
Amino Acid-Derived Metabolites from the Ascidian Aplidium sp.


Abstract
:1. Introduction
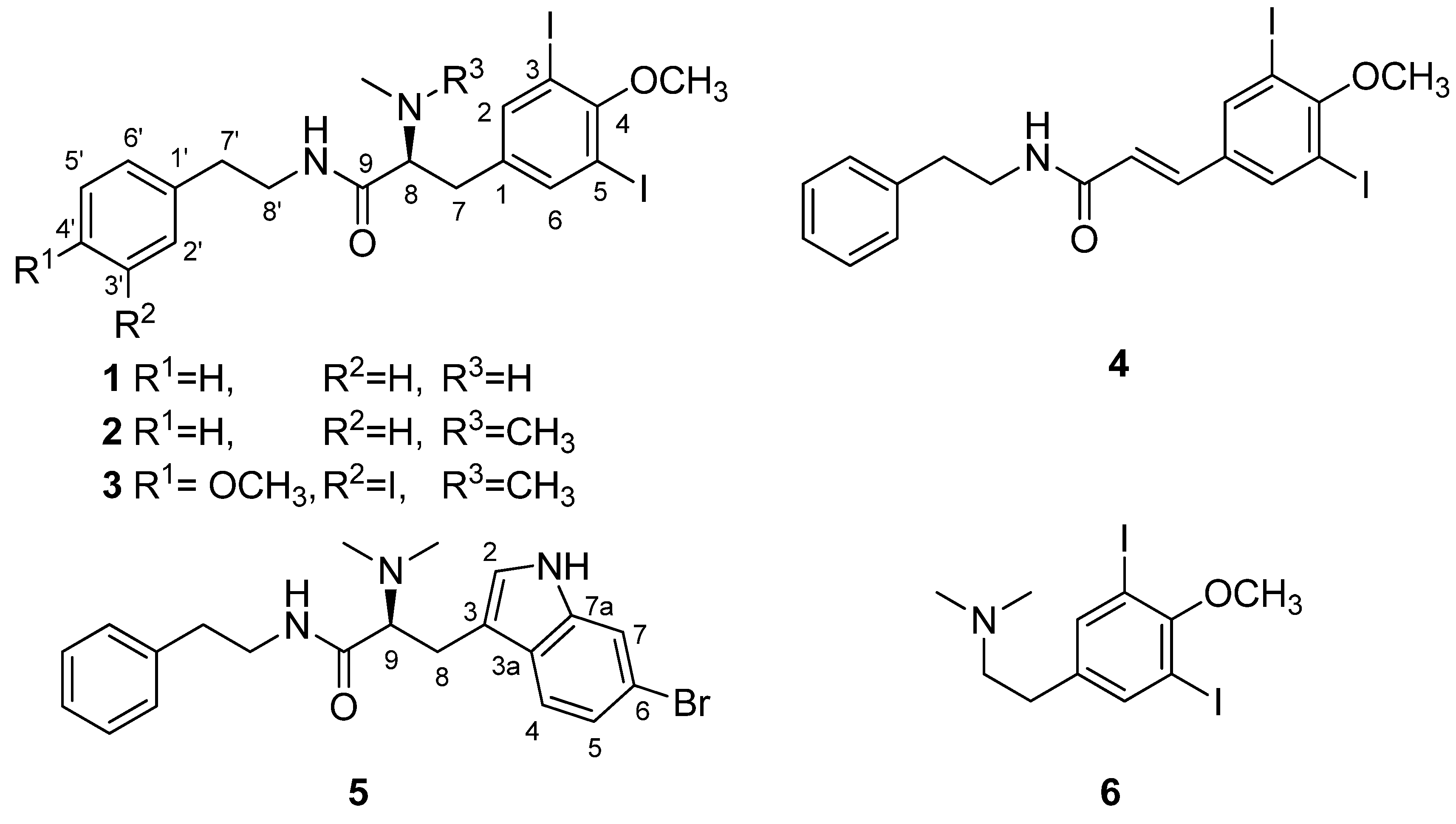
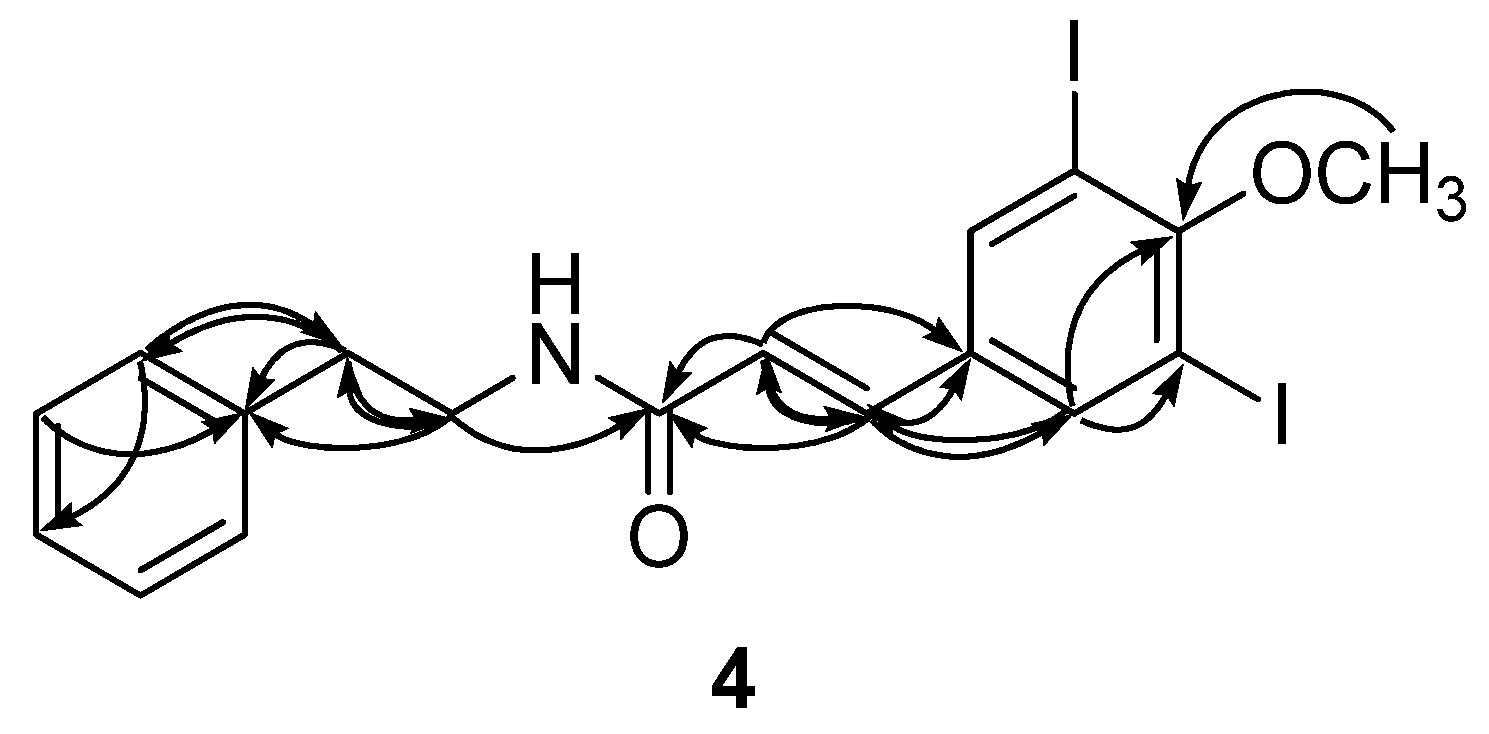
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |||
|---|---|---|---|---|
| δC, Type | δH, mult (J in Hz) | δC, Type | δH, mult (J in Hz) | |
| 1 | 138.2, C | 139.8, C | ||
| 2/6 | 142.0, CH | 7.66, s | 142.0, CH | 7.67, s |
| 3/5 | 91.1, C | 90.9, C | ||
| 4 | 159.5, C | 159.1, C | ||
| 7 | 37.9, CH2 | 2.76, dd (13.5, 8.5) | 34.9, CH2 | 2.78, dd (12.0, 5.0) |
| 2.82, dd (13.5, 6.5) | 2.89, dd (12.0, 11.0) | |||
| 8 | 66.0, CH | 3.23, dd (8.5, 6.5) | 71.8, CH | 3.02, dd (11.0, 5.0) |
| 9 | 173.3, C | 172.2, C | ||
| 1′ | 140.2, C | 140.3, C | ||
| 2′/6′ | 129.7, CH | 7.11, d (8.0) | 129.7, CH | 7.05, d (7.5) |
| 3′/5′ | 129.6, CH | 7.24, t (8.0) | 129.5, CH | 7.22, t (7.5) |
| 4′ | 127.4, CH | 7.16, t (8.0) | 127.3, CH | 7.14, t (7.5) |
| 7′ | 36.7, CH2 | 2.57, ddd (14.0, 7.5, 6.5) | 36.8, CH2 | 2.51, ddd (14.0, 7.5, 7.0) |
| 2.67, ddd (14.0, 8.0, 7.5) | 2.64, ddd (14.0, 7.5, 7.0) | |||
| 8′ | 41.7, CH2 | 3.25, ddd (14.0, 7.5, 7.5) | 41.6, CH2 | 3.17, ddd (13.5, 7.0, 7.0) |
| 3.44, ddd (14.0, 8.0, 6.5) | 3.43, ddd (13.5, 7.5, 7.5) | |||
| 4-OMe | 61.1, CH3 | 3.77, s | 61.1, CH3 | 3.76, s |
| 8-NMe | 34.0, CH3 | 2.28, s | 42.5, CH3 (2C) | 2.29, s (6H) |

| 3 | 4 | |||
|---|---|---|---|---|
| δC, Type | δH, mult (J in Hz) | δC, Type | δH, mult (J in Hz) | |
| 1 | 135.4, C | 134.8, C | ||
| 2/6 | 142.3, CH | 7.72, s | 138.9, CH | 7.86, s |
| 3/5 | 91.5, C | 90.8, C | ||
| 4 | 160.2, C | 159.8, C | ||
| 7 | 33.9, CH2 | 2.94, dd (11.5, 11.5) | 138.7, CH | 7.41, d (15.0) |
| 3.27, dd (11.5, 5.0) | ||||
| 8 | 70.8, CH | 3.73, dd (11.5, 5.0) | 122.1, CH | 6.21, d (15.0) |
| 9 | 167.1, C | 165.0, C | ||
| 1′ | 133.9, C | 137.3, C | ||
| 2′ | 131.0, CH | 7.51, d (2.0) | 128.8, CH | 7.22, d (7.5) |
| 3′ | 86.5, C | 128.7, CH | 7.33, t (7.5) | |
| 4′ | 158.5, C | 126.7, CH | 7.25, d (7.5) | |
