Pinnatifidenyne-Derived Ethynyl Oxirane Acetogenins from Laurencia viridis

 , ,
, , 
Abstract
:1. Introduction
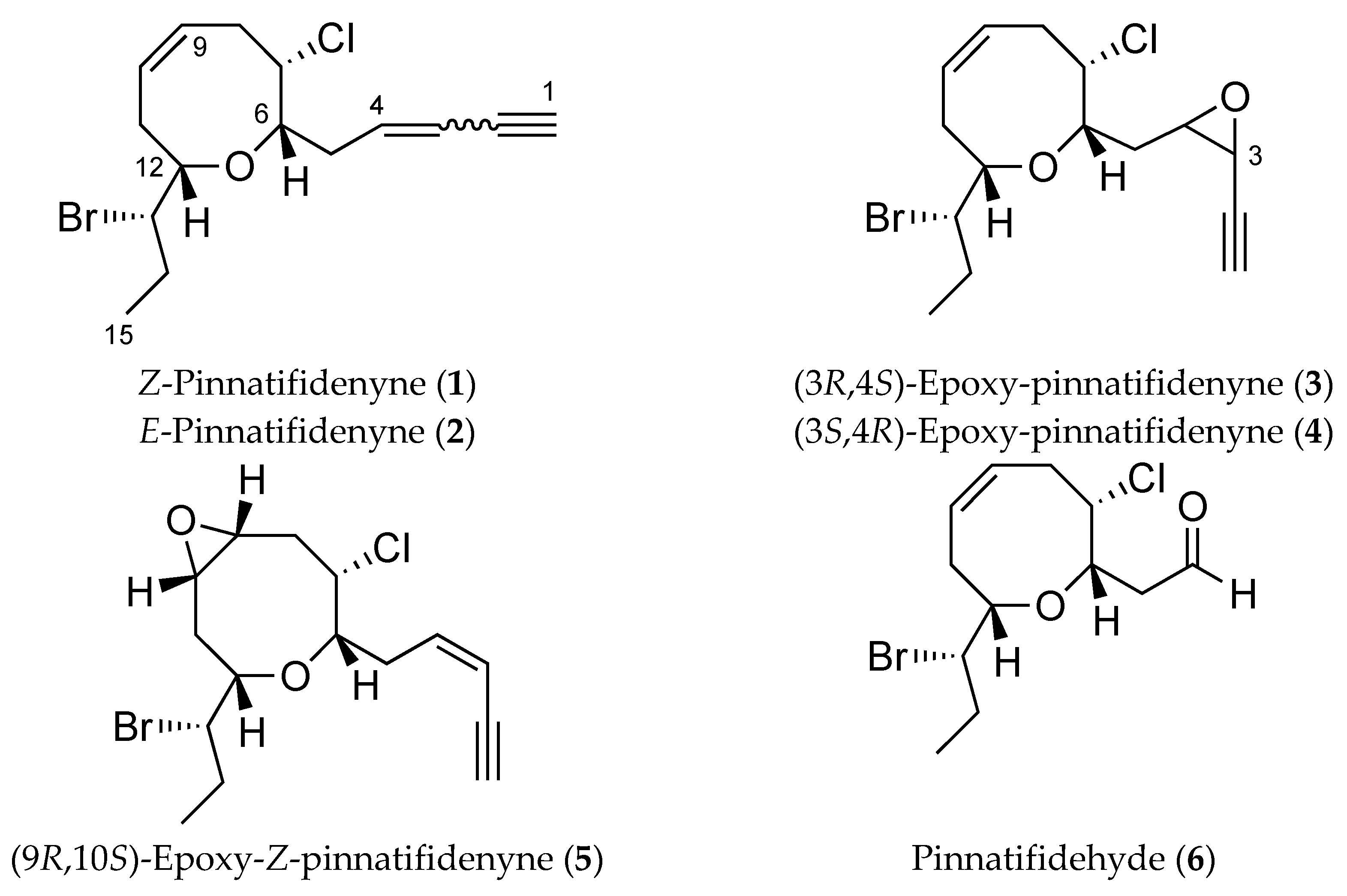
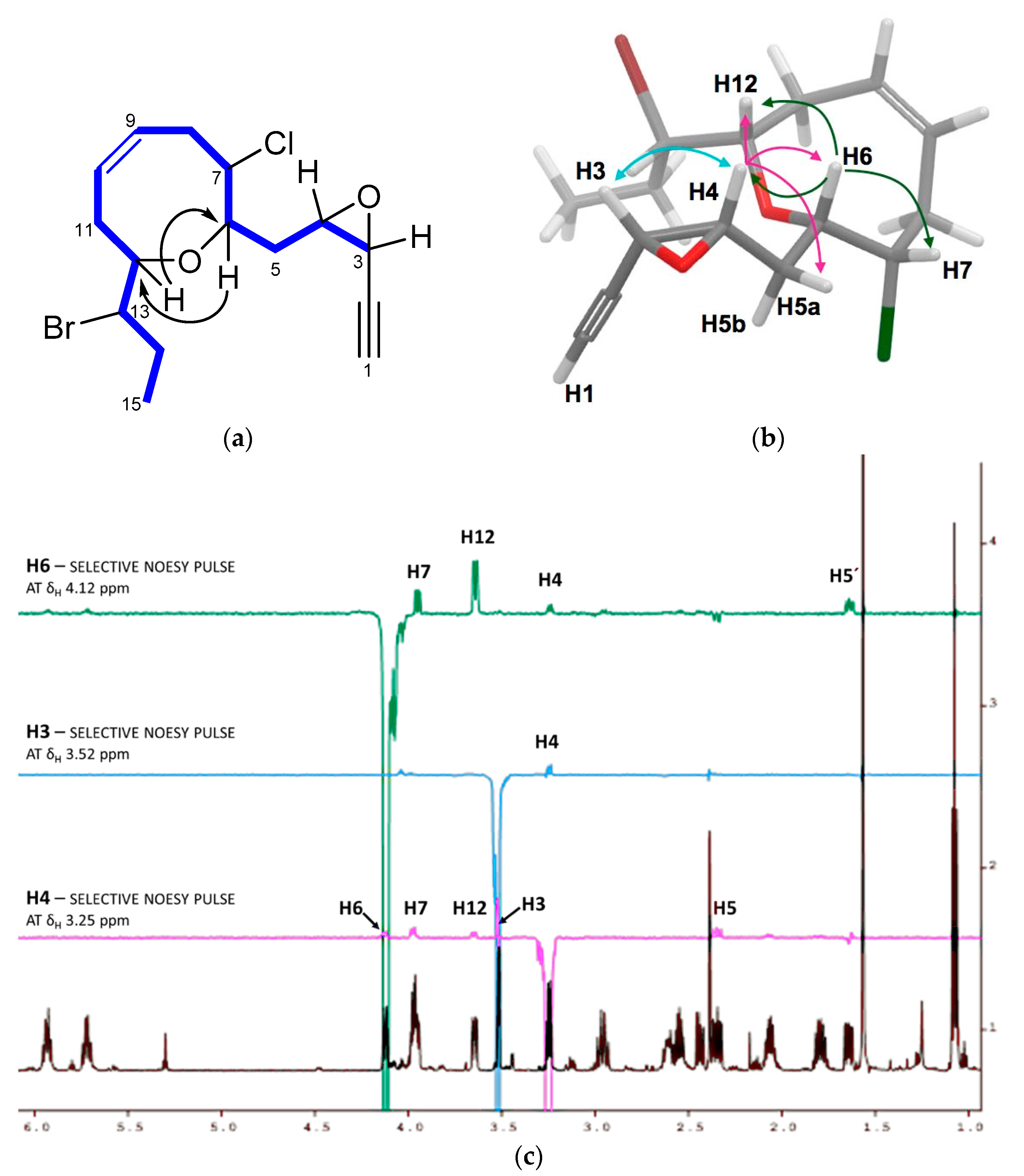
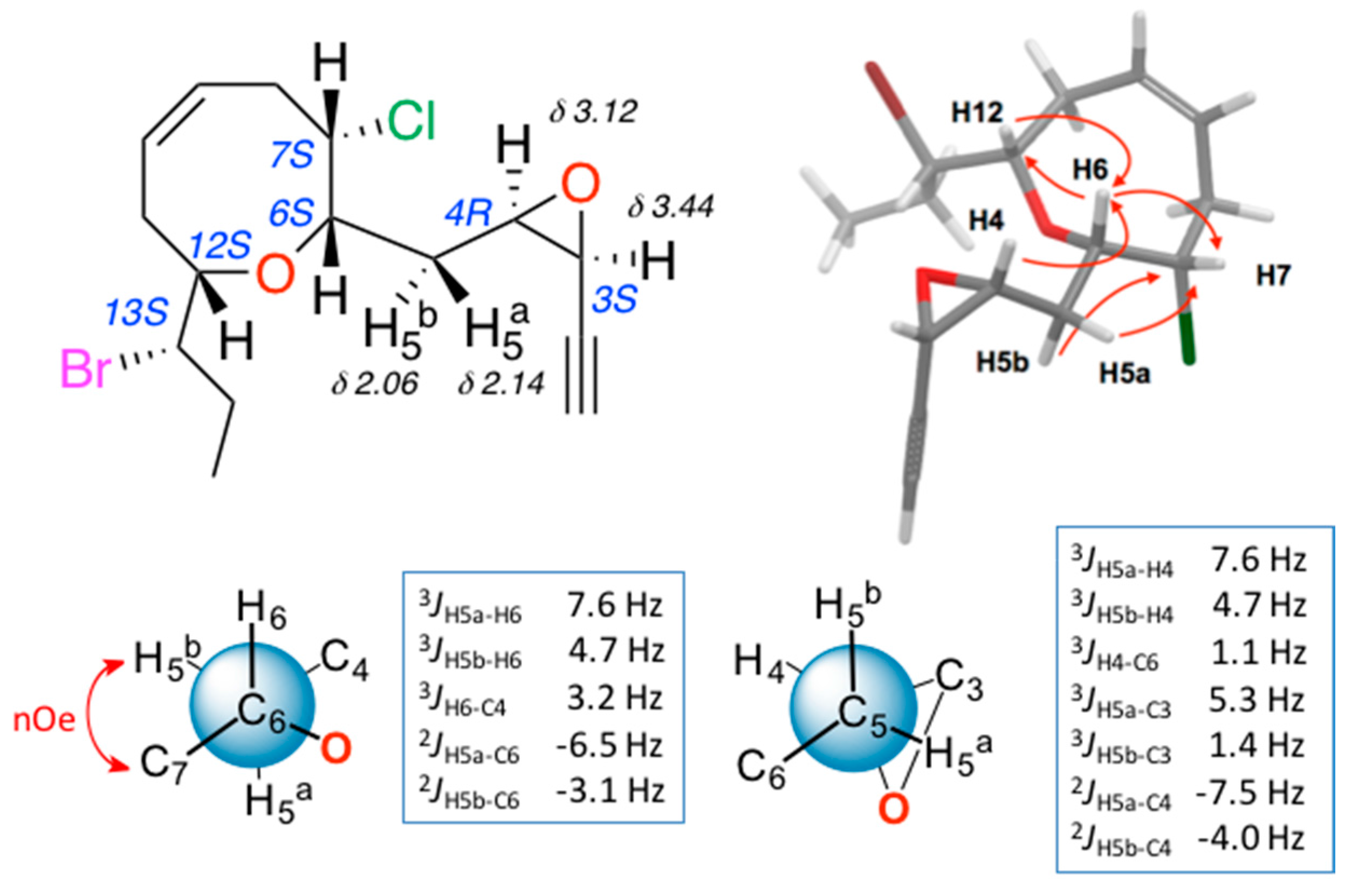
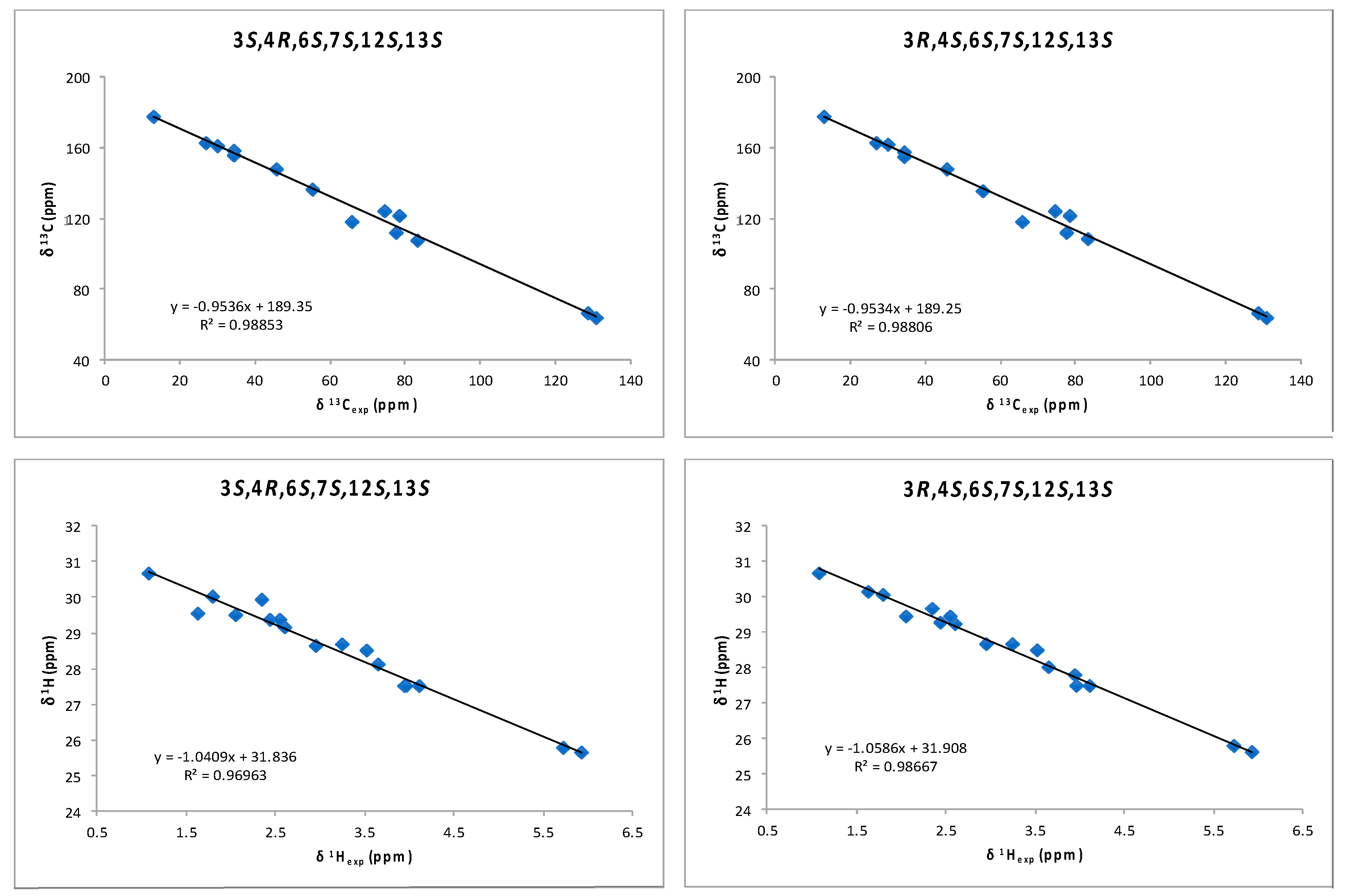
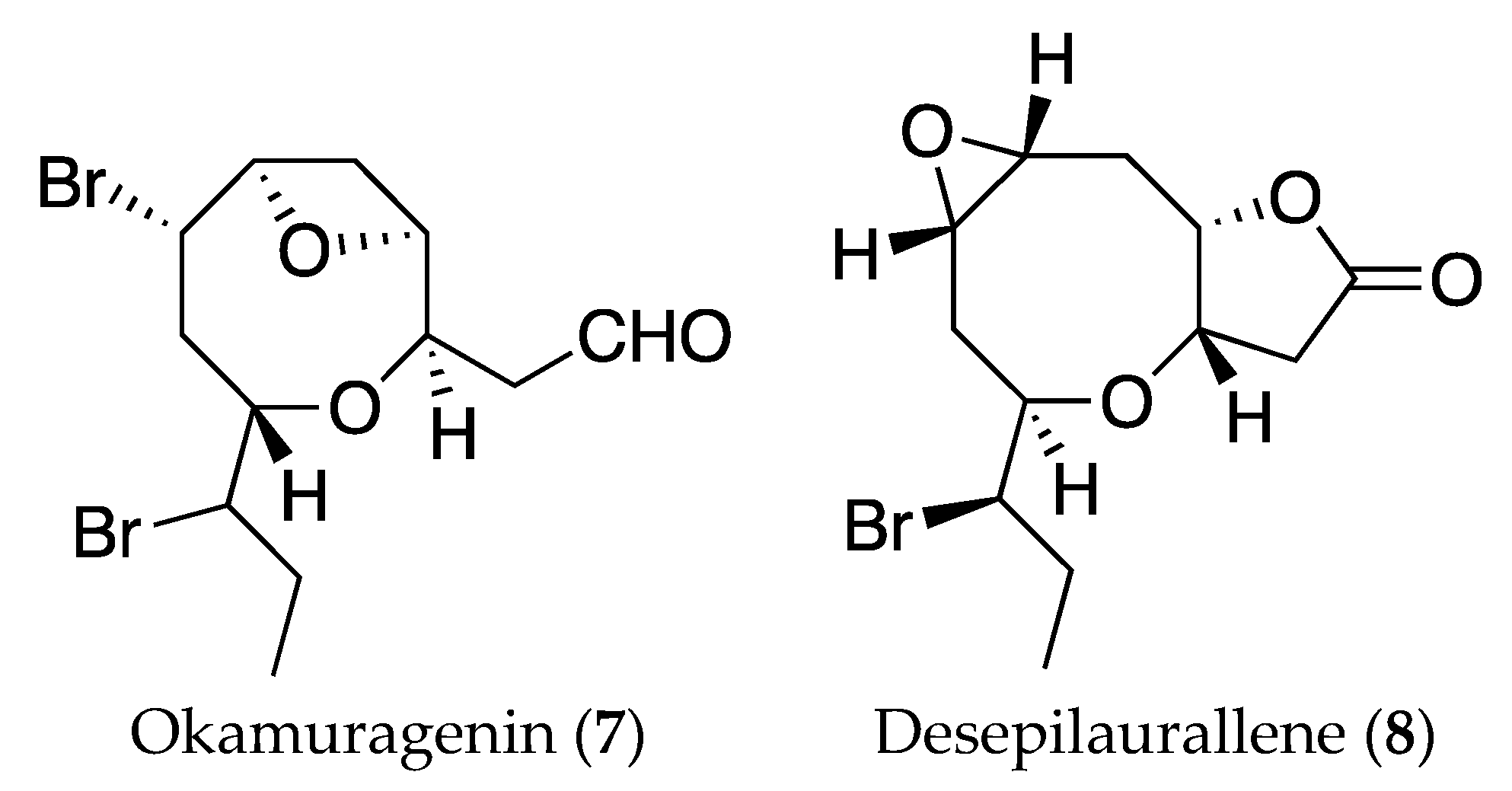
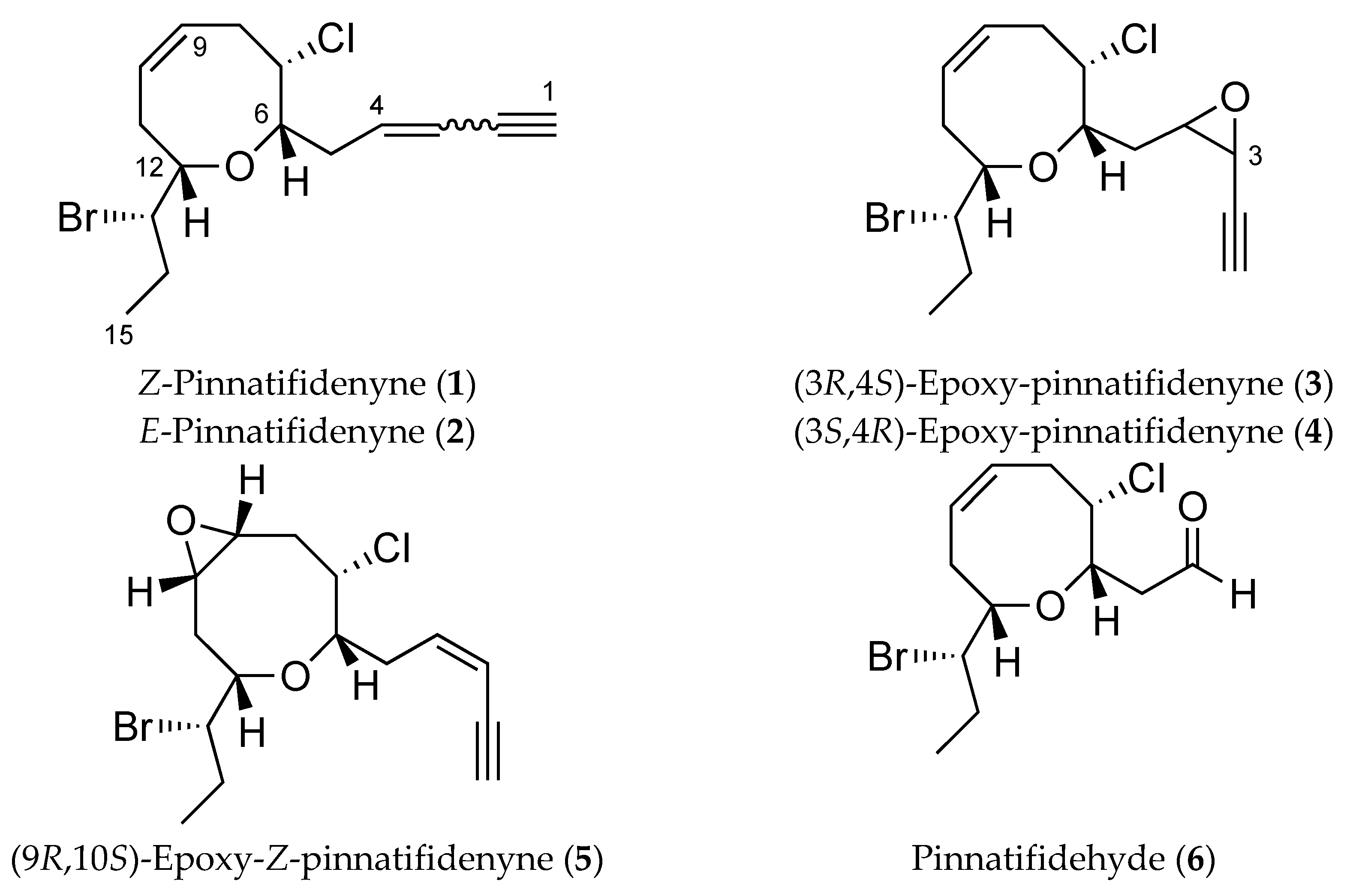
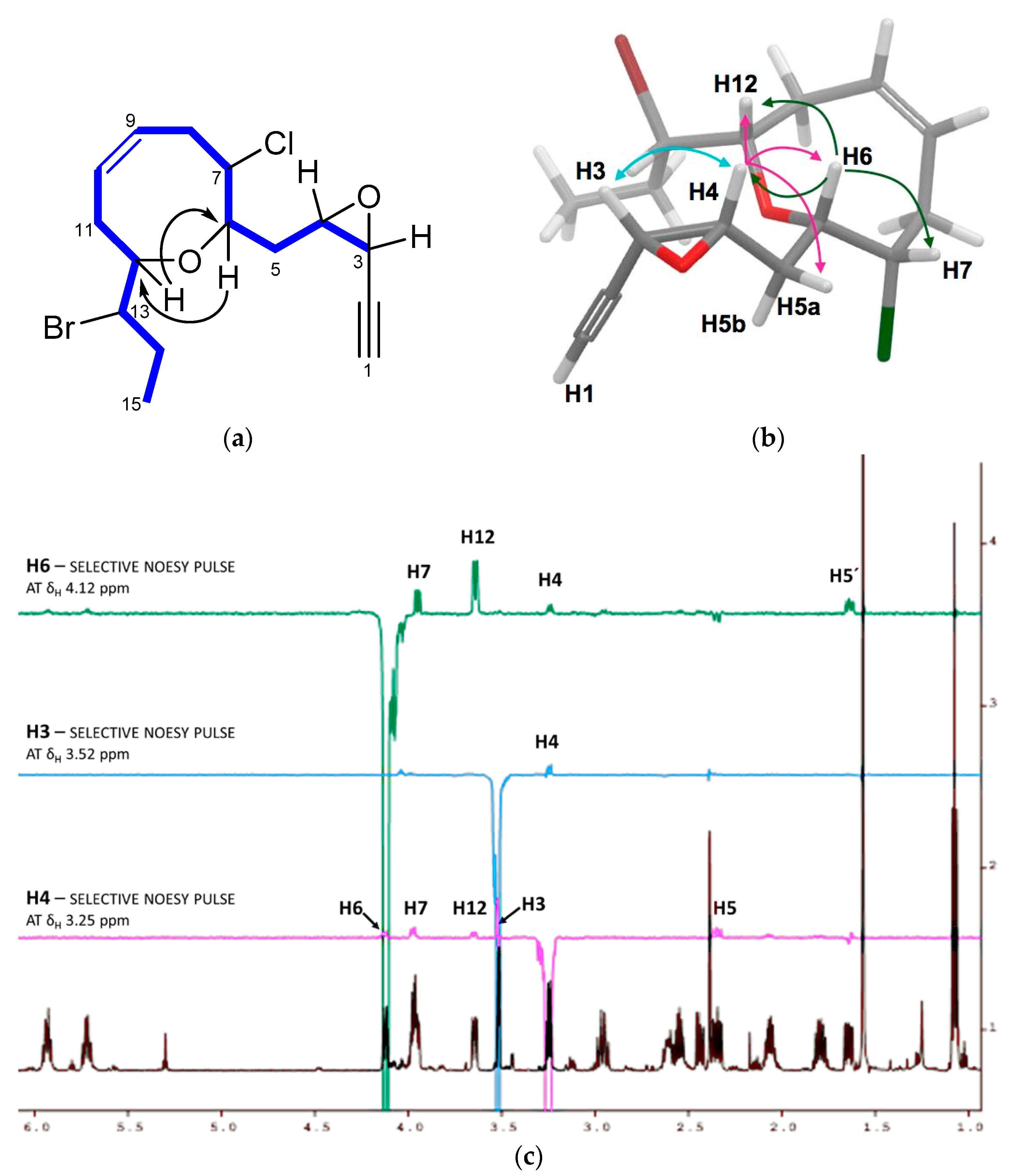
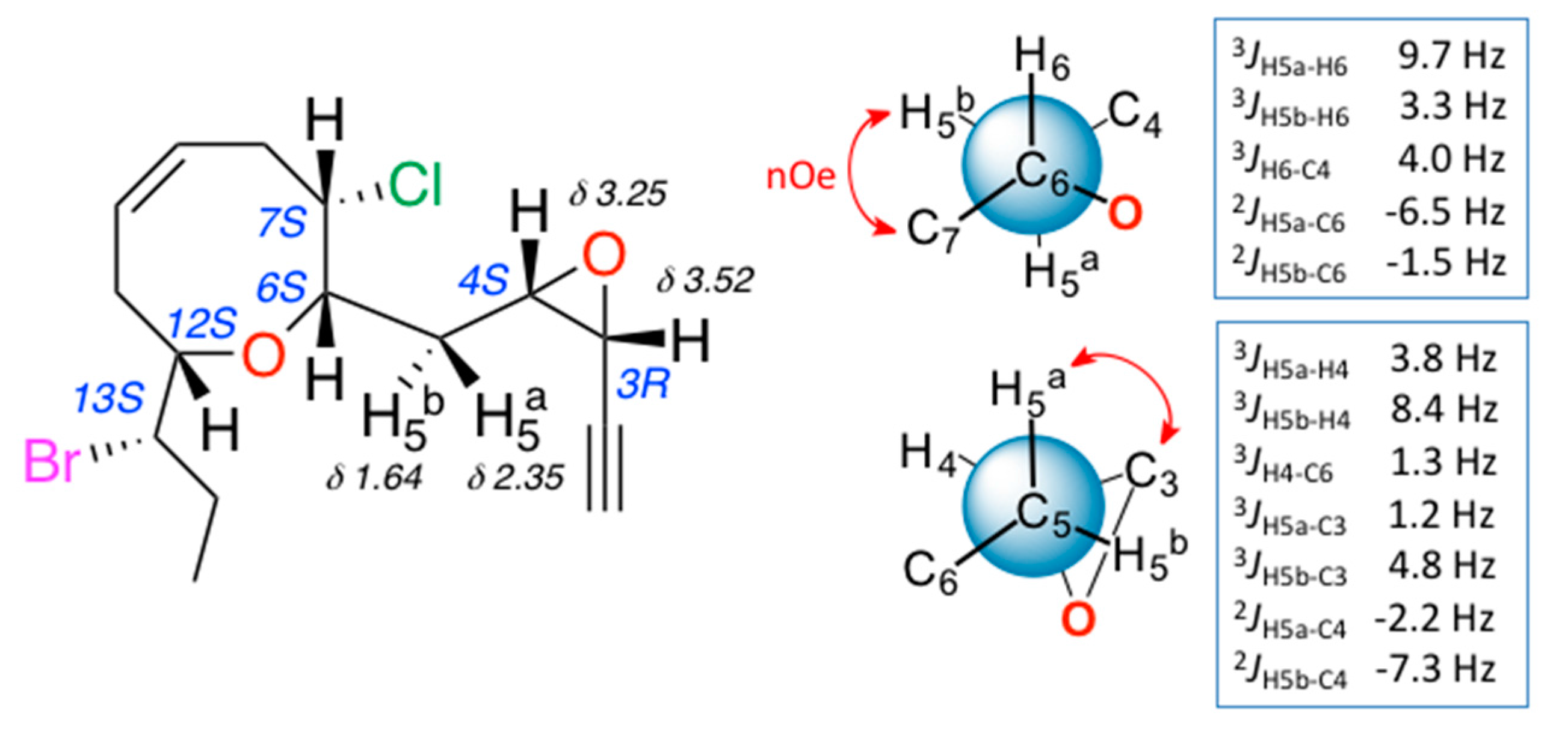
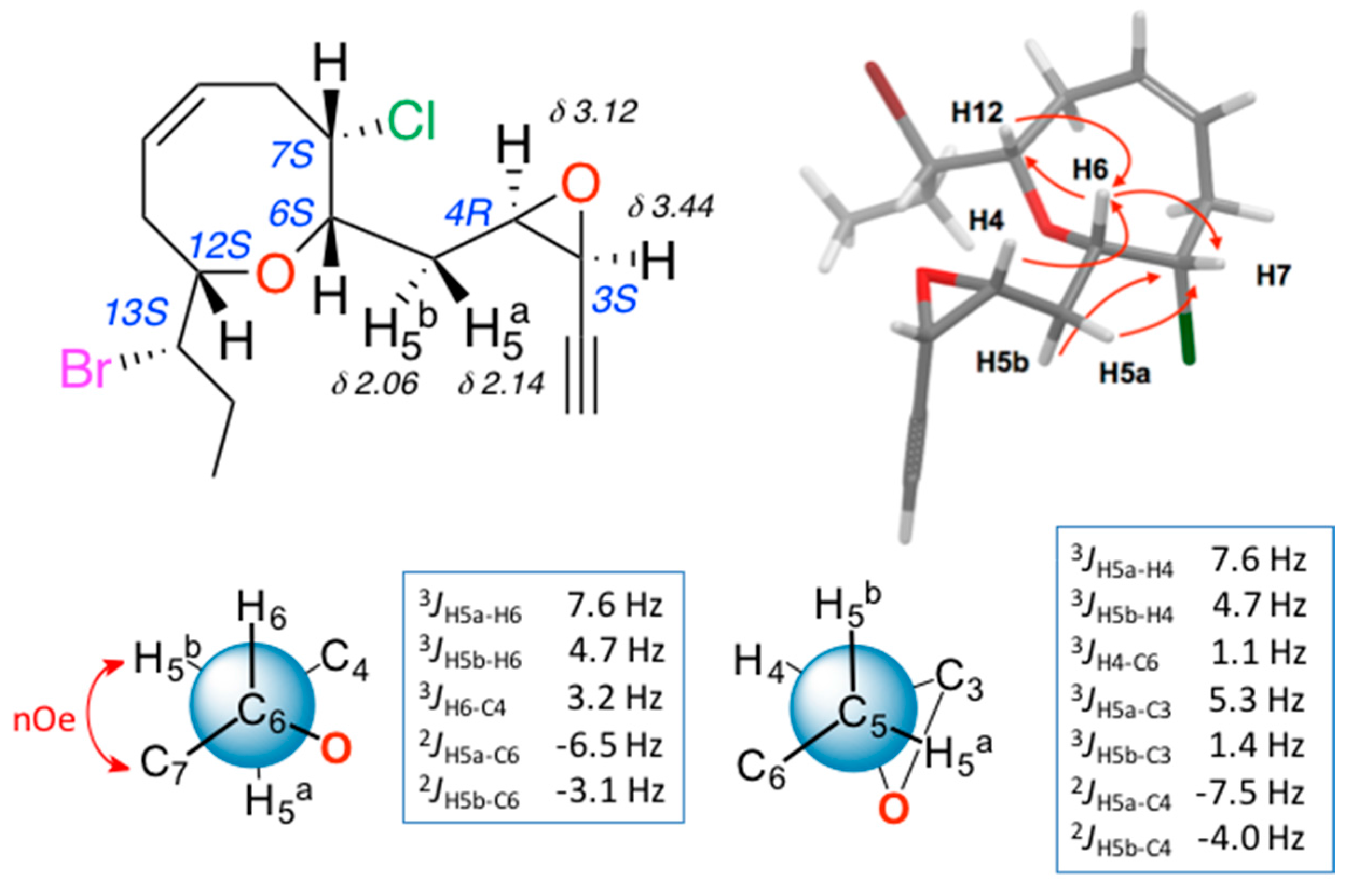
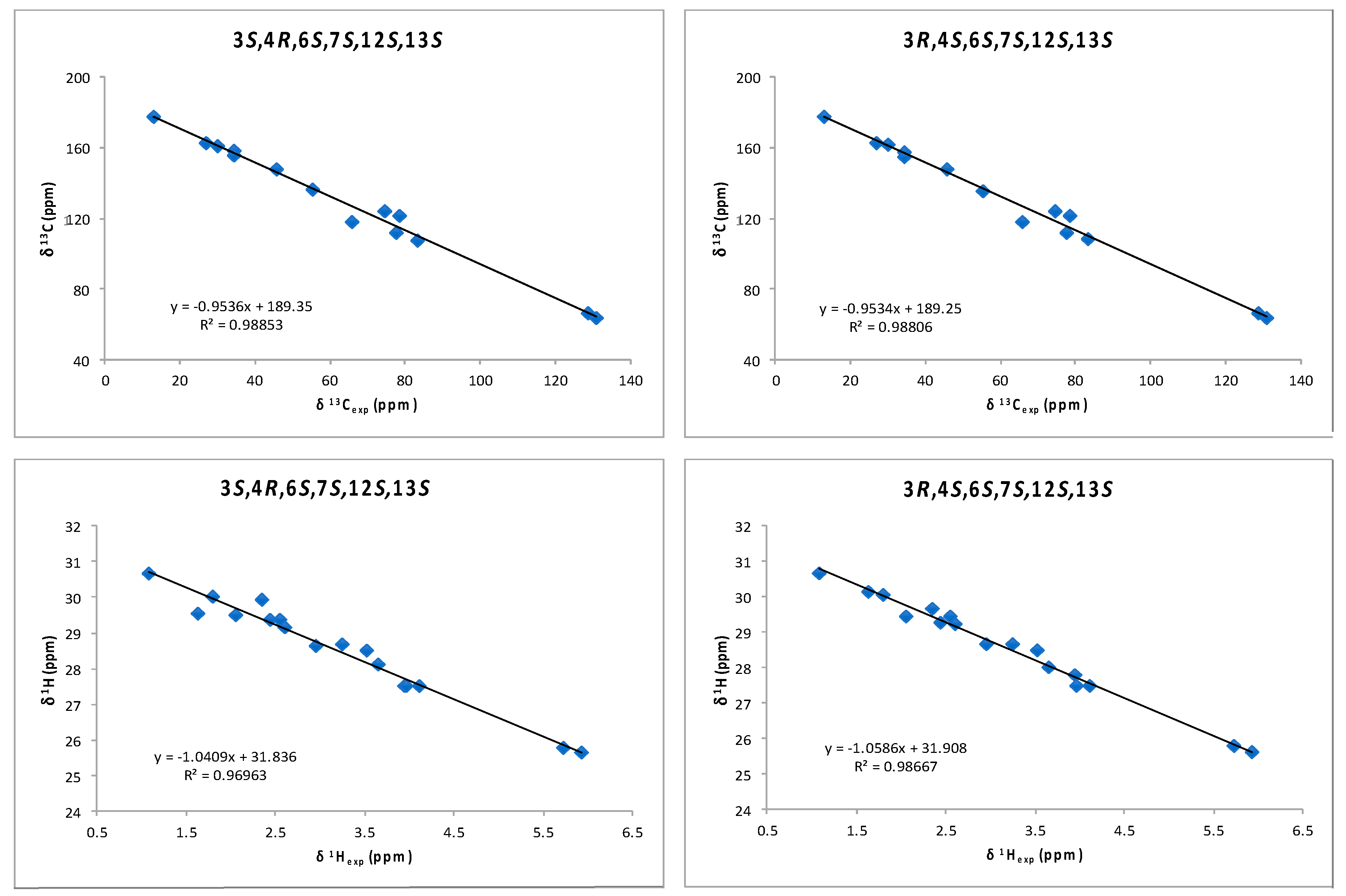
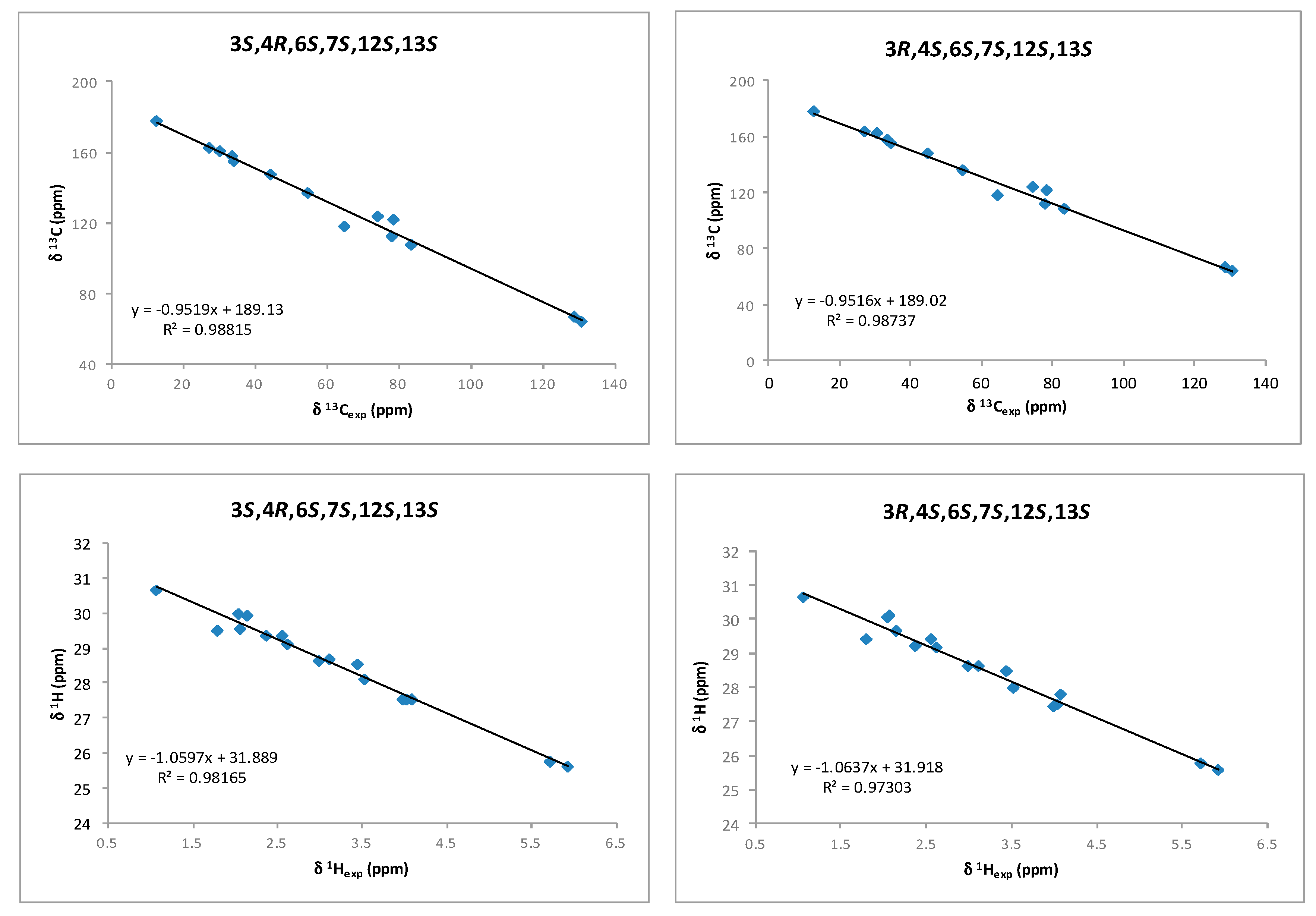
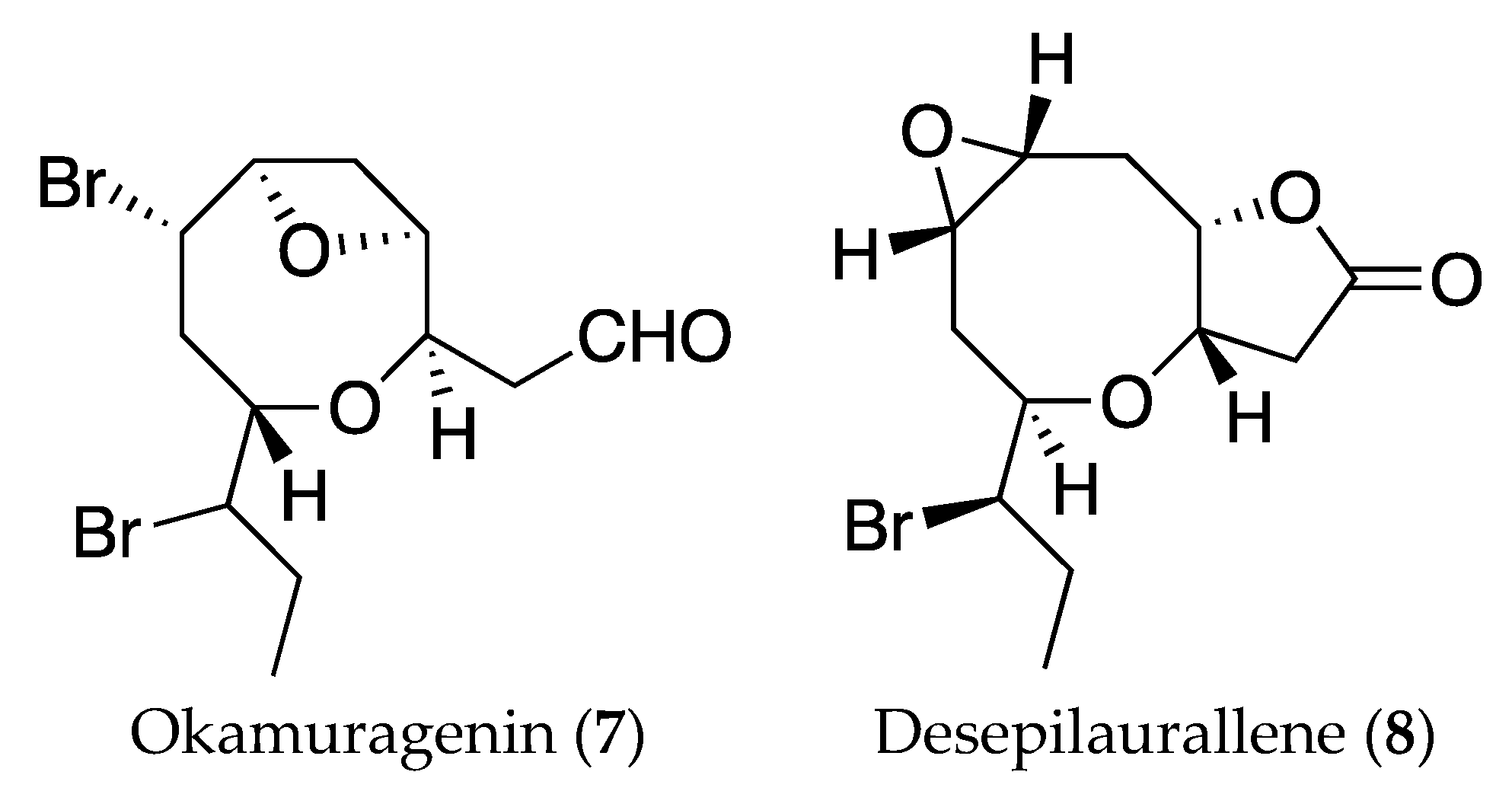
2. Results and Discussion
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Computational Methods
3.3. Biological Material
3.4. Extraction and Isolation
3.5. Chemical Conversion of Z-Pinnatifidenyne (1) to (9R,10S)-Epoxy-Z-Pinnatifidenyne (5)
3.6. Antiproliferative Activity
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinset, M.R. Marine natural products. Nat. Prod. Rep. 2017, 34, 235–294, (and earlier reviews in this series). [Google Scholar] [CrossRef] [PubMed]
- Harizani, M.; Ioannou, E.; Roussis, V. The Laurencia paradox: An endless source of chemodiversity. In Progress in the Chemistry of Organic Natural Products; Kinghorn, A.D., Galk, H., Gibbons, S., Kobayashi, J., Eds.; Springer: Berlin, Germany, 2016; Volume 102, pp. 91–252. [Google Scholar]
