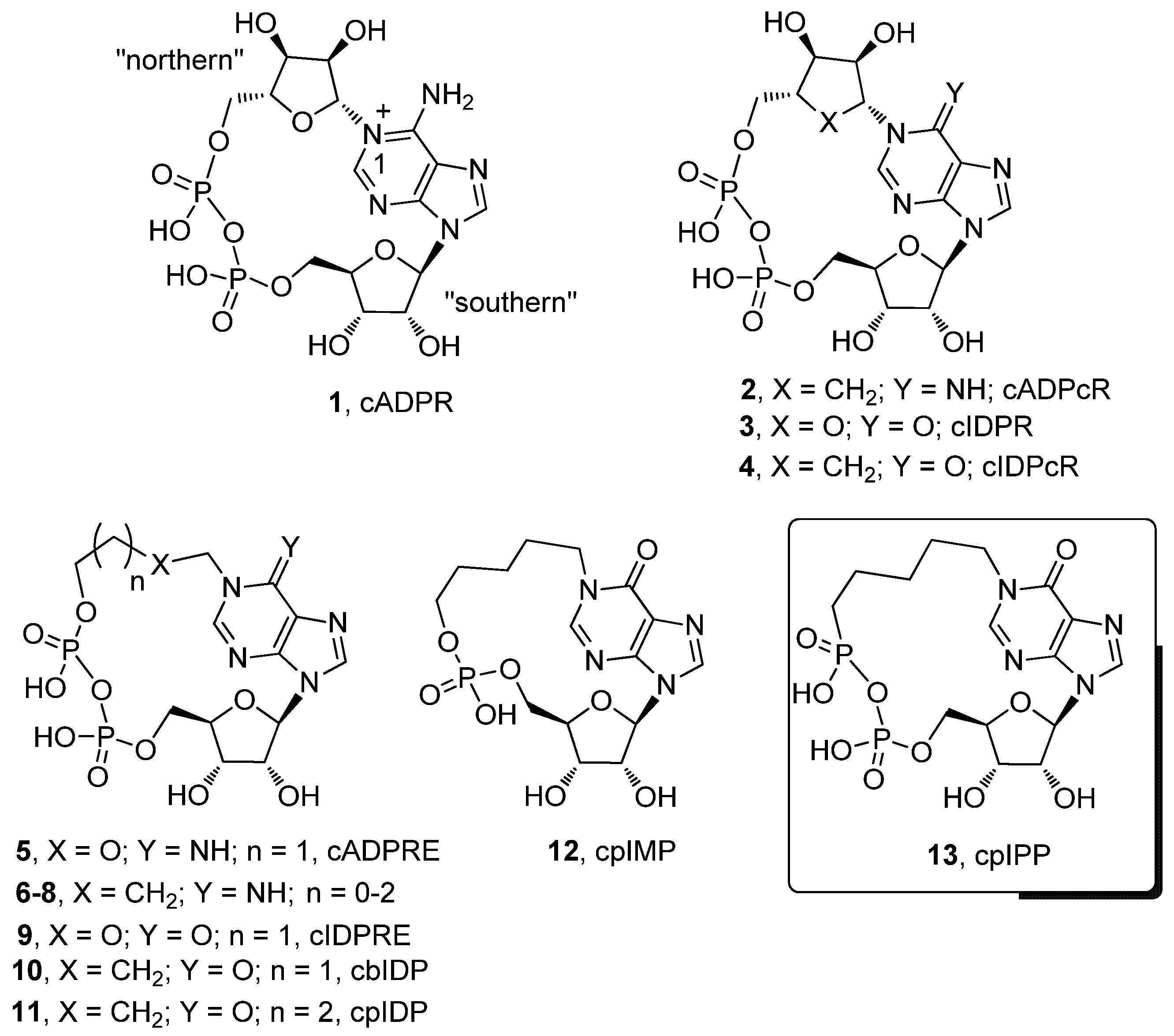{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
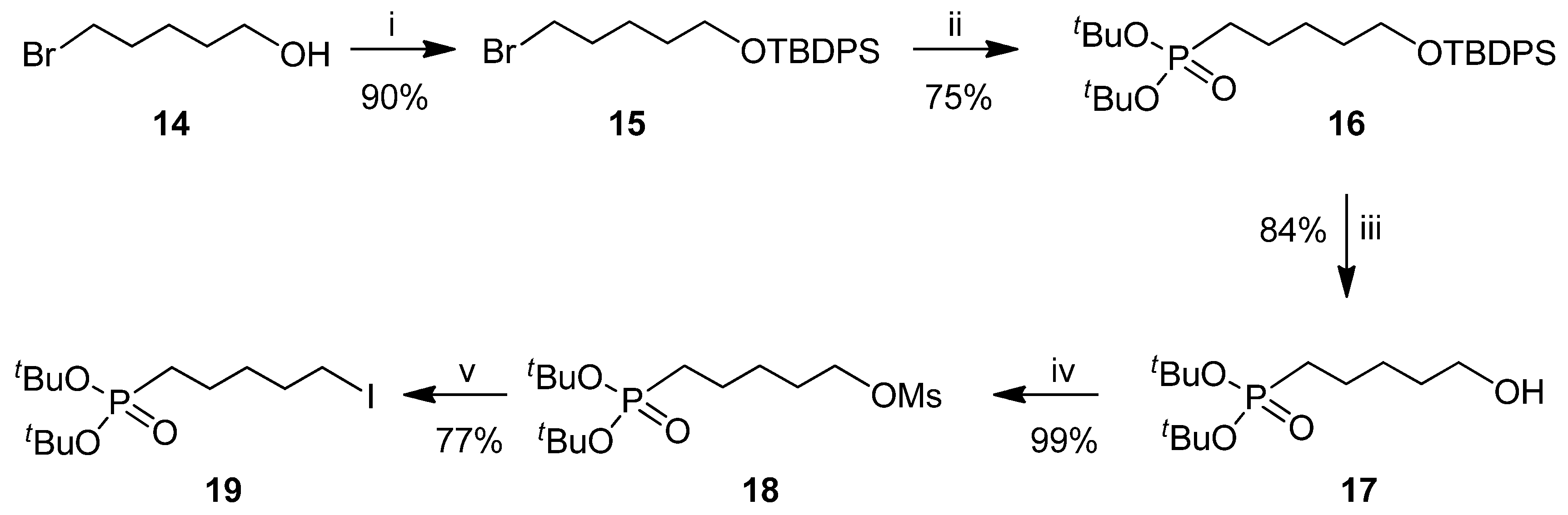
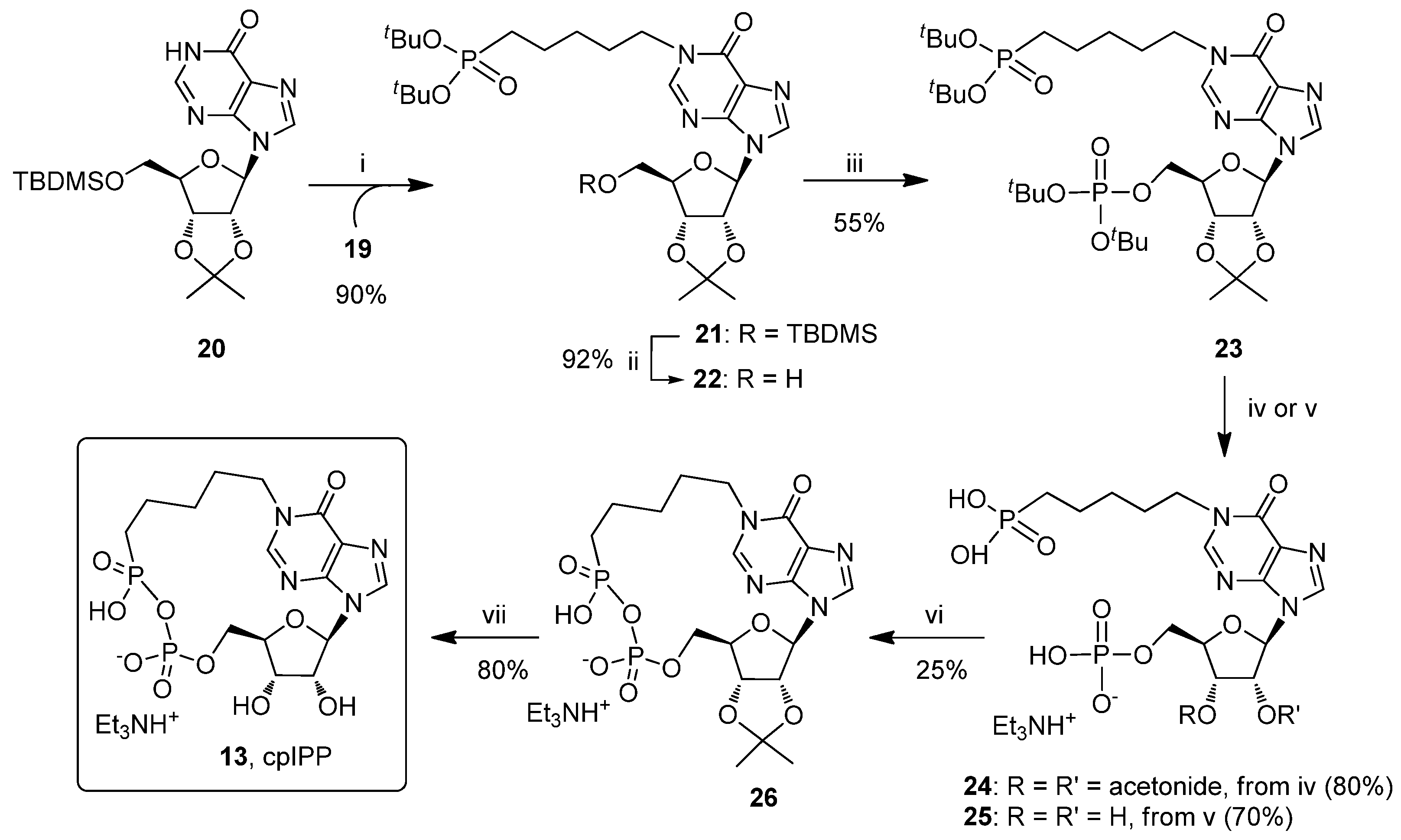
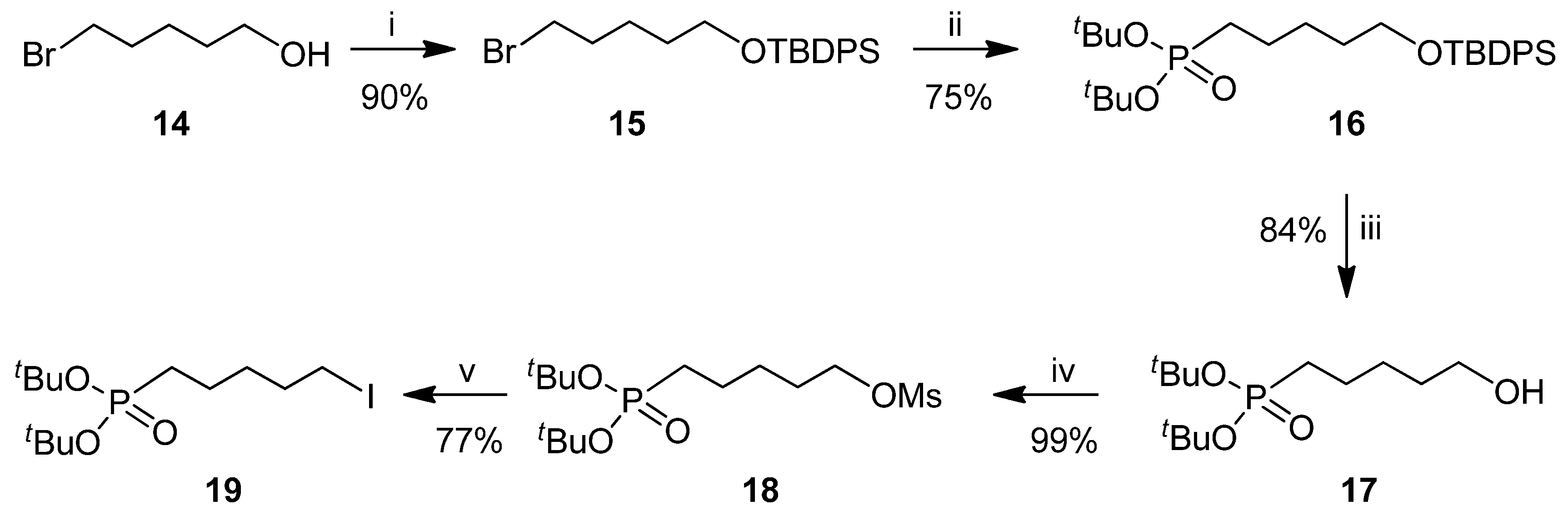
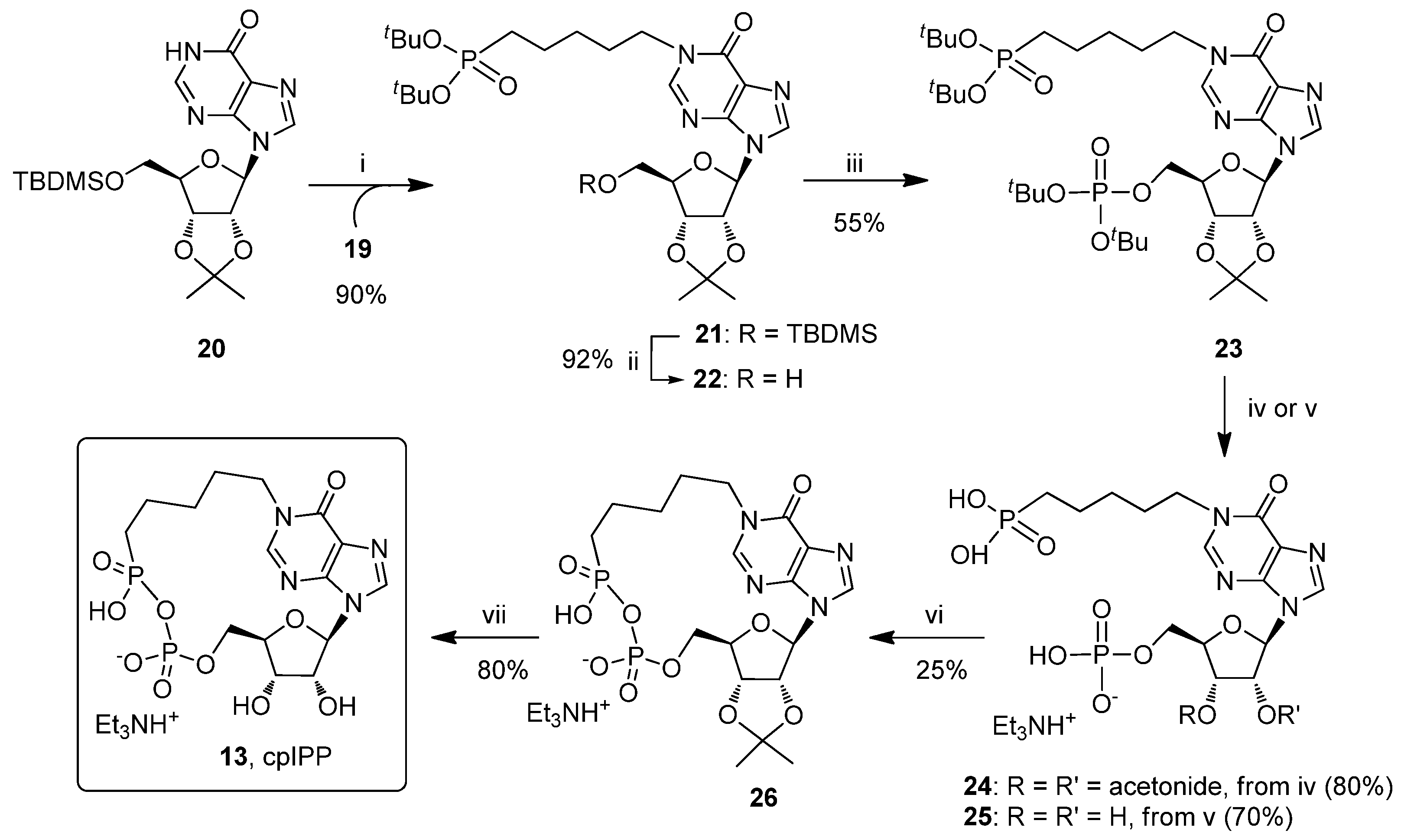
2.1. Chemistry
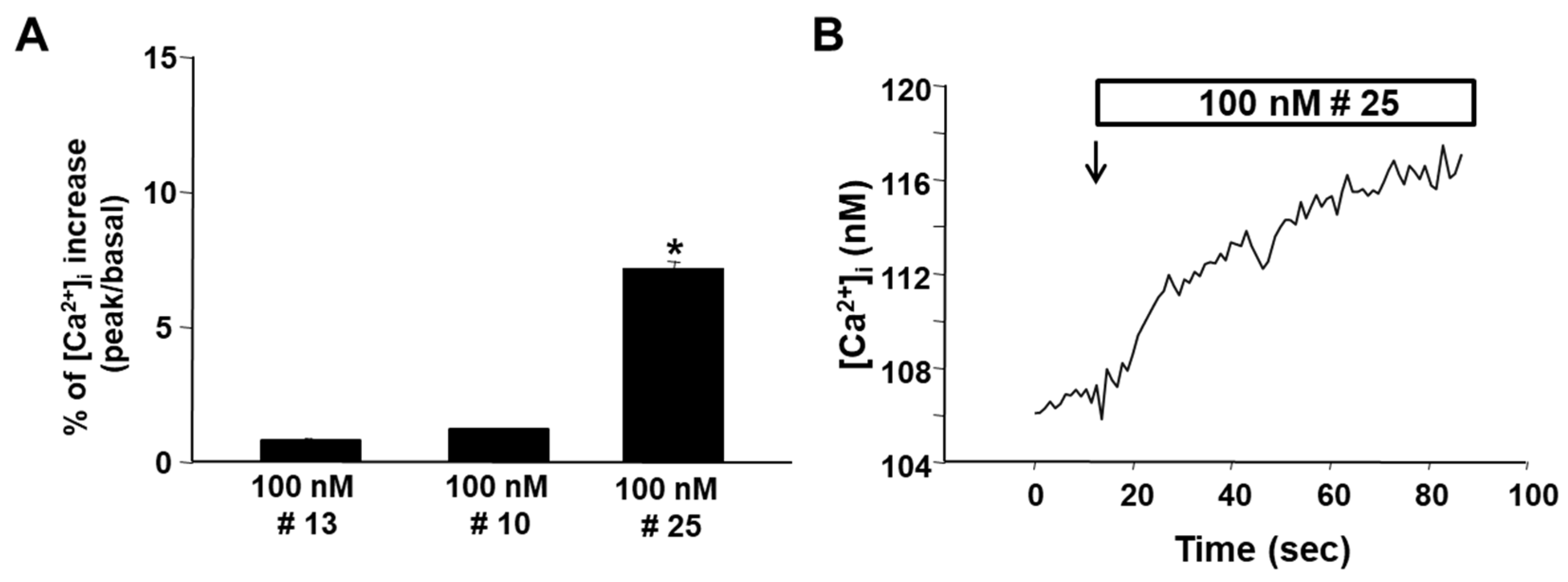
2.2. Biology
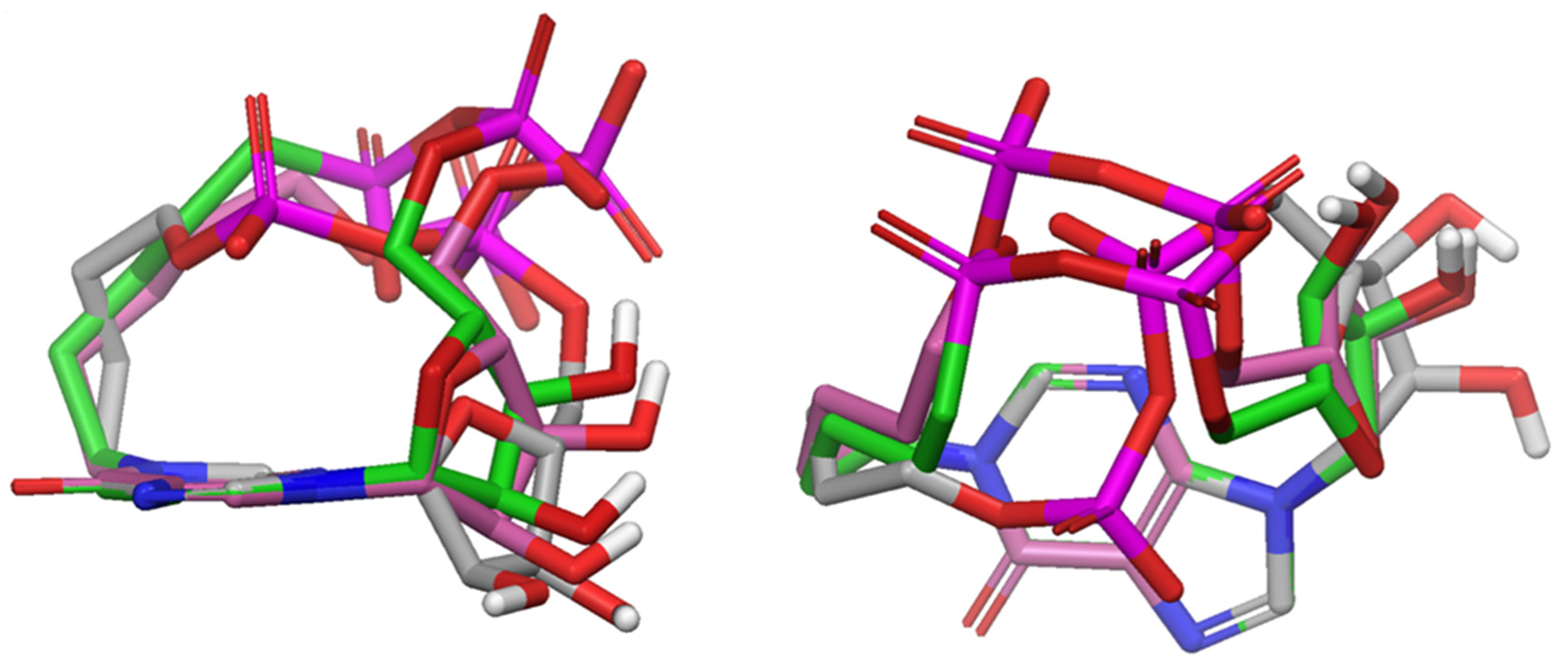
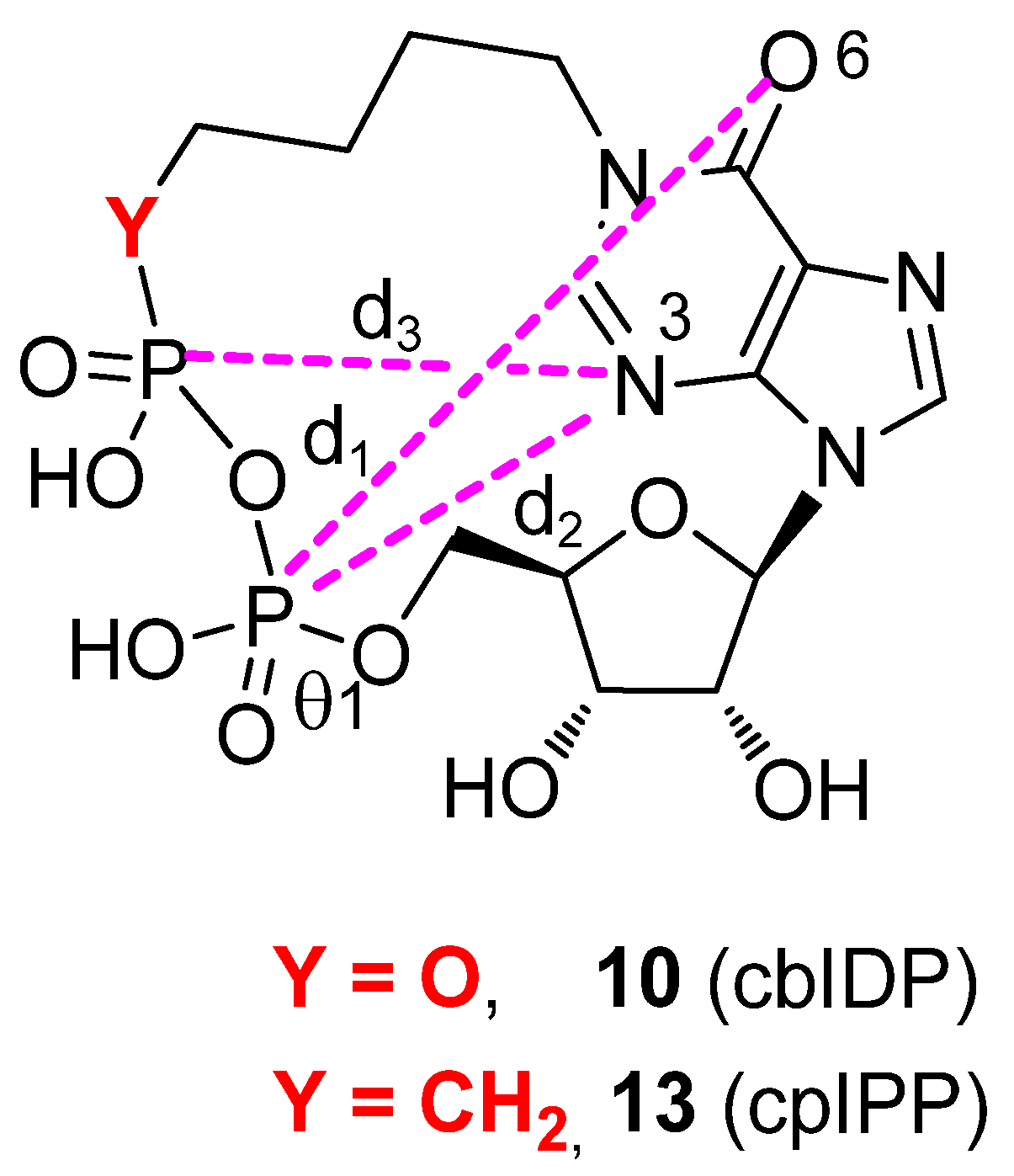
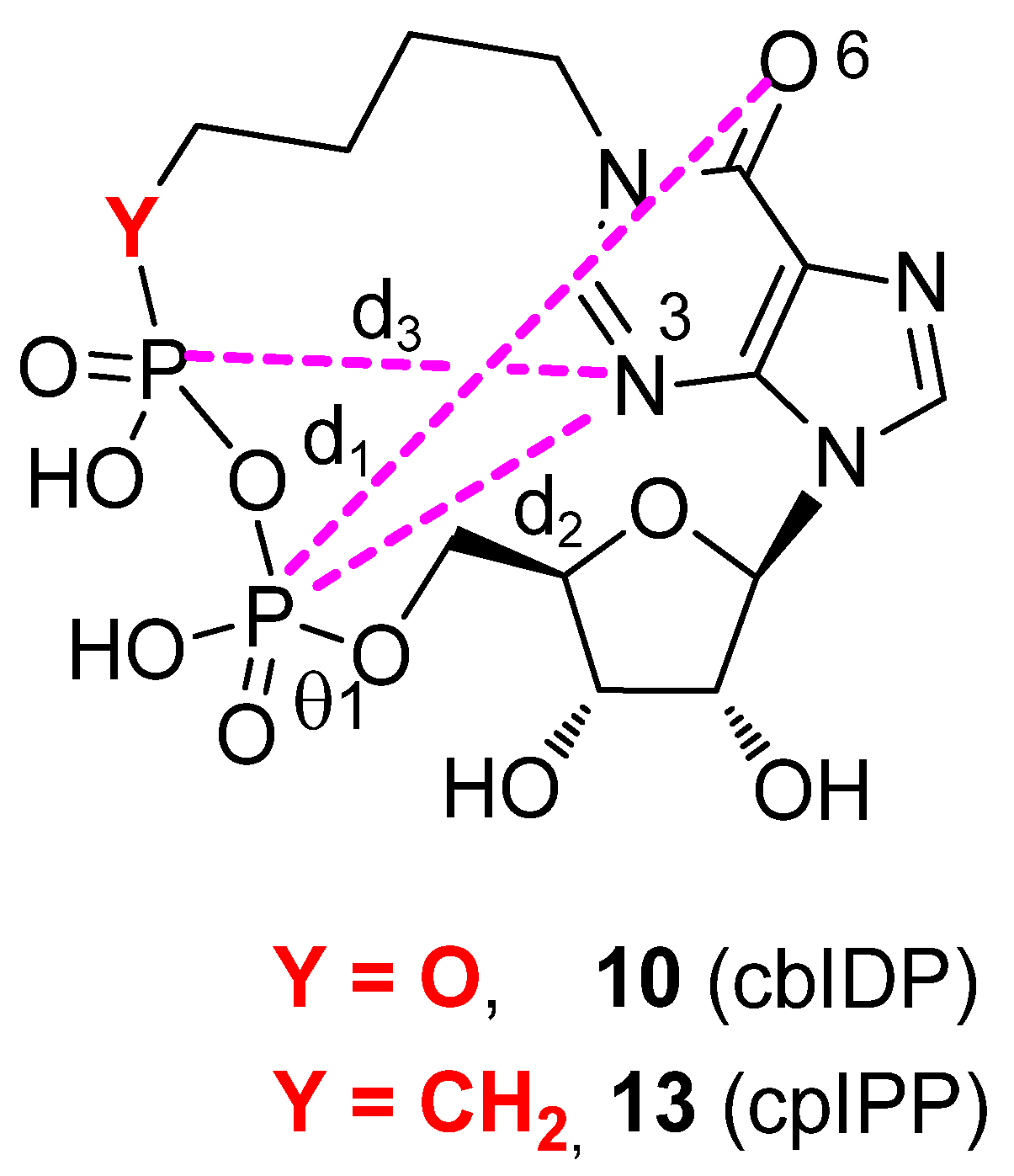
2.3. Conformational Analyses
3. Experimental Section
3.1. General Experimental Procedures
3.2. Synthesis and Charcterization of Compounds 13, 16, 17, 19 and 21–26
3.3. Cell Cultures and Intracellular [Ca2+]i Measurement for Compounds 10, 13 and 25
3.4. Conformational Search
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clapper, D.L.; Walseth, T.F.; Dargie, P.J.; Lee, H.C. Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J. Biol. Chem. 1987, 262, 9561–9568. [Google Scholar] [PubMed]
- Lee, H.C.; Walseth, T.F.; Bratt, G.T.; Hayes, R.N.; Clapper, D.L. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 1989, 264, 1608–1615. [Google Scholar] [PubMed]
- Guse, A.H. Second messenger function and the structure-activity relationship of cyclic adenosine diphosphoribose (cADPR). FEBS J. 2005, 272, 4590–4597. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J. Biol. Chem. 2012, 287, 31633–31640. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.Y.; Li, P.L. Vascular physiology of a Ca2+ mobilizing second messenger - Cyclic ADP-ribose. J. Cell. Mol. Med. 2006, 10, 407–422. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Physiological Functions of Cyclic ADP-Ribose and NAADP as Calcium Messengers. Annu. Rev. Pharmacol. Toxicol. 2001, 41, 317–345. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Liu, H.-X.; Hirai, H.; Torashima, T.; Nagai, T.; Lopatina, O.; Shnayder, N.A.; Yamada, K.; Noda, M.; Seike, T.; et al. CD38 is critical for social behaviour by regulating oxytocin secretion. Nature 2007, 446, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Guse, A.H. Calcium mobilizing second messengers derived from NAD. Biochim. Biophys. Acta 2015, 1854, 1132–1137. [Google Scholar] [CrossRef] [PubMed]
- Higashida, H.; Hashii, M.; Yokoyama, S.; Hoshi, N.; Asai, K.; Kato, T. Cyclic ADP-ribose as a potential second messenger for neuronal Ca2+ signaling. J. Neurochem. 2001, 76, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C. Cyclic ADP-ribose and NAADP: Fraternal twin messengers for calcium signaling. Sci. China Life Sci. 2011, 54, 699–711. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Harde, M.; Empson, R.; Potter, B.V.; Galione, A.; Stanton, P.K. Evidence of a role for cyclic ADP-ribose in long-term synaptic depression in hippocampus. Proc. Natl. Acad. Sci. USA 1999, 96, 4061–4066. [Google Scholar] [CrossRef] [PubMed]
- Guse, A. Biochemistry, Biology, and Pharmacology of Cyclic Adenosine Diphosphoribose (cADPR). Curr. Med. Chem. 2004, 11, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Chini, E.N.; Nagamune, K.; Wetzel, D.M.; Sibley, L.D. Evidence that the cADPR signalling pathway controls calcium-mediated microneme secretion in Toxoplasma gondii. Biochem. J. 2005, 389, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Takasawa, S.; Sugawara, A. The CD38-Cyclic ADP-Ribose System in Mammals: Historical Background, Pathophysiology and Perspective. Messenger 2014, 3, 27–34. [Google Scholar] [CrossRef]