| 5′ | 112.2, CH | 6.82, d (8.0) | 128.7, CH | 7.33, d (7.5) |
| 6′ | 140.6, CH | 6.97, dd (8.0, 2.0) | 128.8, CH | 7.22, d (7.5) |
| 7′ | 34.9, CH2 | 2.36, ddd (14.0, 7.5, 7.5) | 35.6, CH2 | 2.89, t (7.0) |
| 2.53, ddd (14.0, 7.5, 5.5) | ||||
| 8′ | 41.7, CH2 | 3.22, ddd (13.5, 7.5, 5.5) | 40.8, CH2 | 3.67, dt (7.0, 6.5) |
| 3.34, ddd (13.5, 7.5, 7.5) | ||||
| 4-OMe | 61.2, CH3 | 3.79, s | 60.8, CH3 | 3.86, s |
| 4′-OMe | 56.9, CH3 | 3.82, s | ||
| 8-NMe | 42.5, CH3 (2C) | 3.87, s (6H) | ||
| 8′-NH | ND | 5.57, t (6.5) | ||
| 5 | ||
|---|---|---|
| δC, Type | δH, mult (J in Hz) | |
| 2 | 126.5, CH | 7.16, s |
| 3 | 108.1, C | |
| 3a | 127.3, C | |
| 4 | 120.8, CH | 7.47, d (8.5) |
| 5 | 123.4, CH | 7.18, d (8.5, 1.5) |
| 6 | 116.3, C | |
| 7 | 115.5, CH | 7.55, d (1.5) |
| 7a | 138.9, C | |
| 8 | 25.7, CH2 | 3.29, dd (13.5, 5.0) |
| 3.43, dd (13.5, 10.0) | ||
| 9 | 70.5, CH | 3.83, dd (10.0, 5.0) |
| 10 | 167.9, C | |
| 1′ | 139.8, C | |
| 2′/6′ | 129.7, CH | 6.90, d (7.5) |
| 3′/5′ | 129.5, CH | 7.15, t (7.5) |
| 4′ | 127.4, CH | 7.12, t (7.5) |
| 7′ | 35.7, CH2 | 2.25, ddd (14.0, 7.0, 7.0) |
| 2.34, ddd (14.0, 7.0, 7.0) | ||
| 8′ | 41.8, CH2 | 3.19, ddd (13.5, 7.5, 7.0) |
| 3.22, ddd (13.5, 7.5, 7.0) | ||
| 9-NMe | 42.4, CH3 (2C) | 2.87, s (6H) |
| 6 | ||
|---|---|---|
| δC, Type | δH, mult (J in Hz) | |
| 1 | 137.4, C | |
| 2/6 | 141.6, CH | 7.79, s |
| 3/5 | 91.4, C | |
| 4 | 159.8, C | |
| 7 | 29.8, CH2 | 2.95, t (7.5) |
| 8 | 59.3, CH2 | 3.31, t (7.5) |
| 4-OMe | 61.2, CH3 | 3.78, s |
| 8-NMe | 44.0, CH3 (2C) | 2.91, s (6H) |
| K562 | A549 | MRC5 | Na+/K+-ATPase | |
|---|---|---|---|---|
| IC50 (µM) | ||||
| 1 | 14.3 | >100 | 59.2 | 163.0 |
| 2 | 21.1 | 10.8 | 73.7 | >200 |
| 3 | 10.4 | 13.4 | >100 | >200 |
| 4 | 18.2 | >100 | >100 | 3.2 |
| 5 | 7.8 | 22.8 | 37.0 | >200 |
| 6 | 19.7 | 8.3 | >100 | 189.2 |
| Doxorubicin | 1.2 | 1.4 | 9.8 | |
| Ouabain | 6.5 | |||
3. Experimental Section
3.1. General Experimental Procedures
3.2. Animal Materials
