Marine-Derived 2-Aminoimidazolone Alkaloids. Leucettamine B-Related Polyandrocarpamines Inhibit Mammalian and Protozoan DYRK & CLK Kinases

 ,
,
Abstract
: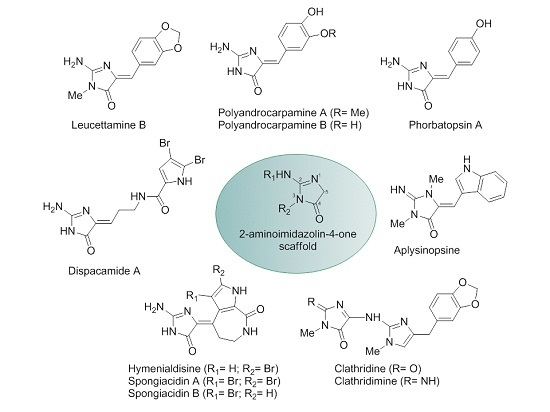
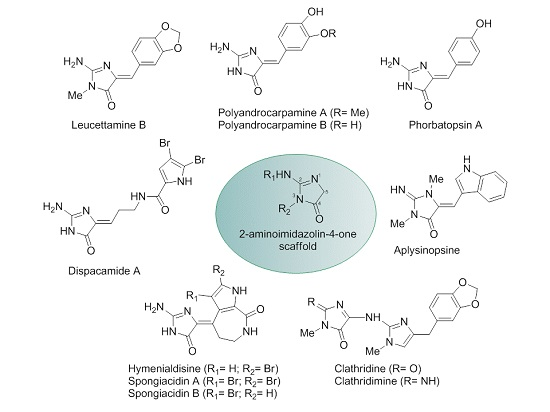
1. Introduction
2. Results
2.1. Phylogeny of Calcareous Sponges with Special Emphasis on Leucetta and Clathrina
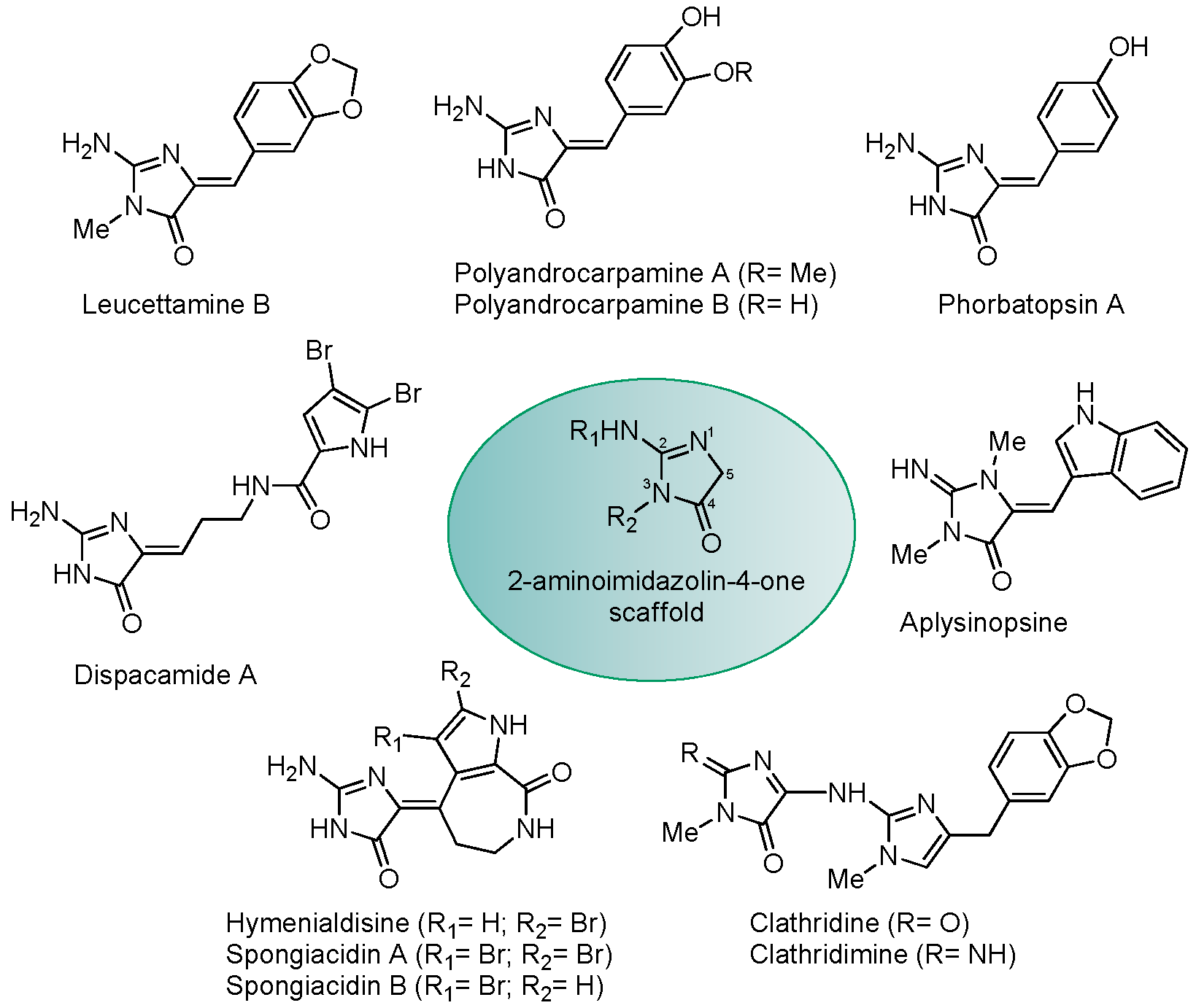
2.2. Polyandrocarpamines Are Potent Inhibitors of the DYRK and CLK Kinases
3. Discussion
4. Experimental Section
4.1. DNA Sequencing, Alignment and Phylogenetic Analyses
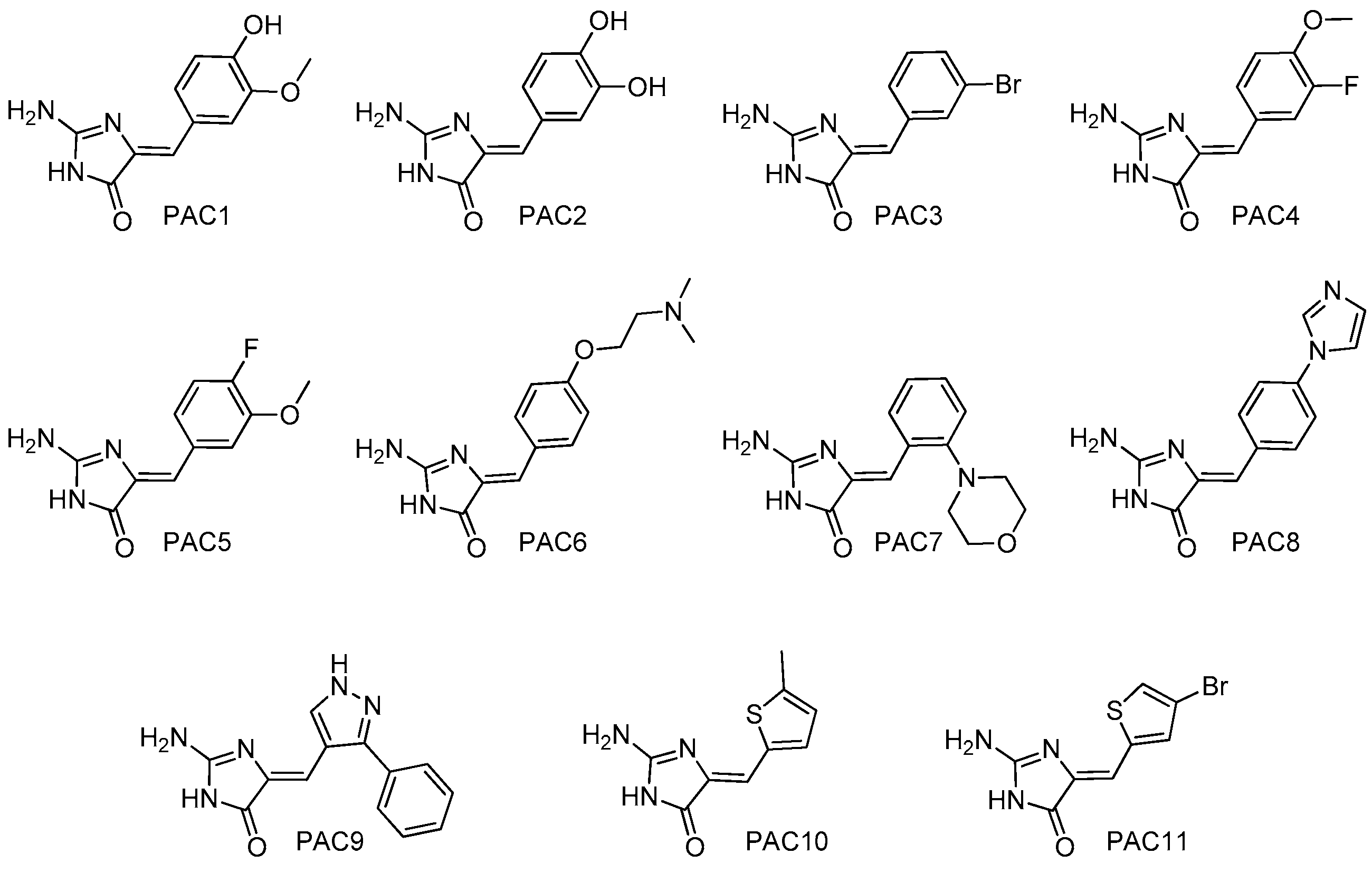
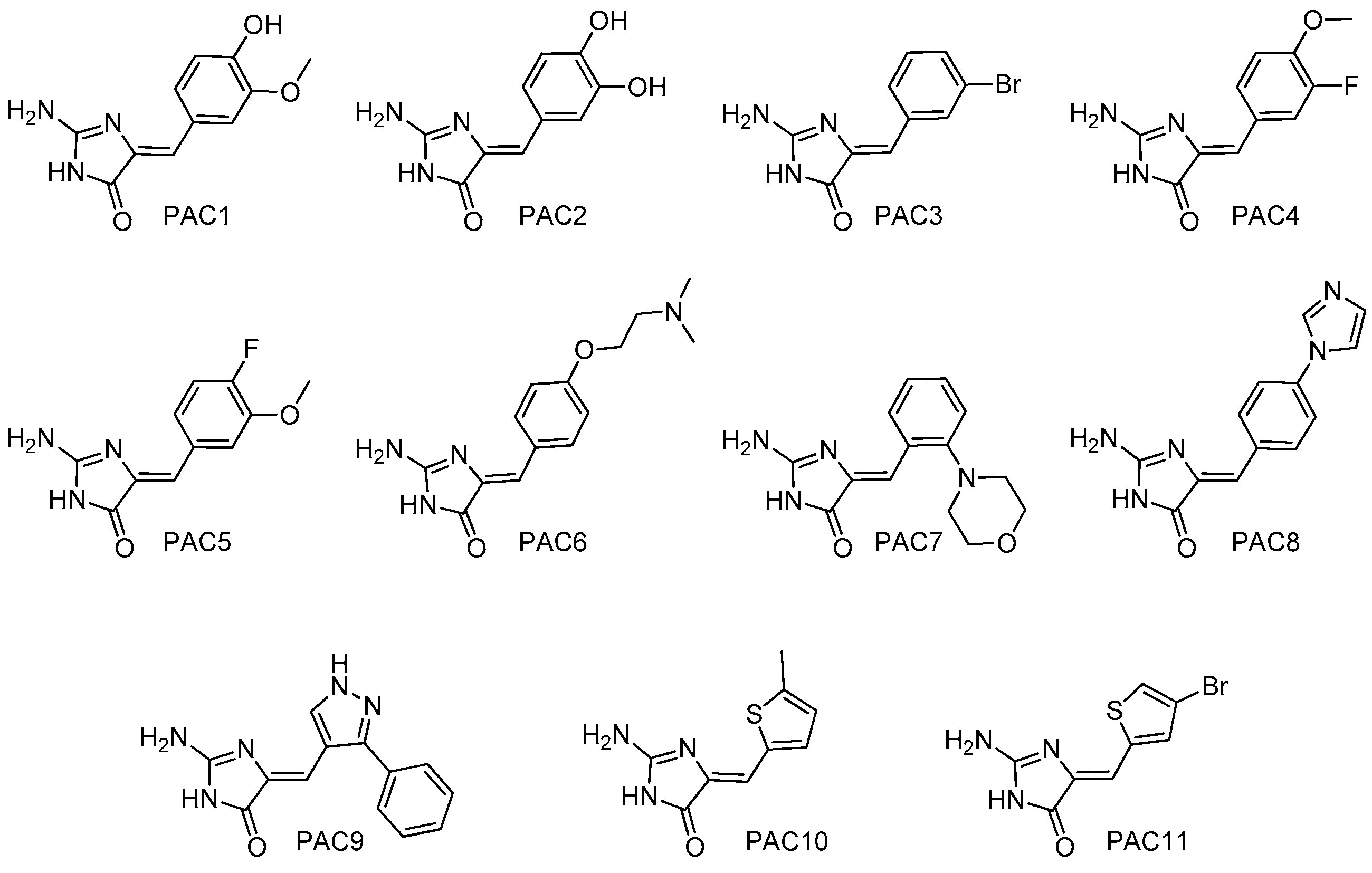
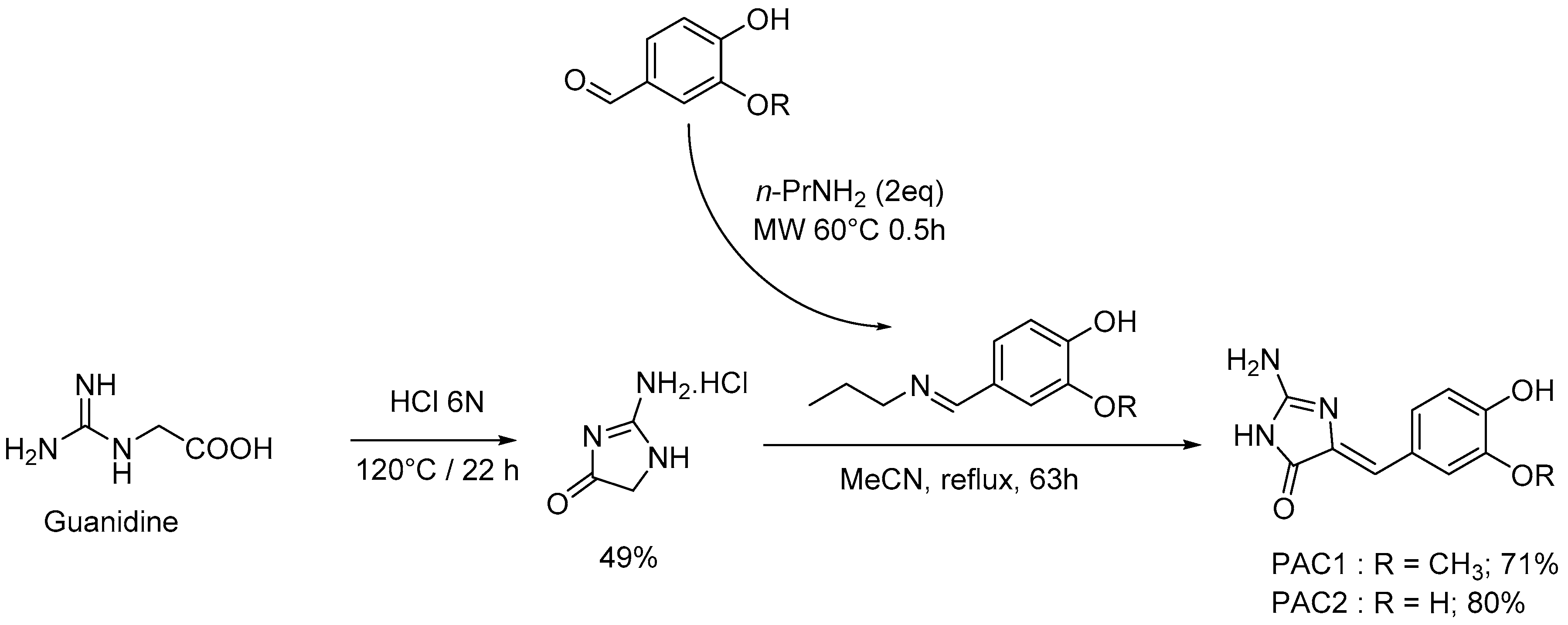
4.2. Chemistry: Purification or Synthesis of Polyandrocarpamines A and B, and Analogues
4.3. Biology
4.3.1. Buffers
4.3.2. Production and Purification of the Parasitic Kinases
4.3.3. Protein Kinase Assays
4.3.4. Kinase Interaction Panel (Ambit Biosciences/DiscoveRx)
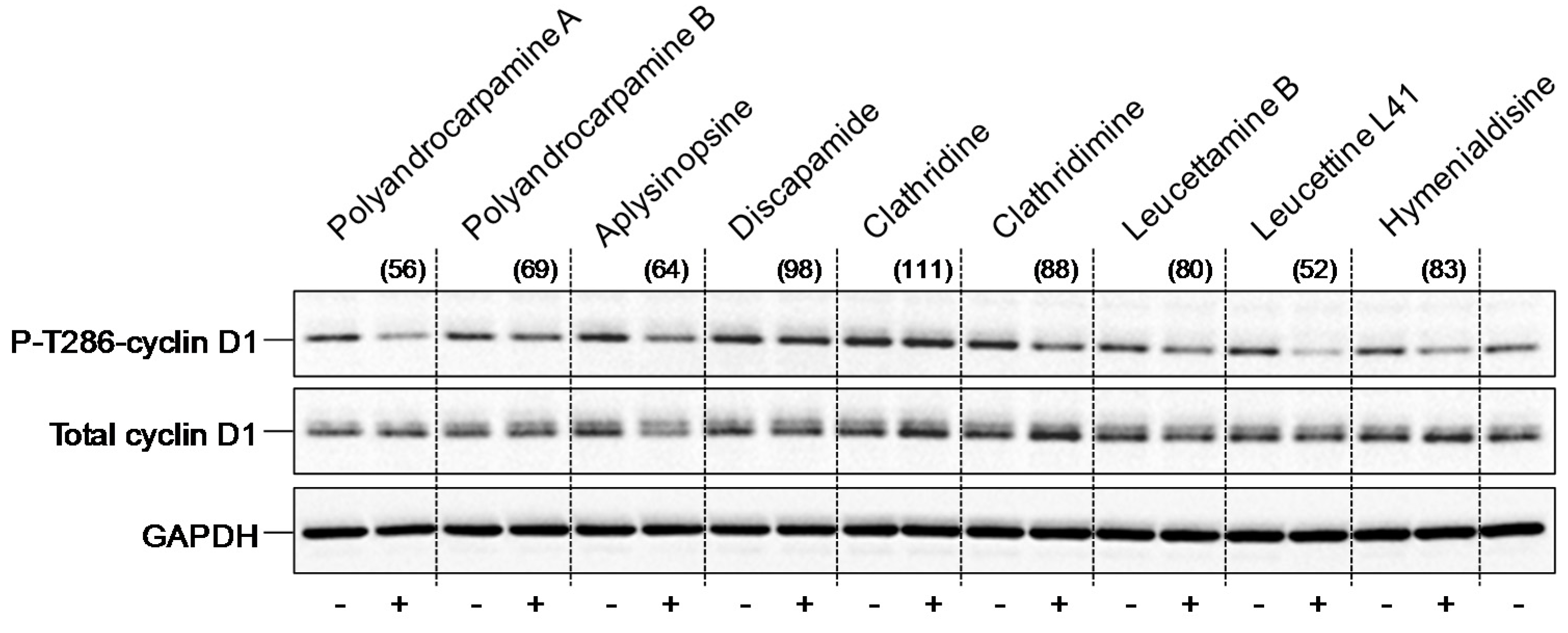
4.3.5. Thr286-Cyclin D1 Phosphorylation in SH-SY5Y Cells
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
References
- Roskoski, R., Jr. A historical overview of protein kinases and their targeted small molecule inhibitors. Pharmacol. Res. 2015, 100, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.H.; Krebs, E.G. Conversion of phosphorylase b to phosphorylase a in muscle extracts. J. Biol. Chem. 1955, 216, 121–132. [Google Scholar] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. Small-molecule kinase inhibitors: An analysis of FDA-approved drugs. Drug Discov. Today 2016, 21, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Nielsen, T.E.; Clausen, M.H. FDA-approved small-molecule kinase inhibitors. Trends Pharmacol. Sci. 2015, 36, 422–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debdab, M.; Renault, S.; Eid, S.; Lozach, O.; Meijer, L.; Carreaux, F.; Bazureau, J.-P. An efficient method for the preparation of new analogs of leucettamine B under solvent-free microwave irradiation. Heterocycles 2009, 78, 1191–1203. [Google Scholar]
- Debdab, M.; Carreaux, F.; Renault, S.; Soundararajan, M.; Fedorov, O.; Filippakopoulos, P.; Lozach, O.; Babault, L.; Tahtouh, T.; Baratte, B.; et al. Leucettines, a class of potent inhibitors of cdc2-like kinases and dual specificity, tyrosine phosphorylation regulated kinases derived from the marine sponge leucettamine B: Modulation of alternative pre-RNA splicing. J. Med. Chem. 2011, 54, 4172–4186. [Google