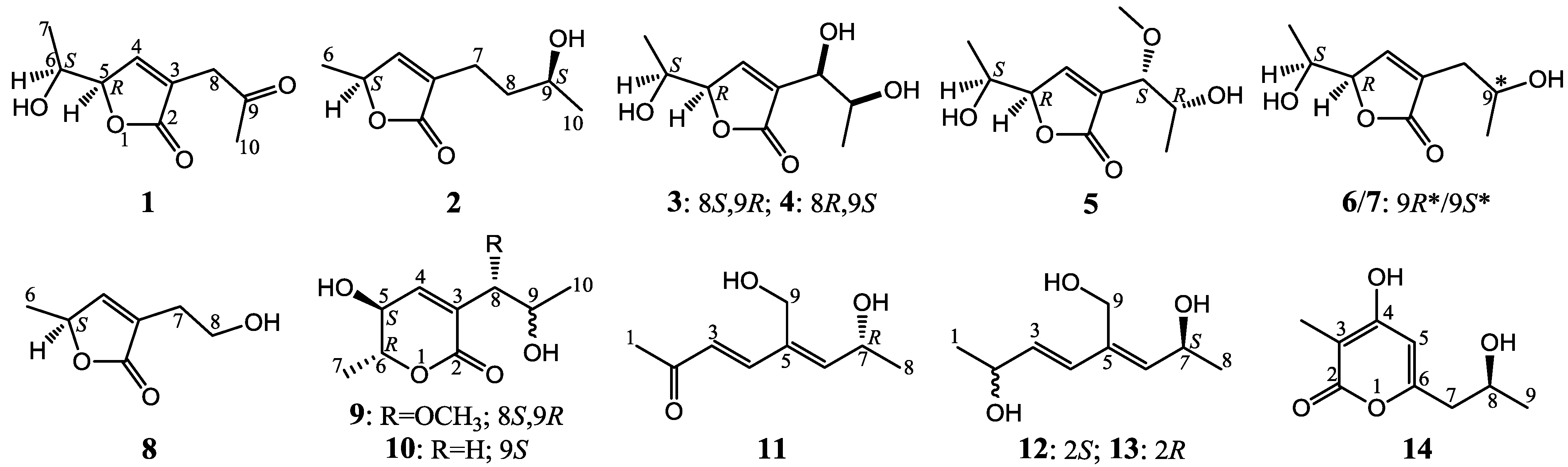
Nine New and Five Known Polyketides Derived from a Deep Sea-Sourced Aspergillus sp. 16-02-1

 and
and
Abstract
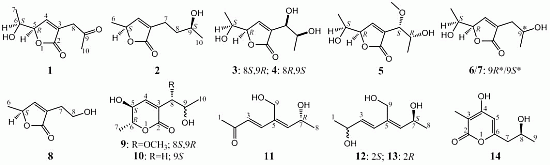
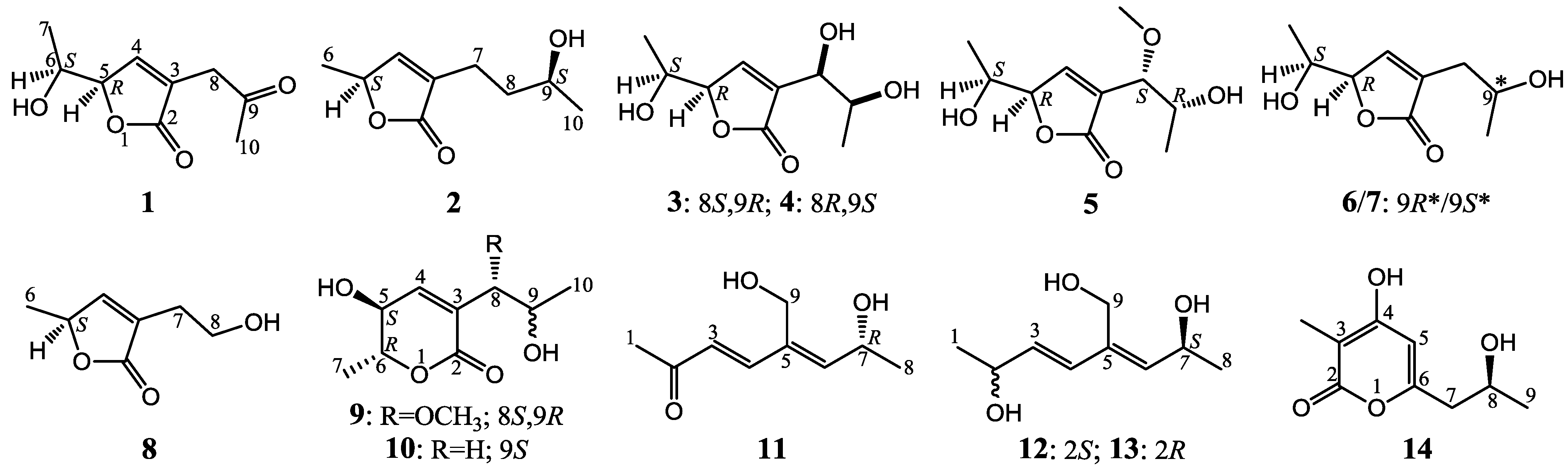
:
1. Introduction
2. Results and Discussion
2.1. Fermentation, Isolation, and Identification of Known Compounds
2.2. Structure Determination of New Compounds
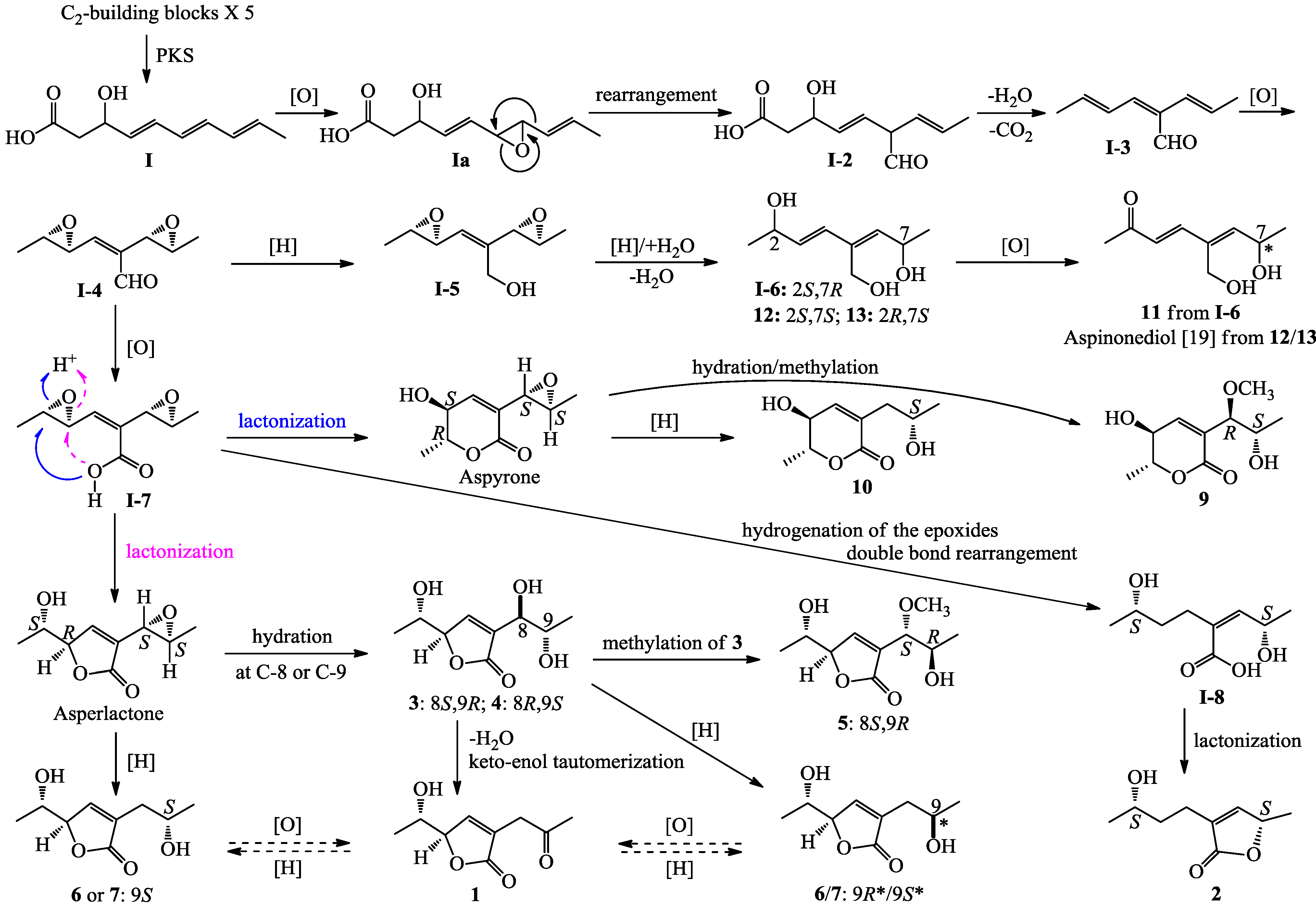
 +10.5 (c 0.12, MeOH), was assigned the molecular formula C9H12O4 by HRESIMS (measured 185.0811 [M + H]+, calculated for C9H13O4 [M + H]+ 185.0814). It showed end UV absorption, and the IR spectrum showed the absorptions due to OH (3417 cm−1), CH3/CH2 (2980, 2938, 2906, 1422 and 1360 cm−1), α,β-unsaturated γ-lactone (1752 and 1656 cm−1) [21,22] and keto carbonyl (1722 cm−1) groups. The olefinic proton and carbon signals at the lower field (δH 7.43 and δC 149.4) [22] of 1H and 13C NMR spectra (Table 1 and Table 2) and the ester carbonyl carbon signal at δC 173.5 in the 13C NMR spectrum (Table 2) supported the presence of the α,β-unsaturated γ-lactone moiety. The carbonyl carbon signal at δC 203.6 in the 13C NMR spectrum further supported the presence of one keto carbonyl in 1. Interpretation of the 1H–1H COSY, HMQC and HMBC data (Table S1 in the Supplementary Information) established the planar structure. The structural part related to the 1H spin system, C-7–C-4(via quaternary sp2 C-3)–C-8, was deduced from the 1H–1H COSY and HMQC data, including the allylic coupling between H-4/H2-8, which was confirmed by the HMBC correlations of H-4, H-5 and H2-8 with C-3. The acetyl group that consisted of the C-9 keto carbonyl and the C-10 methyl was linked to C-8 by the HMBCs on H2-8/C-9, H3-10/C-8 and H3-10/C-9. The ester carbonyl carbon (C-2, δC 173.5) was linked to C-3 by the HMBC correlations of H-4 and H2-8 with C-2. Then, C-2 was further linked to C-5 by an ester linkage to form the α,β-unsaturated γ-lactone ring according to the IR absorption at 1752 cm−1.
