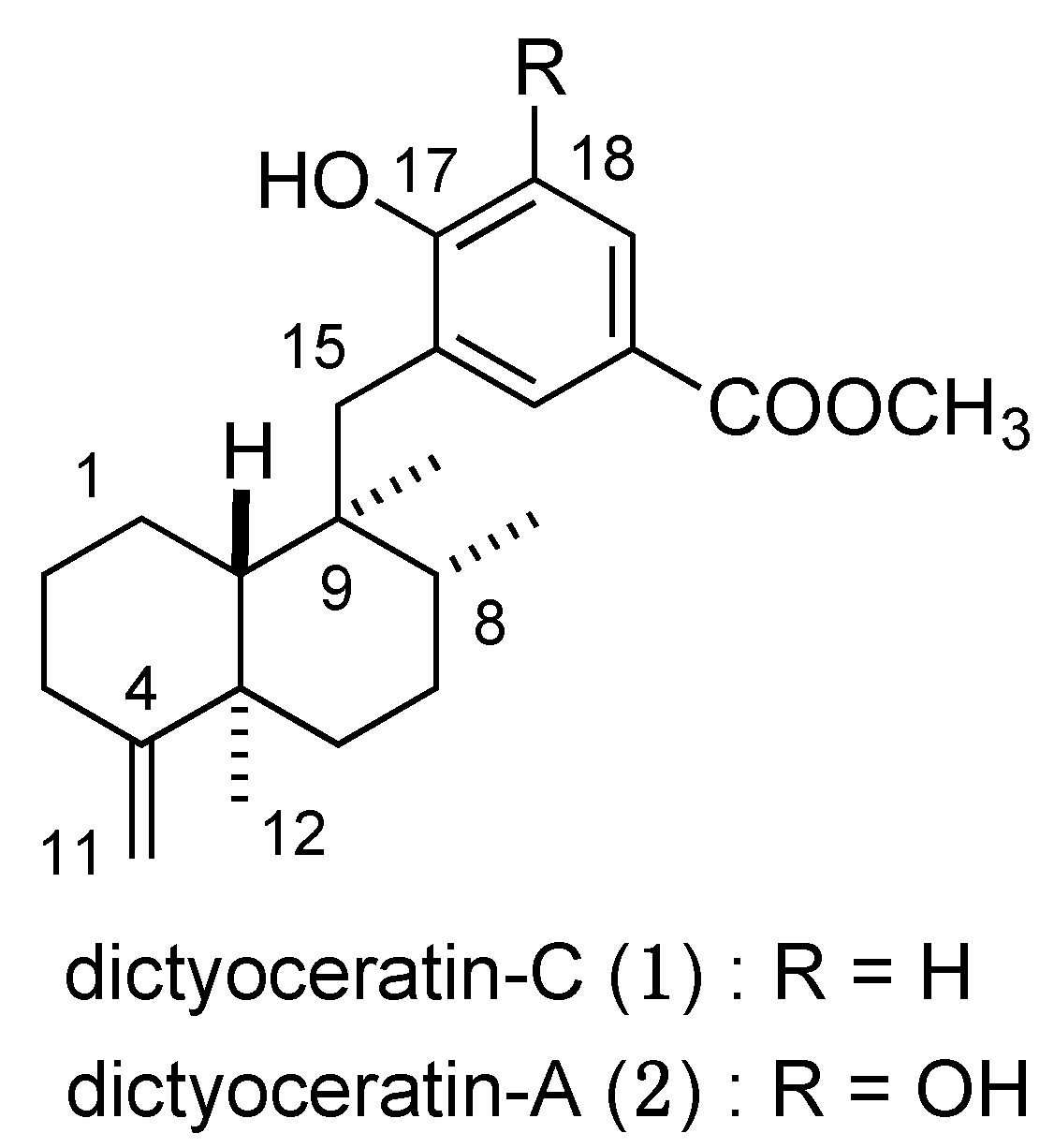
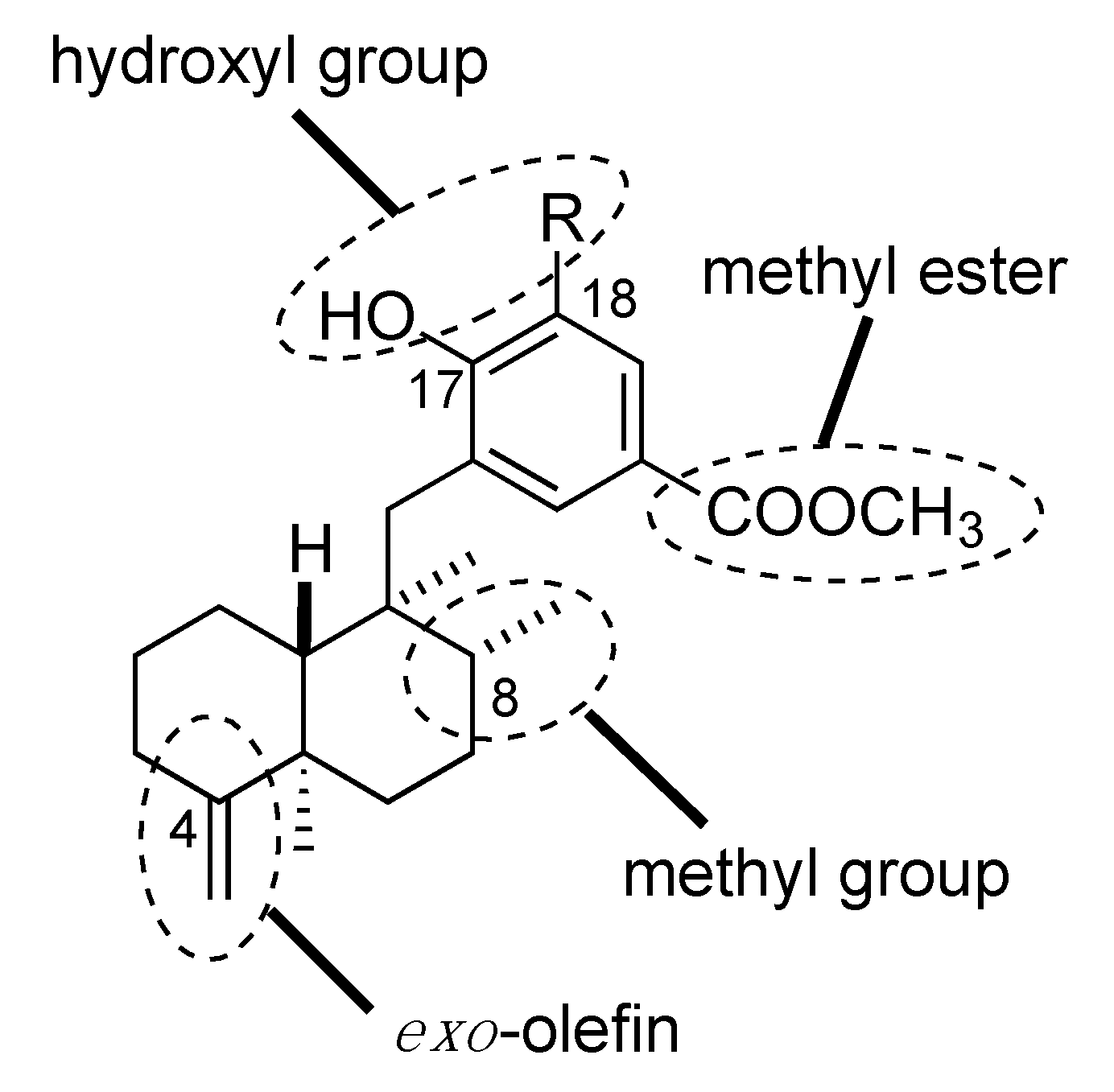
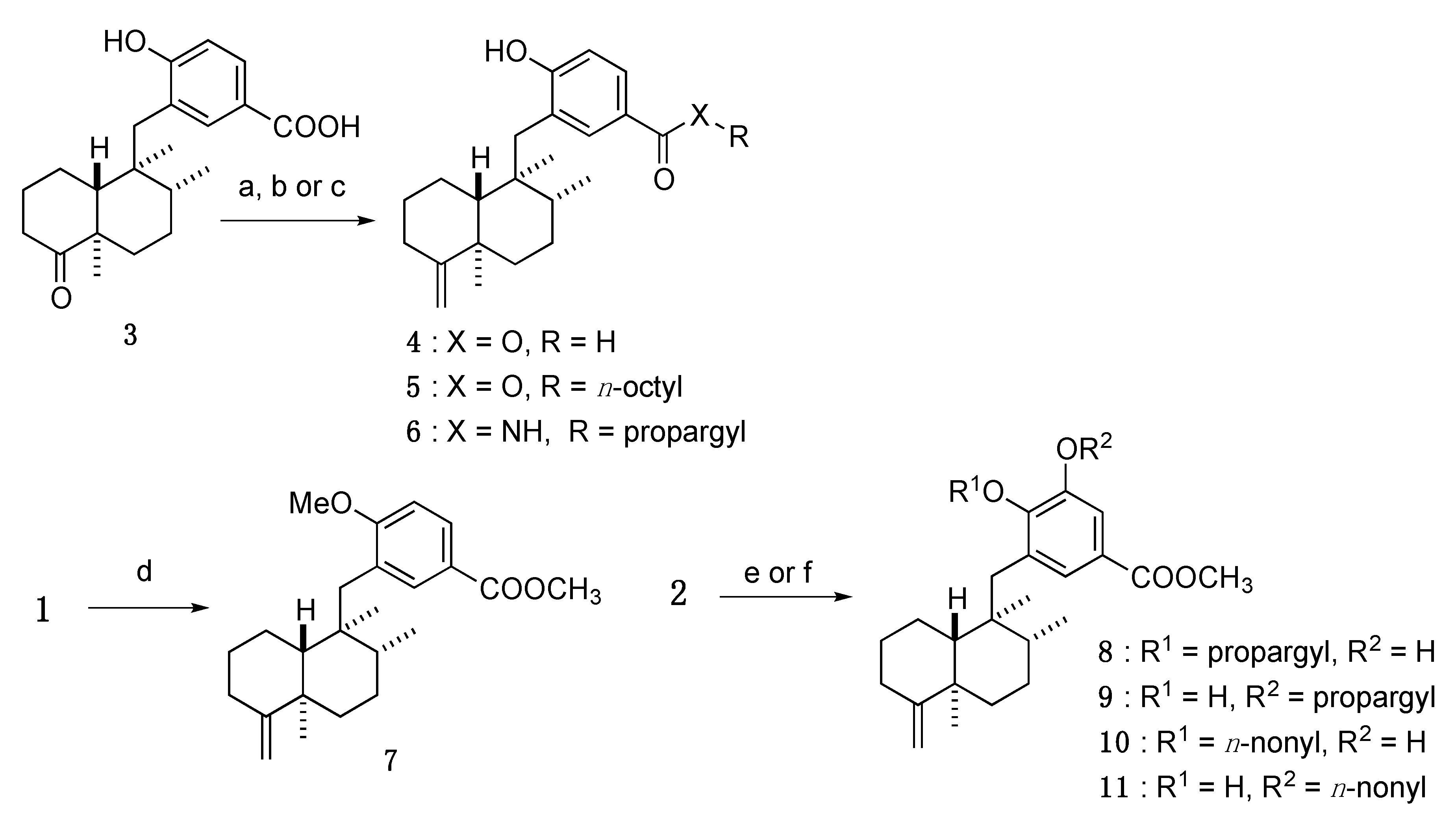
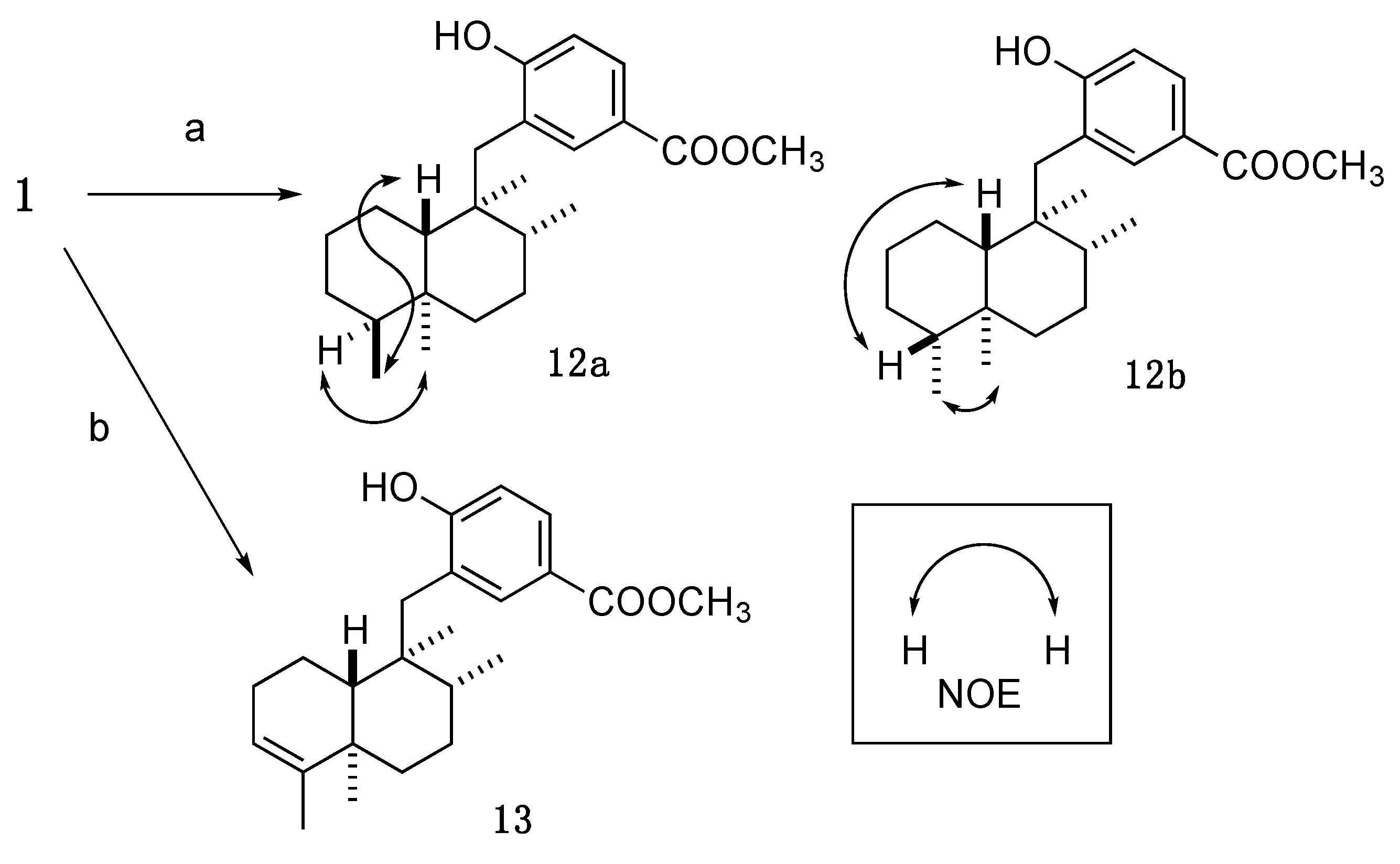
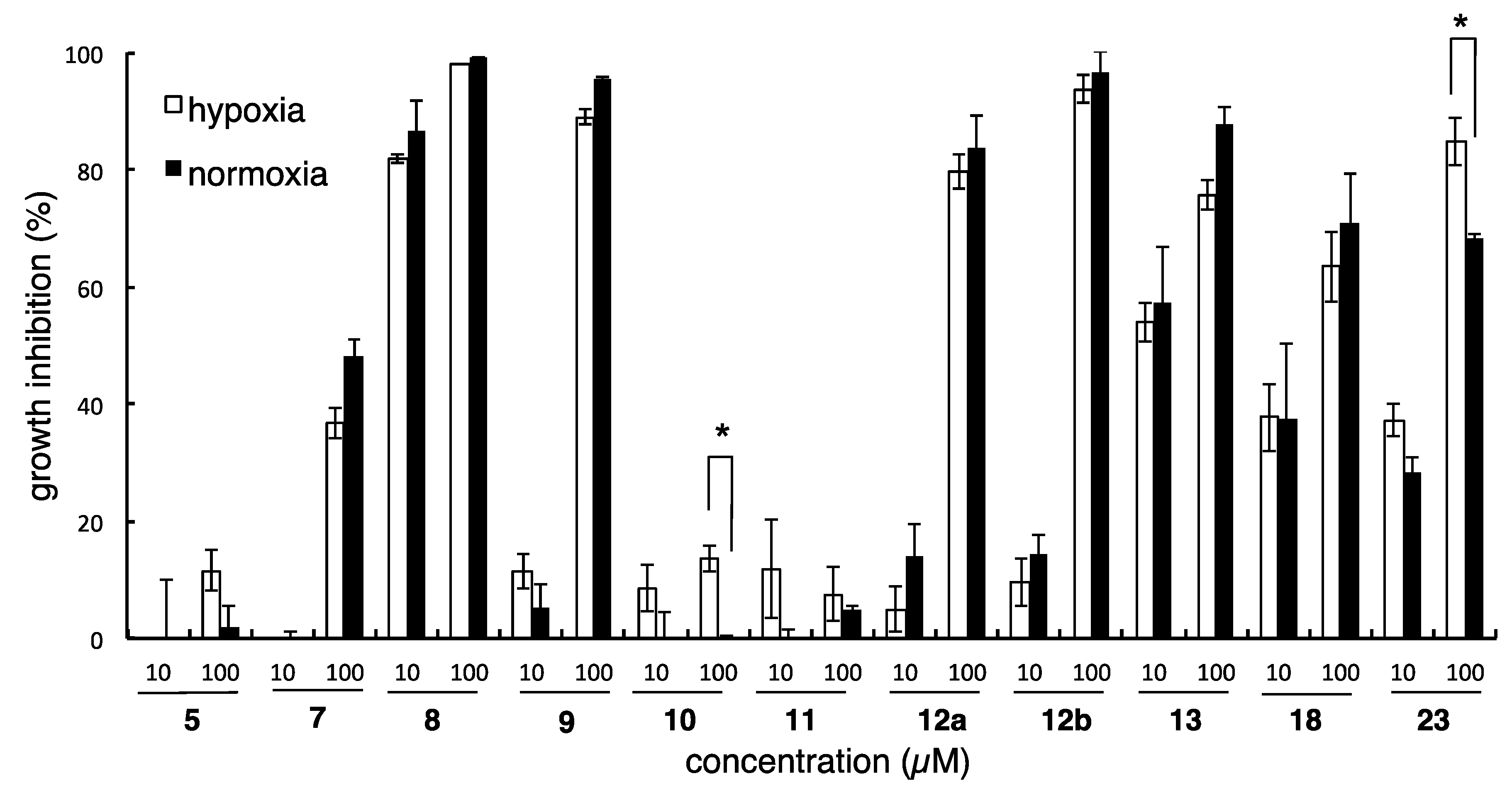
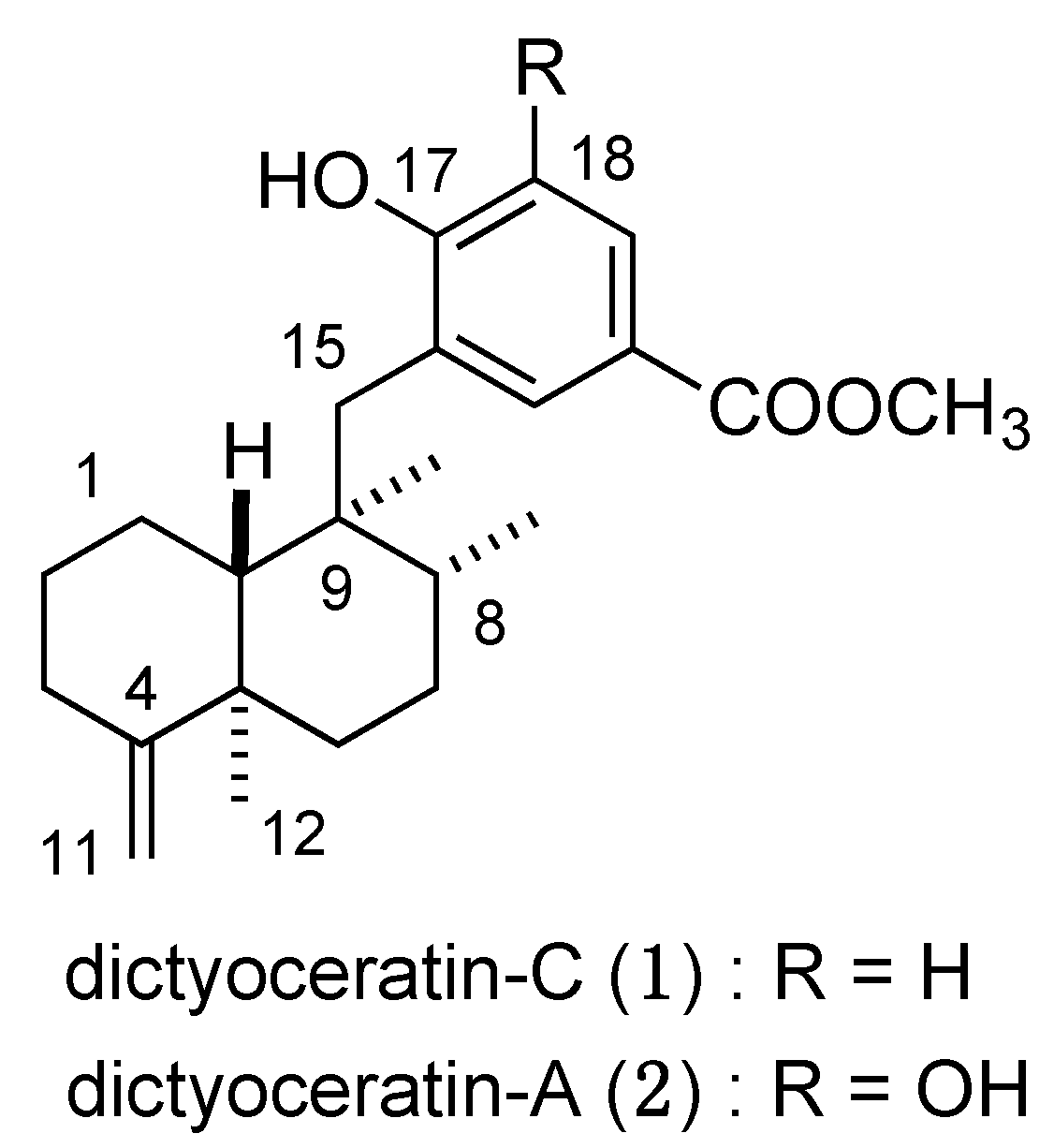
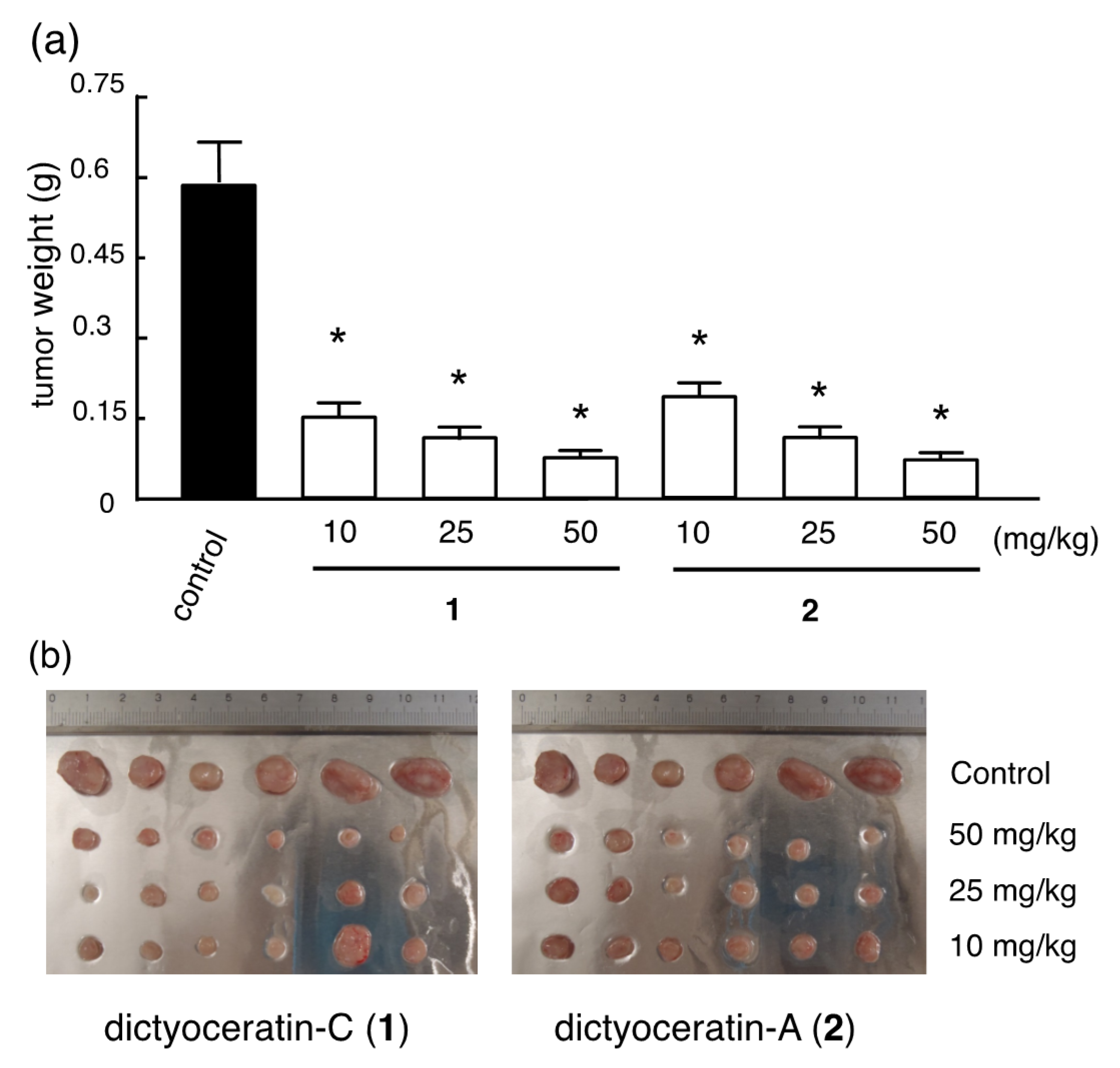
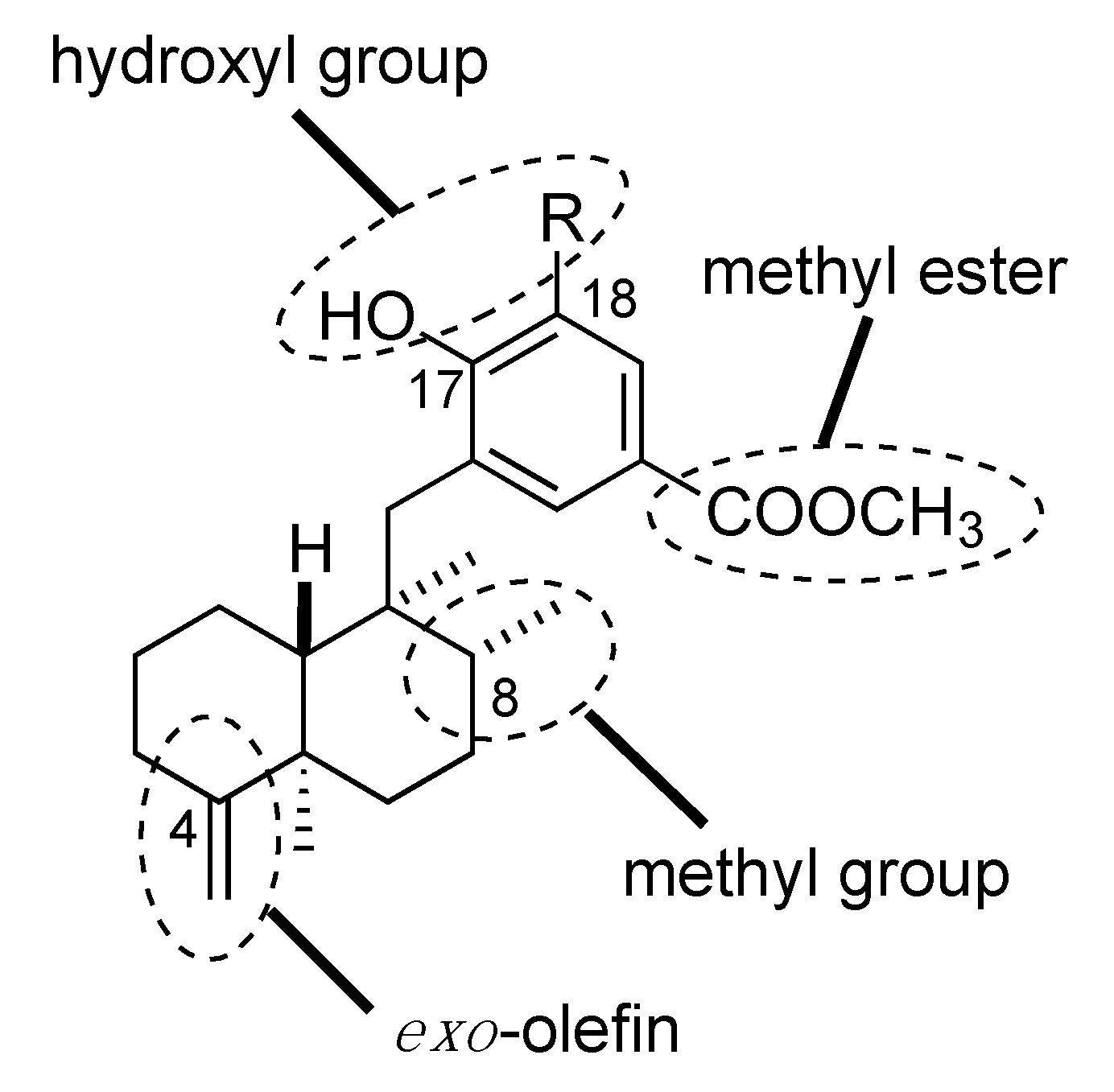