Scholar] [CrossRef] [PubMed]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.W.; Mong, S.; Hemling, M.E.; Freyer, A.J.; Offen, P.M.; De Brosse, C.W.; Sarau, H.M.; Westley, J.W. New leukotriene B4 receptor antagonist: Leucettamine A and related imidazole alkaloids from the marine sponge Leucetta microraphis. J. Nat. Prod. 1993, 56, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.D.; Giles, R.L.; Looper, R.E. 2-Aminoimidazoles from Leucetta Sponges: Synthesis and biology of an important pharmacophore. Curr. Bioact. Compd. 2009, 5, 39–78. [Google Scholar] [CrossRef]
- Koswatta, P.B.; Lovely, C.J. Structure and synthesis of 2-aminoimidazole alkaloids from Leucetta and Clathrina sponges. Nat. Prod. Rep. 2011, 28, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Roué, M.; Quévrain, E.; Domart-Coulon, I.; Bourguet-Kondracki, M.L. Assessing calcareous sponges and their associated bacteria for the discovery of new bioactive natural products. Nat. Prod. Rep. 2012, 29, 739–751. [Google Scholar] [CrossRef] [PubMed]
- Žula, A.; Kikelj, D.; Ilaš, J. 2-Aminoimidazoles in medicinal chemistry. Mini Rev. Med. Chem. 2013, 13, 1921–1943. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Zhu, M.; Davidson, E.H.; Bottjer, D.J.; Zhao, F.; Tafforeau, P. Sponge grade body fossil with cellular resolution dating 60 Myr before the Cambrian. Proc. Natl. Acad. Sci. USA 2015, 112, E1453–E1460. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.L.; Campos, A.P.C.; Barroso, M.M.S.; Klautau, M.; Archanjo, B.S.; Borojevic, R.; Farina, M.; Werckmann, J. Long-range crystalline order in spicules from the calcareous sponge Paraleucilla magna (Porifera, Calcarea). Acta Biomater. 2014, 10, 3875–3884. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.A.; Aalbersberg, W.; Meo, S.; Moreira da Rocha, R.; Ireland, C.M. The isolation and synthesis of polyandrocarpamines A and B. Two new 2-aminoimidazolone compounds from the Fijian ascidian, Polyandrocarpa sp. Tetrahedron Lett. 2002, 58, 3263–3269. [Google Scholar] [CrossRef]
- Davis, R.A.; Baron, P.S.; Neve, J.E.; Cullinane, C. A microwave-assisted stereoselective synthesis of polyandrocarpamines A and B. Tetrahedron Lett. 2008, 50, 880–882. [Google Scholar] [CrossRef]
- Lindel, T.; Hoffmann, H. Synthesis of dispacamide from the marine sponge Agelas dispar. Tetrahedron Lett. 1997, 38, 8935–8938. [Google Scholar] [CrossRef]
- Fresneda, P.M.; Molina, P.; Sanz, M.A. A convergent approach to midpacamide and dispacamide pyrrole-imidazole marine alkaloids. Tetrahedron Lett. 2001, 42, 851–854. [Google Scholar] [CrossRef]
