
Sulfated Galactofucan from the Brown Alga Saccharina latissima—Variability of Yield, Structural Composition and Bioactivity


Abstract
:
1. Introduction
2. Results
2.1. Extraction of Crude Sulfated Polysaccharides of Saccharina latissima (S.l.-SP) from Four S.l. Batches
2.1.1. Composition of Crude S.l.-SP

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.l.-SP Batches | Glycans b (%) | Sulfate b (%) | Protein b (%) | DS c | IC50 (Elastase) d (μg/mL) |
|---|---|---|---|---|---|
| B05-SP | 72.7 ± 2.2 | 14.4 ± 2.8 | 12.8 ± 1.0 | 0.28 ± 0.06 | 0.89 ± 0.13 |
| B06-SP | 74.2 ± 3.3 | 15.5 ± 4.0 | 10.3 ± 1.5 | 0.31 ± 0.09 | 0.66 ± 0.12 |
| A05-SP | 66.4 ± 1.0 | 19.0 ± 2.1 | 14.6 ± 2.7 | 0.42 ± 0.04 | 0.51 ± 0.05 |
| A09-SP | 67.6 ± 2.5 | 22.6 ± 2.6 | 9.8 ± 1.9 | 0.49 ± 0.07 | 0.41 ± 0.06 |
| B05 v. B06 | n.s. | n.s. | p = 0.017 | n.s. | p = 0.020 |
| B05 v. A05 | p < 0.001 | p = 0.012 | n.s. | p = 0.003 | p = 0.010 |