- Zhang, F.J.; Gu, Q.M.; Sih, C.J. Bioorganic chemistry of cyclic ADP-ribose (cADPR). Bioorg. Med. Chem. 1999, 7, 653–664. [Google Scholar] [CrossRef]
- Cakir-Kiefer, C.; Muller-Steffner, H.; Schuber, F. Unifying mechanism for Aplysia ADP-ribosyl cyclase and CD38/NAD(+) glycohydrolases. Biochem. J. 2000, 349, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Moreau, C.; Kirchberger, T.; Zhang, B.; Thomas, M.P.; Weber, K.; Guse, A.H.; Potter, B.V.L. Aberrant cyclization affords a C-6 modified cyclic adenosine 5′-diphosphoribose analogue with biological activity in Jurkat T cells. J. Med. Chem. 2012, 55, 1478–1489. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.C.; Aarhus, R. ADP-ribosyl cyclase: An enzyme that cyclizes NAD+ into a calcium-mobilizing metabolite. Cell Regul. 1991, 2, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Walseth, T.F.; Lee, H.C. Synthesis and characterization of antagonists of cyclic-ADP-ribose-induced Ca2+ release. BBA Mol. Cell Res. 1993, 1178, 235–242. [Google Scholar] [CrossRef]
- Guse, A.H.; Cakir-Kiefer, C.; Fukuoka, M.; Shuto, S.; Weber, K.; Bailey, V.C.; Matsuda, A.; Mayr, G.W.; Oppenheimer, N.; Schuber, F.; et al. Novel hydrolysis-resistant analogues of cyclic ADP-ribose: Modification of the “northern” ribose and calcium release activity. Biochemistry 2002, 41, 6744–6751. [Google Scholar] [CrossRef] [PubMed]
- Bailey, V.C.; Fortt, S.M.; Summerhill, R.J.; Galione, A.; Potter, B.V.L. Cyclic aristeromycin diphosphate ribose: A potent and poorly hydrolysable Ca2+ -mobilising mimic of cyclic adenosine diphosphate ribose. FEBS Lett. 1996, 379, 227–230. [Google Scholar] [CrossRef]
- Shuto, S.; Matsuda, A. Chemistry of Cyclic ADP-Ribose and Its Analogs. Curr. Med. Chem. 2004, 11, 827–845. [Google Scholar] [CrossRef] [PubMed]