- Travert, N.; Al-Mourabit, A. A likely biogenetic gateway linking 2-aminoimidazolinone metabolites of sponges to proline: Spontaneous oxidative conversion of the pyrrole-proline-guanidine pseudo-peptide to dispacamide A. J. Am. Chem. Soc. 2004, 126, 10252–10253, Erratum in 2005, 127, 10454. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeak, K.H.; Schmitz, F.J. Aplysinopsin: Antineoplastic tryptophan derivative from the marine sponge Verongia spengelii. Lloydia 1977, 40, 479–481. [Google Scholar] [PubMed]
- Bialonska, D.; Zjawiony, J.K. Aplysinopsins—Marine indole alkaloids: Chemistry, bioactivity and ecological significance. Mar. Drugs 2009, 7, 166–183. [Google Scholar] [CrossRef] [PubMed]
- Roué, M.; Domart-Coulon, I.; Ereskovsky, A.; Djediat, C.; Perez, T.; Bourguet-Kondracki, M.L. Cellular localization of clathridimine, an antimicrobial 2-aminoimidazole alkaloid produced by the Mediterranean calcareous sponge Clathrina clathrus. J. Nat. Prod. 2010, 73, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Cimino, G.; de Rosa, S.; de Stefano, S.; Mazzarella, L.; Puliti, R.; Sodano, G. Isolation & X-ray crystal structure of a novel bromo-compound from two marine sponges. Tetrahedron Lett. 1982, 23, 767–768. [Google Scholar]
- Sharma, G.M.; Buyer, J.; Pomerantz, M.W. Characterization of a yellow compound isolated from the marine sponge Phakellia flabellata. J. Chem. Soc. Chem. Commun. 1980, 10, 435–436. [Google Scholar] [CrossRef]
- Williams, D.H.; Faulkner, J. Isomers and tautomers of hymenialdisine and debromohymenialdisine. Nat. Prod. Lett. 1996, 9, 57–64. [Google Scholar] [CrossRef]
- Xu, Y.Y.; Yakushijin, K.; Horne, D.A. Synthesis of C(11)N(5) Marine sponge alkaloids: (+/−)-hymenin, stevensine, hymenialdisine, and debromohymenialdisine. J. Org. Chem. 1997, 62, 456–464. [Google Scholar] [CrossRef]
- Papeo, G.; Posteri, H.; Borghi, D.; Varasi, M. A new glycociamidine ring precursor: Syntheses of (Z)-hymenialdisine, (Z)-2-debromohymenialdisine, and (+/−)-endo-2-debromohymenialdisine. Org. Lett. 2005, 7, 5641–5644. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.N.; Tepe, J.J. Preparation of hymenialdisine, analogues and their evaluation as kinase inhibitors. Curr. Med. Chem. 2009, 16, 3122–3143. [Google Scholar] [CrossRef] [PubMed]
- Meijer, L.; Thunnissen, A.M.W.H.; White, A.; Garnier, M.; Nikolic, M.; Tsai, L.H.; Walter, J.; Cleverley, K.E.; Salinas, P.C.; Wu, Y.Z.; et al. Inhibition of cyclin-dependent kinases, GSK-3β and casein kinase 1 by hymenialdisine, a marine sponge constituent. Chem. Biol. 2000, 7, 51–63. [Google Scholar] [CrossRef]