3.3. Extraction and Isolation
3.4. Advanced Marfey’s Analysis of Compound 1
3.5. Biological Assays
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2015, 32, 116–211, and earlier reports in the series. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Marine natural products and related compounds in clinical and advanced preclinical trials. J. Nat. Prod. 2004, 67, 1216–1238. [Google Scholar] [CrossRef] [PubMed]
- Simmons, T.L.; Andrianasolo, E.; McPhail, K.; Flatt, P.; Gerwick, W.H. Marine natural products as anticancer drugs. Mol. Cancer Ther. 2005, 4, 333–342. [Google Scholar] [PubMed]
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug development from marine natural products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Renaissance of marine natural product drug discovery and development. J. Mar. Sci. Res. Dev. 2012, 2, e106. [Google Scholar] [CrossRef]
- Ali, H.A.J.; Tamilselvi, M. Marine ascidians: A promising resource for bioactive compounds. In Marine Pharmacognosy: Trends and Applications; Kim, S.-K., Ed.; CRC Press: Boca Raton, FL, USA, 2013; pp. 173–189. [Google Scholar]
- Menna, M. Important classes of bioactive alkaloids from marine ascidians: Structures, isolation and bioactivity. Curr. Top. Med. Chem. 2014, 14, 207–223. [Google Scholar] [CrossRef] [PubMed]
- Wright, A.E.; Forleo, D.A.; Gunawardana, G.P.; Gunasekera, S.P.; Koehn, F.E.; McConnell, O.J. Antitumor tetrahydroisoquinoline alkaloids from the colonial ascidian Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4508–4512. [Google Scholar] [CrossRef]
- Rinehart, K.L.; Holt, T.G.; Fregeau, N.L.; Stroh, J.G.; Keifer, P.A.; Sun, F.; Li, L.H.; Martin, D.G. Ecteinascidins 729, 743, 745, 759A, 759B, and 770: Potent antitumor agents from the Caribbean tunicate Ecteinascidia turbinata. J. Org. Chem. 1990, 55, 4512–4515. [Google Scholar] [CrossRef]
- Rinehart, K.L., Jr.; Lithgow-Bertelloni, A.M. Dehydrodidemnin B. WO9104985 A1, 19 April 1991. [Google Scholar]