- Lin, R.; Cao, L.; West, F.G. Medium-sized cyclic ethers via Stevens [1,2]-shift of mixed monothioacetal-derived sulfonium ylides: Application to formal synthesis of (+)-laurencin. Org. Lett. 2017, 19, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.-Y.; Zhang, W.; Schienebeck, C.M.; Bennett, S.R.; Tang, W. Intermolecular bromoesterification of conjugated enynes: An efficient synthesis of bromoallenes. Org. Chem. Front. 2014, 1, 386–390. [Google Scholar] [CrossRef]
- Bonney, K.J.; Braddock, D.C. A unifying stereochemical analysis for the formation of halogenated C15-acetogenin medium-ring ethers from Laurencia species via intramolecular bromonium ion assisted epoxide ring-opening and experimental corroboration with a model epoxide. J. Org. Chem. 2012, 77, 9574–9584, (and references cited therein). [Google Scholar] [CrossRef] [PubMed]
- Braddock, D.C.; Sbircea, D.-T. Proof-of-principle direct double cyclisation of a linear C15-precursor to a brominated bicyclic medium-ring ether relevant to Laurencia species. Chem. Commun. 2014, 507, 12691–12693. [Google Scholar] [CrossRef] [PubMed]
- González, A.G.; Martín, J.D.; Martín, V.S.; Norte, M.; Pérez, R.; Ruano, J.Z.; Drexler, S.A.; Clardy, J. Non-terpenoid C-15 metabolites from the red seaweed Laurencia pinnatifida. Tetrahedron 1982, 38, 1009–1014. [Google Scholar] [CrossRef]