- Wan, Y.; Hur, W.; Cho, C.Y.; Liu, Y.; Adrian, F.J.; Lozach, O.; Bach, S.; Mayer, T.; Fabbro, D.; Meijer, L.; et al. Synthesis and target identification of hymenialdisine analogs. Chem. Biol. 2004, 11, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.D.; Nguyen, X.C.; Longeon, A.; Keryhuel, A.; Le, M.H.; Kim, Y.H.; Chau, V.M.; Bourguet-Kondracki, M.L. Antioxidant benzylidene 2-aminoimidazolones from the Mediterranean sponge Phorbas topsenti. Tetrahedron 2012, 68, 9256–9259. [Google Scholar] [CrossRef]
- Ling, Y.; Wang, Z.Q.; Xiao, Y.A.; Zhu, C.; Shen, L.; Wang, X.M.; Hui, Y.; Wang, X.Y. Benzylidene 2-aminoimidazolones derivatives: Synthesis and in vitro evaluation of anti-tumor carcinoma activity. Chem. Pharm. Bull. (Tokyo) 2013, 61, 1081–1084. [Google Scholar] [CrossRef] [PubMed]
- Droucheau, E.; Primot, A.; Thomas, V.; Mattei, D.; Knockaert, M.; Richardson, C.; Sallicandro, P.; Alano, P.; Jafarshad, A.; Baratte, B.; et al. Plasmodium falciparum glycogen synthase kinase-3: Molecular model, expression, intracellular localisation and selective inhibitors. Biochim. Biophys. Acta 2004, 1697, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Rachidi, N.; Taly, J.F.; Durieu, E.; Leclercq, O.; Aulner, N.; Prina, E.; Pescher, P.; Notredame, C.; Meijer, L.; Späth, G.F. Pharmacological assessment defines the Leishmania donovani casein kinase 1 as a drug target and reveals important functions in parasite viability and intracellular infection. Antimicrob. Agents Chemother. 2014, 58, 1501–1515. [Google Scholar] [CrossRef] [PubMed]
- Karaman, M.W.; Herrgard, S.; Treiber, D.K.; Gallant, P.; Atteridge, C.E.; Campbell, B.T.; Chan, K.W.; Ciceri, P.; Davis, M.I.; Edeen, P.T.; et al. A quantitative analysis of kinase inhibitor selectivity. Nat. Biotechnol. 2008, 26, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Soppa, U.; Schumacher, J.; Florencio Ortiz, V.; Pasqualon, T.; Tejedor, F.J.; Becker, W. The Down syndrome-related protein kinase DYRK1A phosphorylates p27(Kip1) and cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle 2014, 13, 2084–2100. [Google Scholar] [CrossRef] [PubMed]