| B05 v. A09 | p = 0.008 | p < 0.001 | p = 0.019 | p = 0.002 | p < 0.001 |
| B06 v. A05 | p < 0.001 | n.s. | p = 0.005 | p = 0.021 | p = 0.041 |
| B06 v. A09 | p = 0.002 | p = 0.003 | n.s. | p = 0.002 | p < 0.001 |
| A05 v. A09 | n.s. | p = 0.016. | p = 0.003 | p = 0.040 | p = 0.013 |
2.1.2. Monosaccharide Composition of Crude S.l.-SP
| Crude S.l.-SP Batches | l-fucose | d-galactose | d-xylose | d-mannose | d-rhamnose | d-glucose |
|---|---|---|---|---|---|---|
| B05-SP | 45.2 ± 14.4 | 8.3 ± 2.5 | 5.4 ± 1.5 | 8.3 ± 2.5 | 1.1 ± 0.2 | 32.7 ± 17.4 |
| B06-SP | 42.9 ± 11.3 | 6.9 ± 4.3 | 4.6 ± 1.1 | 6.8 ± 0.4 | 1.6 ± 0.2 | 37.5 ± 11.5 |
| A05-SP | 62.8 ± 8.2 | 14.9 ± 9.9 | 8.0 ± 0.9 | 8.9 ± 2.0 | 1.8 ± 0.5 | 4.7 ± 1.7 |
| A09-SP | 63.9 ± 6.1 | 10.6 ± 3.9 | 7.1 ± 0.7 | 7.0 ± 1.6 | 1.3 ± 0.5 | 10.1 ± 4.9 |
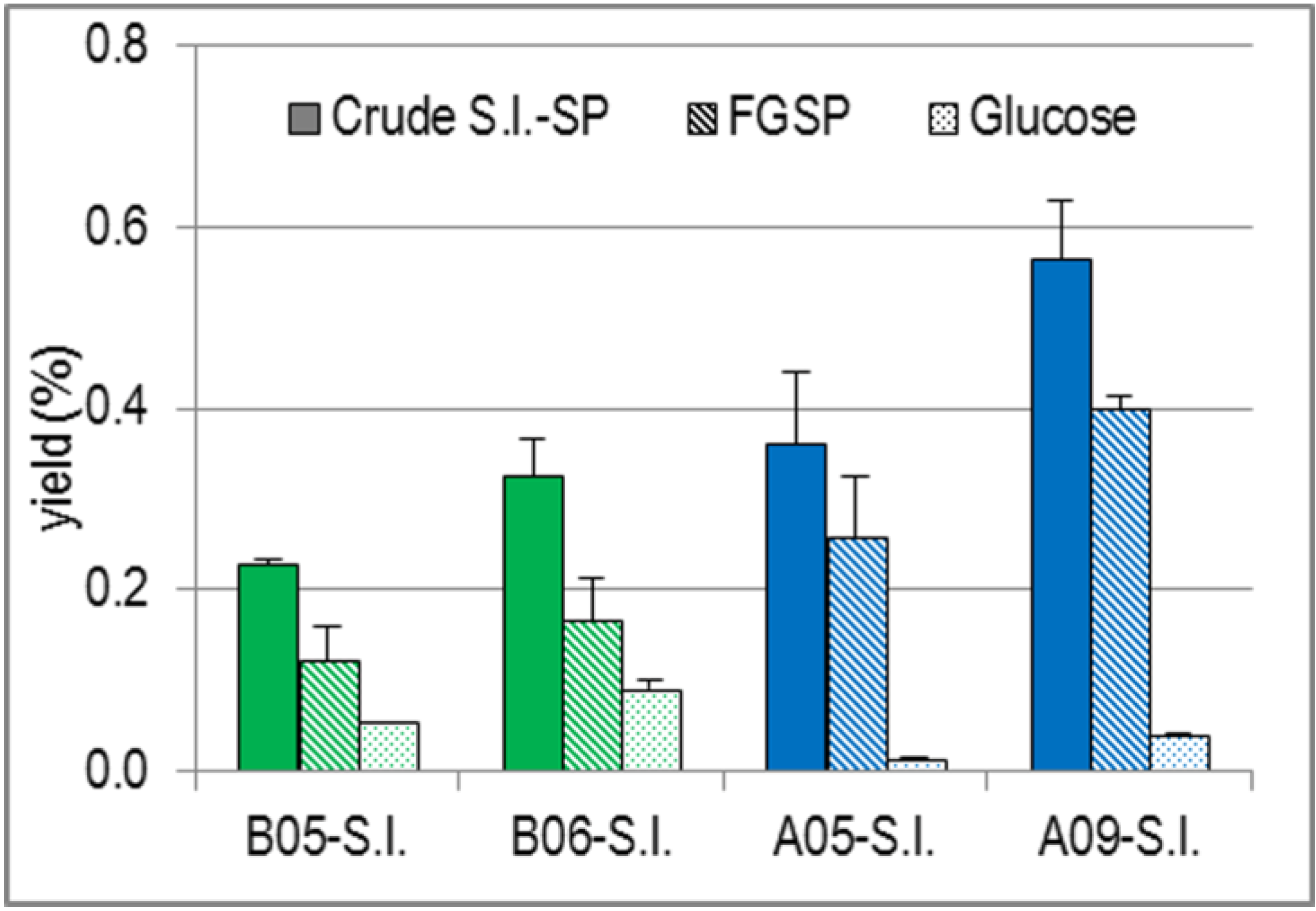