3.3. Design and Synthesis of Structure-Modified Analogues
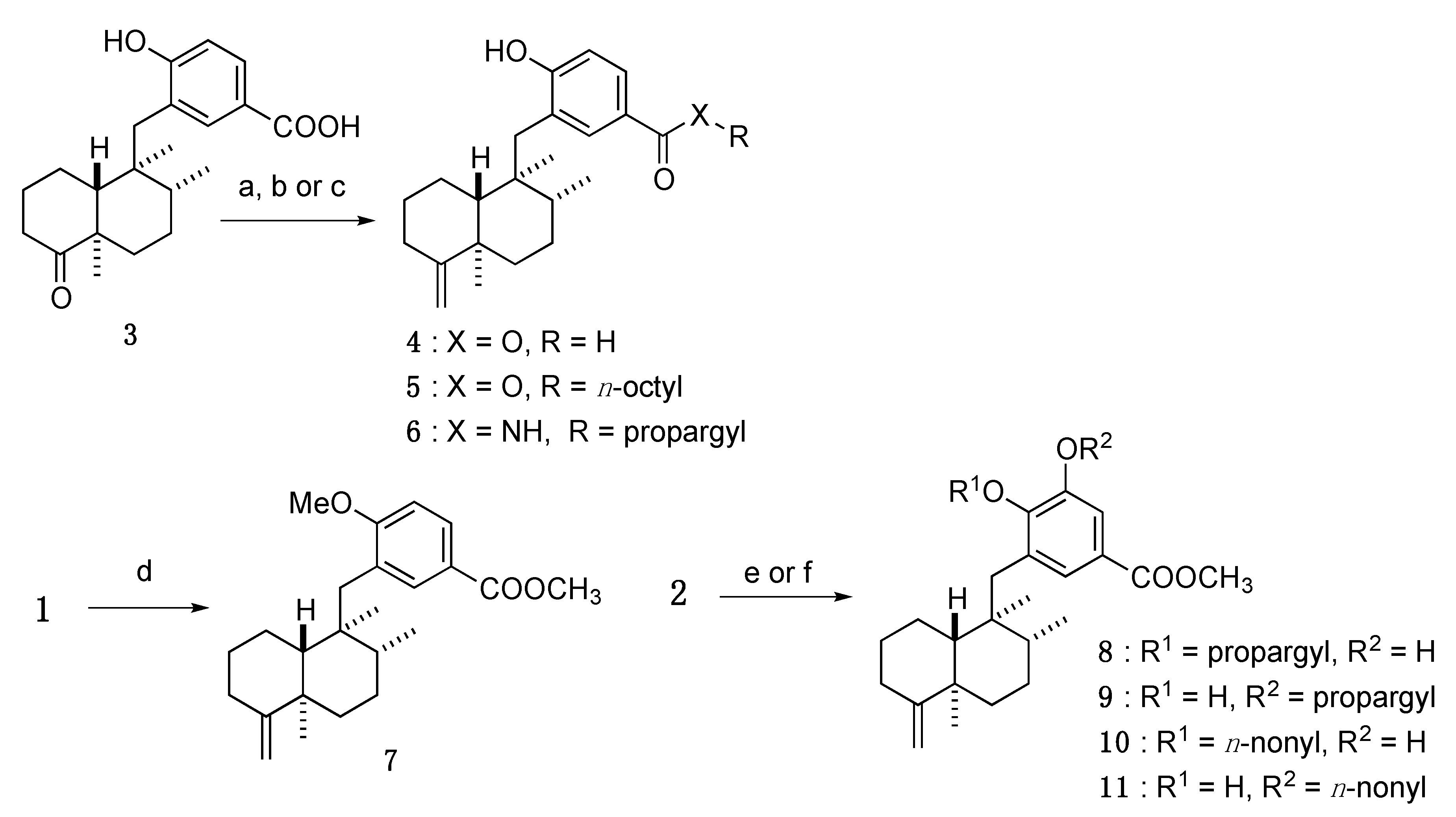
3.3.1. 4-Hydroxy-3-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoic acid (4)
Under an Ar atmosphere, KHMDS (5.9 mL, of a 0.5 M solution in toluene, 3.0 mmol) was added to a solution of Ph3PCH3Br (1.05 g, 3.0 mmol) in anhydrous THF (1.9 mL), and the whole mixture was stirred for 1 h at rt. The above solution (7.2 mL, about 2.7 mmol) was added dropwise to a solution of 3 (93.5 mg, 0.27 mmol) in anhydrous THF (0.9 mL) at 0 °C, and the whole mixture was stirred for 20 h at rt. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 3:2) to give 4 (87.3 mg, 94%) as a white solid.