- Rinehart, K.L., Jr.; Gloer, J.B.; Cook, J.C., Jr.; Mizsak, S.A.; Scahill, T.A. Structures of the didemnins, antiviral and cytotoxic depsipeptides from a Caribbean tunicate. J. Am. Chem. Soc. 1981, 103, 1857–1859. [Google Scholar] [CrossRef]
- Lindquist, N.; Fenical, W.; van Duyne, G.D.; Clardy, J. Isolation and structure determination of diazonamide A and B, unusual cytotoxic metabolites from the marine ascidian Diazona chinensis. J. Am. Chem. Soc. 1991, 113, 2303–2304. [Google Scholar] [CrossRef]
- Murphy, C.D. New frontiers in biological halogenation. J. Appl. Microbiol. 2003, 94, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Sesin, D.F.; Ireland, C.M. Iodinated phenethylamine products from a didemnid tunicate. Tetrahedron Lett. 1984, 25, 403–404. [Google Scholar] [CrossRef]
- Carroll, A.R.; Bowden, B.F.; Coll, J.C. Studies of Australian ascidians. II. Novel cytotoxic iodotyrosine-based alkaloids from colonial ascidians, Aplidium sp. Aust. J. Chem. 1993, 46, 825–832. [Google Scholar] [CrossRef]
- Smith, C.J.; Venables, D.A.; Hopmann, C.; Salomon, C.E.; Jompa, J.; Tahir, A.; Faulkner, D.J.; Ireland, C.M. Plakinidine D, a new pyrroloacridine alkaloid from two ascidians of the genus Didemnum. J. Nat. Prod. 1997, 60, 1048–1050. [Google Scholar] [CrossRef] [PubMed]
- Ford, P.W.; Davidson, B.S. Plakinidine D, a new pyrroloacridine alkaloid from the ascidian Didemnum rubeum. J. Nat. Prod. 1997, 60, 1051–1053. [Google Scholar] [CrossRef] [PubMed]
- Solano, G.; Motti, C.; Jaspars, M. New iodotyramine derivatives from Didemnum rubeum. Tetrahedron 2009, 65, 7482–7486. [Google Scholar] [CrossRef]
- Won, T.H.; Jeon, J.-E.; Lee, S.-H.; Rho, B.J.; Oh, K.-B.; Shin, J. Beta-carboline alkaloids derived from the ascidian Synoicum sp. Bioorg. Med. Chem. 2012, 20, 4082–4087. [Google Scholar] [CrossRef] [PubMed]