2.1.3. Yields and Degree of Sulfation (DS) of Crude S.l.-SP Compared with Those of the Sulfated Fucose and Galactose Residues of S.l.-SP (FGSP) and Glucose Proportions
| S.l.-SP Batches | Yield (%) a | DS a | |||
|---|---|---|---|---|---|
| Crude S.l.-SP | FGSP b | Glucose c | Crude S.l.-SP d | FGSP e | |
| B05-SP | 1.75 ± 0.01 | 0.94 ± 0.29 | 0.42 ± 0.00 | 0.28 ± 0.06 | 0.55 ± 0.11 |
| B06-SP | 2.27 ± 0.29 | 1.16 ± 0.31 | 0.63 ± 0.08 | 0.31 ± 0.09 | 0.60 ± 0.05 |
| A05-SP | 4.24 ± 0.95 | 3.05 ± 0.79 | 0.13 ± 0.04 | 0.42 ± 0.04 | 0.53 ± 0.04 |
| A09-SP | 3.75 ± 0.44 | 2.66 ± 0.10 | 0.26 ± 0.03 | 0.49 ± 0.07 | 0.67 ± 0.10 |
| B05 v. B06 | n.s. | n.s. | p = 0.037 | n.s. | n.s. |
| B05 v. A05 | p = 0.025 | p = 0.025 | p = 0.002 | p = 0.003 | n.s. |
| B05 v. A09 | p = 0.004 | p < 0.001 | p = 0.002 | p = 0.002 | n.s. |
| B06 v. A05 | p = 0.019 | p = 0.012 | p < 0.001 | p = 0.021 | n.s. |
| B06 v. A09 | p = 0.004 | p < 0.001 | p < 0.001 | p = 0.002 | n.s. |
| A05 v. A09 | n.s. | n.s. | p = 0.004 | p = 0.040 | p = 0.011 |