- Shuto, S.; Fukuoka, M.; Manikowsky, A.; Ueno, Y.; Nakano, T.; Kuroda, R.; Kuroda, H.; Matsuda, A. Total synthesis of cyclic ADP-carbocyclic-ribose, a stable mimic of Ca2+-mobilizing second messenger cyclic ADP-ribose. J. Am. Chem. Soc. 2001, 123, 8750–8759. [Google Scholar] [CrossRef] [PubMed]
- Kudoh, T.; Fukuoka, M.; Ichikawa, S.; Murayama, T.; Ogawa, Y.; Hashii, M.; Higashida, H.; Kunerth, S.; Weber, K.; Guse, A.H.; et al. Synthesis of stable and cell-type selective analogues of cyclic ADP-ribose, a Ca2+-mobilizing second messenger. Structure-activity relationship of the N1-ribose moiety. J. Am. Chem. Soc. 2005, 127, 8846–8855. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, T.; Sakaguchi, N.; Kudoh, T.; Takano, S.; Uehara, M.; Murayama, T.; Sakurai, T.; Hashii, M.; Higashida, H.; Weber, K.; et al. Design and Synthesis of Cyclic ADP-4-Thioribose as a Stable Equivalent of Cyclic ADP-Ribose, a Calcium Ion-Mobilizing Second Messenger. Angew. Chem. Int. Ed. 2013, 52, 6633–6637. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.K.; Guse, A.H.; Potter, B.V.L. Rapid synthetic route toward structurally modified derivatives of cyclic adenosine 5′-diphosphate ribose. J. Org. Chem. 2005, 70, 4810–4819. [Google Scholar] [CrossRef] [PubMed]
- Shuto, S.; Shirato, M.; Sumita, Y.; Ueno, Y.; Matsuda, A. Nucleosides and Nucleotides. 173. Synthesis of Cyclic IDP-carbocyclic-ribose, a Stable Mimic of Cyclic ADP-ribose. Significant Facilitation of the Intramolecular Condensation Reaction of N-1-(Carbocyclic-ribosyl)inosine 5′,6′-Diphosphate Derivatives by an. J. Org. Chem. 1998, 63, 1986–1994. [Google Scholar] [CrossRef]
