
Synthesis of Phthalimide Derivatives as Potential PPAR-γ Ligands

 ,
, 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
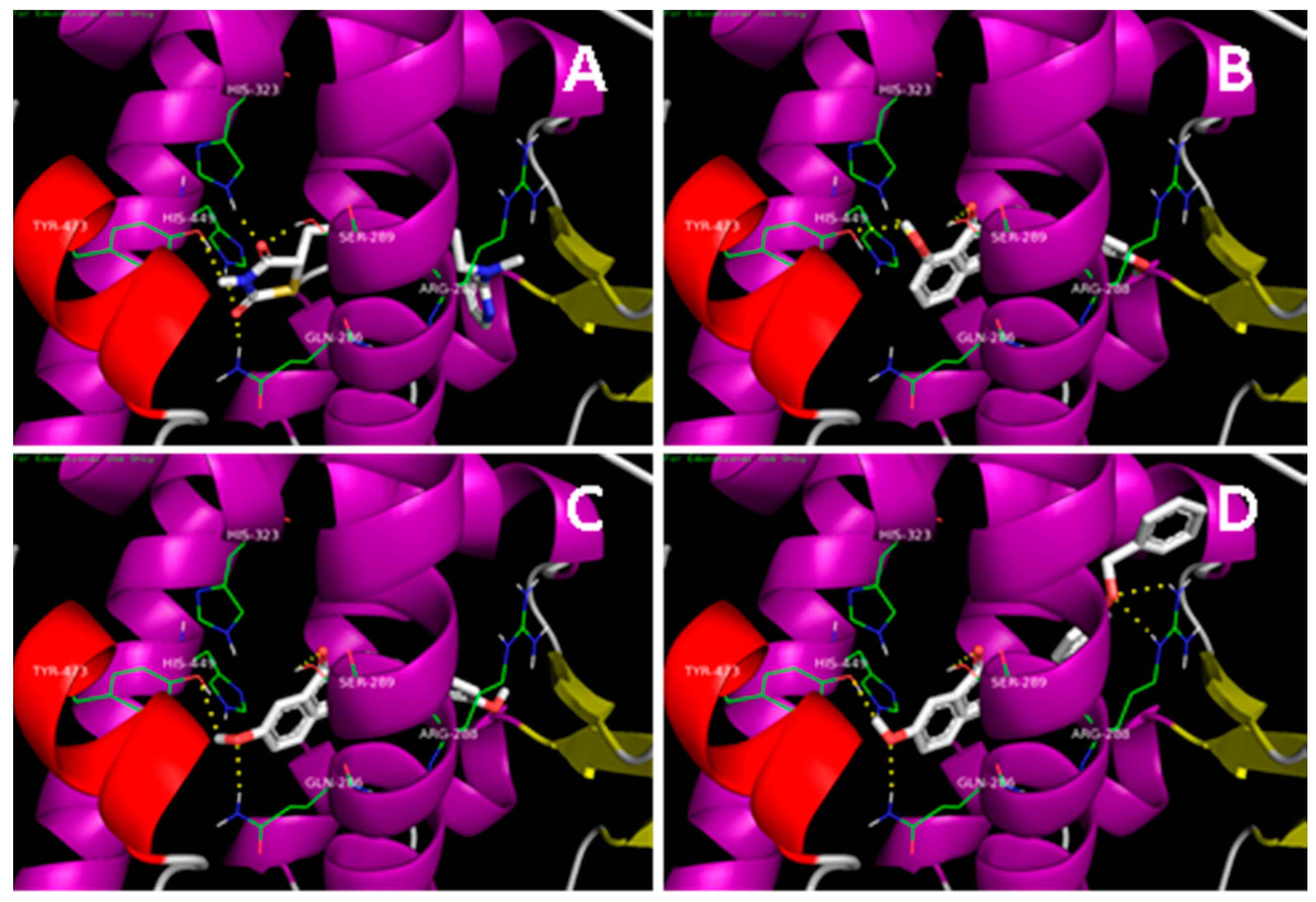
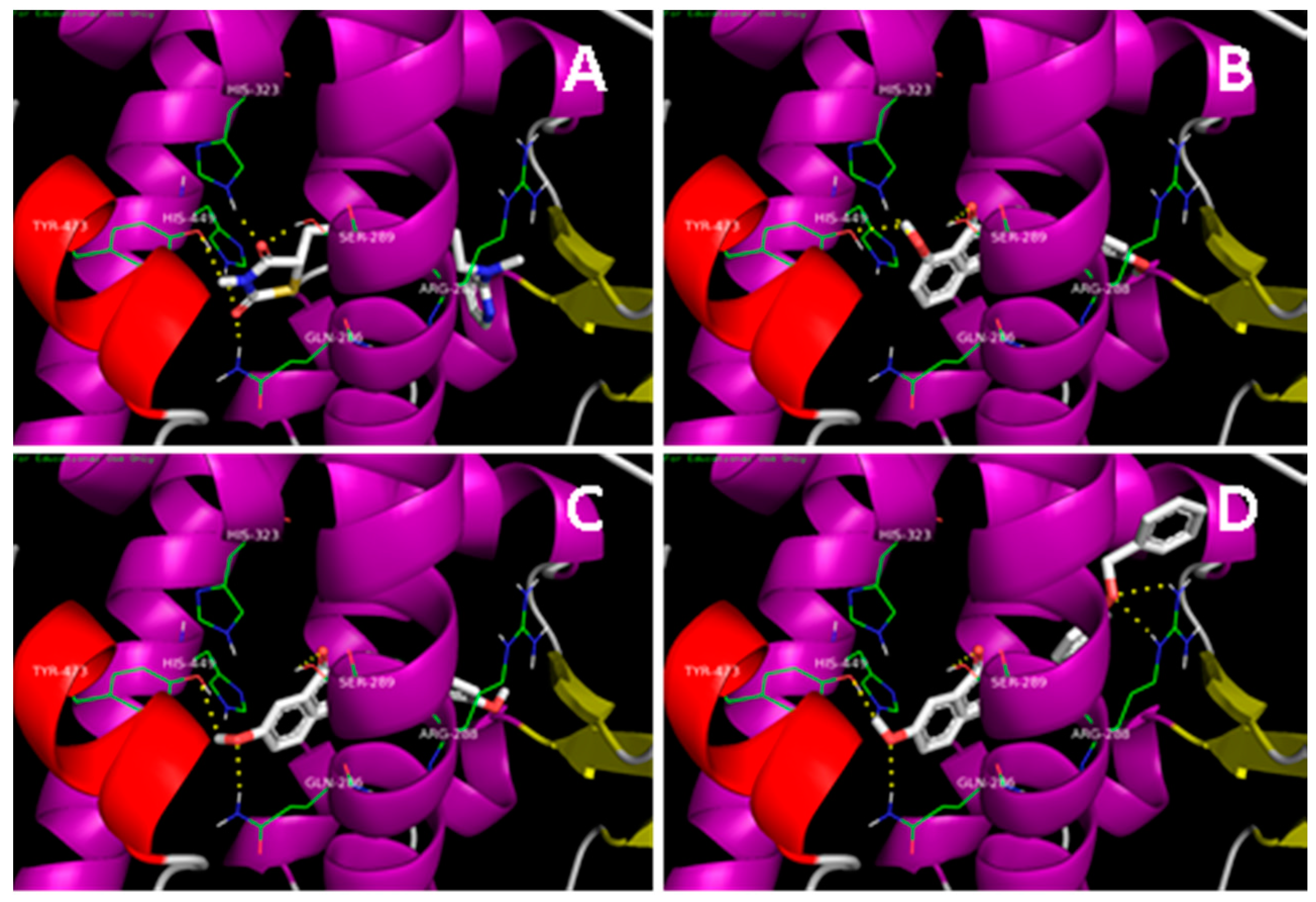
2.1. Docking Simulation
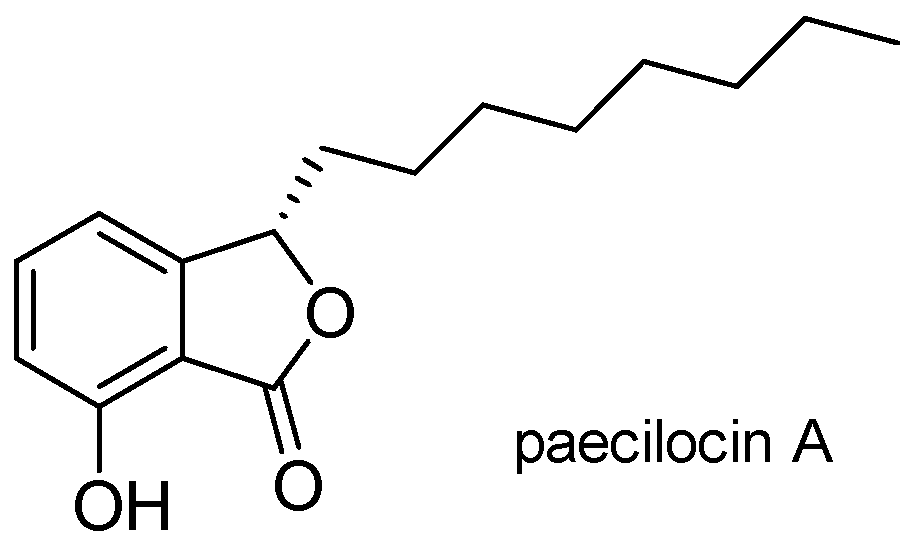
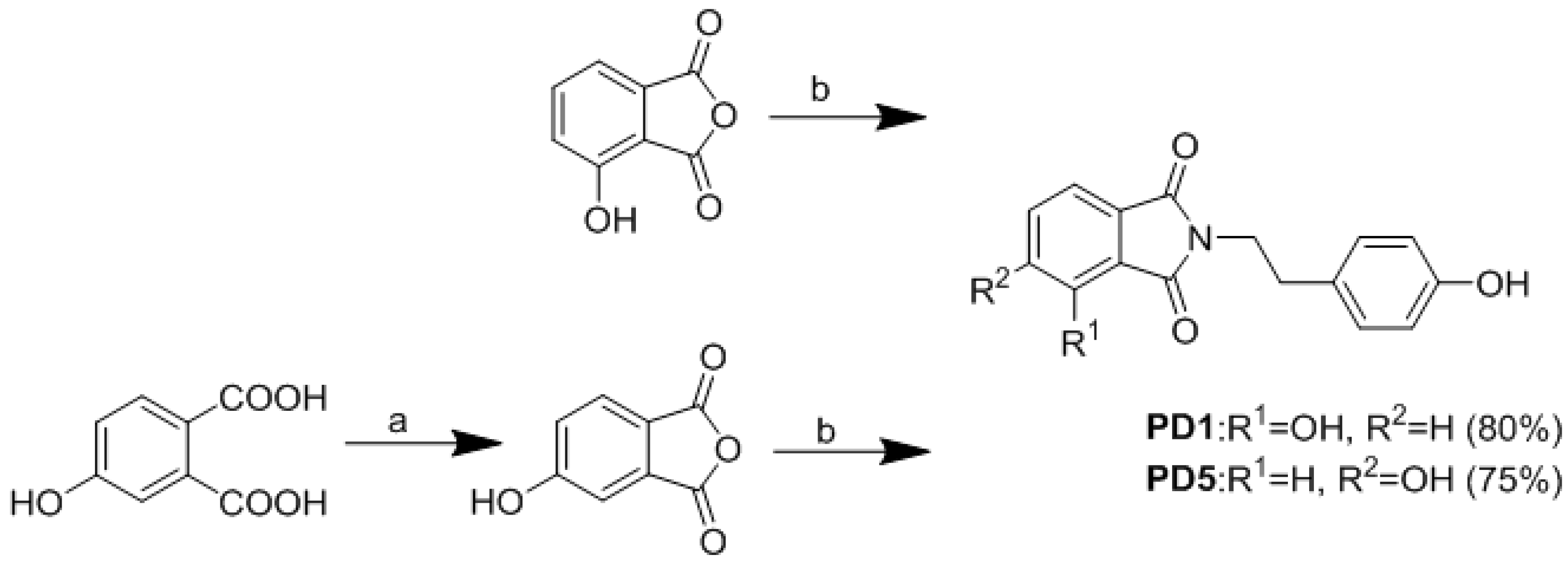
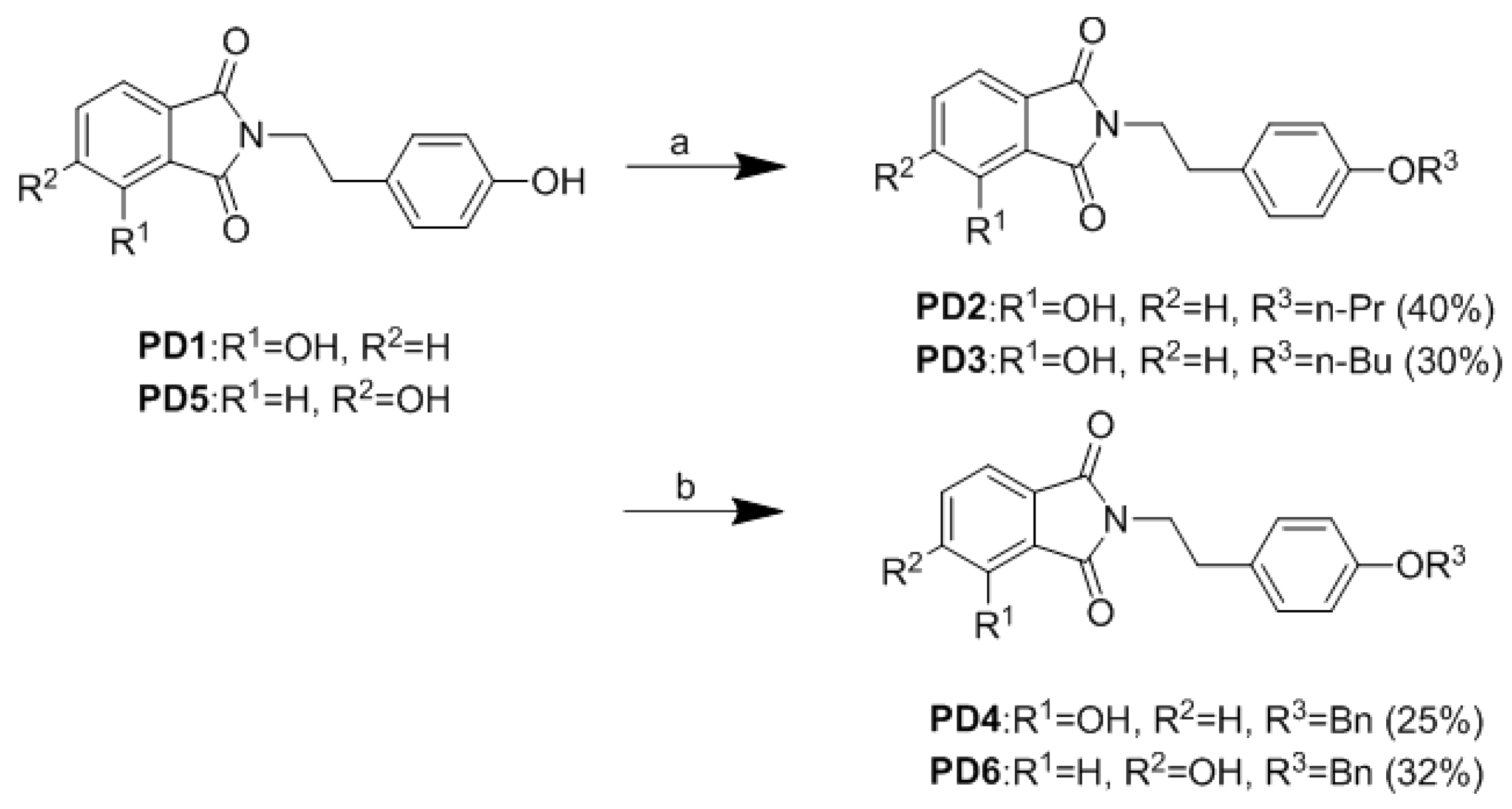
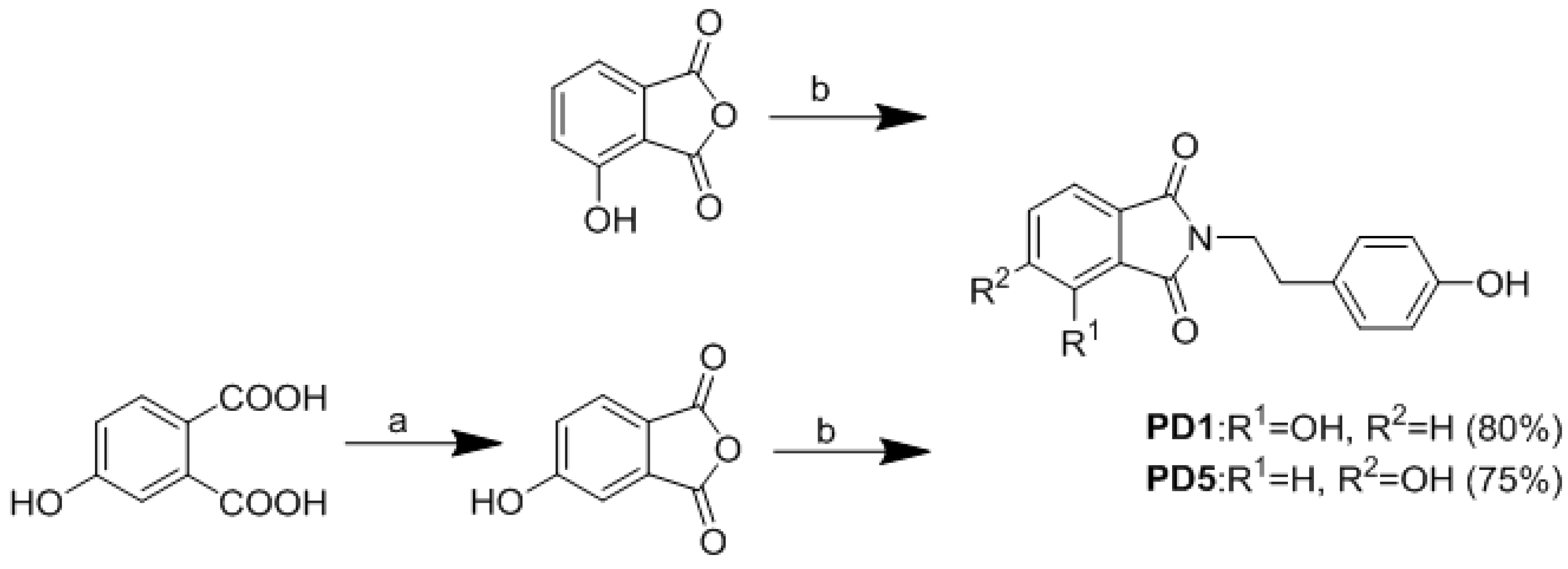
2.2. Synthesis of Analogues
2.3. Biological Evaluation
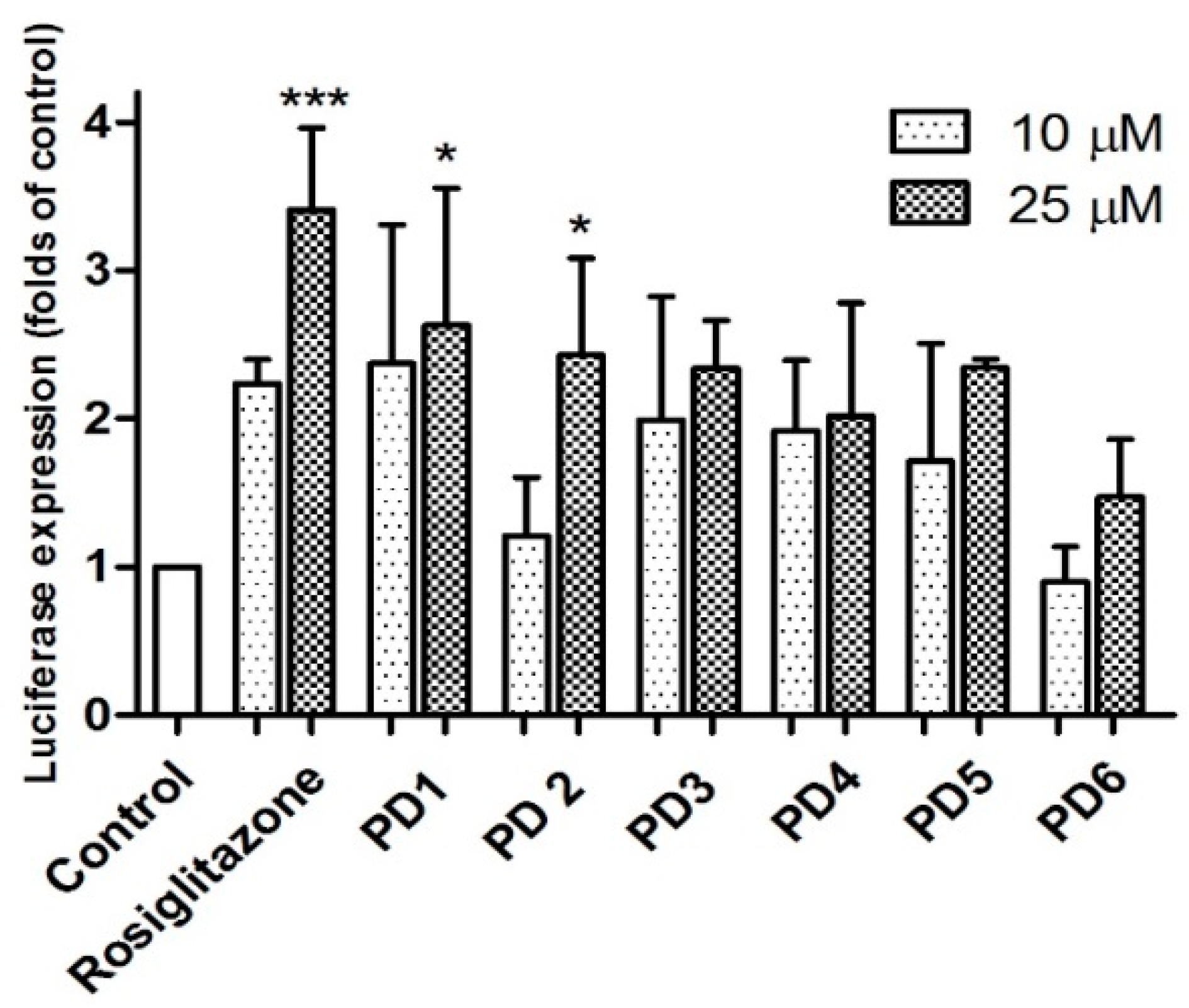
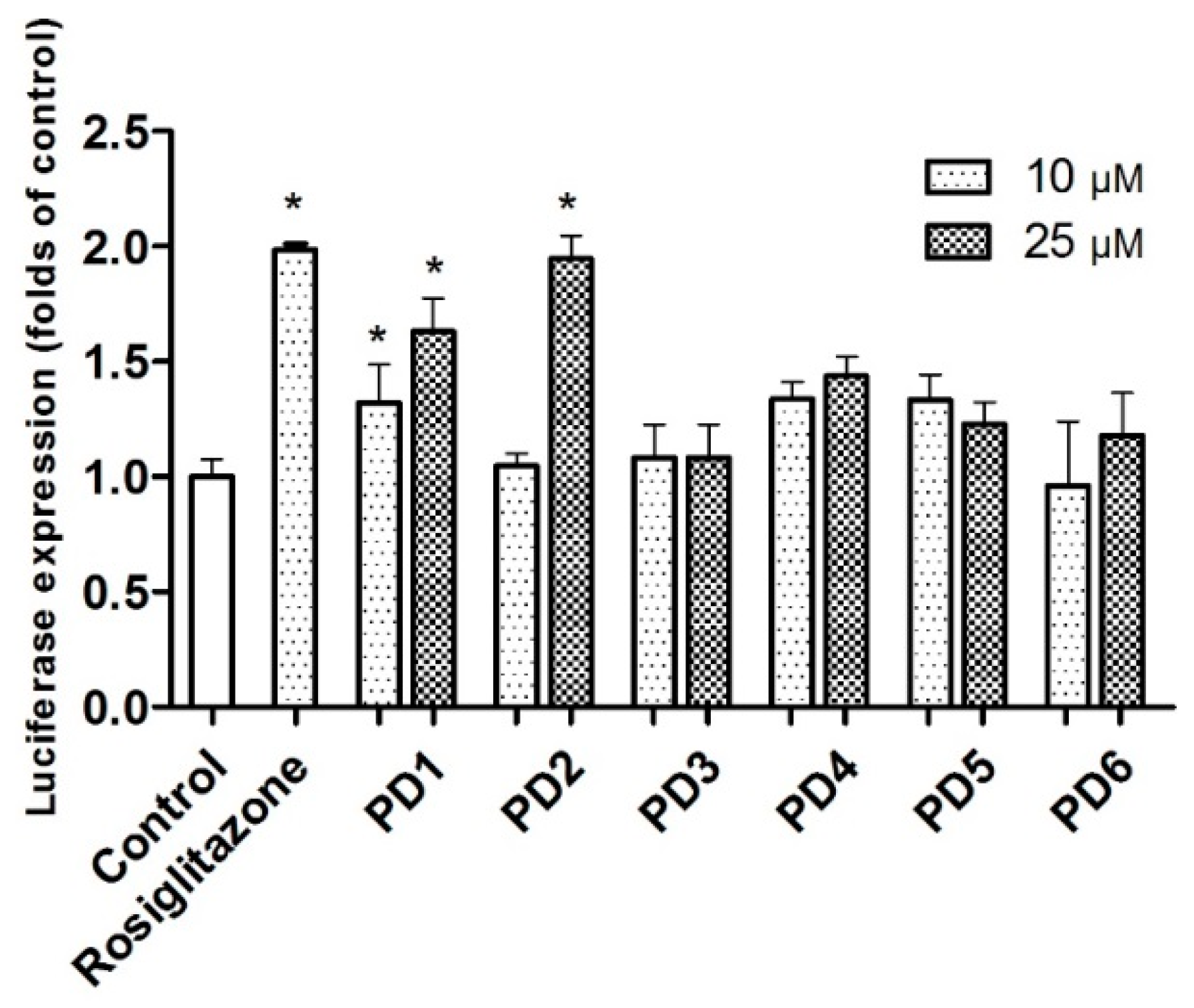
2.3.1. PPAR-γ Activation
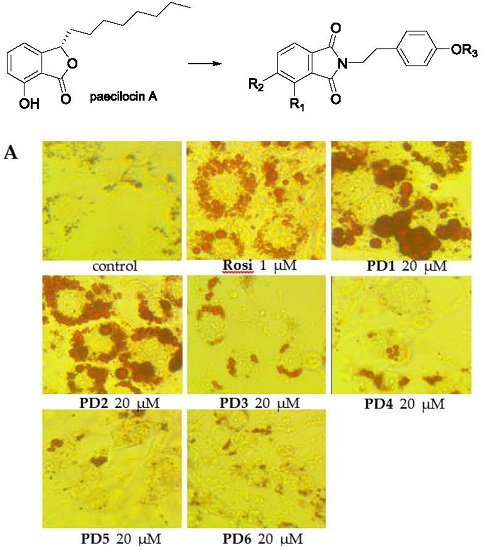
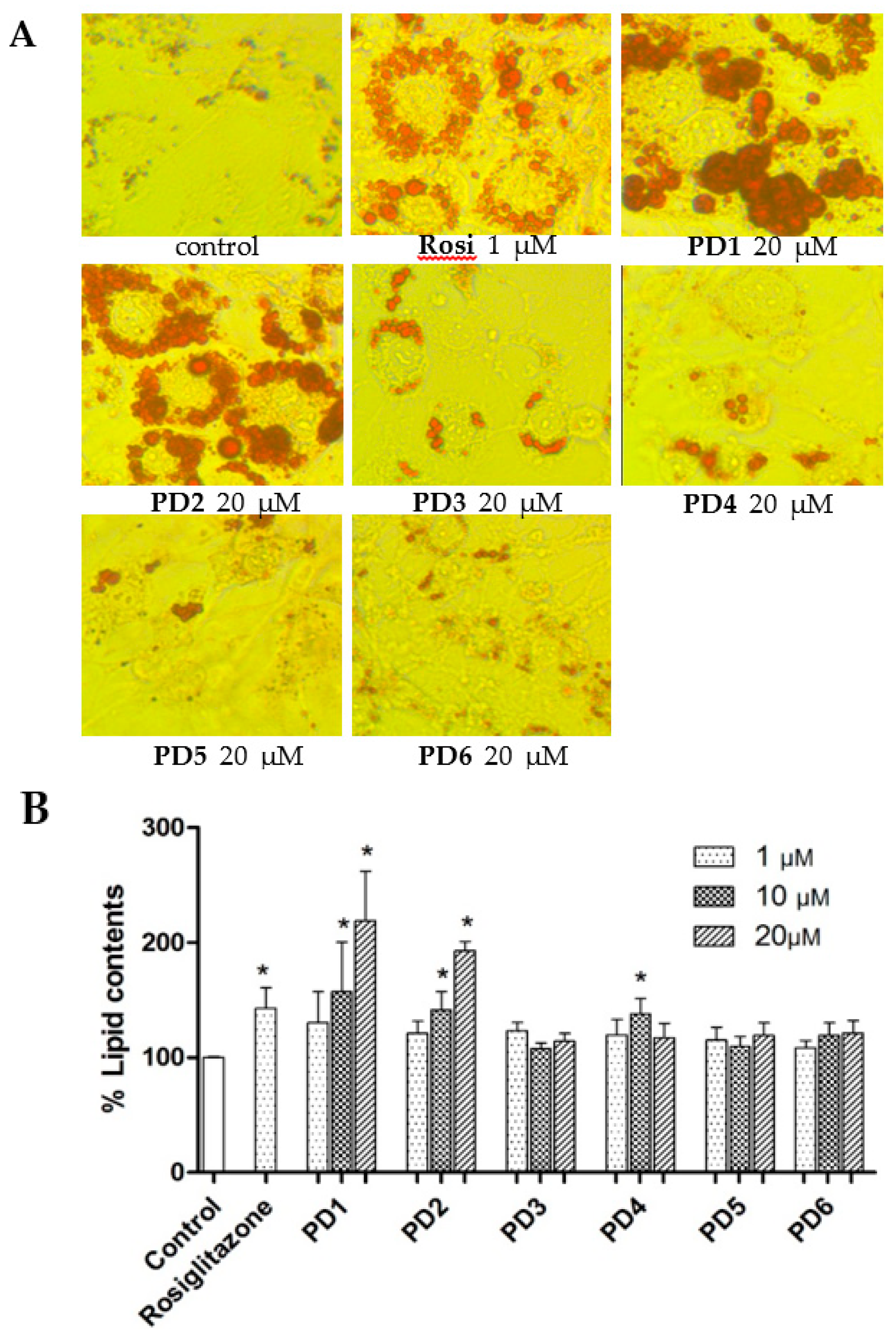