- Norte, M.; González, A.G.; Cataldo, F.; Rodríguez, M.L.; Brito, I. New examples of acyclic and cyclic C-15 acetogenins from Laurencia pinnatifida. Reassignment of the absolute configuration for E and Z pinnatifidienyne. Tetrahedron 1991, 47, 9411–9418. [Google Scholar] [CrossRef]
- Kim, D. Intramolecular enolate alkylation from steroids through cladiellins to isolaurallene. Synlett 2014, 25, 33–57. [Google Scholar] [CrossRef]
- Snyder, S.A.; Brucks, A.P.; Treitler, D.S.; Moga, I. Concise synthetic approaches for the Laurencia family: Formal total syntheses of (±)-laurefucin and (±)-E- and (±)-Z-pinnatifidenyne. J. Am. Chem. Soc. 2012, 134, 17714–17721. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Cepeda, A.; Fernández, J.J.; Norte, M.; López-Rodríguez, M.; Brito, I.; Souto, M.L. Additional insights into the obtusallene family: Components of Laurencia marilzae. J. Nat. Prod. 2016, 79, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Cen-Pacheco, F.; Santiago-Benítez, A.J.; García, C.; Álvarez-Méndez, S.J.; Martín-Rodríguez, A.J.; Norte, M.; Martín, V.S.; Gavín, J.A.; Fernández, J.J.; Daranas, A.H. Oxasqualenoids from Laurencia viridis: Combined spectroscopic–computational analysis and antifouling potential. J. Nat. Prod. 2015, 78, 712–721. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Cepeda, A.; Hernández Daranas, A.; Fernández, J.J.; Norte, M.; Souto, M.L. Stereochemical determination of five-membered cyclic ether acetogenins using a spin-spin coupling constant approach and DFT calculations. Mar. Drugs 2014, 12, 4031–4044. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Cepeda, A.; Fernández, J.J.; Norte, M.; Souto, M.L. New bicyclotridecane C15 nonterpenoid bromoallenes from Laurencia marilzae. Org. Lett. 2011, 13, 2690–2693. [Google Scholar] [CrossRef] [PubMed]
- Bifulco, G.; Dambruoso, P.; Gomez-Paloma, L.; Riccio, R. Determination of relative configuration in organic compounds by NMR spectroscopy and computational methods. Chem. Rev. 2007, 107, 3744–3779. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, A.G.; Reddy, D.S. High-throughput in silico structure validation and revision of halogenated natural products is enabled by parametric corrections to DFT-computed 13C NMR chemical shifts and spin-spin coupling constants. J. Org. Chem. 2017, 82, 3368–3381. [Google Scholar] [CrossRef] [PubMed]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational prediction of 1H and 13C chemical shifts: A useful tool for natural product, mechanistic and synthetic organic chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef] [PubMed]
- Cen-Pacheco, F.; Rodríguez, J.; Norte, M.; Fernández, J.J.; Hernández Daranas, A. Connecting discrete stereoclusters by using DFT and NMR spectroscopy: The case of nivariol. Chem. Eur. J. 2013, 19, 8525–8532. [Google Scholar] [CrossRef] [PubMed]