- Najas, S.; Arranz, J.; Lochhead, P.A.; Ashford, A.L.; Oxley, D.; Delabar, J.M.; Cook, S.J.; Barallobre, M.J.; Arbonés, M.L. DYRK1A-mediated cyclin D1 degradation in neural stem cells contributes to the neurogenic cortical defects in Down syndrome. EBioMedicine 2015, 2, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Baron, P.S.; Neve, J.E.; Camp, D.; Suraweera, L.; Lam, A.; Lai, J.; Jovanovic, L.; Nelson, C.; Davis, R.A. Design, synthesis and spectroscopic characterisation of a focused library based on the polyandrocarpamine natural product scaffold. Magn. Reson. Chem. 2013, 51, 358–363. [Google Scholar] [CrossRef] [PubMed]
- Carmely, S.; Ilan, M.; Kashman, Y. 2-Aminoimidazole alkaloids from the marine sponge Leucetta chagosensis. Tetrahedron 1989, 45, 2193–2200. [Google Scholar] [CrossRef]
- Alvi, K.A.; Crews, P.; Loughhead, D.G. Structures and total synthesis of 2-aminoimidazoles from a Notodoris nudibranch. J. Nat. Prod. 1991, 54, 1509–1515. [Google Scholar] [CrossRef]
- Alvi, K.A.; Peters, B.M.; Hunter, L.M.; Crews, P. 2-Aminoimidazoles and their zinc complexes from indo-pacific Leucetta sponges and Notodoris nudibranchs. Tetrahedron 1993, 49, 329–336. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Fruit, C.; Hérault, Y.; Meijer, L.; Besson, T. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) inhibitors: A survey of recent patent literature. Expert Opin. Ther. Pat. 2017, in press. [Google Scholar]
- Tahtouh, T.; Durieu, E.; Villiers, B.; Bruyere, C.; Nguyen, T.L.; Fant, X.; Ahn, K.H.; Khurana, L.; L’helgoual’ch, J.M.; Limanton, E.; et al. Structure/activity relationship in the leucettine family of kinase inhibitors. J. Med. Chem. 2017. submitted. [Google Scholar]
- Fant, X.; Durieu, E.; Chicanne, G.; Payrastre, B.; Sbrissa, D.; Shisheva, A.; Limanton, E.; Carreaux, F.; Bazureau, J.P.; Meijer, L. Cdc-like/dual-specificity tyrosine phosphorylation-regulated kinases Leucettine L41 induces mTOR-dependent autophagy: Implication for Alzheimer’s disease. Mol. Pharmacol. 2014, 85, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Naert, G.; Ferré, V.; Meunier, J.; Keller, E.; Malmström, S.; Givalois, L.; Carreaux, F.; Bazureau, J.P.; Maurice, T. Leucettine L41, a DYRK1A-preferential DYRKs/CLKs inhibitor, prevents memory impairments and neurotoxicity induced by oligomeric Aβ25–35 peptide administration in mice. Eur. Neuropsychopharmacol. 2015, 25, 2170–2182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorson, M.K.; Van Wagoner, R.M.; Harper, M.K.; Ireland, C.M.; Majtan, T.; Kraus, J.P.; Barrios, A.M. Marine natural products as inhibitors of cystathionine beta-synthase activity. Bioorg. Med. Chem. Lett. 2015, 25, 1064–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhang, Z.; Wong, I.L.; Wan, S.; Chow, L.M.; Jiang, T. 4,5-Di-substituted benzyl-imidazol-2-substituted amines as the structure template for the design and synthesis of reversal agents against P-gp-mediated multidrug resistance breast cancer cells. Eur. J. Med. Chem. 2014, 83, 74–83. [Google Scholar] [CrossRef] [PubMed]