+10.5 (c 0.12, MeOH), was assigned the molecular formula C9H12O4 by HRESIMS (measured 185.0811 [M + H]+, calculated for C9H13O4 [M + H]+ 185.0814). It showed end UV absorption, and the IR spectrum showed the absorptions due to OH (3417 cm−1), CH3/CH2 (2980, 2938, 2906, 1422 and 1360 cm−1), α,β-unsaturated γ-lactone (1752 and 1656 cm−1) [21,22] and keto carbonyl (1722 cm−1) groups. The olefinic proton and carbon signals at the lower field (δH 7.43 and δC 149.4) [22] of 1H and 13C NMR spectra (Table 1 and Table 2) and the ester carbonyl carbon signal at δC 173.5 in the 13C NMR spectrum (Table 2) supported the presence of the α,β-unsaturated γ-lactone moiety. The carbonyl carbon signal at δC 203.6 in the 13C NMR spectrum further supported the presence of one keto carbonyl in 1. Interpretation of the 1H–1H COSY, HMQC and HMBC data (Table S1 in the Supplementary Information) established the planar structure. The structural part related to the 1H spin system, C-7–C-4(via quaternary sp2 C-3)–C-8, was deduced from the 1H–1H COSY and HMQC data, including the allylic coupling between H-4/H2-8, which was confirmed by the HMBC correlations of H-4, H-5 and H2-8 with C-3. The acetyl group that consisted of the C-9 keto carbonyl and the C-10 methyl was linked to C-8 by the HMBCs on H2-8/C-9, H3-10/C-8 and H3-10/C-9. The ester carbonyl carbon (C-2, δC 173.5) was linked to C-3 by the HMBC correlations of H-4 and H2-8 with C-2. Then, C-2 was further linked to C-5 by an ester linkage to form the α,β-unsaturated γ-lactone ring according to the IR absorption at 1752 cm−1. +24.6 (c 0.23, MeOH), was assigned the molecular composition C9H14O3 by HRESIMS (measured 171.1015 [M + H]+, calculated for C9H15O3 [M + H]+ 171.1021). The IR spectrum showed absorptions ascribable to the OH, CH3/CH2 and α,β-unsaturated γ-lactone groups (Experimental Section). Similar to 1, the olefinic proton and carbon signals at the lower field (δH 7.04, H-4 and δC 149.8, C-4) [22] of the 1H and 13C NMR spectra (Table 1 and Table 2) together with the ester carbonyl (δC 174.3 C-2) and the sp2 carbon (δC 134.0, C-3) signals in the 13C NMR spectrum (Table 2) supported the presence of the α,β-unsaturated γ-lactone moiety in 2. Then, the planar structure was established by interpretation of the 1H–1H COSY, HMQC and HMBC data (Table S2 in the Supplementary Information). The carbon backbone chain related to the 1H spin system, C-6–C-4(via quaternary sp2 C-3)–C-7–C-10, could be derived from the 1H–1H COSY and HMQC data (Table S2 in the Supplementary Information), including the allylic coupling between H-4/H2-7, which was confirmed by the HMBC correlations of H-4, H-5, H2-7 and H2-8 with C-3. The carbonyl carbon C-2 (δC 174.3) was linked to C-5 and C-3 by the HMBC correlations between H-5/C-2, H-4/C-2 and H2-7/C-2 to form the α,β-unsaturated γ-lactone ring with the ester linkage between C-5 and C-2.
+24.6 (c 0.23, MeOH), was assigned the molecular composition C9H14O3 by HRESIMS (measured 171.1015 [M + H]+, calculated for C9H15O3 [M + H]+ 171.1021). The IR spectrum showed absorptions ascribable to the OH, CH3/CH2 and α,β-unsaturated γ-lactone groups (Experimental Section). Similar to 1, the olefinic proton and carbon signals at the lower field (δH 7.04, H-4 and δC 149.8, C-4) [22] of the 1H and 13C NMR spectra (Table 1 and Table 2) together with the ester carbonyl (δC 174.3 C-2) and the sp2 carbon (δC 134.0, C-3) signals in the 13C NMR spectrum (Table 2) supported the presence of the α,β-unsaturated γ-lactone moiety in 2. Then, the planar structure was established by interpretation of the 1H–1H COSY, HMQC and HMBC data (Table S2 in the Supplementary Information). The carbon backbone chain related to the 1H spin system, C-6–C-4(via quaternary sp2 C-3)–C-7–C-10, could be derived from the 1H–1H COSY and HMQC data (Table S2 in the Supplementary Information), including the allylic coupling between H-4/H2-7, which was confirmed by the HMBC correlations of H-4, H-5, H2-7 and H2-8 with C-3. The carbonyl carbon C-2 (δC 174.3) was linked to C-5 and C-3 by the HMBC correlations between H-5/C-2, H-4/C-2 and H2-7/C-2 to form the α,β-unsaturated γ-lactone ring with the ester linkage between C-5 and C-2.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proton | 1 | 2 | 3 | 4 | 5 | 6/7 b |