- Domínguez, H.J.; Crespín, G.D.; Santiago Benítez, A.J.; Gavín, J.A.; Norte, M.; Fernández, J.J.; Hernández Daranas, A. Stereochemistry of complex marine natural products by quantum mechanical calculations of NMR chemical shifts: Solvent and conformational effects on okadaic acid. Mar. Drugs 2014, 12, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.G.; Channon, J.A.; Paterson, I.; Goodman, J.M. The stereochemical assignment of acyclic polyols: A computational study of the NMR data of a library of stereopentad sequences from polyketide natural products. Tetrahedron 2010, 66, 6437–6444. [Google Scholar] [CrossRef]
- Rassolov, V.A.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6–31G* basis set for third-row atoms. J. Comp. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Smith, S.G.; Goodman, J.M. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, W.J.; Jung, J.; Kim, S.; Kim, D. Construction of eight-membered ether rings by olefin geometry-dependent internal alkylation: First asymmetric total synthesis of (+)-3-(E)- and (+)-3-(Z)-pinnatifidenyne. J. Am. Chem. Soc. 2003, 125, 10238–10240. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Li, X.-M.; Cui, C.-M.; Li, C.-S.; Sun, H.; Wang, B.-G. Sesquiterpene and acetogenin derivatives from the marine red alga Laurencia okamurai. Mar. Drugs 2012, 10, 2817–2825. [Google Scholar] [CrossRef] [PubMed]