- Zhang, L.; Guse, A.H. Cyclic ADP-Ribose Analogues with Minimal Structure: Synthesis and Calcium-Release Activity. In Drug Discovery Research; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 186–202. ISBN 9780470131862. [Google Scholar]
- Xu, J.; Yang, Z.; Dammermann, W.; Zhang, L.; Guse, A.H.; Zhang, L.H. Synthesis and agonist activity of cyclic ADP-ribose analogues with substitution of the northern ribose by ether or alkane chains. J. Med. Chem. 2006, 49, 5501–5512. [Google Scholar] [CrossRef] [PubMed]
- Gu, X.; Yang, Z.; Zhang, L.; Kunerth, S.; Fliegert, R.; Weber, K.; Guse, A.H.; Zhang, L. Synthesis and biological evaluation of novel membrane-permeant cyclic ADP-ribose mimics: N1-[(5″-O-phosphorylethoxy)methyl]-5′-O-phosphorylinosine 5′,5″-cyclicpyrophosphate (cIDPRE) and 8-substituted derivatives. J. Med. Chem. 2004, 47, 5674–5682. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Dong, M.; Liu, J.; Zhang, K.; Yang, Z.; Zhang, L.; Zhang, L. Concise syntheses of trifluoromethylated cyclic and acyclic analogues of cADPR. Molecules 2010, 15, 8689–8701. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wagner, G.K.; Weber, K.; Garnham, C.; Morgan, A.J.; Galione, A.; Guse, A.H.; Potter, B.V.L. 2′-deoxy cyclic adenosine 5′-diphosphate ribose derivatives: Importance of the 2′-hydroxyl motif for the antagonistic activity of 8-substituted cADPR derivatives. J. Med. Chem. 2008, 51, 1623–1636. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Kirchberger, T.; Huang, X.; Yang, Z.J.; Zhang, L.R.; Guse, A.H.; Zhang, L.H. Trifluoromethylated cyclic-ADP-ribose mimic: Synthesis of 8-trifluoromethyl-N(1)-[(5″-O-phosphorylethoxy)methyl]-5′-O-phosphorylino sine-5′,5″-cyclic pyrophosphate (8-CF(3)-cIDPRE) and its calcium release activity in T cells. Org. Biomol. Chem. 2010, 8, 4705–4715. [Google Scholar] [CrossRef] [PubMed]