|---|---|---|---|---|---|---|
| 4 | 7.43 q (1.4) | 7.04 q (1.6) | 7.54 dd (3.0, 1.5) | 7.56 dd (2.8, 1.6) | 7.55 dd (1.5, 0.9) | 7.41/7.32, dd (2.8, 1.6) |
| 5 | 4.88 dq (4.7, 1.4) | 5.01 qq (6.9,1.6) | 4.96–4.92 m | 4.92 ddd (5.0, 2.8, 1.6) | 4.98 dt (4.5, 1.5) | 4.87 dd (4.4, 2.8, 1.6) |
| 6 | 4.02 qd (6.5, 4.7) | 1.41 3H d (6.9) | 3.95 qd (6.5, 4.6) | 3.92 qd (6.4, 5.0) | 3.97 qd (6.4, 4.5) | 3.92 qd (6.4, 4.4) |
| 7 | 1.28 3H d (6.5) | 2.28–2.57 2H m | 1.25 3H d (6.5) | 1.26 3H d (6.4) | 1.26 3H d (6.4) | 1.25 3H d (6.4) |
| 8 | 3.46 2H t (1.4) | 1.60–1.78 2H m | 4.36 dt (4.6, 1.5) | 4.34 br d (4.9) | 3.96 br d (4.7) | 2.41 2H br d (6.2) |
| 9 | - | 3.80 sext (6.4) | 4.03 qd (6.4, 4.6) | 3.99 qd (6.4, 4.9) | 4.04 qd (6.4, 4.7) | 4.02 sext (6.2) |
| 10 | 2.24 3H s | 1.21 3H d (6.4) | 1.12 3H d (6.4) | 1.12 3H d (6.4) | 1.12 3H d (6.4 ) | 1.19 3H d (6.2) |
| OCH3 | - | - | - | - | 3.35 3H s | - |
| Position | 1 | 2 | 3 | 4 | 5 | 6/7 b |
|---|---|---|---|---|---|---|
| 2 | 173.5 s | 174.3 s | 174.5 s | 174.5 s | 174.6 s | 176.25 s/176.18 s |
| 3 | 128.2 s | 134.0 s | 136.9 s | 137.0 s | 133.9 s | 133.08 s/133.04 s |
| 4 | 149.4 d | 149.8 d | 150.3 d | 150.5 d | 152.0 d | 150.49 d/150.03 d |
| 5 | 85.5 d | 77.8 d | 87.1 d | 87.0 d | 87.4 d | 86.85 d/86.82 d |
| 6 | 67.8 d | 19.3 q | 68.5 d | 68.6 d | 68.5 d | 68.57 d/68.33 d |
| 7 | 18.9 q | 21.6 t | 19.0 q | 19.1 q | 19.0 q | 19.15 q/19.02 q |
| 8 | 39.1 t | 37.2 t | 71.7 d | 71.7 d | 81.6 d | 35.72 t/35.67 t |
| 9 | 203.6 s | 67.0 d | 69.8 d | 70.1 d | 69.0 d | 66.49 d/66.47 d |
| 10 | 30.3 q | 23.6 q | 17.7 q | 17.9 q | 18.2 q | 23.26 q |
| OCH3 | - | - | - | - | 58.1 q | - |
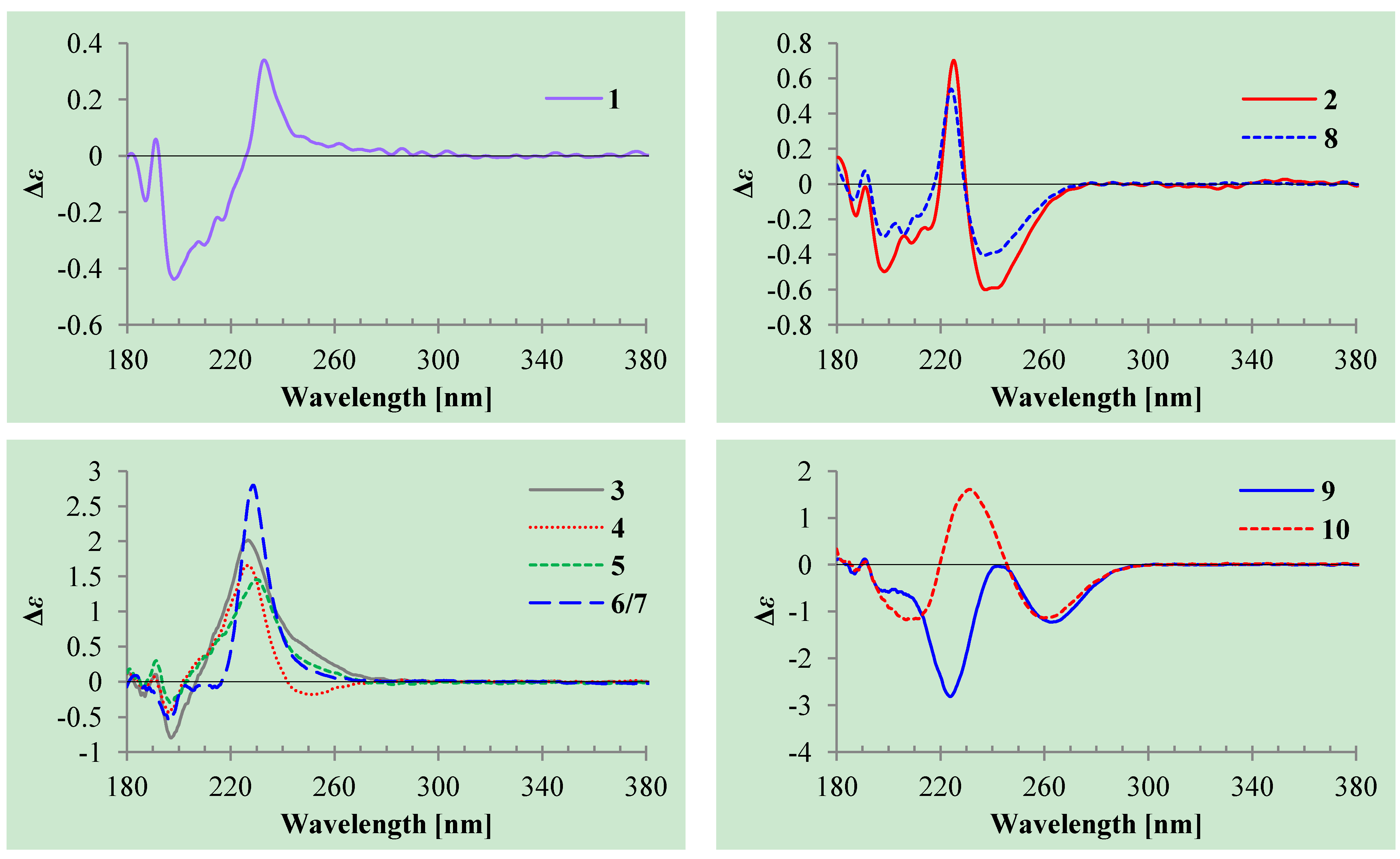
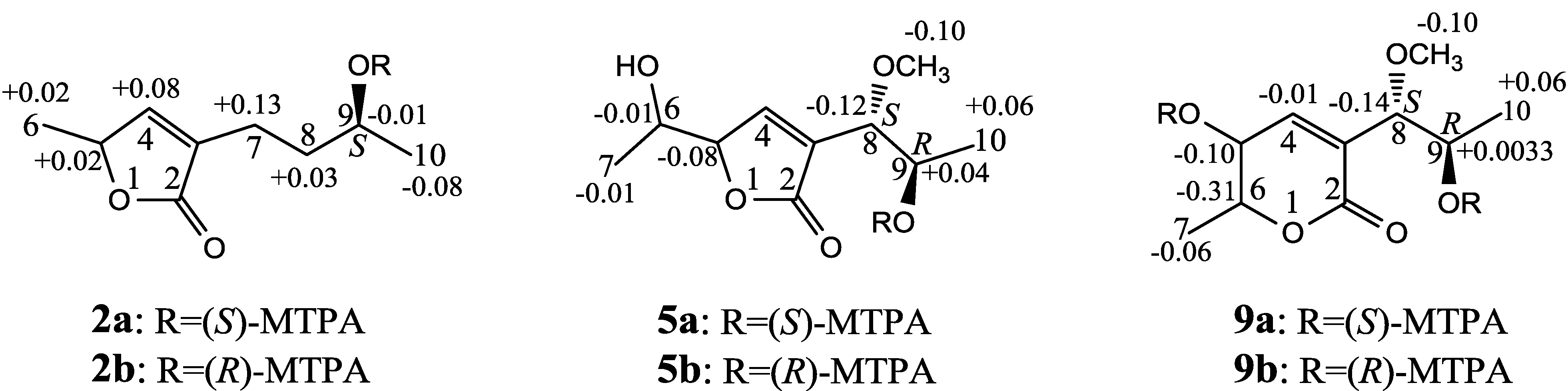
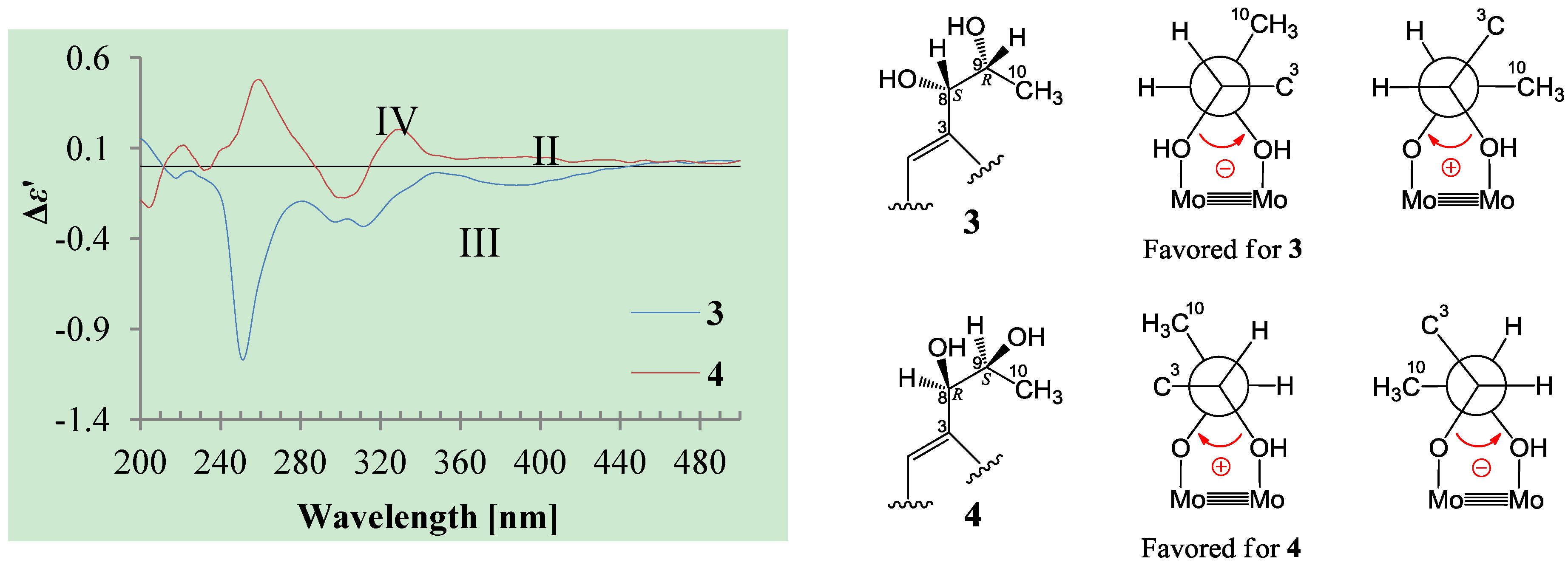
 +27.2 (c 0.32, MeOH), and aspilactonol C (4), +34.5 (c 0.41, MeOH), colorless oils (MeOH), were assigned the molecular composition C9H14O5 by HRESIMS (measured 203.0919 [M + H]+ for 3 and 203.0910 [M + H]+ for 4, calculated for C9H15O4 [M + H]+ 203.0919). Both they showed end UV absorptions, and similar to 1 and 2, their IR spectra revealed the presence of α,β-unsaturated γ-lactone moieties (around 1740 and 1652 cm−1) [21,22]. This was supported by the lower field olefinic proton and carbon signals (around δH 7.55, H-4; around δC 150.4, C-4) [22] in the 1H and 13C NMR spectra (Table 1 and Table 2) and the ester carbonyl (δC 174.5, C-2) and the sp2 carbon (around δC 137, C-3) signals in the 13C NMR spectra (Table 2). Their IR spectra also indicated the presence of OH and CH3/CH2 groups, and the strong OH signals around 3384 cm−1 further revealed more OH groups in 3 and 4 than in 1 and 2 (the IR spectra in the Supplementary Information). Interpretation of the 1H–1H COSY and HMQC data (Tables S3 and S4 in the Supplementary Information) established the carbon backbone chain related to the proton spin system, C-7–C-4(via quaternary sp2 C-3)–C-8–C-10. The allylic couplings between H-4 and H-8 in 3 and 4 indicated the connection of C-4 and C-8 via the quaternary sp2 carbon C-3, and the C-3 carbon was assigned by the HMBC correlations of H-4, H-5, H-8 and H-9 with C-3 (Tables S3 and S4 in the Supplementary Information). The C-2 carbonyl carbons in 3 and 4 were linked to C-5 and C-3 by the HMBC correlations between H-5/C-2, H-4/C-2 and H-8/C-2 to form the α,β-unsaturated γ-lactone rings with the ester linkage between C-5 and C-2.