- Li, X.D.; Miao, F.P.; Li, K.; Ji, N.Y. Sesquiterpenes and acetogenins from the marine red alga Laurencia okamurai. Fitoterapia 2012, 83, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Castillo, Q.A.; Triana, J.; Eiroa, J.L.; Calcul, L.; Rivera, E.; Wojtas, L.; Padrón, J.M.; Boberieth, L.; Keramane, M.; Abel-Santos, E.; et al. ent-Labdane diterpenoids from the aerial parts of Eupatorium obtusissmum. J. Nat. Prod. 2016, 79, 907–913. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | (3R,4S)-Epoxy-Pinnatifidenyne (3) | (3S,4R)-Epoxy-Pinnatifidenyne (4) |
|---|---|---|
| δH (J in Hz) | δH (J in Hz) | |
| 1 | 2.39, d (1.7) | 2.38, d (1.7) |
| 3 | 3.52, dd (1.7, 4.0) | 3.44, dd (1.7, 4.0) |
| 4 | 3.25, ddd (3.8, 4.0, 8.4) | 3.12, ddd (4.0, 4.7, 7.6) |
| 5 | 1.64, ddd (3.3, 8.4, 14.6) 2.35, ddd (3.8, 9.7, 14.6) | 2.06, ddd (4.7, 7.0, 14.5) 2.14, ddd (7.6, 7.6, 14.5) |
| 6 | 4.12, ddd (2.5, 3.3, 9.7) | 4.03, ddd (2.6, 4.7, 7.6) |
| 7 | 3.95, ddd (2.5, 5.0, 11.6) | 4.08, ddd (2.6, 5.1,11.7) |
| 8 | 2,55, ddd (5.0, 6.6, 11.8) 2.96, ddd (1.7, 11.6, 11.8) | 2.56, ddd (5.8, 6.1, 12.5) 2.99, dddd (1.8, 11.6, 12.5) |
| 9 | 5.72, dddd (1.7, 6.6, 10.1) | 5.72, ddd (1.8, 6.1, 10.3) |
| 10 | 5.93, br.dd (8.4, 10.1) | 5.93, br.dd (8.5, 10.3) |
| 11 | 2.44, ddd (1.3, 8.4, 14.0) 2.61, br.dd (3.7, 14.0) | 2.37, ddd (1.0, 8.5, 13.9) 2.62, br.dd (3.8, 13.9) |
| 12 | 3.65, ddd (1.3, 3.7, 10.0) | 3.53, ddd (1.0, 3.8, 10.1) |
| 13 | 3.97, ddd (3.0, 3.3, 10.0) | 3.97, ddd (2.8, 3.5, 10.1) |