- Galeone, A.; Mayol, L.; Oliviero, G.; Piccialli, G.; Varra, M. Synthesis of a New N1-Pentyl Analogue of Cyclic Inosine Diphosphate Ribose (cIDPR) as a Stable Potential Mimic of Cyclic ADP Ribose (cADPR). Eur. J. Org. Chem. 2002, 2002, 4234–4238. [Google Scholar] [CrossRef]
- Oliviero, G.; D’Errico, S.; Borbone, N.; Amato, J.; Piccialli, V.; Varra, M.; Piccialli, G.; Mayol, L. A solid-phase approach to the synthesis of N-1-alkyl analogues of cyclic inosine-diphosphate-ribose (cIDPR). Tetrahedron 2010, 66, 1931–1936. [Google Scholar] [CrossRef]
- Chang, S.H.; Han, J.L.; Tseng, S.Y.; Lee, H.Y.; Lin, C.W.; Lin, Y.C.; Jeng, W.Y.; Wang, A.H.J.; Wu, C.Y.; Wong, C.H. Glycan array on aluminum oxide-coated glass slides through phosphonate chemistry. J. Am. Chem. Soc. 2010, 132, 13371–13380. [Google Scholar] [CrossRef] [PubMed]
- De Napoli, L.; Di Fabio, G.; Messere, A.; Montesarchio, D.; Piccialli, G.; Varra, M. Synthetic studies on the glycosylation of the base residues of inosine and uridine. J. Chem. Soc. Perkin Trans. 1 1999, 3489–3493. [Google Scholar] [CrossRef]
- Swamy, K.C.K.; Kumar, N.N.B.; Balaraman, E.; Kumar, K.V.P.P. Mitsunobu and Related Reactions: Advances and Applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef] [PubMed]
- Zaragoza Dörwald, F. Side Reactions in Organic Synthesis: A Guide to Successful Synthesis Design; WILEY-VCH Verlag GmbH & Co.: Weinheim, Germany, 2005; ISBN 3527310215. [Google Scholar]
- Oliviero, G.; Amato, J.; Borbone, N.; D’Errico, S.; Piccialli, G.; Mayol, L. Synthesis of N-1 and ribose modified inosine analogues on solid support. Tetrahedron Lett. 2007, 48, 397–400. [Google Scholar] [CrossRef]