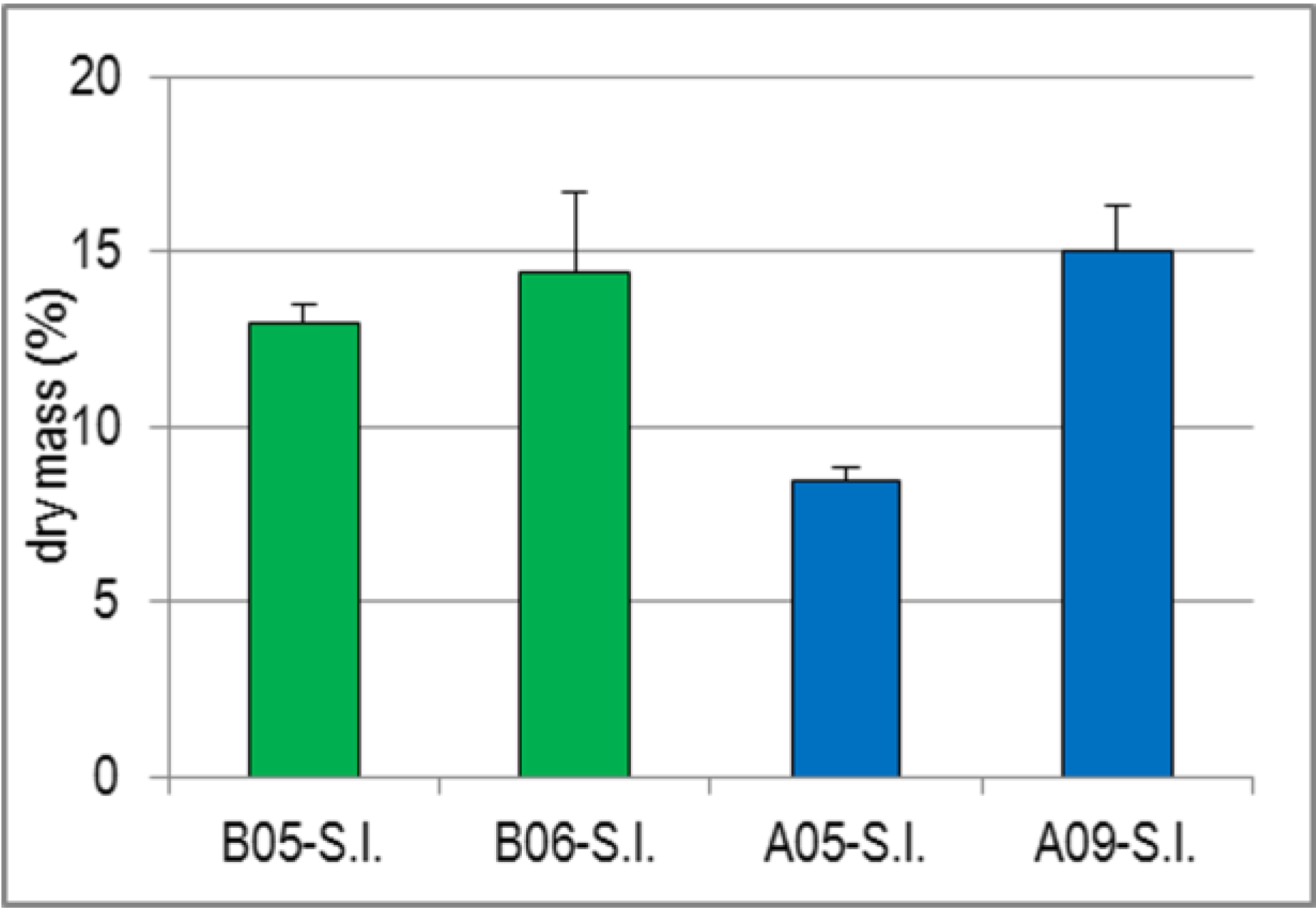
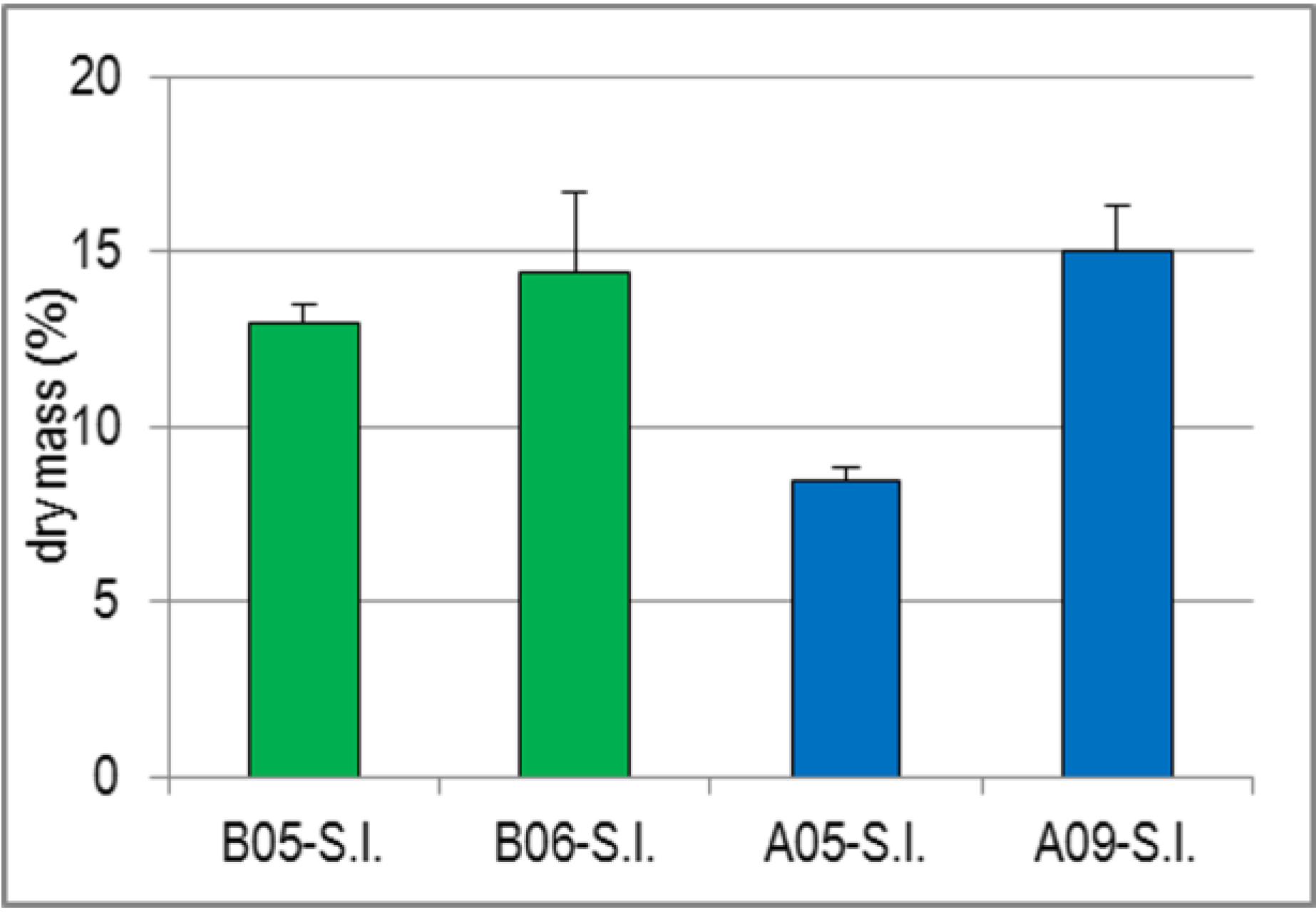
2.1.4. Variability of Dry Mass of S.l. and Its Impact on the Yields
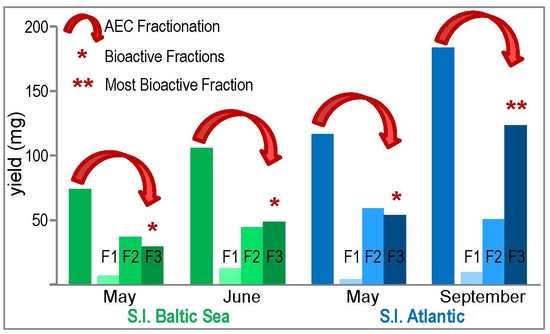
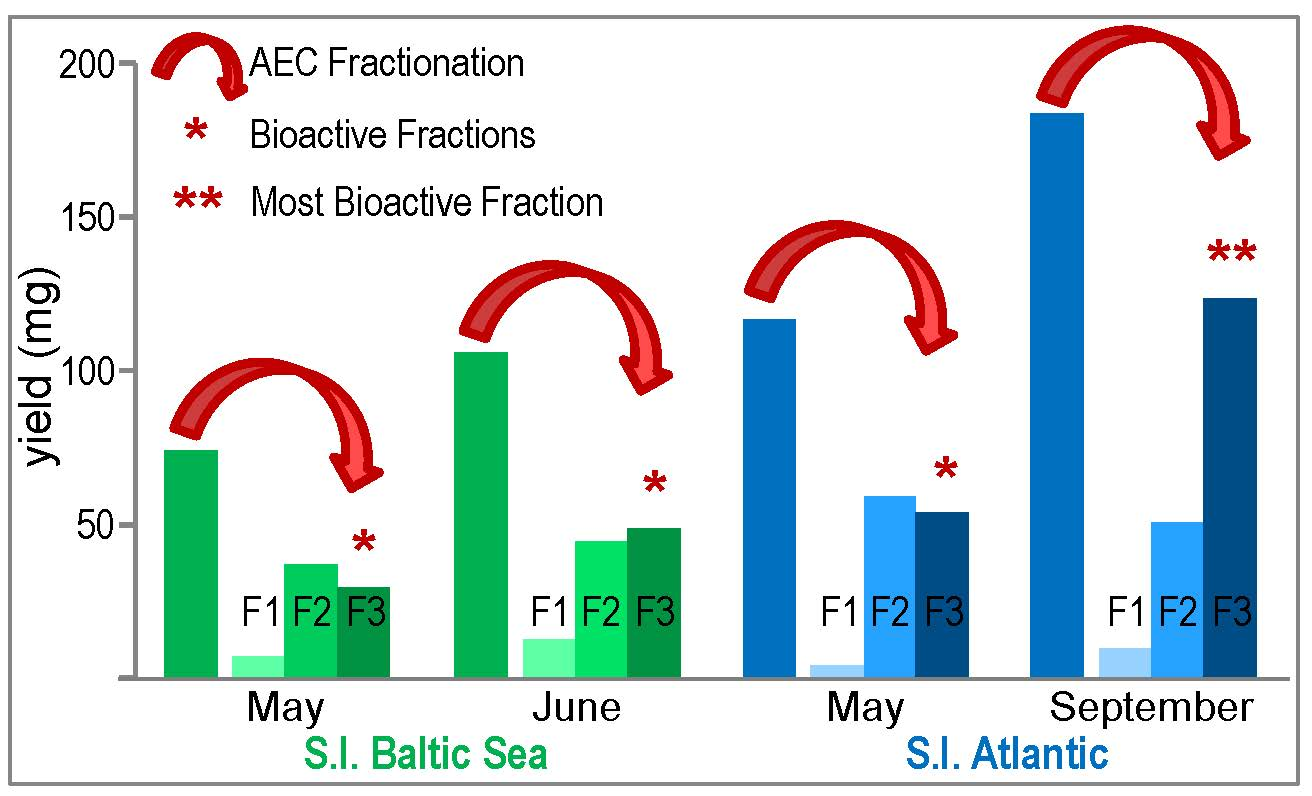
2.2. Fractionation of Crude S.l.-SP
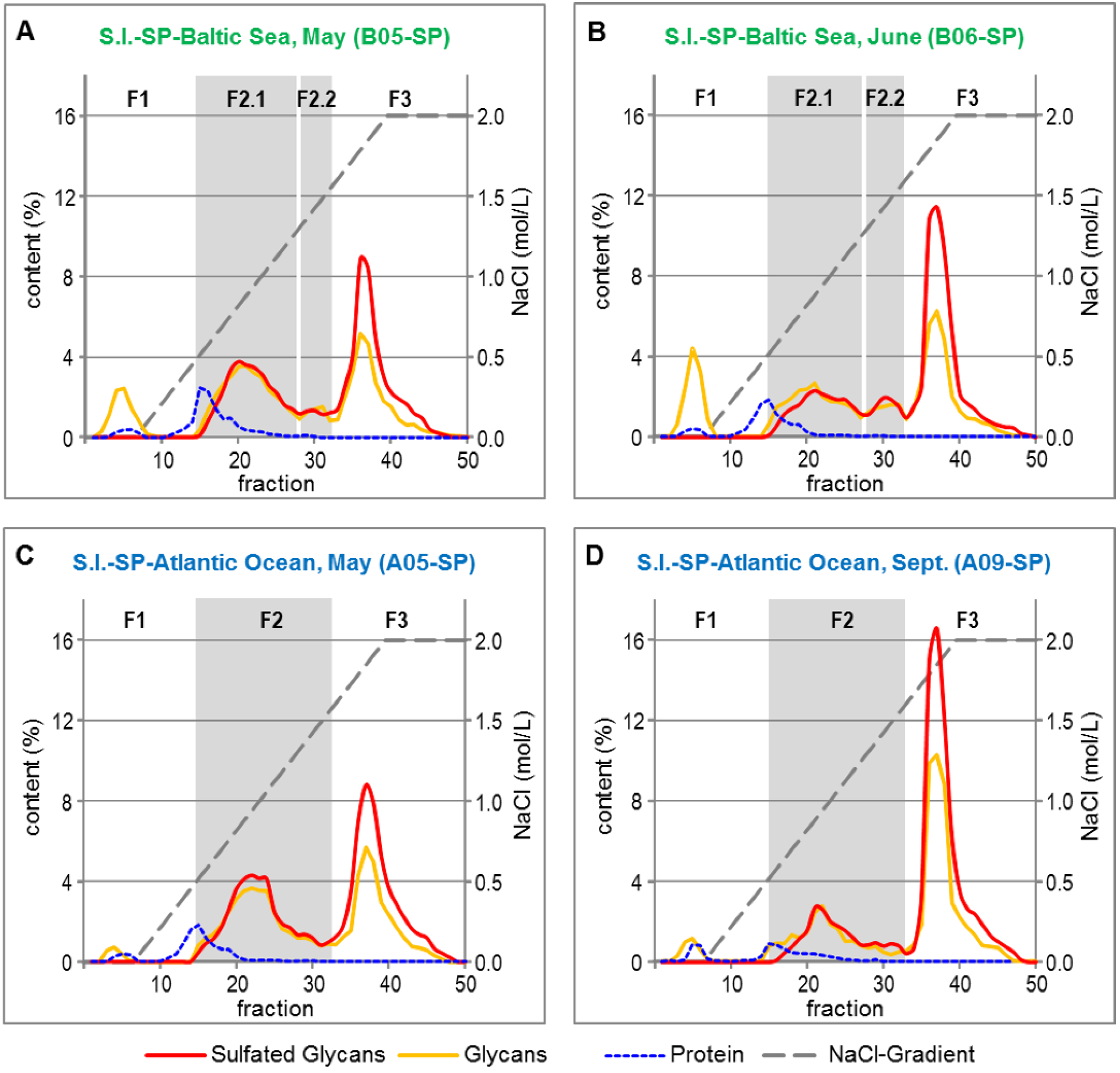
| S.l.-SP | Yield (%) 1 | Glycans (%) 2,3 | Sulfate (%) 2,4 | Protein (%) 2,5 | Sulfated Glycans (%) 6 | Monosaccharide Composition (mol %) 7 | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Fuc | Gal | Xyl | Man | Glc | |||||||
| B05-SP | |||||||||||
| F11–14 | 9.8 | 8.5 | - | 1.3 | - | - | - | - | 2.1 | 97.9 | |
| F2.115–27 | 50.3 | 30.1 | 5.7 | 12 | 30.0 | 54.7 | 10.4 | 13.7 | 16.2 | 3.3 | |
| F2.128–32 | 5.9 | - | 6.2 | ||||||||
| F333–50 | 39.9 | 25.5 | 13.8 | - | 42.0 | 67.0 | 26.5 | 2.8 | 2.4 | 1.3 | |
| 100% 8 | |||||||||||
| B06-SP | |||||||||||