+33 (c = 1.22, MeOH). 1H-NMR (500 MHz, CDCl3) δ: 7.85–7.83 (2H, m), 6.76 (1H, d, J = 8.6 Hz), 4.42 (1H, s), 4.37 (1H, s), 2.70 (1H, d, J = 14.4 Hz), 2.63 (1H, d, J = 14.4 Hz), 2.34 (1H, td, J = 13.6, 5.1 Hz), 2.12–2.06 (2H, m), 1.95–1.91 (1H, m), 1.64–1.56 (1H, m), 1.48 (1H, dt, J = 12.4, 3.1 Hz), 1.44–1.19 (5H, m), 1.07 (3H, s), 1.04 (3H, d, J = 6.7 Hz), 0.95 (1H, dd, J = 12.2, 1.8 Hz), 0.89 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 171.9, 159.9, 159.4, 135.7, 130.1, 125.2, 121.2, 115.5, 102.9, 48.1, 42.1, 40.2, 37.1, 36.5, 36.3, 32.9, 27.9, 27.7, 23.3, 20.5, 17.62, 17.57. MS (ESI-TOF) m/z: 365 [M + Na]+. HRMS (ESI-TOF) m/z: 365.2093 calcd for C22H30O3Na; Found: 365.2083.
3.3.2. Octyl 4-hydroxy-3-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (5)
The components SOCl2 (6.0 µL, 0.083 mmol) and DMF (1 drop) were added to a solution of 4 (4.8 mg, 0.014 mmol) in anhydrous THF (0.2 mL), and the whole mixture was stirred for 1 h at rt. n-Octanol (6.0 µL, 0.083 mmol) was added to the mixture, and the whole mixture was stirred for 7 h at 50 °C. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (CHCl3/MeOH/H2O = 200:3:1, lower phase) to give 5 (0.8 mg, 13%) as a white solid.
+14 (c = 0.31, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.78 (1H, dd, J = 8.2, 1.9 Hz), 7.75 (1H, d, J = 1.9 Hz), 6.73 (1H, d, J = 8.2 Hz), 5.17 (1H, s), 4.41 (1H, s), 4.37 (1H, s), 4.31–4.20 (2H, m), 2.67 (1H, d, J = 14.3 Hz), 2.62 (1H, d, J = 14.3 Hz), 2.34 (1H, td, J = 13.7, 5.2 Hz), 2.08 (2H, t, J = 11.7 Hz), 1.94–1.91 (1H, m), 1.76–1.70 (2H, m), 1.60 (2H, qd, J = 13.2, 6.6 Hz), 1.47–1.41 (5H, m), 1.31–1.23 (10H, m), 1.07 (3H, s), 1.03 (3H, d, J = 6.9 Hz), 0.96 (1H, dd, J = 12.0, 1.7 Hz), 0.88 (3H, t, J = 6.9 Hz), 0.88 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 166.6, 159.9, 158.5, 134.8, 129.3, 124.8, 122.5, 115.3, 102.9, 64.8, 47.9, 42.0, 40.2, 37.0, 36.5, 36.2, 33.0, 31.8, 29.3, 29.2, 28.8, 27.9, 27.7, 26.1, 23.3, 22.7, 20.5, 17.63, 17.60, 14.1. IR (KBr) : 2926, 1672, 1599, 1279 cm−1. MS (ESI-TOF) m/z: 477 [M + Na]+. HRMS (ESI-TOF) m/z: 477.3345 calcd for C30H46O3Na; Found: 477.3348.
3.3.3. 4-Hydroxy-N-(prop-2-yn-1-yl)-3-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzamide (6)
Propargylamine (7.0 µL, 0.10 mmol), EDCI·HCl (18.0 mg, 0.094 mmol) and HOBt (14.2 mg, 0.11 mmol) were added to a solution of 4 (10.8 mg, 0.032 mmol) in anhydrous DMF (0.3 mL), and the whole mixture was stirred for 6 h at rt. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (CHCl3/MeOH/H2O = 30:3:1, lower phase) to give 6 (7.2 mg, 60%) as a white solid.
+22.0 (c = 0.58, MeOH). 1H-NMR (500 MHz, acetone-d6) δ: 8.89 (1H, d, J = 4.0 Hz), 7.79 (1H, brs), 7.71 (1H, d, J = 2.3 Hz), 7.61 (1H, dd, J = 8.6, 2.3 Hz), 6.86 (1H, d, J = 8.6 Hz), 4.37 (1H, s), 4.33 (1H, s), 4.20–4.09 (2H, m), 2.72 (1H, d, J = 14.3 Hz), 2.68 (1H, d, J = 14.3 Hz), 2.63 (1H, t, J = 2.6 Hz), 2.35 (1H, td, J = 13.7, 5.2 Hz), 2.22 (1H, d, J = 13.2 Hz), 2.08–2.04 (1H, m), 1.90–1.85 (1H, m), 1.62–1.54 (1H, m), 1.51–1.36 (4H, m), 1.21–1.15 (1H, m), 1.07 (3H, s), 1.05 (3H, d, J = 5.7 Hz), 1.01 (1H, dd, J = 11.7, 2.0 Hz), 0.89 (3H, s). 13C-NMR (125 MHz, acetone-d6) δ: 166.8, 160.7, 160.0, 133.6, 127.0, 126.0, 125.8, 115.4, 103.2, 81.8, 71.6, 48.9, 42.7, 40.9, 37.6, 37.5, 37.1, 33.7, 29.2, 28.8, 28.4, 23.7, 20.9, 18.1, 18.0. IR (KBr): 3299, 2921, 1630, 1588, 1490, 1268 cm−1. MS (ESI-TOF) m/z: 402 [M + Na]+. HRMS (ESI-TOF) m/z: 402.2409 calcd for C25H33NO2Na; Found: 402.2418.
3.3.4. Methyl 4-methoxy-3-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (7)
K2CO3 (7.5 mg, 0.054 mmol) and MeI (6.0 µL, 0.096 mmol) were added to a solution of 1 (6.8 mg, 0.018 mmol) in anhydrous DMF (0.2 mL), and the whole mixture was stirred for 12.5 h at rt. Sat. Na2S2O3 aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 8:1) to give 7 (7.6 mg, quantitative yield) as a white solid.
+23 (c = 0.68, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.87 (1H, dd, J = 8.9, 1.7 Hz), 7.74 (1H, d, J = 1.7 Hz), 6.82 (1H, d, J = 8.9 Hz), 4.40 (1H, s), 4.35 (1H, s), 3.87 (3H, s), 3.81 (3H, s), 2.68 (1H, d, J = 14.3 Hz), 2.65 (1H, d, J = 14.3 Hz), 2.33 (1H, td, J = 13.5, 5.2 Hz), 2.11–2.08 (2H, m), 1.95–1.92 (1H, m), 1.58–1.53 (2H, m), 1.45 (1H, dt, J = 12.4, 3.3 Hz), 1.41–1.30 (3H, m), 1.25–1.15 (1H, m), 1.06 (3H, s), 1.01 (3H, d, J = 6.3 Hz), 0.88 (1H, d, J = 12.6 Hz), 0.86 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 167.2, 162.1, 160.1, 134.1, 129.4, 127.4, 121.5, 109.6, 102.7, 55.1, 51.8, 48.0, 42.1, 40.2, 36.7, 36.6, 36.3, 33.1, 27.9, 27.7, 23.2, 20.6, 17.6, 17.5. IR (KBr): 2927, 1719, 1437, 1298, 1269 cm−1. MS (ESI-TOF) m/z: 393 [M + Na]+. HRMS (ESI-TOF) m/z: 393.2406 calcd for C24H34O3Na; Found: 393.2414.
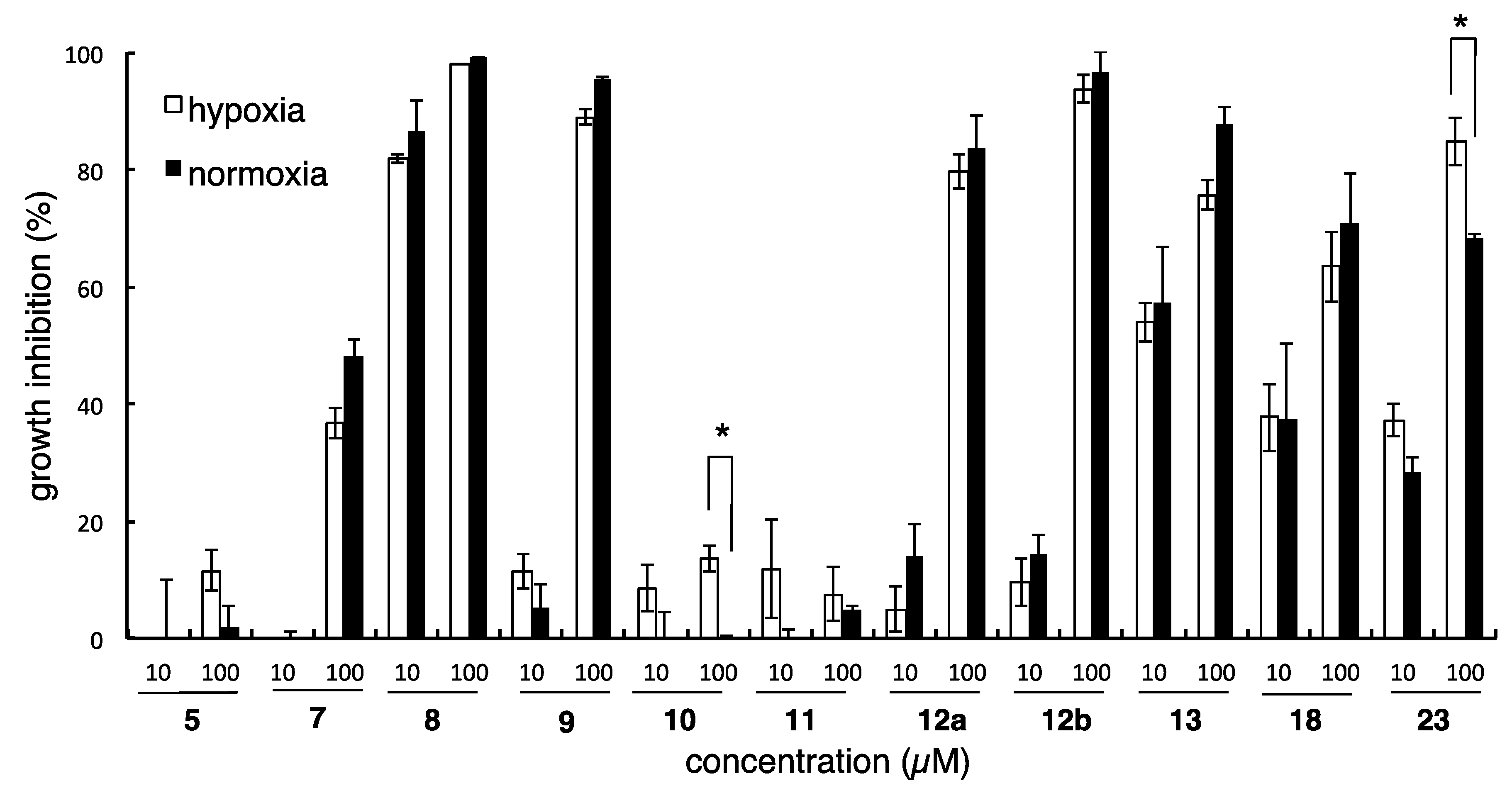
3.3.5. Methyl 3-hydroxy-4-(prop-2-yn-1-yloxy)-5-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (8); Methyl 4-hydroxy-3-(prop-2-yn-1-yloxy)-5-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (9)
K2CO3 (13.3 mg, 0.096 mmol) and propargyl bromide (9.0 µL, of a 80 wt% in toluene, 0.10 mmol) were added to a solution of 2 (30.6 mg, 0.082 mmol) in anhydrous DMF (1.65 mL), and the whole mixture was stirred for 3 h at 0 °C and for 13 h at rt. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 5:1) to give 8 (11.9 mg, 35%) as a colorless amorphous and 9 (4.3 mg, 13%) as a colorless amorphous.