- Won, T.H.; Jeon, J.-E.; Kim, S.-H.; Lee, S.-H.; Rho, B.J.; Oh, D.-C.; Oh, K.-B.; Shin, J. Brominated aromatic furanones and related esters from the ascidian Synoicum sp. J. Nat. Prod. 2012, 75, 2055–2061. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.-H.; Won, T.H.; Kim, H.; Shin, J.; Oh, K.-B. Inhibition of Candida albicans isocitrate lyase activity by cadiolides and synoilides from the ascidian Synoicum sp. Bioorg. Med. Chem. Lett. 2013, 23, 4099–4101. [Google Scholar] [CrossRef] [PubMed]
- Won, T.H.; You, M.; Lee, S.-H.; Rho, B.J.; Oh, D.-C.; Oh, K.-B.; Shin, J. Amino alcohols from the ascidian Pseudodistoma sp. Mar. Drugs 2014, 12, 3754–3769. [Google Scholar] [CrossRef] [PubMed]
- Fujii, K.; Ikai, Y.; Mayumi, T.; Oka, H.; Suzuki, M.; Harada, K.-I. A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: Elucidation of limitations of Marfey’s method and of its separation mechanism. Anal. Chem. 1997, 69, 3346–3352. [Google Scholar] [CrossRef]
- Fujii, K.; Ikai, Y.; Oka, H.; Suzuki, M.; Harada, K.-I. A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: Combination of Marfey’s method with mass spectrometry and its practical application. Anal. Chem. 1997, 69, 5146–5151. [Google Scholar] [CrossRef]
- Gao, H.; Kelly, M.; Hamann, M.T. Bromotyrosine-derived Metabolites from the Sponge Aiolochroia crassa. Tetrahedron 1999, 55, 9717–9726. [Google Scholar] [CrossRef]
- Rasmussen, T.; Jensen, J.; Anthoni, U.; Christophersen, C.; Nielsen, P.H. Structure and synthesis of bromoindoles from the marine sponge Pseudosuberites hyalinus. J. Nat. Prod. 1993, 56, 1553–1558. [Google Scholar] [CrossRef]
- Morel, A.F.; Flach, A.; Zanatta, N.; Ethur, E.M.; Mostardeiro, M.A.; Gehrke, I.T.S. A new cyclopeptide alkaloid from the bark of Waltheria douradinha. Tetrahedron Lett. 1999, 40, 9205–9209. [Google Scholar] [CrossRef]
- Segraves, N.L.; Crews, P. Investigation of brominated tryptophan alkaloids from two Thorectidae sponges: Thorectandra and Smenospongia. J. Nat. Prod. 2005, 68, 1484–1488. [Google Scholar] [CrossRef] [PubMed]