2.3.2. Effect on Adipocyte Differentiation in 3T3-L1 Cells
3. Experimental Section
3.1. Chemistry
3.2. Cell Culture
3.3. Luciferase Assay
3.4. Molecular Docking Study
3.5. Adipocyte Differentiation Assay
3.6. Quantification of Lipid Contents in 3T3-L1 Cells
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dragica, S.J.; Željko, M. PPARγ—A new concept of treatment. Diabetol. Croat. 2009, 38, 23–28. [Google Scholar]
- Hannele, Y.J. Pathogenesis of non-insulin-dependent diabetes mellitus. Lancet 1994, 343, 91–95. [Google Scholar]
- Rosen, E.D. PPARγ: A nuclear regulator of metabolism, differentiation, and cell growth. J. Biol. Chem. 2001, 276, 37731–37734. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M.; Barish, G.D.; Wang, Y.X. PPARs and the complex journey to obesity. Nat. Med. 2004, 10, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Willson, T.M.; Wahli, W. Peroxisome proliferator-activated receptor agonists. Chem. Biol. 1997, 1, 235–241. [Google Scholar] [CrossRef]
- Willson, T.M.; Cobb, J.E.; Cowan, D.J.; Wiethe, R.W.; Correa, I.D.; Prakash, S.R.; Beck, K.D.; Moore, L.B.; Kliewer, S.A.; Lehmann, J.M. The structure-activity relationship between peroxisome proliferator-activated receptor γ agonism and the antihyperglycemic activity of thiazolidinediones. J. Med. Chem. 1996, 39, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Hannele, Y.J. Thiazolidinediones. N. Engl. J. Med. 2004, 351, 1106–1118. [Google Scholar]
- Guan, Y.F.; Hao, C.; Cha, D.R.; Rao, R.; Lu, W.; Kohan, D.E.; Magnuson, M.A.; Redha, R.; Zhang, Y.; Breyer, M.D. Thiazolidinediones expand body fluid volume through PPARγ stimulation of ENaC-mediated renal salt absorption. Nat. Med. 2005, 11, 861–866. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Park, J.Y.; Lee, H.J.; Kim, D.H.; Park, D.U.; Jeong, H.O.; Park, C.H.; Chun, P.; Moon, H.R.; Chung, H.Y. Potent anti-diabetic effects of MHY908, a newly synthesized PPAR α/γ dual agonist in db/db mice. PLoS ONE 2013, 8, e78815. [Google Scholar] [CrossRef] [PubMed]
- Yumuk, V.D. Targeting components of the stress system as potential therapies for the metabolic syndrome: The Peroxisome-Proliferator-Activated Receptors. Ann. N. Y. Acad. Sci. 2006, 1083, 306–318. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, F.; Kim, E.L.; Li, J.L.; Hong, J.K.; Bae, K.S.; Chung, H.Y.; Kim, H.S.; Jung, J.H. Antibacterial polyketides from the jellyfish-derived fungus Pacecilomyces variotii. J. Nat. Prod. 2011, 74, 1826–1829. [Google Scholar] [CrossRef] [PubMed]
- D’Orazio, D.; Saizieu, A.D.; Schueler, G.; Raederstorff, D.; Teixeira, S.R.; Schmidt, Y.W.; Weber, P.; Wolfram, S. Use of Phthalide Derivatives for the Treatment and Prevention of Diabetes Mellitus. U.S. Patent 2007/0082947 A1, 12 April 2007. [Google Scholar]