8: +22 (c = 0.94, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.48 (1H, d, J = 2.0 Hz), 7.38 (1H, d, J = 2.0 Hz), 5.74 (1H, s), 4.59 (1H, dd, J = 15.5, 2.5 Hz), 4.53 (1H, dd, J = 15.5, 2.5 Hz), 4.40 (1H, s), 4.35 (1H, s), 3.87 (3H, s), 2.66 (1H, d, J = 13.7 Hz), 2.62 (1H, d, J = 13.7 Hz), 2.58 (1H, t, J = 2.5 Hz), 2.37–2.28 (1H, m), 2.10–2.07 (1H, m), 1.99 (1H, d, J = 13.2 Hz), 1.94–1.88 (1H, m), 1.57 (1H, qd, J = 13.0, 3.3 Hz), 1.45 (1H, dt, J = 12.4, 3.3 Hz), 1.42–1.33 (2H, m), 1.27–1.13 (3H, m), 1.05 (3H, s), 1.00 (3H, d, J = 6.3 Hz), 0.87 (3H, s), 0.84 (1H, dd, J = 12.0, 1.7 Hz). 13C-NMR (150 MHz, CDCl3) δ: 166.7, 159.8, 149.0, 148.8, 132.8, 126.2, 125.9, 115.2, 102.9, 78.2, 76.8, 61.0, 52.1, 47.8, 42.4, 40.1, 37.7, 36.4, 36.1, 32.9, 27.7, 27.5, 23.2, 20.6, 17.8, 17.6. IR (KBr): 3302, 2928, 1718, 1591, 1437, 1262 cm−1. MS (ESI-TOF) m/z: 433 [M + Na]+. HRMS (ESI-TOF) m/z: 433.2355 calcd for C26H34O4Na; Found: 433.2372.
9: +14 (c = 0.36, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.493 (1H, d, J = 2.0 Hz), 7.486 (1H, d, J = 2.0 Hz), 6.12 (1H, s), 4.78 (2H, d, J = 2.2 Hz), 4.41 (1H, s), 4.36 (1H, s), 3.87 (3H, s), 2.68 (2H, s), 2.58 (1H, t, J = 2.3 Hz), 2.34 (1H, td, J = 13.7, 5.2 Hz), 2.14–2.07 (2H, m), 1.92 (1H, m), 1.66–1.52 (1H, m), 1.48–1.36 (3H, m), 1.32–1.17 (3H, m), 1.06 (3H, s), 1.03 (3H, d, J = 6.9 Hz), 0.94 (1H, dd, J = 11.7, 2.0 Hz), 0.87 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 167.0, 160.2, 149.7, 143.9, 128.5, 125.0, 120.4, 111.1, 102.6, 77.6, 76.5, 57.3, 51.9, 48.0, 42.1, 40.2, 36.9, 36.6, 36.3, 33.0, 27.9, 27.7, 23.1, 20.6, 17.7, 17.6. IR (KBr): 3295, 2926, 1713, 1437, 1300 cm−1. MS (ESI-TOF) m/z: 433 [M + Na]+. HRMS (ESI-TOF) m/z: 433.2355 calcd for C26H34O4Na; Found: 433.2364.
3.3.6. Methyl 3-hydroxy-4-(nonyloxy)-5-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (10); Methyl 4-hydroxy-3-(nonyloxy)-5-(((1S,2R,4aR,8aR)-1,2,4a-trimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (11)
K2CO3 (9.2 mg, 0.067 mmol) and n-nonyl bromide (8.0 µL, 0.042 mmol) were added to a solution of 2 (12.6 mg, 0.034 mmol) in anhydrous DMF (0.34 mL), and the whole mixture was stirred for 3 h at rt. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 30:1 to 5:1) to give 10 (3.6 mg, 21%) as a colorless amorphous and 11 (2.3 mg, 14%) as a colorless amorphous.
10: +15 (c = 0.20, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.43 (1H, d, J = 1.7 Hz), 7.36 (1H, d, J = 1.7 Hz), 6.16 (1H, s), 4.41 (1H, s), 4.36 (1H, s), 4.07 (2H, t, J = 6.6 Hz), 3.86 (3H, s), 2.69 (1H, d, J = 14.0 Hz), 2.66 (1H, d, J = 14.0 Hz), 2.34 (1H, td, J = 13.5, 5.2 Hz), 2.13–2.07 (2H, m), 1.94–1.90 (1H, m), 1.85–1.79 (2H, m), 1.62–1.52 (3H, m), 1.48–1.19 (16H, m), 1.06 (3H, s), 1.03 (3H, d, J = 6.3 Hz), 0.96 (1H, dd, J = 12.0, 1.7 Hz), 0.89 (3H, t, J = 6.9 Hz), 0.87 (3H, s). 13C-NMR (150 MHz, CDCl3) δ: 167.2, 160.3, 149.4, 145.3, 127.4, 124.2, 120.3, 109.9, 102.6, 69.2, 51.9, 48.0, 42.1, 40.2, 36.9, 36.6, 36.4, 33.1, 31.9, 29.5, 29.4, 29.2, 29.1, 27.9, 27.7, 26.0, 23.1, 22.7, 20.7, 17.7, 17.6, 14.1. IR (KBr): 2925, 1716, 1437, 1301 cm−1. MS (ESI-TOF) m/z: 521 [M + Na]+. HRMS (ESI-TOF) m/z: 521.3607 calcd for C32H50O4Na; Found: 521.3616.
11: +20 (c = 0.61, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.46 (1H, d, J = 2.0 Hz), 7.36 (1H, d, J = 2.0 Hz), 5.57 (1H, s), 4.40 (1H, s), 4.35 (1H, s), 3.87 (3H, s), 3.84–3.74 (2H, m), 2.66 (1H, d, J = 14.0 Hz), 2.58 (1H, d, J = 14.0 Hz), 2.31 (1H, td, J = 13.6, 5.5 Hz), 2.08 (1H, dd, J = 14.0, 3.2 Hz), 2.01 (1H, d, J = 12.6 Hz), 1.91–1.87 (1H, m), 1.83–1.77 (2H, m), 1.69–1.51 (3H, m), 1.47–1.11 (16H, m), 1.05 (3H, s), 0.99 (3H, d, J = 6.9 Hz), 0.89 (3H, t, J = 7.2 Hz), 0.87 (3H, s), 0.84 (1H, dd, J = 12.0, 1.7 Hz). 13C-NMR (150 MHz, CDCl3) δ: 166.8, 159.8, 149.9, 148.9, 132.2, 125.8, 125.5, 114.4, 102.9, 74.6, 52.0, 47.9, 42.3, 40.1, 37.3, 36.5, 36.1, 33.0, 31.8, 30.2, 29.5, 29.4, 29.2, 27.9, 27.6, 26.0, 23.1, 22.7, 20.6, 17.8, 17.5, 14.1. IR (KBr): 3394, 2926, 1722, 1437, 1335 cm−1. MS (ESI-TOF) m/z: 521 [M + Na]+. HRMS (ESI-TOF) m/z: 521.3607 calcd for C32H50O4Na; Found: 521.3630.