- Boehm, J.C.; Gleason, J.G.; Pendrak, I.; Sarau, H.M.; Schmidt, D.B.; Foley, J.J.; Kingsbury, W.D. Synthesis and LTB4 receptor antagonist activities of the naturally occurring LTB4 receptor antagonist Leucettamine A and related analogues. J. Med. Chem. 1993, 36, 3333–3340. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Current status of marine-derived compounds as warheads in anti-tumor drug candidates. Mar Drugs 2017, 15, 99. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Drugs and drug candidates from marine sources: An assessment of the current “state of play”. Planta Med. 2016, 82, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Djura, P.; Faulkner, D.J. Metabolites of the marine sponge Dercitus sp. J. Org. Chem. 1980, 45, 735–737. [Google Scholar] [CrossRef]
- Roué, N.; Bergman, J. Synthesis of the marine alkaloid Leucettamine B. Tetrahedron 1999, 55, 14729–14738. [Google Scholar] [CrossRef]
- Guihéneuf, S.; Paquin, L.; Carreaux, F.; Durieu, E.; Meijer, L.; Bazureau, J.P. An efficient approach to dispacamide A and its derivatives. Org. Biomol. Chem. 2012, 10, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Primot, A.; Baratte, B.; Gompel, M.; Borgne, A.; Liabeuf, S.; Romette, J.L.; Costantini, F.; Meijer, L. Purification of GSK-3 by affinity chromatography on immobilised axin. Protein Expr. Purif. 2000, 20, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, J.; Ferandin, Y.; Meijer, L. Purification CK1 by affinity chromatography on immobilised axin. Protein Expr. Purif. 2007, 54, 101–109. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinases | Leucettamine B | Leucettine L41 | Polyandrocarpamine A | Polyandrocarpamine B | Aplysinopsine | Dispacamide | Hymenialdisine | Spongiacidin B | Clathridine | Clathridimine |
|---|---|---|---|---|---|---|---|---|---|---|
| CDK1/cyclin B | >10 | >10 | >10 | >10 | >10 | >10 | 0.005 | 0.031 | >10 | >10 |
| CDK2/cyclin A | >10 | >10 | >10 | >10 | >10 | >10 | 0.011 | 0.068 | >10 | >10 |
| CDK5/p25 | >10 | >10 | >10 | >10 | >10 | >10 | 0.0061 | 0.042 | >10 | >10 |
| CDK9/cyclin T | >10 | >10 | 0.93 | 5.2 | >10 | >10 | 0.011 | 0.2 | >10 | >10 |
| CK1δ/ε | >10 | 4.2 | 3.3 | >10 | >10 | >10 | 0.0037 | 0.031 | >10 | >10 |