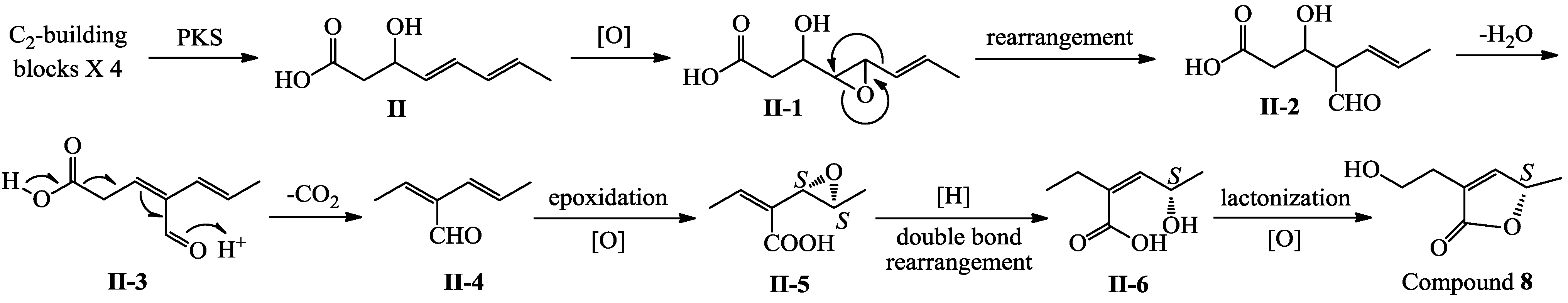
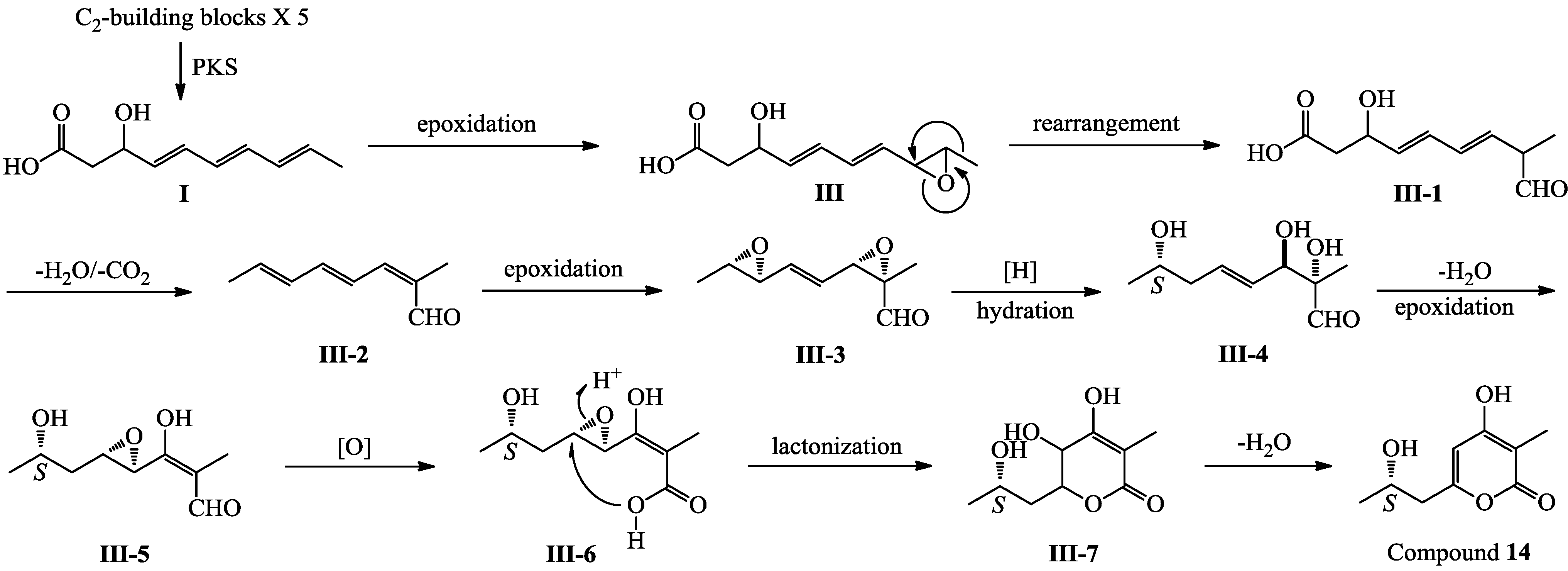
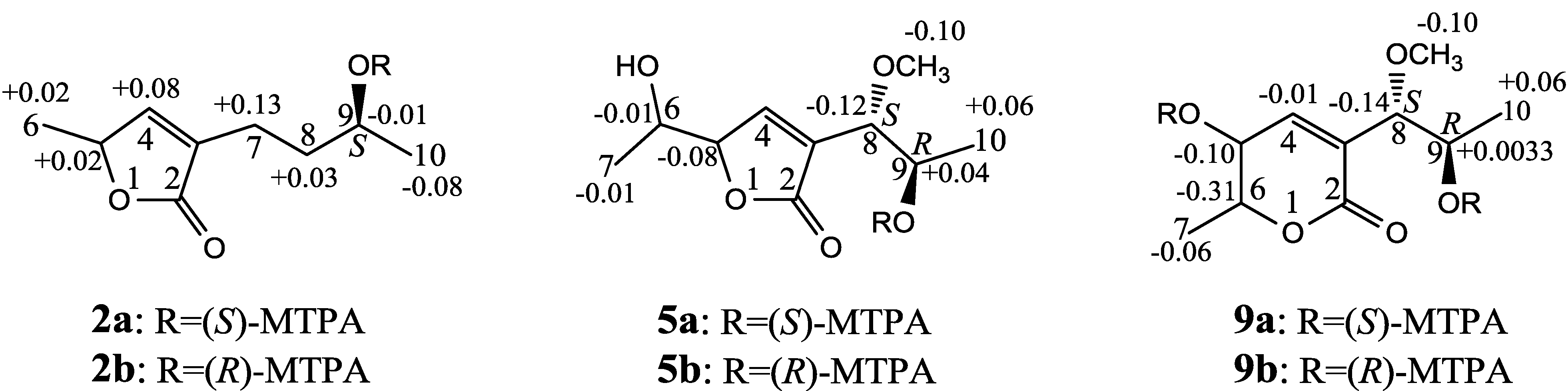
+27.2 (c 0.32, MeOH), and aspilactonol C (4), +34.5 (c 0.41, MeOH), colorless oils (MeOH), were assigned the molecular composition C9H14O5 by HRESIMS (measured 203.0919 [M + H]+ for 3 and 203.0910 [M + H]+ for 4, calculated for C9H15O4 [M + H]+ 203.0919). Both they showed end UV absorptions, and similar to 1 and 2, their IR spectra revealed the presence of α,β-unsaturated γ-lactone moieties (around 1740 and 1652 cm−1) [21,22]. This was supported by the lower field olefinic proton and carbon signals (around δH 7.55, H-4; around δC 150.4, C-4) [22] in the 1H and 13C NMR spectra (Table 1 and Table 2) and the ester carbonyl (δC 174.5, C-2) and the sp2 carbon (around δC 137, C-3) signals in the 13C NMR spectra (Table 2). Their IR spectra also indicated the presence of OH and CH3/CH2 groups, and the strong OH signals around 3384 cm−1 further revealed more OH groups in 3 and 4 than in 1 and 2 (the IR spectra in the Supplementary Information). Interpretation of the 1H–1H COSY and HMQC data (Tables S3 and S4 in the Supplementary Information) established the carbon backbone chain related to the proton spin system, C-7–C-4(via quaternary sp2 C-3)–C-8–C-10. The allylic couplings between H-4 and H-8 in 3 and 4 indicated the connection of C-4 and C-8 via the quaternary sp2 carbon C-3, and the C-3 carbon was assigned by the HMBC correlations of H-4, H-5, H-8 and H-9 with C-3 (Tables S3 and S4 in the Supplementary Information). The C-2 carbonyl carbons in 3 and 4 were linked to C-5 and C-3 by the HMBC correlations between H-5/C-2, H-4/C-2 and H-8/C-2 to form the α,β-unsaturated γ-lactone rings with the ester linkage between C-5 and C-2.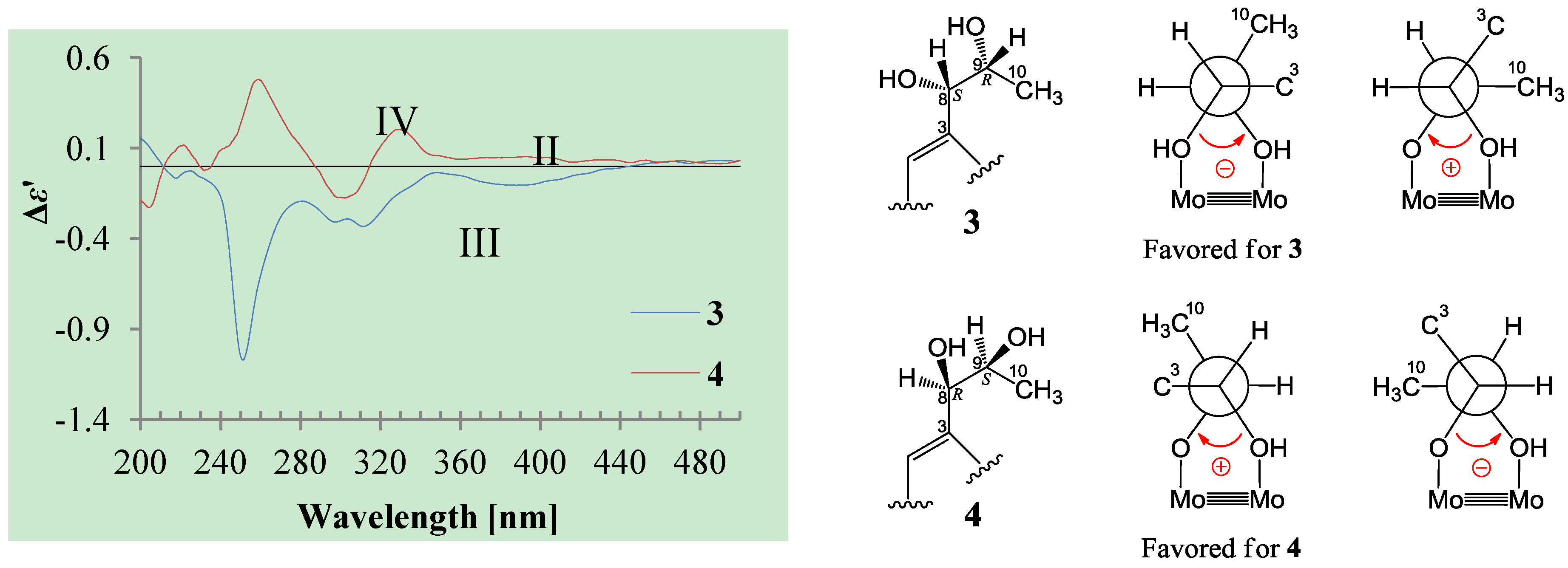 −1.7 (c 0.38, MeOH), was assigned the molecular formula C10H16O5 by HRESIMS (measured 217.1071 [M + H]+, calculated for C10H17O5 [M + H]+ 217.1076), which had a CH2 composition more than in 3 and 4. It showed UV and IR absorptions similar to 3 and 4, and the IR absorptions revealed the presence of OH, CH3/CH2 and α,β-unsaturated γ-lactone groups (Experimental Section). The 1H and 13C NMR spectra of 5 in CD3OD showed signals similar to 3 and 4 except additional signals ascribable to an O-methyl group were detected (Table 1 and Table 2). These NMR data indicated that 5 was an O-methylated. Analyses of the 1H–1H COSY, HMQC and HMBC spectra (Table S5 in the Supplementary Information) established its planar structure. The α,β-unsaturated γ-lactone ring was confirmed by the HMBC correlations between H-5/C-2, H-4/C-2 and H-8/C-2. The O-methyl group was located at C-8 by the HMBC correlations of the O-methyl protons with C-8. The absolute configuration at C-5 in 5 was assigned to be R by the positive π→π* Cotton effect at 231.5 nm in the CD spectrum (Figure 2) [26,27]. The coupling of H-5/H-6 (4.5 Hz) indicated the erythro relative configuration of 5,6-diol in 5 [23,24,25], and thus the absolute configuration at C-6 was assigned to be S. The R absolute configuration of C-9 was determined by the modified Mosher’s method [30,31] on the basis of the Δδ (δS–δR) values from the (S)- and (R)-MTRA esters (Figure 3). Since the coupling of H-8/H-9 (4.7 Hz) indicated the erythro relative configuration of 8,9-diol in 5 [32,33,34,35,36], the absolute configuration at C-8 was assigned to be S. +78.1 (c 1.00, MeOH), and the molecular formula C9H14O4 was determined by HRESIMS (measured 187.0967 [M + H]+, calculated for C9H15O5 [M + H]+ 187.0970). In the UV spectrum, 6/7 showed the end absorption, and the IR spectrum showed absorptions due to the OH, CH3/CH2 and α,β-unsaturated γ-lactone