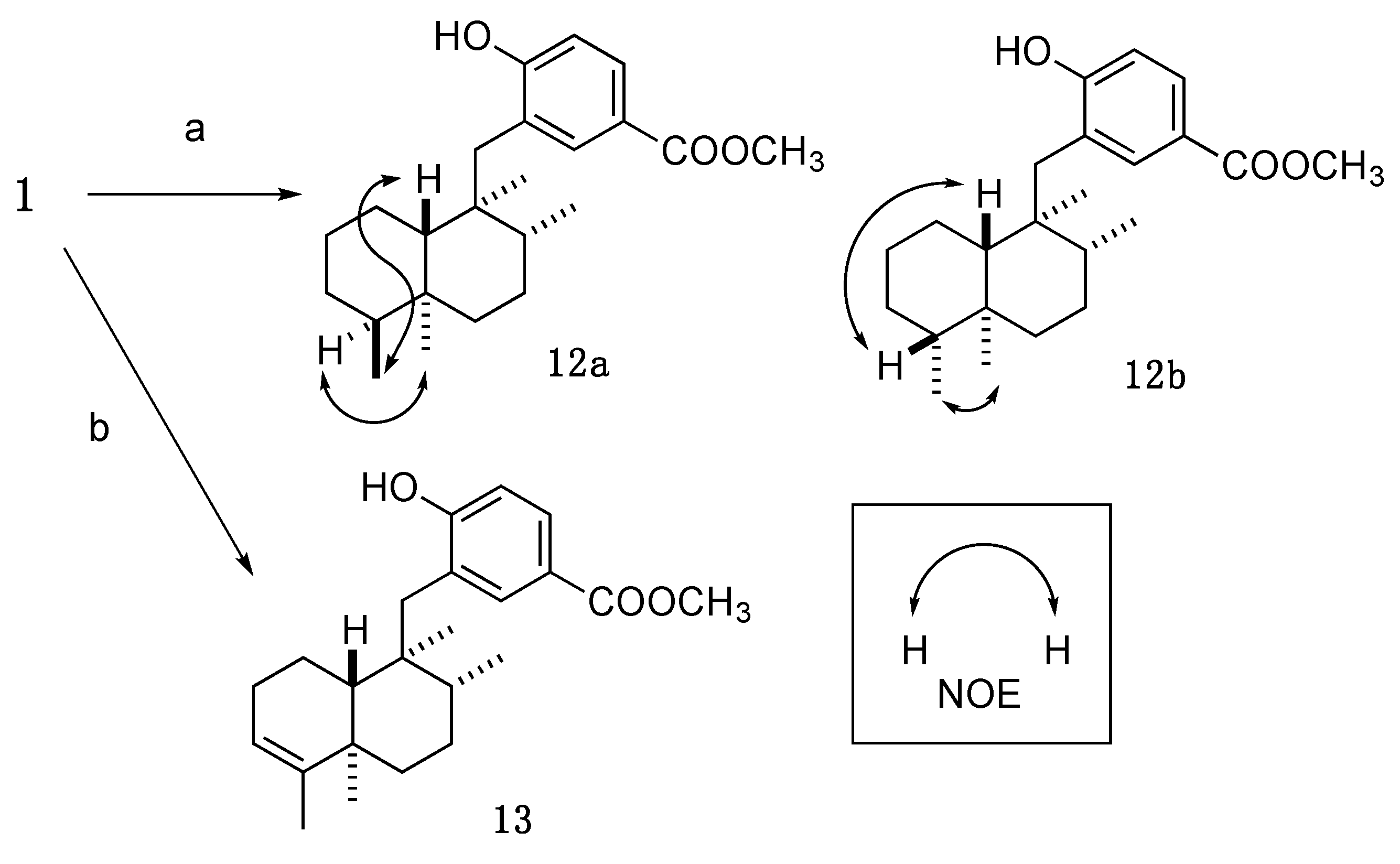
3.3.7. Methyl 4-hydroxy-3-(((1S,2R,4aS,5S,8aS)-1,2,4a,5-tetramethyldecahydronaphthalen-1-yl)methyl)benzoate (12a); Methyl 4-hydroxy-3-(((1S,2R,4aS,5R,8aS)-1,2,4a,5-tetramethyldecahydronaphthalen-1-yl)methyl)benzoate (12b)
An amount of 10% Pd-C (6.8 mg) was added to a solution of 1 (15.4 mg, 0.041 mmol) in anhydrous MeOH (2.0 mL), and the whole mixture was stirred for 16 h under H2 atmosphere (baloon). The mixture was filtered through a short pad of Celite. Removal of the solvent from the filtrate under reduced pressure gave a crude product, which was purified by reversed-phase HPLC (Cosmosil AR-II, 85% aq. CH3CN, flow rate: 3 mL/min, UV: 220 nm) to give 12a (7.5 mg, 51%) and 12b (7.0 mg, 47%) as a white amorphous solid.
12a: −3.8 (c = 0.66, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.83 (1H, d, J = 2.2 Hz), 7.79 (1H, dd, J = 8.5, 2.2 Hz), 6.78 (1H, d, J = 8.5 Hz), 5.22 (1H, s), 3.88 (3H, s), 2.63 (2H, s), 1.91–1.83 (2H, m), 1.69–1.55 (3H, m), 1.48–1.31 (3H, m), 1.28–1.21 (2H, m), 1.17–1.11 (2H, m), 1.04 (3H, s), 0.99 (3H, d, J = 6.3 Hz), 0.88 (1H, dt, J = 13.0, 3.2 Hz), 0.82 (3H, s), 0.70 (3H, d, J = 6.9 Hz). 13C-NMR (150 MHz, CDCl3) δ: 167.2, 158.8, 135.0, 129.2, 125.1, 121.8, 115.3, 51.8, 42.2, 41.1, 38.8, 36.8, 36.51, 36.48, 36.4, 29.1, 27.6, 23.5, 22.1, 21.2, 17.7, 17.5, 14.6. IR (KBr): 3291, 2934, 1680, 1601, 1420, 1285 cm−1. MS (ESI-TOF) m/z: 381 [M + Na]+. HRMS (ESI-TOF) m/z: 381.2406 calcd for C23H34O3Na; Found: 381.2413.
12b: −6.5 (c = 0.67, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.80 (1H, d, J = 2.0 Hz), 7.78 (1H, dd, J = 8.4, 2.0 Hz), 6.76 (1H, d, J = 8.4 Hz), 5.25 (1H, s), 3.88 (3H, s), 2.63 (2H, s), 1.93 (1H, d, J = 12.0 Hz), 1.84–1.80 (1H, m), 1.67–1.42 (5H, m), 1.40–1.13 (5H, m), 1.01 (3H, d, J = 6.3 Hz), 0.90 (1H, dd, J = 12.3, 2.6 Hz), 0.84 (3H, s), 0.80 (3H, s), 0.65 (3H, d, J = 6.9 Hz). 13C-NMR (125 MHz, CDCl3) δ: 167.1, 158.7, 135.1, 129.2, 125.0, 122.0, 115.3, 51.8, 48.9, 45.4, 41.6, 38.3, 37.3, 36.9, 36.1, 30.8, 27.5, 26.4, 23.1, 17.73, 17.68, 15.0, 13.2. IR (KBr): 2928, 1717, 1692, 1283 cm−1. MS (ESI-TOF) m/z: 381 [M + Na]+. HRMS (ESI-TOF) m/z: 381.2406 calcd for C23H34O3Na; Found: 381.2413.
3.3.8. Methyl 4-hydroxy-3-(((1S,2R,4aR,8aR)-1,2,4a,5-tetramethyl-1,2,3,4,4a,7,8,8a-octahydronaphthalen-1-yl)methyl)benzoate (13)
Compound 1 (7.6 mg, 0.020 mmol) and RhCl3·3H2O (2.6 mg, 0.010 mmol) were mixed and dried. EtOH (1.0 mL) was added to the mixture, and the whole mixture was stirred for 12 h under reflux. Removal of the solvent from the mixture under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 5:1) and reversed-phase HPLC (Cosmosil AR-II, 85% aq. CH3CN, flow rate: 3 mL/min, UV: 220 nm) to give 13 (4.0 mg, 61%) as a white amorphous solid.
−27 (c = 0.39, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.80 (1H, d, J = 2.0 Hz), 7.77 (1H, dd, J = 8.6, 2.0 Hz), 6.74 (1H, d, J = 8.6 Hz), 5.27 (1H, s), 5.15 (1H, s), 3.87 (3H, s), 2.72 (1H, d, J = 14.6 Hz), 2.68 (1H, d, J = 14.6 Hz), 2.27–2.19 (1H, m), 2.13–2.08 (1H, m), 2.04 (1H, dd, J = 13.5, 6.6 Hz), 1.66–1.55 (2H, m), 1.50 (3H, d-like, J = 1.7 Hz), 1.38–1.25 (3H, m), 1.18 (1H, dd, J = 12.0, 1.1 Hz), 1.03 (3H, d, J = 6.3 Hz), 1.02 (3H, s), 0.93–0.88 (1H, m), 0.88 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 167.1, 158.7, 144.1, 135.2, 129.3, 125.0, 122.1, 120.6, 115.3, 51.8, 45.5, 41.7, 38.3, 37.2, 36.0, 35.7, 27.7, 26.0, 20.0, 19.9, 18.1, 17.7, 17.6. IR (KBr): 2944, 1688, 1603, 1433, 1285 cm−1. MS (ESI-TOF) m/z: 379 [M + Na]+. HRMS (ESI-TOF) m/z: 379.2249 calcd for C23H32O3Na; Found: 379.2252.
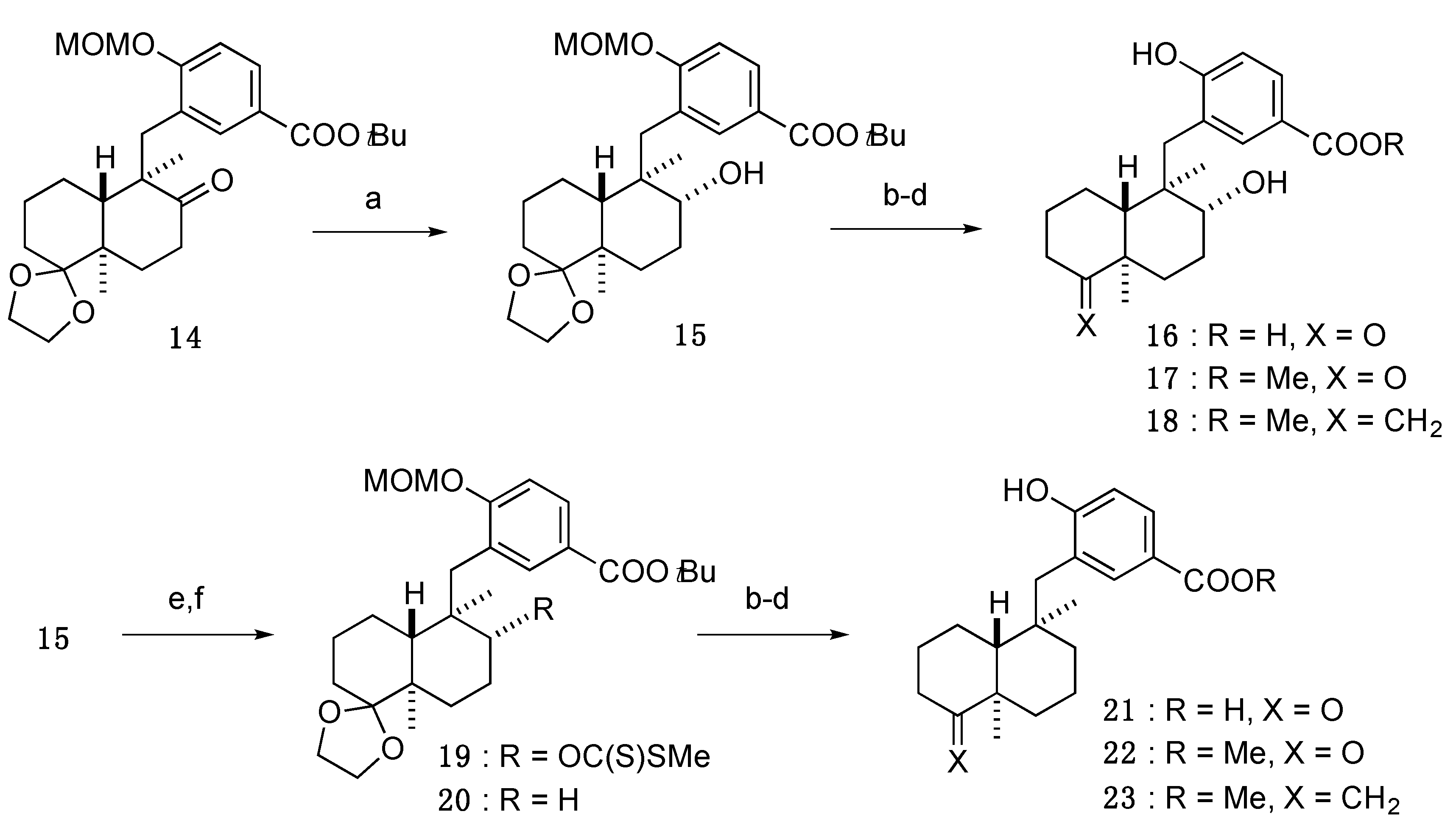
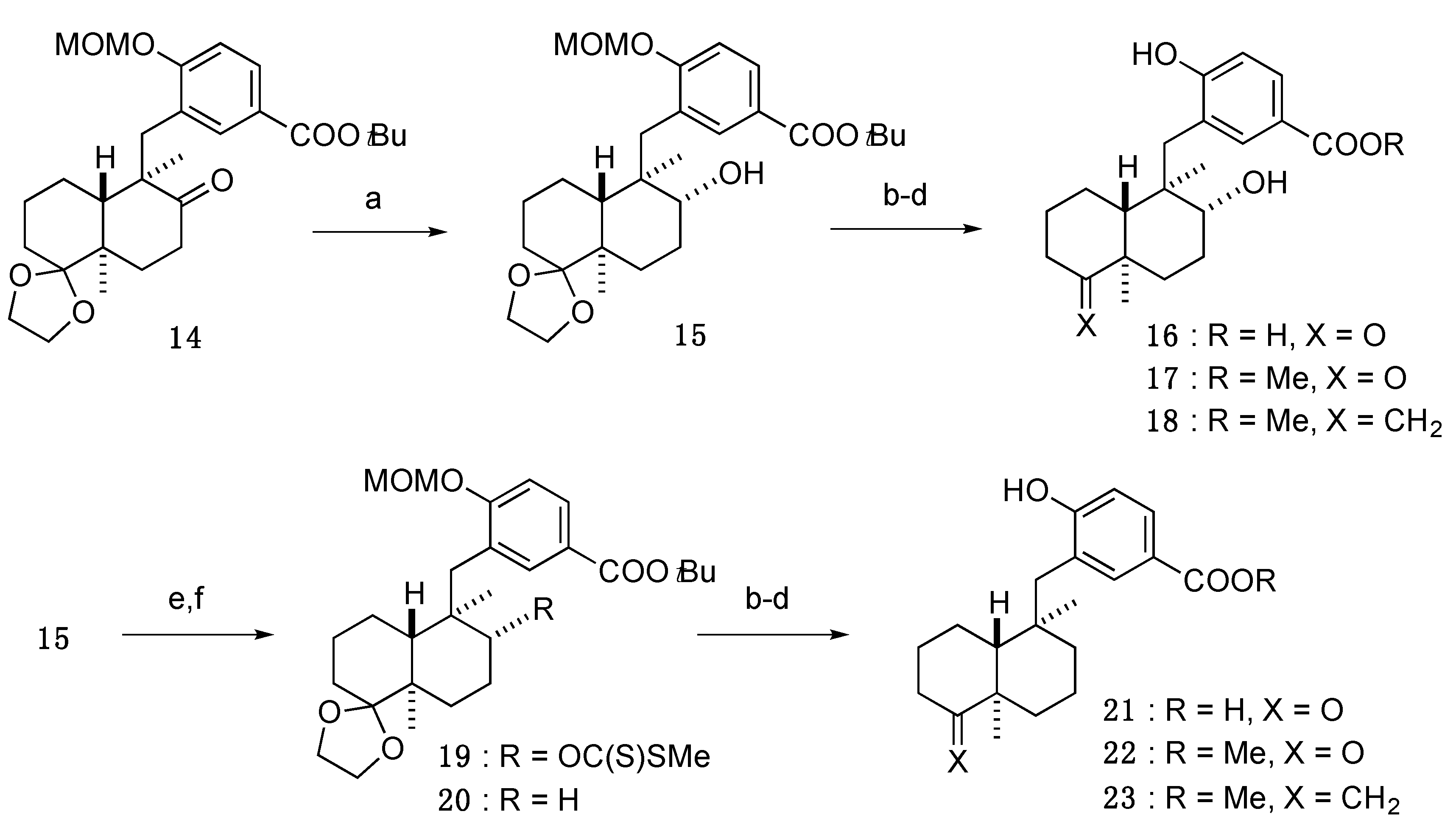
3.3.9. tert-Butyl 3-(((4a′R,5′R,6′R,8a′R)-6′-hydroxy-5′,8a′-dimethyloctahydro-2′H-spiro[[1,3]dioxolane-2,1′-naphthalen]-5′-yl)methyl)-4-(methoxymethoxy)benzoate (15)
CeCl
3·7H
2O (545 mg, 1.46 mmol) was added to a solution of
14 (285 mg, 0.58 mmol) [
9] in MeOH (5.8 mL), and the whole mixture was stirred for 5 min. NaBH
4 (110 mg, 2.9 mmol) was added to the mixture at 0 °C, and the whole mixture was stirred for 2.5 h at 0 °C. Sat. NH