| CLK1 | 0.1 | 0.039 | 1.0 | 2.7 | >10 | >10 | 0.003 | 0.022 | 6.9 | 7.2 |
| CLK2 | 0.91 | 0.21 | 1.2 | 4.9 | >10 | >10 | 0.0041 | 0.0073 | 6.0 | 6.1 |
| CLK3 | 9.7 | 2.4 | 2.6 | 8.6 | >10 | >10 | 0.0053 | 0.049 | >10 | >10 |
| CLK4 | 0.12 | 0.031 | 0.32 | 0.82 | >10 | >10 | 0.0029 | 0.0036 | 3.9 | 3.5 |
| DYRK1A | 0.42 | 0.032 | 0.27 | 0.47 | >10 | >10 | 0.0033 | 0.78 | 7.8 | 5.2 |
| DYRK1B | 1.8 | 0.05 | 0.43 | 1.2 | >10 | >10 | 0.0032 | 0.0058 | >10 | 5.6 |
| DYRK2 | 0.49 | 0.088 | 0.42 | 0.76 | >10 | >10 | 0.0031 | 0.021 | 7.1 | 6.2 |
| DYRK3 | 0.6 | 0.42 | 0.17 | 0.88 | >10 | >10 | 0.0049 | 0.044 | >10 | 6.1 |
| GSK-3α/β | 7.3 | 1.1 | >10 | >10 | >10 | >10 | 0.0032 | 0.044 | >10 | >10 |
| PfGSK-3 | >10 | 8.5 | >10 | >10 | >10 | >10 | 0.0041 | 0.038 | >10 | >10 |
| PfCLK1 | 0.79 | 0.42 | 0.65 | 3.1 | >10 | >10 | 0.011 | 0.085 | >10 | >10 |
| LmCK1 | >10 | >10 | >10 | >10 | >10 | >10 | 0.0025 | 0.025 | >10 | >10 |
| LmCLK | >10 | >10 | >10 | >10 | >10 | >10 | 0.4 | 5.2 | >10 | >10 |
| LmDYRK2 | 4.2 | 2.9 | 5.9 | >10 | >10 | >10 | 0.38 | 9.0 | >10 | >10 |
| LdDYRK1B | 6.9 | 0.82 | 1.1 | >10 | >10 | >10 | 0.042 | 0.5 | >10 | >10 |
| LdDYRK3 | >10 | >10 | >10 | >10 | >10 | >10 | 0.021 | 0.3 | >10 | 7.9 |
| LdDYRK4 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| TbCLK1 | >10 | >10 | >10 | >10 | >10 | >10 | 0.53 | 0.71 | >10 | >10 |
| TcCLK1 | >10 | >10 | >10 | >10 | >10 | >10 | 0.51 | 1.1 | >10 | >10 |
| CpLAMMER | 1.8 | 0.1 | 0.17 | 0.2 | >10 | >10 | 0.016 | 0.13 | >10 | >10 |
| GlCLK | 3.1 | >10 | 0.95 | 2.2 | >10 | >10 | 0.012 | 0.12 | >10 | >10 |
| TgCLK | 4.1 | >10 | >10 | >10 | >10 | >10 | 0.041 | 0.48 | >10 | >10 |
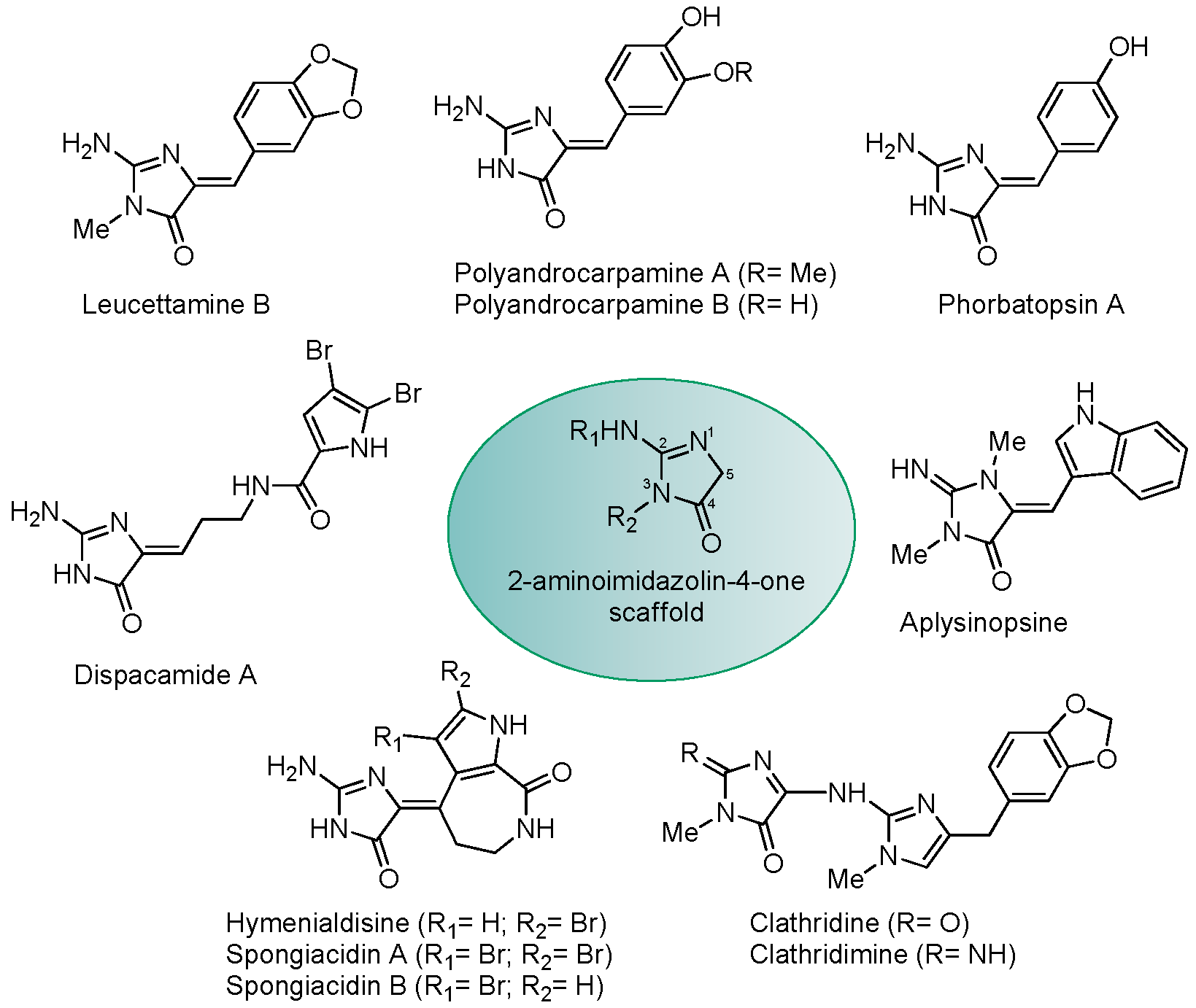
| Kinases | Abbreviation | Score |
|---|---|---|
| Dual specificity tyrosine-phosphorylation-regulated kinase 1A | DYRK1A | 3 |
| Casein kinase 2 α | CSNK2A1 | 12 |
| Homeodomain interacting protein kinase 3 | HIPK3 | 19 |
| Proviral Integrations of Moloney virus 2 | PIM2 | 21 |
| Ribosomal protein S6 kinase alpha-4 (Kin.Dom.2-C-terminal) | RPS6KA4 | 22 |
| Dual specificity tyrosine-phosphorylation-regulated kinase 2 | DYRK2 | 24 |
| Dual specificity tyrosine-phosphorylation-regulated kinase 1B | DYRK1B | 25 |
| Casein kinase 2 α’ | CSNK2A2 | 26 |
| Interleukin-1 receptor-associated kinase 1 | IRAK1 | 28 |
| Cdc2-like kinase 4 | CLK4 | 30 |
| Cdc2-like kinase 1 | CLK1 | 32 |
| Kinases | Polyandrocarpamines (PAC) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | |
| CDK1/cyclin B | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| CDK2/cyclin A | >10 | >10 | ≥10 | ≥10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| CDK5/p25 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| CDK9/cyclin T | 1.1 | 2 | >10 | >10 | ≥10 | >10 | >10 | 0.51 | >10 | >10 | >10 | 0.9 | 1.8 |
| CK1δ/ε | 5.2 | >10 | ≥10 | ≥10 | >10 | >10 | >10 | >10 | 3.6 | >10 | >10 | 5 | >10 |
| CLK1 | 0.43 | 1 | 2.2 | ≥10 | 2.2 | >10 | >10 | >10 | ≥10 | >10 | 4.2 | 0.47 | 0.59 |
| CLK2 | 1.2 | 2.5 | 6.3 | >10 | 7.5 | >10 | >10 | >10 | >10 | >10 | ≥10 | 0.64 | 2.42 |
| CLK3 | 2.3 | 9.2 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | 2.7 | 10 |
| CLK4 | 0.4 | 0.95 | 2.3 | ≥10 | 2.4 | >10 | >10 | >10 | >10 | >10 | 3.2 | 0.47 | 0.91 |
| DYRK1A | 0.23 | 0.75 | 2.1 | 4 | 1.3 | >10 | >10 | >10 | >10 | 3 | 2.1 | 0.3 | 0.59 |
| DYRK1B | 0.4 | 1.1 | 8.2 | >10 | 6.1 | >10 | >10 | >10 | >10 | >10 | 6.2 | 0.47 | 0.96 |
| DYRK2 | 0.14 | 0.48 | 2.2 | 4.5 | 0.64 | >10 | >10 | >10 | >10 | 3.4 | 1.3 | 0.24 | 0.38 |
| DYRK3 | 0.28 | 0.9 | >10 | 5.1 | 5.7 | >10 | >10 | >10 | >10 | >10 | 2.4 | 0.34 | 0.68 |
| GSK-3α/β | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | 2 | >10 | >10 |
| PfGSK-3 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| LmCK1 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | 0.63 | >10 | >10 | >10 | >10 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Loaëc, N.; Attanasio, E.; Villiers, B.; Durieu, E.; Tahtouh, T.; Cam, M.; Davis, R.A.; Alencar, A.; Roué, M.; Bourguet-Kondracki, M.-L.; et al. Marine-Derived 2-Aminoimidazolone Alkaloids. Leucettamine B-Related Polyandrocarpamines Inhibit Mammalian and Protozoan DYRK & CLK Kinases. Mar. Drugs 2017, 15, 316. https://doi.org/10.3390/md15100316
Loaëc N, Attanasio E, Villiers B, Durieu E, Tahtouh T, Cam M, Davis RA, Alencar A, Roué M, Bourguet-Kondracki M-L, et al. Marine-Derived 2-Aminoimidazolone Alkaloids. Leucettamine B-Related Polyandrocarpamines Inhibit Mammalian and Protozoan DYRK & CLK Kinases. Marine Drugs. 2017; 15(10):316. https://doi.org/10.3390/md15100316
Chicago/Turabian StyleLoaëc, Nadège, Eletta Attanasio, Benoît Villiers, Emilie Durieu, Tania Tahtouh, Morgane Cam, Rohan A. Davis, Aline Alencar, Mélanie Roué, Marie-Lise Bourguet-Kondracki, and et al. 2017. "Marine-Derived 2-Aminoimidazolone Alkaloids. Leucettamine B-Related Polyandrocarpamines Inhibit Mammalian and Protozoan DYRK & CLK Kinases" Marine Drugs 15, no. 10: 316. https://doi.org/10.3390/md15100316