groups (Experimental Section). The 1H and 13C NMR spectra of 6/7 in CD3OD showed signals similar to 3–5, but they were characterized by the appearance of methylene signals instead of the signals from an oxygenated methine in 3–5 and an O-methyl group in 5 (Table 1 and Table 2). These NMR data indicated the same skeletal structures in 3–7. The appearance of the proton H-4 and the carbon signals except for the C-10 signal as pairs in an approximate 1:1 ratio indicated that 6/7 was a 1:1 mixture of stereoisomers. The planar structure of 6/7 was deduced by the 1H–1H COSY, HMQC and HMBC data (Table S6 in the Supplementary Information), coupled with the IR absorptions at 1748 and 1651 cm−1 from an α,β-unsaturated γ-lactone ring [21,22]. The absolute configuration at C-5 in 6/7 was assigned both to be R by the strong positive π→π* Cotton effect at 228.5 nm in the CD spectrum (Figure 2) [26,27]. Since the coupling of H-5/H-6 (4.4 Hz) indicated the erythro relative configuration of 5,6-diols in 6/7 [23,24,25], the absolute configuration at C-6 was assigned to be S for both 6/7. Thus, 6/7 was a 1:1 mixture of epimers at C-9. Although a Doctor’s Thesis has recorded the same planar structure of 6/7, its stereochemistry was not elucidated [40]. We therefore named 6/7 as aspilactonols E/F as new compounds. −41.6 (c 0.22, MeOH), was assigned the molecular formula C10H16O5 by HRESIMS (measured 217.1073 [M + H]+, calculated for C10H17O5 [M + H]+ 217.1076). It showed end UV absorption and the IR absorptions due to OH (3398 cm−1), CH3/CH2 (2982, 2938, 2905, 2835, 1451 and 1384 cm−1), C=O (1714 cm−1) and C=C (1649 cm−1) groups. The 1H and 13C NMR spectra (Table 3) resembled those of dihydroaspyrone (10) except additional signals from a methoxy and an oxygenated methine groups were detected instead of the methylene signals in 10. There were also slight changes in several 1H and 13C signals. These NMR data suggested that 9 was a methoxylated derivative of 10, and this was confirmed by analysis of the 1H–1H COSY, HMQC and HMBC spectra (Table S7 in the Supplementary Information) to complete the planar structure. The carbon chain related to the proton spin system, C-7–C-4(via quaternary sp2 C-3)–C-8–C-10, was deduced by interpretation of the 1H–1H COSY and HMQC data (Table S7 in the Supplementary Information). The allylic coupling between H-4 and H-8 suggested the connectivity of C-4 and C-8 via a quaternary sp2 carbon C-3, and C-3 was assigned by the HMBC correlations between H-8/C-3 and H-9/C-3. The OCH3 group was located at C-8 by the HMBCs of the methoxy protons with the carbon C-8. The carbonyl carbon C-2 was linked to C-3 by the HMBCs on H-4/C-2 and H-8/C-2. The ester linkage of the C-2 carbonyl was then linked to C-6 to form a δ-lactone ring by the coupling of H-5/H-6 (9.4 Hz), which requires C-5/C-6 fixed in a six-membered ring with trans orientated H-5/H-6. This was supported by the chemical shift of C-6, δC 80.0 in 9 and δC 79.6 in 10. The R absolute configuration at C-9 was determined by the modified Mosher’s method [30,31] on the basis of the Δδ (δS–δR) values from the (S)- and (R)-MTRA esters of 9 (Figure 3), and the absolute configuration at C-8 was assigned to be S because the coupling of H-8/H-9 (4.6 Hz) indicated the erythro relative configuration of 8,9-diol in 9[32,33,34,35,36]. The absolute configuration at C-5 and C-6 in 9 was determined to be 5S,6R, the same as 10, according to the negative signs of the Cotton effects around 260 nm both from the chiral α,β-unsaturated δ-lactone units in 9 and 10 (Figure 2) [28]. This was also supported by the co-generation of 9 and 10 by the same Aspergillus sp. 16-02-1 strain from a biogenetic consideration.