4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO
2 column (
n-hexane/AcOEt = 4:1) to give
15 (270 mg, 94%) as a white amorphous solid.
−8 (c = 1.07, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.87 (1H, dd, J = 8.9, 2.0 Hz), 7.68 (1H, d, J = 2.0 Hz), 7.14 (1H, d, J = 8.9 Hz), 5.28 (2H, s), 3.86 (1H, q, J = 6.1 Hz), 3.79–3.76 (2H, m), 3.71 (1H, q, J = 6.5 Hz), 3.52 (3H, s), 3.37 (1H, d, J = 3.4 Hz), 3.00 (1H, ddd, J = 10.3, 5.2, 3.4 Hz), 2.82 (1H, d, J = 14.3 Hz), 2.59 (1H, d, J = 14.3 Hz), 1.93 (1H, d, J = 13.2 Hz), 1.76–1.61 (2H, m), 1.58 (9H, s), 1.57–1.45 (5H, m), 1.31–1.22 (2H, m), 1.10 (3H, s), 0.97 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 165.6, 158.9, 133.6, 129.4, 126.7, 125.5, 113.2, 113.0, 95.1, 80.7, 73.0, 65.0, 64.7, 56.8, 43.7, 43.1, 41.4, 35.8, 30.4, 28.3, 28.2, 25.0, 22.6, 21.3, 17.2, 16.6. MS (ESI-TOF) m/z: 513 [M + Na]+. HRMS (ESI-TOF) m/z: 513.2828 calcd for C28H42O7Na; Found: 513.2829.
3.3.10. 4-Hydroxy-3-(((1R,2R,4aR,8aR)-2-hydroxy-1,4a-dimethyl-5-oxodecahydronaphthalen-1-yl)methyl)benzoic acid (16)
An amount of 80% TFA (3.6 mL) was added to a solution of 15 (51.5 mg, 0.11 mmol) in THF (1.2 mL) at 0 °C, and the whole mixture was stirred for 3.5 h at 50 °C. Removal of the solvent from the mixture under reduced pressure gave a crude product, which was purified by SiO2 column (CHCl3/MeOH/H2O = 15:3:1, lower phase) to give 16 (36.6 mg, 97%) as a white amorphous solid.
+14 (c = 1.02, MeOH). 1H-NMR (500 MHz, acetone-d6) δ: 7.77 (1H, dd, J = 8.6, 2.0 Hz), 7.69 (1H, d, J = 2.0 Hz), 6.88 (1H, d, J = 8.6 Hz), 3.06–3.03 (1H, m), 2.81 (1H, d, J = 14.3 Hz), 2.76 (1H, d, J = 14.3 Hz), 2.65 (1H, td, J = 14.0, 7.1 Hz), 2.27 (1H, dd, J = 14.0, 2.0 Hz), 2.18–2.13 (1H, m), 2.08–2.07 (1H, m), 1.96 (1H, qd, J = 13.0, 3.6 Hz), 1.76–1.59 (3H, m), 1.44 (1H, dt, J = 14.1, 3.3 Hz), 1.35–1.25 (2H, m), 1.20 (3H, s), 1.15 (1H, dd, J = 12.3, 2.0 Hz), 1.10 (3H, s). 13C-NMR (125 MHz, acetone-d6) δ: 213.7, 167.5, 162.0, 134.8, 130.7, 124.5, 122.6, 117.1, 73.9, 49.0, 47.2, 44.3, 37.7, 37.3, 32.0, 26.1, 26.0, 21.7, 19.6, 17.2. MS (ESI-TOF) m/z: 369 [M + Na]+. HRMS (ESI-TOF) m/z: 369.1678 calcd for C20H26O5Na; Found: 369.1680.
3.3.11. Methyl 4-hydroxy-3-(((1R,2R,4aR,8aR)-2-hydroxy-1,4a-dimethyl-5-oxodecahydronaphthalen-1-yl)methyl)benzoate (17)
SOCl2 (0.02 mL, 0.28 mmol) was added to a solution of 16 (23.6 mg, 0.068 mmol) in anhydrous MeOH (0.7 mL), and the whole mixture was stirred for 6.5 h at 45 °C. Removal of the solvent from the mixture under reduced pressure gave a crude product, which was purified by SiO2 column (CHCl3/MeOH/H2O = 60:3:1, lower phase) to give 17 (23.9 mg, 98%) as a white solid.
+12 (c = 0.82, MeOH). 1H-NMR (500 MHz, acetone-d6) δ: 7.74 (1H, dd, J = 8.2, 1.9 Hz), 7.67 (1H, d, J = 1.9 Hz), 6.89 (1H, d, J = 8.2 Hz), 3.80 (3H, s), 3.04 (1H, dd, J = 10.3, 5.7 Hz), 2.88 (1H, br), 2.82 (1H, d, J = 14.9 Hz), 2.77 (1H, d, J = 14.9 Hz), 2.66 (1H, td, J = 14.0, 7.0 Hz), 2.27 (1H, dd, J = 14.0, 2.0 Hz), 2.22–2.17 (1H, m), 2.10–2.06 (1H, m), 2.03–1.97 (1H, m), 1.75–1.55 (3H, m), 1.44 (1H, dt, J = 14.3, 3.4 Hz), 1.33–1.23 (2H, m), 1.21 (3H, s), 1.16–1.13 (1H, dd, J = 12.3, 2.3 Hz), 1.11 (3H, s). 13C-NMR (125 MHz, acetone-d6) δ: 213.6, 167.0, 162.1, 134.5, 130.3, 124.6, 122.3, 117.1, 73.9, 51.9, 49.0, 47.2, 44.3, 37.7, 37.3, 32.0, 26.1, 26.0, 21.7, 19.6, 17.2. MS (ESI-TOF) m/z: 383 [M + Na]+. HRMS (ESI-TOF) m/z: 383.1834 calcd for C21H28O5Na; Found: 383.1847.
3.3.12. Methyl 4-hydroxy-3-(((1R,2R,4aR,8aS)-2-hydroxy-1,4a-dimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)benzoate (18)
Under an Ar atmosphere, KHMDS (0.9 mL, of a 0.5 M solution in toluene, 0.45 mmol) was added to a solution of Ph3PCH3Br (165 mg, 0.46 mmol) in anhydrous THF (0.22 mL), and the whole mixture was stirred for 1 h at rt. The above solution was added dropwise to a solution of 17 (10.9 mg, 0.030 mmol) in anhydrous THF (0.1 mL) at 0 °C, and the whole mixture was stirred for 1 h at rt. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 3:1) to give 18 (5.6 mg, 39%) as a white solid.