| 14 | 1.79, ddq (3.3, 7.2, 14.5) 2.06, ddq (3.0, 7.2, 14.5) | 1.80, ddq (2.8, 7.2, 14.5) 2.04, ddq (3.5, 7.2, 14.5) |
| 15 | 1.08, t (7.2) | 1.08, t (7.2) |
| Position | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|
| Multi. | δC | δC | δC | δC | |
| 1 | CH | 74.5 | 74.3 | 83.1 | |
| 2 | C | 78.6 | 78.6 | 80.5 | |
| 3 | CH | 45.6 | 44.7 | 111.7 | |
| 4 | CH | 55.3 | 54.8 | 140.2 | 200.2 |
| 5 | CH2 | 34.3 | 33.7 | 35.1 | 49.1 |
| 6 | CH | 77.5 | 78.1 | 81.5 | 74.2 |
| 7 | CH | 65.8 | 64.8 | 60.3 | 65.4 |
| 8 | CH2 | 34.5 | 34.4 | 34.5 | 34.4 |
| 9 | CH | 128.9 | 128.9 | 52.6 | 128.7 |
| 10 | CH | 131.0 | 131.0 | 52.9 | 131.3 |
| 11 | CH2 | 30.0 | 30.4 | 32.1 | 31.2 |
| 12 | CH | 83.1 | 83.5 | 81.8 | 83.3 |
| 13 | CH | 61.1 | 61.2 | 59.9 | 61.8 |
| 14 | CH2 | 27.1 | 27.3 | 26.8 | 27.7 |
| 15 | CH3 | 12.8 | 12.9 | 12.9 | 12.8 |
| Position | (9R,10S)-Epoxy-Z-Pinnatifidenyne (5) | Pinnatifidehyde (6) |
|---|---|---|
| δH (J in Hz) | δH (J in Hz) | |
| 1 | 3.17, br.s | |
| 3 | 5.63, br.d (10.8) | |
| 4 | 6.05, ddd (7.6, 7.7, 10.8) | 9.80, br.s |
| 5 | 2.58, dd (5.5, 7.7, 14.2) 2.87, ddd (7.6, 7.6, 14.2) | 2.71, dd (4.0, 18.7) 3.10, br.dd (8.0, 18.7) |
| 6 | 3.79, ddd (2.3, 5.5, 7.6) | 4.46, ddd (2.9, 4.0, 8.0) |
| 7 | 4.11, ddd (2.3, 4.8, 11.7) | 4.00, ddd (2.9, 5.1, 11.8) |
| 8 | 1.94, ddd (11.6, 11.7, 13.2) 2.67, ddd (3.5, 4.8, 13.2) | 2.53, ddd (2.2, 5.1, 12.0) 2.96, ddd (5.1, 6.6, 12.0) |