- Xiao, B.; Yin, J.; Park, M.; Liu, J.; Li, J.L.; Kim, E.L.; Hong, J.; Chung, H.Y.; Jung, J.H. Design and synthesis of marine fungal phthalide derivatives as PPAR-γ agonists. Bioorg. Med. Chem. 2012, 20, 4954–4961. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Su, M.J.; Kim, E.L.; Hong, J.K.; Chung, H.Y.; Kim, H.S.; Yin, J.; Jung, J.H. Synthesis of PPAR-γ activators inspired by the marine natural product, paecilocin A. Mar. Drugs 2014, 12, 926–939. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPARγ2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Pluempanup at, W.; Adisakwattana, S.; Yibchok-Anun, S.; Chavasiri, W. Synthesis of N-pheylphthalimide derivatives as alpha-glucosidase inhibitors. Arch. Pharm. Res. 2007, 30, 1501–1506. [Google Scholar] [CrossRef]
- McDonnell, D.P.; Vegeto, E.; Gleeson, M.A.G. Nuclear hormone receptors as targets for new drug discovery. Nat. Biotechnol. 1993, 11, 1256–1261. [Google Scholar] [CrossRef]
- Wang, J.Q.; Satoh, M. Preparation and salt-resistivity of poly(vinyl alcohol)-4-hydroxyl phthalate hydrogels. Macromol. Res. 2011, 19, 468–475. [Google Scholar] [CrossRef]





© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eom, S.H.; Liu, S.; Su, M.; Noh, T.H.; Hong, J.; Kim, N.D.; Chung, H.Y.; Yang, M.H.; Jung, J.H. Synthesis of Phthalimide Derivatives as Potential PPAR-γ Ligands. Mar. Drugs 2016, 14, 112. https://doi.org/10.3390/md14060112
Eom SH, Liu S, Su M, Noh TH, Hong J, Kim ND, Chung HY, Yang MH, Jung JH. Synthesis of Phthalimide Derivatives as Potential PPAR-γ Ligands. Marine Drugs. 2016; 14(6):112. https://doi.org/10.3390/md14060112
Chicago/Turabian StyleEom, So Hyeon, Sen Liu, Mingzhi Su, Tae Hwan Noh, Jongki Hong, Nam Deuk Kim, Hae Young Chung, Min Hye Yang, and Jee H. Jung. 2016. "Synthesis of Phthalimide Derivatives as Potential PPAR-γ Ligands" Marine Drugs 14, no. 6: 112. https://doi.org/10.3390/md14060112