−1.7 (c 0.38, MeOH), was assigned the molecular formula C10H16O5 by HRESIMS (measured 217.1071 [M + H]+, calculated for C10H17O5 [M + H]+ 217.1076), which had a CH2 composition more than in 3 and 4. It showed UV and IR absorptions similar to 3 and 4, and the IR absorptions revealed the presence of OH, CH3/CH2 and α,β-unsaturated γ-lactone groups (Experimental Section). The 1H and 13C NMR spectra of 5 in CD3OD showed signals similar to 3 and 4 except additional signals ascribable to an O-methyl group were detected (Table 1 and Table 2). These NMR data indicated that 5 was an O-methylated. Analyses of the 1H–1H COSY, HMQC and HMBC spectra (Table S5 in the Supplementary Information) established its planar structure. The α,β-unsaturated γ-lactone ring was confirmed by the HMBC correlations between H-5/C-2, H-4/C-2 and H-8/C-2. The O-methyl group was located at C-8 by the HMBC correlations of the O-methyl protons with C-8. The absolute configuration at C-5 in 5 was assigned to be R by the positive π→π* Cotton effect at 231.5 nm in the CD spectrum (Figure 2) [26,27]. The coupling of H-5/H-6 (4.5 Hz) indicated the erythro relative configuration of 5,6-diol in 5 [23,24,25], and thus the absolute configuration at C-6 was assigned to be S. The R absolute configuration of C-9 was determined by the modified Mosher’s method [30,31] on the basis of the Δδ (δS–δR) values from the (S)- and (R)-MTRA esters (Figure 3). Since the coupling of H-8/H-9 (4.7 Hz) indicated the erythro relative configuration of 8,9-diol in 5 [32,33,34,35,36], the absolute configuration at C-8 was assigned to be S. +78.1 (c 1.00, MeOH), and the molecular formula C9H14O4 was determined by HRESIMS (measured 187.0967 [M + H]+, calculated for C9H15O5 [M + H]+ 187.0970). In the UV spectrum, 6/7 showed the end absorption, and the IR spectrum showed absorptions due to the OH, CH3/CH2 and α,β-unsaturated γ-lactone groups (Experimental Section). The 1H and 13C NMR spectra of 6/7 in CD3OD showed signals similar to 3–5, but they were characterized by the appearance of methylene signals instead of the signals from an oxygenated methine in 3–5 and an O-methyl group in 5 (Table 1 and Table 2). These NMR data indicated the same skeletal structures in 3–7. The appearance of the proton H-4 and the carbon signals except for the C-10 signal as pairs in an approximate 1:1 ratio indicated that 6/7 was a 1:1 mixture of stereoisomers. The planar structure of 6/7 was deduced by the 1H–1H COSY, HMQC and HMBC data (Table S6 in the Supplementary Information), coupled with the IR absorptions at 1748 and 1651 cm−1 from an α,β-unsaturated γ-lactone ring [21,22]. The absolute configuration at C-5 in 6/7 was assigned both to be R by the strong positive π→π* Cotton effect at 228.5 nm in the CD spectrum (Figure 2) [26,27]. Since the coupling of H-5/H-6 (4.4 Hz) indicated the erythro relative configuration of 5,6-diols in 6/7 [23,24,25], the absolute configuration at C-6 was assigned to be S for both 6/7. Thus, 6/7 was a 1:1 mixture of epimers at C-9. Although a Doctor’s Thesis has recorded the same planar structure of 6/7, its stereochemistry was not elucidated [40]. We therefore named 6/7 as aspilactonols E/F as new compounds. −41.6 (c 0.22, MeOH), was assigned the molecular formula C10H16O5 by HRESIMS (measured 217.1073 [M + H]+, calculated for C10H17O5 [M + H]+ 217.1076). It showed end UV absorption and the IR absorptions due to OH (3398 cm−1), CH3/CH2 (2982, 2938, 2905, 2835, 1451 and 1384 cm−1), C=O (1714 cm−1) and C=C (1649 cm−1) groups. The 1H and 13C NMR spectra (Table 3) resembled those of dihydroaspyrone (10) except additional signals from a methoxy and an oxygenated methine groups were detected instead of the methylene signals in 10. There were also slight changes in several 1H and 13C signals. These NMR data suggested that 9 was a methoxylated derivative of 10, and this was confirmed by analysis of the 1H–1H COSY, HMQC and HMBC spectra (Table S7 in the Supplementary Information) to complete the planar structure. The carbon chain related to the proton spin system, C-7–C-4(via quaternary sp2 C-3)–C-8–C-10, was deduced by interpretation of the 1H–1H COSY and HMQC data (Table S7 in the Supplementary Information). The allylic coupling between H-4 and H-8 suggested the connectivity of C-4 and C-8 via a quaternary sp2 carbon C-3, and C-3 was assigned by the HMBC correlations between H-8/C-3 and H-9/C-3. The OCH3 group was located at C-8 by the HMBCs of the methoxy protons with the carbon C-8. The carbonyl carbon C-2 was linked to C-3 by the HMBCs on H-4/C-2 and H-8/C-2. The ester linkage of the C-2 carbonyl was then linked to C-6 to form a δ-lactone ring by the coupling of H-5/H-6 (9.4 Hz), which requires C-5/C-6 fixed in a six-membered ring with trans orientated H-5/H-6. This was supported by the chemical shift of C-6, δC 80.0 in 9 and δC 79.6 in 10. The R absolute configuration at C-9 was determined by the modified Mosher’s method [30,31] on the basis of the Δδ (δS–δR) values from the (S)- and (R)-MTRA esters of 9 (Figure 3), and the absolute configuration at C-8 was assigned to be S because the coupling of H-8/H-9 (4.6 Hz) indicated the erythro relative configuration of 8,9-diol in 9[32,33,34,35,36]. The absolute configuration at C-5 and C-6 in 9 was determined to be 5S,6R, the same as 10, according to the negative signs of the Cotton effects around 260 nm both from the chiral α,β-unsaturated δ-lactone units in 9 and 10 (Figure 2) [28]. This was also supported by the co-generation of 9 and 10 by the same Aspergillus sp. 16-02-1 strain from a biogenetic consideration.| NO. | in CD3OD | in CDCl3 | ||
|---|---|---|---|---|
| δC | δH (J in Hz) | δH (J in Hz) | NOE | |
| 2 | 165.7 s | - | - | - |
−5.4 (c 0.14, MeOH), was assigned the molecular formula C9H14O3 by HRESIMS (measured 193.0842 [M + Na]+, calculated for C9H14O3Na [M + Na]+ 193.0841). The 1H and 13C NMR data of 11 in CD3OD were identical with those of aspinonediol in CD3OD [19], indicating the same planar structures of both compounds. Aspinonediol had the absolute configuration 7S and showed +2.4 (c 0.63, MeOH) [19]. In contrast, 11 showed the opposite optical rotation. Thus, the epiaspinonediol (11) was determined to be the epimer of aspinonediol at C-7 with the 7R absolute configuration.2.3. Inhibitory Effects of 1–14 on Several Human Cancer Cell Lines
2.4. Discussions
3. Experimental Section
3.1. General Experimental
3.2. Producing Strain, Fermentation and Extraction
3.3. Isolation of 1–14
3.4. Physicochemical and Spectroscopic Data of 1–14