−32 (c = 0.55, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.81 (1H, dd, J = 8.5, 1.9 Hz), 7.63 (1H, d, J = 1.9 Hz), 6.89 (1H, d, J = 8.5 Hz), 4.45 (1H, s), 4.37 (1H, s), 3.85 (3H, s), 3.12 (1H, dd, J = 11.7, 4.3 Hz), 2.71 (2H, s), 2.31 (1H, td, J = 13.7, 5.7 Hz), 2.16–2.09 (2H, m), 2.04–2.00 (1H, m), 1.78 (1H, qd, J = 12.6, 3.2 Hz), 1.71–1.56 (4H, m), 1.48–1.40 (1H, m), 1.36 (1H, td, J = 13.3, 3.8 Hz), 1.11 (3H, s), 1.03 (3H, s), 1.00 (1H, dd, J = 12.6, 2.3 Hz). 13C-NMR (150 MHz, CDCl3) δ: 167.1, 160.9, 158.5, 133.9, 129.8, 123.2, 121.7, 116.6, 103.2, 74.7, 51.8, 46.2, 43.1, 39.5, 36.6, 34.8, 32.7, 27.4, 26.9, 22.1, 21.2, 16.3. IR (KBr): 3210, 2938, 1721, 1435, 1289 cm−1. MS (ESI-TOF) m/z: 381 [M + Na]+. HRMS (ESI-TOF) m/z: 381.2042 calcd for C22H30O4Na; Found: 381.2032.
3.3.13. tert-Butyl 3-(((4a′R,5′R,6′R,8a′R)-5′,8a′-dimethyl-6′-(((methylthio)carbonothioyl)oxy)octahydro-2′H-spiro[[1,3]dioxolane-2,1′-naphthalen]-5′-yl)methyl)-4-(methoxymethoxy)benzoate (19)
Under an Ar atmosphere, NaH (91.0 mg, 60% suspension in oil, 2.3 mmol) was added to a solution of 15 (136 mg, 0.28 mmol) in anhydrous THF (2.8 mL) at 0 °C, and the whole mixture was stirred for 30 min at rt. CS2 (0.17 mL, 2.8 mmol) was added dropwise to the mixture, and the whole mixture was stirred for 50 min at rt. MeI (0.21 mL, 3.4 mmol) was added to the mixture, and the whole mixture was stirred for 5 h at 50 °C. Sat. Na2S2O3 aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 5:1) to give 19 (152 mg, 94%) as a white amorphous solid.
−18 (c = 1.17, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.79 (1H, dd, J = 8.6, 2.3 Hz), 7.68 (1H, d, J = 2.3 Hz), 7.02 (1H, d, J = 8.6 Hz), 5.18 (2H, dd, J = 8.0, 6.9 Hz), 5.13 (1H, dd, J = 11.2, 4.3 Hz), 3.92–3.88 (3H, m), 3.79–3.77 (1H, m), 3.43 (3H, s), 2.96 (1H, d, J = 14.0 Hz), 2.64 (1H, d, J = 14.0 Hz), 2.28–2.24 (1H, m), 2.19 (3H, s), 1.88 (1H, d, J = 12.6 Hz), 1.80 (1H, dd, J = 12.0, 2.3 Hz), 1.77–1.61 (3H, m), 1.58 (9H, s), 1.54–1.44 (4H, m), 1.39–1.35 (1H, m), 1.17 (3H, s), 1.15 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 213.4, 165.8, 158.8, 133.6, 129.3, 127.1, 124.6, 113.0, 112.9, 93.9, 88.7, 80.4, 65.2, 64.9, 56.2, 45.7, 43.2, 38.9, 30.4, 28.2 (3C), 27.7, 22.8, 21.6, 21.0, 17.8, 17.0, 16.9. MS (ESI-TOF) m/z: 603 [M + Na]+. HRMS (ESI-TOF) m/z: 603.2426 calcd for C30H44O7S2Na; Found: 603.2449.
3.3.14. tert-Butyl 3-(((4a′R,5′R,8a′R)-5′,8a′-dimethyloctahydro-2′H-spiro[[1,3]dioxolane-2,1′-naphthalen]-5′-yl)methyl)-4-(methoxymethoxy)benzoate (20)
Under an Ar atmosphere, nBu3SnH (0.21 mL, 0.80 mmol) and AIBN (34.5 mg, 0.21 mmol) were added to a solution of 19 (138 mg, 0.24 mmol) in anhydrous toluene (3.4 mL), and the whole mixture was stirred for 3 h at 80 °C. H2O was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 8:1) to give 20 (96.6 mg, 86%) as a white amorphous solid.
+21 (c = 1.26, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.79 (1H, dd, J = 8.6, 2.3 Hz), 7.70 (1H, d, J = 2.3 Hz), 7.07 (1H, d, J = 8.6 Hz), 5.21 (1H, d, J = 6.9 Hz), 5.18 (1H, d, J = 6.9 Hz), 3.94–3.89 (3H, m), 3.83–3.79 (1H, m), 3.46 (3H, s), 2.88 (1H, d, J = 13.0 Hz), 2.40 (1H, d, J = 13.0 Hz), 1.81 (1H, d, J = 13.2 Hz), 1.75–1.64 (2H, m), 1.61–1.58 (1H, m), 1.57 (9H, s), 1.52–1.50 (1H, m), 1.44–1.21 (8H, m), 1.10 (3H, s), 0.85 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 165.9, 159.5, 134.1, 128.9, 127.9, 124.3, 113.5, 112.7, 94.2, 80.4, 65.2, 64.8, 56.2, 47.6, 43.8, 42.3, 37.9, 37.6, 30.6, 30.2, 28.2 (3C), 23.0, 20.9, 19.3, 17.8, 17.1. MS (ESI-TOF) m/z: 497 [M + Na]+. HRMS (ESI-TOF) m/z: 497.2879 calcd for C28H42O6Na; Found: 497.2897.
3.3.15. 3-(((1R,4aR,8aR)-1,4a-Dimethyl-5-oxodecahydronaphthalen-1-yl)methyl)-4-hydroxybenzoic acid (21)
An amount of 80% TFA (4.0 mL) was added to a solution of 20 (74.9 mg, 0.16 mmol) in THF (2.0 mL), and the whole mixture was stirred for 2.5 h at 50 °C. Removal of the solvent from the mixture under reduced pressure gave a crude product, which was purified by SiO2 column (CHCl3/MeOH/H2O = 30:3:1, lower phase) to give 21 (50.0 mg, 93%) as a white amorphous solid.
+46 (c = 1.13, MeOH). 1H-NMR (500 MHz, acetone-d6) δ: 7.75 (1H, dd, J = 8.6, 2.3 Hz), 7.74 (1H, d, J = 2.3 Hz), 6.92 (1H, d, J = 8.6 Hz), 2.77 (1H, d, J = 13.2 Hz), 2.70–2.60 (1H, m), 2.61 (1H, d, J = 13.2 Hz), 2.16–2.07 (2H, m), 1.83 (1H, qd, J = 12.9, 3.7 Hz), 1.69–1.40 (4H, m), 1.37–1.26 (5H, m), 1.20 (3H, s), 1.05 (3H, s). 13C-NMR (125 MHz, acetone-d6) δ: 214.3, 161.1, 135.6, 130.3, 125.8, 122.0, 115.7, 78.1, 51.1, 49.7, 42.5, 39.6, 37.88, 37.86, 33.8, 26.6, 21.8, 21.4, 19.4, 18.2. MS (ESI-TOF) m/z: 353 [M + Na]+. HRMS (ESI-TOF) m/z: 353.1729 calcd for C20H26O4Na; Found: 353.1730.
3.3.16. Methyl 3-(((1R,4aR,8aR)-1,4a-dimethyl-5-oxodecahydronaphthalen-1-yl)methyl)-4-hydroxybenzoate (22)
SOCl2 (0.03 mL, 0.413 mmol) was added to a solution of 21 (30.3 mg, 0.092 mmol) in anhydrous MeOH (0.92 mL), and the whole mixture was stirred for 15 h at 50 °C. Removal of the solvent from the mixture under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 3:1) to give 22 (27.5 mg, 87%) as a white solid.
+46 (c = 0.73, MeOH). 1H-NMR (500 MHz, acetone-d6) δ: 9.10 (1H, br s), 7.72 (1H, dd, J = 8.6, 1.7 Hz), 7.70 (1H, d, J = 1.7 Hz), 6.91 (1H, d, J = 8.6 Hz), 3.81 (3H, s), 2.77 (1H, d, J = 13.2 Hz), 2.66 (1H, td, J = 13.7, 6.9 Hz), 2.60 (1H, d, J = 13.2 Hz), 2.16–2.08 (3H, m), 1.83 (1H, qd, J = 13.2, 3.7 Hz), 1.68–1.25 (7H, m), 1.20 (3H, s), 1.11 (1H, t, J = 7.2 Hz), 1.05 (3H, s). 13C-NMR (125 MHz, acetone-d6) δ: 214.2, 167.1, 161.1, 135.2, 130.0, 125.9, 121.8, 115.8, 51.8, 51.1, 49.7, 42.4, 39.6, 37.89, 37.86, 33.8, 26.6, 21.8, 21.4, 19.4, 18.2. MS (ESI-TOF) m/z: 367 [M + Na]+. HRMS (ESI-TOF) m/z: 367.1885 calcd for C21H28O4Na; Found: 367.1875.
3.3.17. Methyl 3-(((1R,4aR,8aR)-1,4a-dimethyl-5-methylenedecahydronaphthalen-1-yl)methyl)-4-hydroxybenzoate (23)
Under an Ar atmosphere, KHMDS (0.57 mL, of a 0.5 M solution in toluene, 0.29 mmol) was added to Ph3PCH3Br (114 mg, 0.32 mmol), and the whole mixture was stirred for 1 h at rt. The above solution was added dropwise to a solution of 22 (14.5 mg, 0.042 mmol) in anhydrous THF (0.21 mL) at 0 °C, and the whole mixture was stirred for 3.5 h at rt. Sat. NH4Cl aq. was added to the mixture, and the whole mixture was extracted with AcOEt. Removal of the solvent from the AcOEt extract under reduced pressure gave a crude product, which was purified by SiO2 column (n-hexane/AcOEt = 4:1) to give 23 (13.7 mg, 95%) as a white solid.
+60 (c = 0.89, CHCl3). 1H-NMR (500 MHz, CDCl3) δ: 7.78 (1H, dd, J = 8.3, 2.3 Hz), 7.73 (1H, d, J = 2.3 Hz), 6.78 (1H, d, J = 8.3 Hz), 5.28 (1H, br s), 4.47 (1H, s), 4.46 (1H, s), 3.87 (3H, s), 2.68 (1H, d, J = 13.5 Hz), 2.44 (1H, d, J = 13.5 Hz), 2.36 (1H, td, J = 13.7, 5.2 Hz), 2.15–2.11 (1H, m), 1.97–1.92 (2H, m), 1.62–1.46 (4H, m), 1.44–1.23 (4H, m), 1.11 (3H, s), 1.09 (1H, d, J = 12.0 Hz), 0.95 (3H, s). 13C-NMR (125 MHz, CDCl3) δ: 167.0, 160.2, 158.4, 134.9, 129.3, 124.9, 122.1, 115.2, 102.5, 51.9, 42.5, 40.2, 38.7, 38.1, 37.0, 33.0, 29.7, 28.4, 22.3, 20.8, 20.3, 18.4. IR (KBr): 2928, 1688, 1603, 1429, 1283 cm−1. MS (ESI-TOF) m/z: 365 [M + Na]+. HRMS (ESI-TOF) m/z: 365.2093 calcd for C22H30O3Na; Found: 365.2089.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}