| F11–14 | 12.1 | 11.5 | - | 1.1 | - | - | - | - | 2.7 | 97.3 | |
| F2.115–26 | 41.7 | 21.4 | 5.7 | 8.9 | 18.7 | 54.3 | 13.7 | 13.5 | 12.7 | 3.4 | |
| F2.227–32 | 8.2 | - | 9.2 | ||||||||
| F333–50 | 46.2 | 28.4 | 16.1 | - | 50.7 | 75.6 | 19.6 | 3.4 | 1.4 | - | |
| 100% 8 | |||||||||||
| A05-SP | |||||||||||
| F11–14 | 3.4 | 1.9 | - | 1.0 | - | ||||||
| F215–32 | 50.5 | 36.1 | 5.5 | 8.9 | 38,8 | 46.9 | 11.2 | 15.3 | 16.0 | 8.4 | |
| F333–50 | 46.1 | 29.5 | 15.7 | - | 49.5 | 63.4 | 31.8 | 2.6 | 1.4 | 0.9 | |
| 100% 8 | |||||||||||
| A09-SP | |||||||||||
| F11–14 | 5.1 | 3.2 | - | 1.9 | - | ||||||
| F215–32 | 27.6 | 20.6 | 2.8 | 5.5 | 21.3 | 39.7 | 22.5 | 18.4 | 12.2 | 4.7 | |
| F333–50 | 67.3 | 42.9 | 22.8 | - | 68.3 | 86.1 | 11.1 | 2.8 | - | - | |
| 100% 8 | |||||||||||
2.2.1. Common Structural Characteristics of the S.l.-SP Fractions
| Degree of Sulfation (DS) | Fucose/Galactose Ratio | IC50 (Elastase) (μg/mL) a | DC (APTT) (μg/mL) b | ||||