| 9 | 2.88, ddd (3.5, 4.0, 11.6) | 5.72, ddd (2.2, 6.6, 10.3) |
| 10 | 3.05, ddd (4.0, 4.6, 9.4) | 5.96, br.dd, (8.3, 10.3) |
| 11 | 1.65, ddd (9.4, 10.9, 14.0) 2.58, ddd (3.3, 4.6, 14.0) | 2.31, ddd, (1.4, 8.3, 14.0) 2.64, br.dd, (3.9, 14.0) |
| 12 | 3.66, dd (3.3; 10.9) | 3.67, ddd (1.4, 3.9, 10.3) |
| 13 | 4.00, ddd (3.1, 3.1, 10.9) | 3.84, ddd (2.8, 2.9, 10.3) |
| 14 | 1.73, ddq (3.1, 7.3, 14.5) 2.02, ddq (3.1, 7.3, 14.5) | 1.74, ddq (2.9, 7.3, 14.6) 1.90, ddq (2.8, 7.3, 14.6) |
| 15 | 1.08, t (7.3) | 1.04, t (7.3) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morales-Amador, A.; De Vera, C.R.; Márquez-Fernández, O.; Hernández Daranas, A.; Padrón, J.M.; Fernández, J.J.; Souto, M.L.; Norte, M. Pinnatifidenyne-Derived Ethynyl Oxirane Acetogenins from Laurencia viridis. Mar. Drugs 2018, 16, 5. https://doi.org/10.3390/md16010005
Morales-Amador A, De Vera CR, Márquez-Fernández O, Hernández Daranas A, Padrón JM, Fernández JJ, Souto ML, Norte M. Pinnatifidenyne-Derived Ethynyl Oxirane Acetogenins from Laurencia viridis. Marine Drugs. 2018; 16(1):5. https://doi.org/10.3390/md16010005
Chicago/Turabian StyleMorales-Amador, Adrián, Caterina R. De Vera, Olivia Márquez-Fernández, Antonio Hernández Daranas, José M. Padrón, José J. Fernández, María L. Souto, and Manuel Norte. 2018. "Pinnatifidenyne-Derived Ethynyl Oxirane Acetogenins from Laurencia viridis" Marine Drugs 16, no. 1: 5. https://doi.org/10.3390/md16010005