+10.5 (c 1.2, MeOH). Positive ion ESIMS m/z: 185 [M + H]+, 207 [M + Na]+; Negative ion ESIMS m/z: 183 [M − H]−. Positive ion HRESIMS m/z: measured 185.0811 [M + H]+, calculated for C9H13O4 [M + H]+ 185.0814; measured 207.0631 [M + Na]+, calculated for C9H12O4Na [M + Na]+ 207.0633. UV λmax nm in MeOH: 213 nm. IR νmax cm−1 (Diamond ATR crystal): 3417, 3090, 2980, 2938, 2906, 1752, 1722, 1656, 1422, 1360, 1323, 1207, 1166, 1138, 1081, 1024, 982, 935, 896. CD Δε (nm): 0 (192.5), −0.44 (198.0), −0.30 (207.5), −0.32 (209.5), −0.21 (215.0), −0.23 (216.5), 0 (226.0), +0.34 (233.0), +0.07 (244.5), 0 (282). 1H and 13C NMR data: Table 1 and Table 2. +24.6 (c 0.23, MeOH). Positive ion ESIMS m/z: 171 [M + H]+, 193 [M + Na]+. Positive ion HRESIMS m/z: measured 171.1015 [M + H]+, calculated for C9H15O3 [M + H]+ 171.1021; measured 193.0836 [M + Na]+, calculated for C9H14O3Na [M + Na]+ 193.0841; measured 209.0573 [M + K]+, calculated for C9H14O3K [M + Na]+ 209.0580. UV λmax nm in MeOH: 210 nm. IR νmax cm−1 (Diamond ATR crystal): 3436, 3080, 2971, 2932, 1746, 1653, 1449, 1406, 1374, 1320, 1201, 1030, 960, 870, 786. CD Δε (nm): 0 (191.0), −0.49 (198.0), −0.30 (206.0), −0.33 (208.5), −0.25 (214.0), −0.26 (215.5), 0 (219.5), +0.70 (225.0), 0 (229.5), −0.60 (237.5), −0.59 (240.5), 0 (279.5). 1H and 13C NMR data: Table 1 and Table 2. +34.5 (c 0.41, MeOH). Positive ion ESIMS m/z: 203 [M + H]+, 225 [M + Na]+; Negative ion ESIMS m/z: 201 [M − H]−. Positive ion HRESIMS m/z: measured 203.0919 [M + H]+, calculated for C9H15O5 [M + H]+ 203.0919; measured 225.0736 [M + Na]+, calculated for C9H14O5Na [M + Na]+ 225.0739. UV λmax nm in MeOH: 214 nm. IR νmax cm−1 (Diamond ATR crystal): 3388, 3095, 2978, 2936, 2900, 2831, 1742, 1653, 1451, 1406, 1378, 1314, 1200, 1136, 1069, 996, 934, 894, 870, 830, 803, 770, 721, 698. CD Δε (nm): 0 (192.0), −0.78 (197.0), 0 (206.5), +2.02 (226.5), 0 (282.5). 1H and 13C NMR data: Table 1 and Table 2. +27.2 (c 0.32, MeOH). Positive ion ESIMS m/z: 203 [M + H]+, 225 [M + Na]+; Negative ion ESIMS m/z: 201 [M − H]−. Positive ion HRESIMS m/z: measured 203.0910 [M + H]+, calculated for C9H15O5 [M + H]+ 203.0919; measured 225.0732 [M + Na]+, calculated for C9H14O5Na [M + Na]+ 225.0739; measured 241.0476 [M + K]+, calculated for C9H14O5K [M + K]+ 241.0478. UV λmax nm in MeOH: 214 nm. IR νmax cm−1 (Diamond ATR crystal): 3379, 3092, 2979, 2935, 1737, 1651, 1450, 1402, 1377, 1300, 1200, 1133, 1063, 994, 933, 894, 868, 826, 763, 693. CD Δε (nm): 0 (192.0), −0.43 (196.5), −0.39 (197.0), −0.41 (197.5), 0 (201.5), +0.37 (210.5), +1.64 (225.5), +1.65 (226.0), 0 (242.0), −0.19 (251.5), 0 (275.0). 1H and 13C NMR data: Table 1 and Table 2. −1.7 (c 0.38, MeOH). Positive ion ESIMS m/z: 217 [M + H]+, 239 [M + Na]+. Positive ion HRESIMS m/z: measured 217.1071 [M + H]+, calculated for C10H17O5 [M + H]+ 217.1076; measured 239.0890 [M + Na]+, calculated for C10H16O5Na [M + Na]+ 239.0895; measured 255.0635 [M + K]+, calculated for C10H16O5K [M + K]+ 255.0635. UV λmax nm in MeOH: 213 nm. IR νmax cm−1 (Diamond ATR crystal): 3401, 3088, 2978, 2937, 2902, 2832, 1750, 1655, 1451, 1378, 1298, 1256, 1200, 1136, 1108, 1066, 1022, 977, 950, 936, 894, 853, 833, 803, 780, 717. CD Δε (nm): 0 (193.5), −0.29 (196.5), 0 (203.5), +1.41 (231.5), 0 (271.0). 1H and 13C NMR data: Table 1 and Table 2. +78.1 (c 1.00, MeOH). Positive ion ESIMS m/z: 187 [M + H]+, 209 [M + Na]+; Negative ion ESIMS m/z: 185 [M − H]−. Positive ion HRESIMS m/z: measured 187.0967 [M + H]+, calculated for C9H15O5 [M + H]+ 187.0970; measured 209.0789 [M + Na]+, calculated for C9H14O5Na [M + Na]+ 209.0790. UV λmax nm in MeOH: 213 nm. IR νmax cm−1 (Diamond ATR crystal): 3396, 3093, 2975, 2934, 1748, 1651, 1456, 1417, 1377, 1205, 1127, 1070, 990, 942, 882, 841, 677. CD Δε (nm): 0 (185.0), −0.55 (196.5), −0.09 (201.5), −0.05 (202.5), −0.07 (214.0), 0 (216.5), +2.78 (228.5), 0 (280.5). 1H and 13C NMR data: Table 1 and Table 2. −41.6 (c 0.22, MeOH). Positive ion ESIMS m/z: 217 [M + H]+, 239 [M + Na]+. Positive ion HRESIMS m/z: measured 217.1073 [M + H]+, calculated for C10H17O5 [M + H]+ 217.1076; measured 239.0896 [M + Na]+, calculated for C10H16O5Na [M + Na]+ 239.0895; measured 255.0634 [M + K]+, calculated for C10H16O5K [M + K]+ 255.0635. UV λmax nm in MeOH: 208 nm. IR νmax cm−1 (Diamond ATR crystal): 3398, 2982, 2938, 2905, 2835, 1714, 1649, 1451, 1384, 1309, 1216, 1044, 1018, 783, 721, 669. CD Δε (nm): 0 (192.0), −0.53 (196.5), −0.72 (209.5), −2.81 (223.5), 0 (242.0), −1.22 (263.0), 0 (301.0). 1H and 13C NMR data: Table 3. −5.4 (c 0.14, MeOH). Positive ion ESIMS m/z: 193 [M + Na]+, 209 [M + K]+. Positive ion HRESIMS m/z: measured 171.1003 [M + H]+, calculated for C9H15O3 [M + H]+ 171.1021; measured 193.0842 [M + Na]+, calculated for C9H14O3Na [M + Na]+ 193.0841. 1H NMR (400 MHz, CD3OD) δ: 7.20 (1H, d, J = 16.2 Hz, H-4), 6.41 (1H, d, J = 16.2 Hz, H-3), 6.05 (1H, d, J = 8.4 Hz, H-6), 4.77 (1H, dq, J = 8.4, 6.4 Hz, H-7), 4.33 (2H, s, H-9), 2.31 (3H, s, H-1), 1.29 (3H, d, J = 6.4 Hz, H-8). 13C NMR (100 MHz, CD3OD) δ: 201.6 (C-2), 148.3 (C-6), 147.5 (C-4), 137.0 (C-5), 128.4 (C-3), 64.8 (C-7), 56.9 (C-9), 27.3 (C), 23.6 (C-8).3.5. Preparation of (S)- and (R)-MTPA Esters of 1, 5 and 9
3.6. Measurement of ICD Spectra of 3 and 4 Using Mo2(OAc)4
3.7. MTT Assay
4. Conclusions
Supplementary Files
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2014, 31, 160–258. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2013, 30, 237–323. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Keyzers, R.A.; Munro, M.H.G.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2012, 29, 144–222. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2011, 28, 196–268. [Google Scholar] [CrossRef]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2010, 27, 165–237. [Google Scholar]
- Bugni, T.S.; Ireland, C.M. Marine-derived fungi: A chemically and biologically diverse group of microorganisms. Nat. Prod. Rep. 2004, 21, 143–163. [Google Scholar] [CrossRef]
- Saleem, M.; Ali, M.S.; Hussain, S.; Jabbar, A.; Ashraf, M.; Lee, Y.S. Marine natural products of fungal origin. Nat. Prod. Rep. 2007, 24, 1142–1152. [Google Scholar] [CrossRef]
- Bhatnagar, I.; Kim, S.-K. Immense essence of excellence: Marine microbial bioactive compounds. Mar. Drugs 2010, 8, 2673–2701. [Google Scholar] [CrossRef]
- Rateb, M.E.; Ebel, R. Secondary metabolites of fungi from marine habitats. Nat. Prod. Rep. 2011, 28, 290–344. [Google Scholar] [CrossRef]
- Pejin, B.; Jovanović, K.K.; Mojović, M.; Savić, A.G. New and highly potent antitumor natural products from marine-derived fungi: Covering the period from 2003 to 2012. Curr. Top. Med. Chem. 2013, 13, 2745–2766. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Marine-sourced anti-cancer and cancer pain control agents in clinical and late preclinical development. Mar. Drugs 2014, 12, 255–278. [Google Scholar] [CrossRef]
- Skropeta, D. Deep-sea natural products. Nat. Prod. Rep. 2008, 25, 1131–1166. [Google Scholar] [CrossRef]
- Li, D.H.; Cai, S.X.; Zhu, T.J.; Wang, F.P.; Xiao, X.; Gu, Q.Q. Three new sorbicillin trimers, trisorbicillinones B, C, and D, from a deep ocean sediment derived fungus, Phialocephala sp. FL30r. Tetrahedron 2010, 66, 5101–5106. [Google Scholar] [CrossRef]
- Li, Y.; Ye, D.Z.; Shao, Z.Z.; Cui, C.B.; Che, Y.S. A sterol and spiroditerpenoids from a Penicillium sp. isolated from a deep sea sediment sample. Mar. Drugs 2012, 10, 497–508. [Google Scholar] [CrossRef]
- Wang, F.Z.; Huang, Z.; Shi, X.F.; Chen, Y.C.; Zhang, W.M.; Tian, X.P.; Li, J.; Zhang, S. Cytotoxic indole diketopiperazines from the deep sea-derived fungus Acrostalagmus luteoalbus SCSIO F457. Bioorg. Med. Chem. Lett. 2012, 22, 7265–7267. [Google Scholar] [CrossRef]