|---|---|---|---|---|---|---|---|
| Fraction | F2 | F3 | F2 | F3 | F2 * | F3 * | F3 * |
| B05-SP | 0.23 | 0.79 | 5.3 | 2.5 | 2.41 ± 0.12 | n.d. | n.d. |
| B06-SP | 0.28 | 0.82 | 4.0 | 3.9 | 1.87 ± 0.12 | 0.26 ± 0.02 | 7.44 ± 0.18 |
| A05-SP | 0.22 | 0.76 | 4.2 | 2.0 | 2.81 ± 0.21 | 0.28 ± 0.01 | 8.75 ± 0.25 |
| A09-SP | 0.19 | 0.81 | 1.8 | 7.8 | 3.77 ± 0.16 | 0.21 ± 0.01 | 4.89 ± 0.50 |
2.2.2. Composition of the SP Fractions Related to the Harvest Time of S.l.
2.2.3. Composition of the SP Fractions Related to the Two S.l. Habitats Baltic Sea and Atlantic Ocean
2.2.4. Elastase Inhibitory and Anticoagulant Activities of the S.l.-SP Fractions
2.3. Structural Characterization of Fraction F3 from September
| mol % | Glycosidic Linkage | Position of Sulfate | Sulfate (mol %) |
|---|---|---|---|
| 83.1 | Fucose | ||
| 9.3 | Fucp-(1→ | 2 | 9.3 |
| 14.5 | →2)-Fucp-(1→ | - | - |
| 35.3 | →3)-Fucp-(1→ | 4 | 35.3 |
| 15.5 | →3)-Fucp-(1→ | 2 | 15.5 |
| 8.5 | →2,3)-Fucp-(1→ | 4 | 8.5 |
| 16.6 | Galactose | ||
| 1.5 | Galp-(1→ | 3 + 6 | 3.0 |
| 5.0 | →3)-Galp-(1→ | - | - |
| 2.5 | →4)-Galp-(1→ | 3 | 2.5 |
| 5.3 | →6)-Galp-(1→ | 3 | 5.3 |
| 2.3 | →4,6)-Galp-(1→ | 3 | 2.3 |
| 81.7 |
3. Discussion
3.1. Increase of Laminarin as well as FGSP and Sulfated Galactofucan in S.l. from Spring to Autumn
3.2. Inverse Yields of FGSP/Sulfated Galactofucan and Laminarin from Baltic Sea and Atlantic Ocean S.l. Batches
3.3. High DS and High Fucose Content as Determinants of Elastase Inhibitory Activity
3.4. Sulfated Galactofucan as the Most Active Fraction of S.l.-SP
4. Experimental
4.1. Algae Material
4.2. Extraction and Isolation
4.3. Fractionation
4.4. Analytical Testing of AEC Fractions
4.4.1. Sulfated Glycans
4.4.2. Total Carbohydrates
4.4.3. Total Proteins
4.5. Monosaccharide Composition by Acetylation Analysis
4.6. Monosaccharide Linkages by Methylation Analysis
4.7. Chemical Modifications
4.7.1. Desulfation
4.7.2. Reduction of Uronic Acids
4.8. Molecular Mass (Mr)
4.9. Elemental Analysis
4.10. Uronic Acids
4.11. Pharmacological Methods
4.11.1. Elastase Inhibition Assay
4.11.2. Activated Partial Thromboplastin-Time (APTT)
4.12. Statistical Analysis
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Abbreviations
| AEC | anion exchange chromatography |
| A-SP | sulfated polysaccharides from S.l. harvested from the Atlantic Ocean in May (A05-SP) or September (A09-SP) |
| B-SP | sulfated polysaccharides from S.l. harvested from the Baltic Sea in May (B05-SP) or June (B06-SP) |
| D.s. | Delesseria sanguinea |
| D.s.-SP | sulfated polysaccharides extracted from red alga Delesseria sanguinea |
| DS | degree of sulfation |
| FGSP | amount of sulfated fucose and galactose residues of S.l.-SP |
| S.l. | Saccharina latissima |
| S.l.-SP | sulfated polysaccharides extracted from Saccharina latissima |
| SGF | sulfated galactofucan |
| SP | sulfated polysaccharides |
Conflicts of Interest
References
- Sarrazin, S.; Lamanna, W.C.; Esko, J.D. Heparan sulfate proteoglycans. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Tzanakakis, G.N.; Karamanos, N.K. Proteoglycans in health and disease: Novel roles for proteoglycans in malignancy and their pharmacological targeting. FEBS J. 2010, 277, 3904–3923. [Google Scholar] [CrossRef]
- Jiao, G.; Yu, G.; Zhang, J.; Ewart, H.S. Chemical structures and bioactivities of sulfated polysaccharides from marine algae. Mar. Drugs 2011, 9, 196–223. [Google Scholar] [CrossRef]
- Pomin, V.H. Fucanomics and galactanomics: Marine distribution, medicinal impact, conceptions, and challenges. Mar. Drugs 2012, 10, 793–811. [Google Scholar] [CrossRef]
- Aquino, R.S.; Grativol, C.; Mourão, Paulo, A.S. Rising from the sea: Correlations between sulfated polysaccharides and salinity in plants. PLoS One 2011, 6, e18862. [Google Scholar] [CrossRef]
- Algal Polysaccharides, Novel Applications and Outlook; InTech: Rijeka, Croatia, 2012.
- Fitton, J.H. Therapies from fucoidan; multifunctional marine polymers. Mar. Drugs 2011, 9, 1731–1760. [Google Scholar] [CrossRef]
- Vo, T.-S.; Kim, S.-K. Fucoidans as a natural bioactive ingredient for functional foods. J. Funct. Foods 2013, 5, 16–27. [Google Scholar] [CrossRef]
- Wijesinghe, W.A.J.P.; Jeon, Y.-J. Biological activities and potential industrial applications of fucose rich sulfated polysaccharides and fucoidans isolated from brown seaweeds: A review. Carbohydr. Polym. 2012, 88, 13–20. [Google Scholar] [CrossRef]
- Ale, M.T.; Mikkelsen, J.D.; Meyer, A.S. Important determinants for fucoidan bioactivity: A critical review of structure-function relations and extraction methods for fucose-containing sulfated polysaccharides from brown seaweeds. Mar. Drugs 2011, 9, 2106–2130. [Google Scholar] [CrossRef] [PubMed]
- Lühn, S.; Grimm, J.C.; Alban, S. Simple and rapid quality control of sulfated glycans by a fluorescence sensor assay—Exemplarily developed for the sulfated polysaccharides from red algae Delesseria sanguinea. Mar. Drugs 2014, 12, 2205–2227. [Google Scholar] [CrossRef]
- Mohamed, S.; Hashim, S.N.; Rahman, H.A. Seaweeds: A sustainable functional food for complementary and alternative therapy. Trends Food Sci. Technol. 2012, 23, 83–96. [Google Scholar] [CrossRef]