- Oliviero, G.; Amato, J.; Borbone, N.; D’Errico, S.; Piccialli, G.; Bucci, E.; Piccialli, V.; Mayol, L. Synthesis of 4-N-alkyl and ribose-modified AICAR analogues on solid support. Tetrahedron 2008, 64, 6475–6481. [Google Scholar] [CrossRef]
- Oliviero, G.; D’Errico, S.; Borbone, N.; Amato, J.; Piccialli, V.; Piccialli, G.; Luciano, M. Facile solid-phase synthesis of AICAR 5-monophosphate (ZMP) and its 4-N-Alkyl derivatives. Eur. J. Org. Chem. 2010, 1517–1524. [Google Scholar] [CrossRef]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Amato, J.; D’Alonzo, D.; Piccialli, V.; Mayol, L.; Piccialli, G. A facile synthesis of 5′-Fluoro-5′-deoxyacadesine (5′-F-AICAR): A novel non-phosphorylable AICAR Analogue. Molecules 2012, 17, 13036–13044. [Google Scholar] [CrossRef] [PubMed]
- D’Errico, S.; Oliviero, G.; Borbone, N.; Amato, J.; Piccialli, V.; Varra, M.; Mayol, L.; Piccialli, G. Synthesis of new acadesine (AICA-riboside) analogues having acyclic D-ribityl or 4-hydroxybutyl chains in place of the ribose. Molecules 2013, 18, 9420–9431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Errico, S.; Oliviero, G.; Amato, J.; Borbone, N.; Cerullo, V.; Hemminki, A.; Piccialli, V.; Zaccaria, S.; Mayol, L.; Piccialli, G. Synthesis and biological evaluation of unprecedented ring-expanded nucleosides (RENs) containing the imidazo[4,5-d][1,2,6]oxadiazepine ring system. Chem. Commun. 2012, 48, 9310–9312. [Google Scholar] [CrossRef] [PubMed]
- Hyde, R.M.; Broom, A.D.; Buckheit, R.W. Antiviral amphipathic oligo- and polyribonucleotides: Analogue development and biological studies. J. Med. Chem. 2003, 46, 1878–1885. [Google Scholar] [CrossRef] [PubMed]
- Maestro, Schrodinger, Inc. Release 11.3. 2017. Available online: https://www.schrodinger.com/products/macrocycles.