- Van, L.T.K.; Hung, T.M.; Thuong, P.T.; Ngoc, T.M.; Kim, J.C.; Jang, H.-S.; Cai, X.F.; Oh, S.R.; Min, B.-S.; Woo, M.H.; et al. Oleanane-type triterpenoids from Aceriphyllum rossii and their cytotoxic activity. J. Nat. Prod. 2009, 72, 1419–1423. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.; Karlsson, L.; Wennergren, M.; Jansson, T.; Powell, T.L. Activity and protein expression of Na+/K+ ATPase are reduced in microvillous syncytiotrophoblast plasma membranes isolated from pregnancies complicated by intrauterine growth restriction. J. Clin. Endocrinol. Metable 2 2003, 88, 2831–2837. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.-B.; Kim, S.-H.; Lee, J.; Cho, W.-J.; Lee, T.; Kim, S. Discovery of diarylacrylonitriles as a novel series of small molecule sortase A inhibitors. J. Med. Chem. 2004, 47, 2418–2421. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.C.; Jang, K.H.; Park, J.; Ahn, C.-H.; Shin, J.; Oh, K.-B. Actin depolymerizing effect of trisoxazole-containing macrolides. Bioorg. Med. Chem. Lett. 2011, 21, 1958–1961. [Google Scholar] [CrossRef] [PubMed]
- Oh, K.-B.; Lee, J.H.; Chung, S.-C.; Shin, J.; Shin, H.J.; Kim, H.-K.; Lee, H.-S. Antimicrobial activities of the bromophenols from the red alga Odonthalia corymbifera and some synthetic derivatives. Bioorg. Med. Chem. Lett. 2008, 18, 104–108. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Won, T.H.; Kim, C.-K.; Lee, S.-H.; Rho, B.J.; Lee, S.K.; Oh, D.-C.; Oh, K.-B.; Shin, J. Amino Acid-Derived Metabolites from the Ascidian Aplidium sp. Mar. Drugs 2015, 13, 3836-3848. https://doi.org/10.3390/md13063836
Won TH, Kim C-K, Lee S-H, Rho BJ, Lee SK, Oh D-C, Oh K-B, Shin J. Amino Acid-Derived Metabolites from the Ascidian Aplidium sp. Marine Drugs. 2015; 13(6):3836-3848. https://doi.org/10.3390/md13063836
Chicago/Turabian StyleWon, Tae Hyung, Chang-Kwon Kim, So-Hyoung Lee, Boon Jo Rho, Sang Kook Lee, Dong-Chan Oh, Ki-Bong Oh, and Jongheon Shin. 2015. "Amino Acid-Derived Metabolites from the Ascidian Aplidium sp." Marine Drugs 13, no. 6: 3836-3848. https://doi.org/10.3390/md13063836