- Grünewald, N.; Alban, S. Optimized and standardized isolation and structural characterization of anti-inflammatory sulfated polysaccharides from the red alga Delesseria sanguinea (Hudson) Lamouroux (Ceramiales, Delesseriaceae). Biomacromolecules 2009, 10, 2998–3008. [Google Scholar] [CrossRef]
- Grünewald, N.; Groth, I.; Alban, S. Evaluation of seasonal variations of the structure and anti-inflammatory activity of sulfated polysaccharides extracted from the red alga Delesseria sanguinea (Hudson) Lamouroux (Ceramiales, Delesseriaceae). Biomacromolecules 2009, 10, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Berteau, O.; Mulloy, B. Sulfated fucans, fresh perspectives: Structures, functions, and biological properties of sulfated fucans and an overview of enzymes active toward this class of polysaccharide. Glycobiology 2003, 13, 29R–40R. [Google Scholar] [CrossRef]
- Pomin, V.H.; Mourao, Paulo, A.S. Structure, biology, evolution, and medical importance of sulfated fucans and galactans. Glycobiology 2008, 18, 1016–1027. [Google Scholar] [CrossRef]
- Deniaud-Bouet, E.; Kervarec, N.; Michel, G.; Tonon, T.; Kloareg, B.; Herve, C. Chemical and enzymatic fractionation of cell walls from Fucales: Insights into the structure of the extracellular matrix of brown algae. Ann. Bot. 2014. [Google Scholar] [CrossRef]
- Bilan, M.I.; Grachev, A.A.; Shashkov, A.S.; Kelly, M.; Sanderson, C.J.; Nifantiev, N.E.; Usov, A.I. Further studies on the composition and structure of a fucoidan preparation from the brown alga Saccharina latissima. Carbohydr. Res. 2010, 345, 2038–2047. [Google Scholar] [CrossRef]
- Usov, A.I.; Smirnova, G.P.; Bilan, M.I.; Shashkov, A.S. Polysaccharides of Algae. 53. Brown Alga Laminaria saccharina(L.) Lam. as a Source of Fucoidan. Bioorg. Khim. 1998, 24, 437–445. [Google Scholar]
- Croci, D.O.; Cumashi, A.; Ushakova, N.A.; Preobrazhenskaya, M.E.; Piccoli, A.; Totani, L.; Ustyuzhanina, N.E.; Bilan, M.I.; Usov, A.I.; Grachev, A.A.; et al. Fucans, but not fucomannoglucuronans, determine the biological activities of sulfated polysaccharides from Laminaria saccharina brown seaweed. PLoS One 2011, 6, e17283. [Google Scholar] [CrossRef] [PubMed]
- Ustyuzhanina, N.E.; Ushakova, N.A.; Zyuzina, K.A.; Bilan, M.I.; Elizarova, A.L.; Somonova, O.V.; Madzhuga, A.V.; Krylov, V.B.; Preobrazhenskaya, M.E.; Usov, A.I.; et al. Influence of fucoidans on hemostatic system. Mar. Drugs 2013, 11, 2444–2458. [Google Scholar] [CrossRef]
- Cumashi, A.; Ushakova, N.A.; Preobrazhenskaya, M.E.; D'Incecco, A.; Piccoli, A.; Totani, L.; Tinari, N.; Morozevich, G.E.; Berman, A.E.; Bilan, M.I.; et al. A comparative study of the anti-inflammatory, anticoagulant, antiangiogenic, and antiadhesive activities of nine different fucoidans from brown seaweeds. Glycobiology 2007, 17, 541–552. [Google Scholar] [CrossRef]
- Groth, I.; Alban, S. Elastase inhibition assay with peptide substrates—An example for the limited comparability of in vitro results. Planta Med. 2008, 74, 852–858. [Google Scholar] [CrossRef]
- Groth, I.; Grünewald, N.; Alban, S. Pharmacological profiles of animal- and nonanimal-derived sulfated polysaccharides—Comparison of unfractionated heparin, the semisynthetic glucan sulfate PS3, and the sulfated polysaccharide fraction isolated from Delesseria sanguinea. Glycobiology 2009, 19, 408–417. [Google Scholar] [CrossRef]
- McComb, E.A.; McCready, R.M. Determination of Acetyl in Pectin and in Acetylated Carbohydrate Polymers. Anal. Chem. 1957, 29, 819–821. [Google Scholar] [CrossRef]
- Sloneker, J.H.; Orentas, D.G. Pyruvic Acid, a Unique Component of an Exocellular Bacterial Polysaccharide. Nature 1962, 194, 478–479. [Google Scholar] [CrossRef]
- Black, W. The seasonal variation in weight and chemical composition of the common British Laminariaceae. J. Mar. Biol. Assoc. UK 1950, 29, 45–72. [Google Scholar] [CrossRef]
- Michel, G.; Tonon, T.; Scornet, D.; Cock, J.M.; Kloareg, B. The cell wall polysaccharide metabolism of the brown alga Ectocarpus siliculosus. Insights into the evolution of extracellular matrix polysaccharides in Eukaryotes. New Phytol. 2010, 188, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.H.I.; Castaneda, H.G.T. Preparation and chromatographic analysis of phlorotannins. J. Chromatogr. Sci. 2013, 51, 825–838. [Google Scholar] [CrossRef]
- Becker, M.; Franz, G.; Alban, S. Inhibition of PMN-elastase activity by semisynthetic glucan sulfates. Thromb. Haemost. 2003, 89, 915–925. [Google Scholar] [PubMed]
- Usov, A.I. Polysaccharides of the red algae. Adv. Carbohydr. Chem. Biochem. 2011, 65, 115–217. [Google Scholar]
- Black, W. The seasonal variation in the combined l-fucose content of the common British Laminariaceae and Fucaceae. J. Sci. Food Agric. 1954, 5, 445–448. [Google Scholar] [CrossRef]
- Adams, J.M.M.; Ross, A.B.; Anastasakis, K.; Hodgson, E.M.; Gallagher, J.A.; Jones, J.M.; Donnison, I.S. Seasonal variation in the chemical composition of the bioenergy feedstock Laminaria digitata for thermochemical conversion. Bioresour. Technol. 2011, 102, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Dittami, S.M.; Gravot, A.; Goulitquer, S.; Rousvoal, S.; Peters, A.F.; Bouchereau, A.; Boyen, C.; Tonon, T. Towards deciphering dynamic changes and evolutionary mechanisms involved in the adaptation to low salinities in Ectocarpus (brown algae). Plant J. 2012, 71, 366–377. [Google Scholar] [PubMed]