- Oliviero, G.; Borbone, N.; Amato, J.; D’Errico, S.; Galeone, A.; Piccialli, G.; Varra, M.; Mayol, L. Synthesis of quadruplex-forming tetra-end-linked oligonucleotides: Effects of the linker size on quadruplex topology and stability. Biopolymers 2009, 91, 466–477. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Oliviero, G.; Roviello, G.N.; Bucci, E.M.; Piccialli, G. G-quadruplex-forming oligonucleotide conjugated to magnetic nanoparticles: Synthesis, characterization, and enzymatic stability assays. Bioconjug. Chem. 2012, 23, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Piccialli, V.; Borbone, N.; Oliviero, G. Ruthenium-catalyzed oxidative cyclization of 1,7-dienes. A novel diasteroselective synthesis of 2,7-disubstituted trans-oxepane diols. Tetrahedron Lett. 2007, 48, 5131–5135. [Google Scholar] [CrossRef]
- Piccialli, V.; D’Errico, S.; Borbone, N.; Oliviero, G.; Centore, R.; Zaccaria, S. A general synthesis of bis-α-acyloxy-1,4- and -1,5-diketones through catalytic oxidative opening of acylated THF and THP diols. Eur. J. Org. Chem. 2013, 2, 1781–1789. [Google Scholar] [CrossRef]
- Secondo, A.; Staiano, R.I.; Scorziello, A.; Sirabella, R.; Boscia, F.; Adornetto, A.; Valsecchi, V.; Molinaro, P.; Canzoniero, L.M.T.; Di Renzo, G.; et al. BHK cells transfected with NCX3 are more resistant to hypoxia followed by reoxygenation than those transfected with NCX1 and NCX2: Possible relationship with mitochondrial membrane potential. Cell Calcium 2007, 42, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [PubMed]
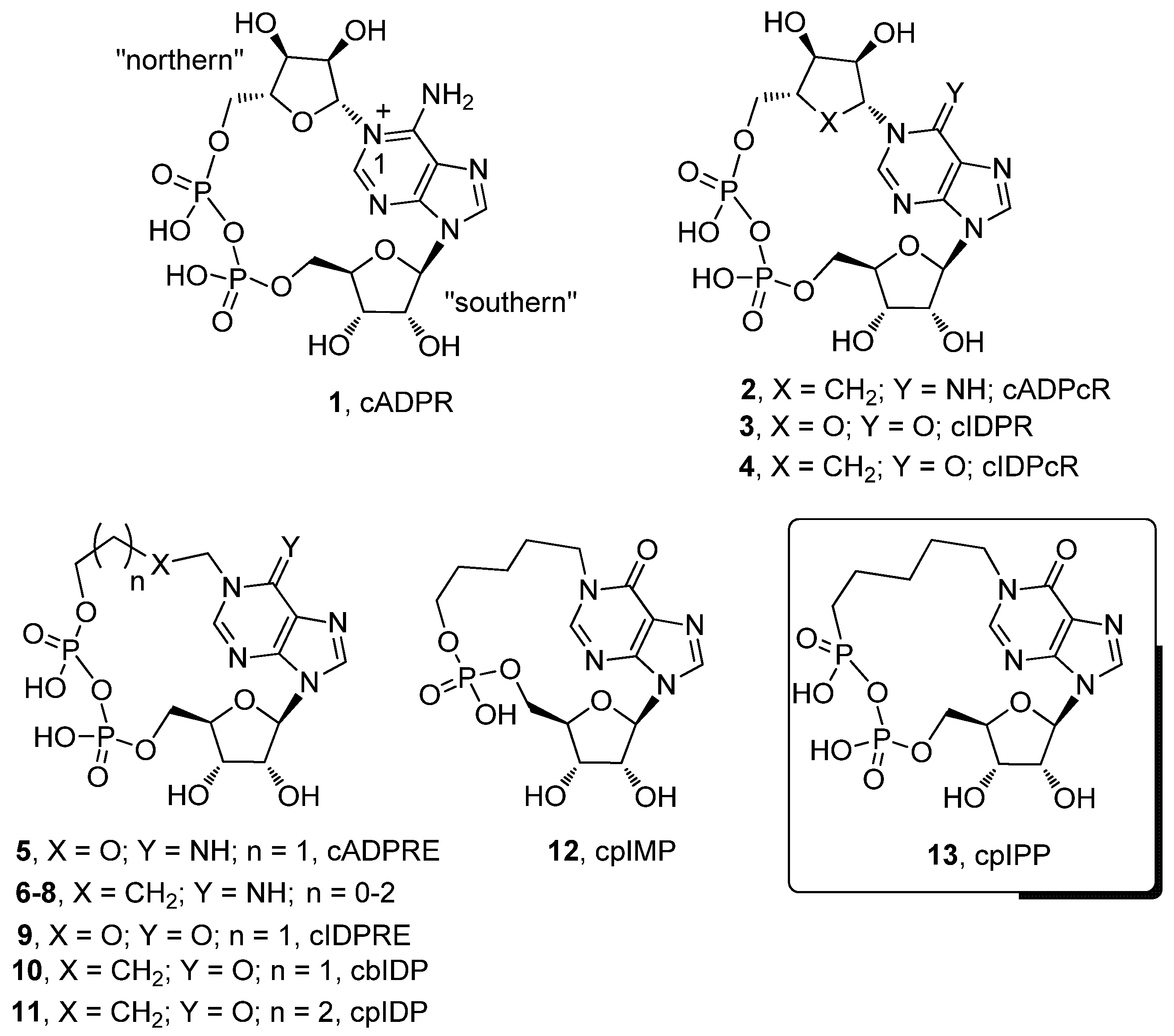


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Errico, S.; Borbone, N.; Catalanotti, B.; Secondo, A.; Petrozziello, T.; Piccialli, I.; Pannaccione, A.; Costantino, V.; Mayol, L.; Piccialli, G.; et al. Synthesis and Biological Evaluation of a New Structural Simplified Analogue of cADPR, a Calcium-Mobilizing Secondary Messenger Firstly Isolated from Sea Urchin Eggs. Mar. Drugs 2018, 16, 89. https://doi.org/10.3390/md16030089
D’Errico S, Borbone N, Catalanotti B, Secondo A, Petrozziello T, Piccialli I, Pannaccione A, Costantino V, Mayol L, Piccialli G, et al. Synthesis and Biological Evaluation of a New Structural Simplified Analogue of cADPR, a Calcium-Mobilizing Secondary Messenger Firstly Isolated from Sea Urchin Eggs. Marine Drugs. 2018; 16(3):89. https://doi.org/10.3390/md16030089
Chicago/Turabian StyleD’Errico, Stefano, Nicola Borbone, Bruno Catalanotti, Agnese Secondo, Tiziana Petrozziello, Ilaria Piccialli, Anna Pannaccione, Valeria Costantino, Luciano Mayol, Gennaro Piccialli, and et al. 2018. "Synthesis and Biological Evaluation of a New Structural Simplified Analogue of cADPR, a Calcium-Mobilizing Secondary Messenger Firstly Isolated from Sea Urchin Eggs" Marine Drugs 16, no. 3: 89. https://doi.org/10.3390/md16030089