- Mabeau, S.; Kloareg, B. Isolation and Analysis of the Cell Walls of Brown Algae: Fucus spiralis, F. ceranoides, F. vesiculosus, F. serratus, Bifurcaria bifurcata and Laminaria digitata. J. Exp. Bot. 1987, 38, 1573–1580. [Google Scholar] [CrossRef]
- Kloareg, B.; Quatrano, R.S. Structure of the cell walls of marine algae and ecophysiological functions of the matrix polysaccharides. Oceanogr. Mar. Biol. 1988, 26, 259–315. [Google Scholar]
- Bartsch, I.; Wiencke, C.; Bischof, K.; Buchholz, C.M.; Buck, B.H.; Eggert, A.; Feuerpfeil, P.; Hanelt, D.; Jacobsen, S.; Karez, R.; et al. The genus Laminaria sensu lato: Recent insights and developments. Eur. J. Phycol. 2008, 43, 1–86. [Google Scholar] [CrossRef]
- Stengel, D.B.; Connan, S.; Popper, Z.A. Algal chemodiversity and bioactivity: Sources of natural variability and implications for commercial application. Biotechnol. Adv. 2011, 29, 483–501. [Google Scholar]
- Olischlager, M.; Iniguez, C.; Gordillo, Francisco Javier Lopez; Wiencke, C. Biochemical composition of temperate and Arctic populations of Saccharina latissima after exposure to increased pCO2 and temperature reveals ecotypic variation. Planta 2014, 240, 1213–1224. [Google Scholar]
- Alban, S.; Franz, G. Partial synthetic glucan sulfates as potential new antithrombotics: A review. Biomacromolecules 2001, 2, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Vilela-Silva, A.C.; Alves, A.P.; Valente, A.P.; Vacquier, V.D.; Mourao, P.A. Structure of the sulfated alpha-l-fucan from the egg jelly coat of the sea urchin Strongylocentrotus franciscanus: Patterns of preferential 2-O- and 4-O-sulfation determine sperm cell recognition. Glycobiology 1999, 9, 927–933. [Google Scholar] [CrossRef]
- Guiry, M.D.; Guiry, G.M. AlgaeBase. World-wide electronic publication, National University of Ireland, Galway. 2014. Available online: http://www.algaebase.org (accessed on 8 July 2014).
- Ale, M.T.; Mikkelsen, J.D.; Meyer, A.S. Designed optimization of a single-step extraction of fucose-containing sulfated polysaccharides from Sargassum sp. J. Appl. Phycol. 2012, 24, 715–723. [Google Scholar] [CrossRef]
- Bilan, M.I.; Grachev, A.A.; Ustuzhanina, N.E.; Shashkov, A.S.; Nifantiev, N.E.; Usov, A.I. Structure of a fucoidan from the brown seaweed Fucus evanescens C.Ag. Carbohydr. Res. 2002, 337, 719–730. [Google Scholar] [CrossRef]
- Farndale, R.W.; Buttle, D.J.; Barrett, A.J. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim. Biophys. Acta (BBA) Gen. Subj. 1986, 883, 173–177. [Google Scholar] [CrossRef]
- Pons, A.; Roca, P.; Aguilo, C.; Garcia, F.J.; Alemany, M.; Palou, A. A method for the simultaneous determination of total carbohydrate and glycerol in biological samples with the anthrone reagent. J. Biochem. Biophys. Methods. 1981, 4, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Ogden, G.; Foldi, P. Amino acid analysis: An overview of current methods. LCGC North Am. 1987, 5, 28–40. [Google Scholar]
- Roth, M. Fluorescence reaction for amino acids. Anal. Chem. 1971, 43, 880–882. [Google Scholar] [CrossRef]
- Albersheim, P.; Nevins, D.J.; English, P.D.; Karr, A. A method for the analysis of sugars in plant cell-wall polysaccharides by gas-liquid chromatography. Carbohydr. Res. 1967, 5, 340–345. [Google Scholar] [CrossRef]
- Blakeney, A.B.; Harris, P.J.; Henry, R.J.; Stone, B.A. A simple and rapid preparation of alditol acetates for monosaccharide analysis. Carbohydr. Res. 1983, 113, 291–299. [Google Scholar] [CrossRef]
- Harris, P.J.; Henry, R.J.; Blakeney, A.B.; Stone, B.A. An improved procedure for the methylation analysis of oligosaccharides and polysaccharides. Carbohydr. Res. 1984, 127, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Inoue, Y.; Kamata, T. Solvolytic desulfation of glycosaminoglycuronan sulfates with dimethyl sulfoxide containing water or methanol. Carbohydr. Res. 1977, 58, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.L.; Conrad, H.E. Stoichiometric depolymerization of polyuronides and glycosaminoglycuronans to monosaccharides following reduction of their carbodiimide-activated carboxyl groups. Biochemistry 1972, 11, 1383–1388. [Google Scholar] [CrossRef] [PubMed]
- Blumenkrantz, N. New method for quantitative determination of uronic acids. Anal. Biochem. 1973, 54, 484–489. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ehrig, K.; Alban, S. Sulfated Galactofucan from the Brown Alga Saccharina latissima—Variability of Yield, Structural Composition and Bioactivity. Mar. Drugs 2015, 13, 76-101. https://doi.org/10.3390/md13010076
Ehrig K, Alban S. Sulfated Galactofucan from the Brown Alga Saccharina latissima—Variability of Yield, Structural Composition and Bioactivity. Marine Drugs. 2015; 13(1):76-101. https://doi.org/10.3390/md13010076
Chicago/Turabian StyleEhrig, Karina, and Susanne Alban. 2015. "Sulfated Galactofucan from the Brown Alga Saccharina latissima—Variability of Yield, Structural Composition and Bioactivity" Marine Drugs 13, no. 1: 76-101. https://doi.org/10.3390/md13010076