- Chen, Y.; Mao, W.J.; Wang, B.F.; Zhou, L.; Gu, Q.Q.; Chen, Y.L.; Zhao, C.Q.; Li, N.; Wang, C.Y.; Shan, J.M.; et al. Preparation and characterization of an extracellular polysaccharide produced by the deep-sea fungus Penicillium griseofulvum. Bioresour. Technol. 2013, 132, 178–182. [Google Scholar] [CrossRef]
- Chen, X.W.; Li, C.W.; Hua, W.; Wu, C.J.; Cui, C.B.; Zhu, T.J.; Gu, Q.Q. Metabolites of Aspergillus sp. 16-02-1 isolated from a deep sea sediment and preliminary test of their antitumor and antifungal activities. Chin. J. Mar. Drugs 2013, 32, 1–10. [Google Scholar]
- Harmange, J.C.; Figadėre, B.; Hocquemiller, R. Enantiospecific preparation of the lactone fragment of murisolin. Tetrahedron: Asymmetry 1991, 2, 347–350. [Google Scholar] [CrossRef]
- Kito, K.; Ookura, R.; Yoshida, S.; Namikoshi, M.; Ooi, T.; Kusumi, T. Pentaketides relating to aspinonene and dihydroaspyrone from a marine-derived fungus, Aspergillus ostianus. J. Nat. Prod. 2007, 70, 2022–2025. [Google Scholar] [CrossRef]
- Fujimoto, H.; Nozawa, M.; Okuyama, E.; Ishibashi, M. Six new constituents from an ascomycete, Cheatomium quadrangulatum, found in a screening study focused on monoamine oxidase inhibitory activity. Chem. Pharm. Bull. 2003, 51, 247–251. [Google Scholar] [CrossRef]
- Garson, M.J.; Staunton, J. New polyketide metabolites from Aspergmus melleus: Structural and stereochemical studies. J. Chem. Soc. Perkin Trans. I 1984, 1021–1026. [Google Scholar] [CrossRef]
- Namikoshi, M.; Negishi, R.; Nagai, H.; Dimitrenok, M.; Kobayashi, H. Three new chlorine containing antibiotics from a marine-derived fungus Aspergillus ostianus collected in Pohnpei. J. Antibot. 2003, 56, 755–761. [Google Scholar] [CrossRef]
- Sy, A.A.; Swenson, D.C.; Gloer, J.B.; Wicklow, D.T. Botryolides A–E, decarestrictine analogues from a fungicolous Botryotrichum sp. (NRRL 38180). J. Nat. Prod. 2008, 71, 415–419. [Google Scholar] [CrossRef]
- Buchanan, M.; Hashimoto, T.; Takaoka, S.; Asakawa, Y. (+)-Osmundalactone, γ-lactones and spiromentins from the fungus Paxillus atrotomentosus. Phytochemistry 1995, 40, 1251–1257. [Google Scholar] [CrossRef]
- Franck, X.; Araujo, M.E.V.; Jullian, J.-C.; Hocquemiller, R.; Figadère, B. Synthesis and structure determination of iso-cladospolide B. Tetrahedron Lett. 2001, 42, 2801–2803. [Google Scholar] [CrossRef]
- Uchida, I.; Kuriyama, K. The π-π* circular dichroism of α,β-unsaturated γ-lactones. Tetrahedron Lett. 1974, 15, 3761–3764. [Google Scholar] [CrossRef]
- Gawronski, J.K.; Oeveren, A.V.; Deen, H.V.D.; Leung, C.W.; Feringa, B.L. Simple circular dicroic method for the determination of absolute configuration of 5-substitutaed 2(5H)-furanones. J. Org. Chem. 1996, 61, 1513–1515. [Google Scholar] [CrossRef]
- Beecham, A.F. The CD of α,β-unsaturated γ-lactones. Tetrahedron 1972, 28, 5543–5554. [Google Scholar] [CrossRef]
- Lee, C.-L.; Chang, F.-R.; Hsieh, P.-W.; Chiang, M.-Y.; Wu, C.-C.; Huang, Z.-Y.; Lan, Y.-H.; Chen, M.; Lee, K.-H.; Yen, H.-F.; et al. Cytotoxic ent-abietane diterpenes from Gelonium aequoreum. Phytochemistry 2008, 69, 276–287. [Google Scholar] [CrossRef]
- Dale, J.A.; Mosher, H.S. Nuclear magnetic resonance enantiomer regents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and α-methoxy-α-trifluoromethylphenylacetate (MTPA) esters. J. Am. Chem. Soc. 1973, 95, 512–519. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-field FT NMR application of Mosher’s method. The absolute configurations of marine terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Jarvis, B.B.; Stahly, G.P.; Pavanasasivam, G.; Midiwo, J.O.; DeSilva, T.; Holmlund, C.E.; Mazzola, E.P.; Geoghegan, R.F., Jr. Isolation and characterization of the trichoverroids and new roridins and verrucarins. J. Org. Chem. 1982, 47, 1117–1124. [Google Scholar] [CrossRef]
- Jarvis, B.B.; Comezoglu, S.N.; Rao, M.M.; Pena, N.B. Isolation of macrocyclic trichothecenes from a large-scale extract of Baccharis megapotamica. J. Org. Chem. 1987, 52, 45–56. [Google Scholar] [CrossRef]
- Takeshita, M.; Sato, T. Synthesis of optically active 1-phenyl-1,2-propanediol by use of Baker’s yeast. Chem. Pharm. Bull. 1989, 37, 1085–1086. [Google Scholar] [CrossRef]
- Ayer, W.A.; Trifonov, L.S. Metabolites of Peniophora polygonia, part 2. Some aromatic compounds. J. Nat. Prod. 1993, 56, 85–89. [Google Scholar] [CrossRef]
- Jarvis, B.B.; Wang, S.; Ammon, H.L. Trichoverroid stereoisomers. J. Nat. Prod. 1996, 59, 254–261. [Google Scholar] [CrossRef]
- Bari, L.D.; Pescitelli, G.; Pratelli, C.; Pini, D.; Salvadori, P. Determination of absolute configuration of acyclic 1,2-diols with Mo2(OAc)4. 1. Snatzke’s method revisited. J. Org. Chem. 2001, 66, 4819–4825. [Google Scholar] [CrossRef]
- Frelek, J.; Ruśkowska, P.; Suszczyńska, A.; Szewczyk, K.; Osuch, A.; Jarosz, S.; Jagodziński, J. Configurational assignment of sugar erythro-1,2-diols from their electronic circular dichroism spectra with dimolybdenum tetraacetate. Tetrahedron Asymmetry 2008, 19, 1709–1713. [Google Scholar] [CrossRef]
- Xia, M.-W.; Cui, C.-B.; Li, C.-W.; Wu, C.-J. Three new and eleven known unusual C25 steroids: Activated production of silent metabolites in a marine-derived fungus by chemical mutagenesis strategy using diethyl sulphate. Mar. Drugs 2014, 12, 1545–1568. [Google Scholar] [CrossRef]
- Zhang, G.J. Studies on the Meroterpenoidal Constituents from Two Marine-Derived Fungal Strains. Ph. D. Thesis, Ocean University of China, Qingdao, China, June 2011. [Google Scholar]
- Staunton, J.; Sutkowski, A.C. Biosynthesis of aspyrone, a metabolite of Aspergillus melleus: Advanced precursor studies to identify the product of the polyketide synthesis. J. Chem. Soc. Chem. Commun. 1991, 1108–1110. [Google Scholar] [CrossRef]
- Staunton, J.; Sutkowski, A.C. The polyketide synthase (PKS) of aspyrone biosynthesis evidence: Evidence for the enzyme bound intermediates from incorporation studies with N-acetylcysteamine thioesters in intact cells of Aspergillus melleus. J. Chem. Soc. Chem. Commun. 1991, 1110–1112. [Google Scholar] [CrossRef]
- Jacobs, A.; Staunton, J.; Sutkowski, A.C. Aspyrone biosynthesis in Aspergillus melleus: Identification of the intermediates formed on the polyketide synthase (PKS) in the first chain extension cycle leading to crotonate. J. Chem. Soc. Chem. Commun. 1991, 1113–1114. [Google Scholar] [CrossRef]
- Brereton, R.; Garson, M.; Staunton, J. Biosynthesis of fungal metabolites: Asperlactone and its relationship to other metabolites of Aspergillus melleus. J. Chem. Soc. Perkin Trans. I 1984, 1027–1033. [Google Scholar] [CrossRef]
- Ahmed, S.A.; Simpson, T.J.; Staunton, J.; Sutkowski, A.C.; Trimble, L.A.; Vederas, J.C. Biosynthesis of aspyrone and asperlactone, petaketide metabolites of Aspergillus melleus. Incorporation studies with [1-13C,18O2] acetate and 18O2 gas. J. Chem. Soc. Chem. Commun. 1985, 1685–1687. [Google Scholar]
- Staunton, J.; Sutkowski, A.C. 17O NMR biosynthetic studies: Aspyrone, asperlactone and isoasperlactone, metabolites of Aspergillus melleus. J. Chem. Soc. Chem. Commun. 1991, 1106–1108. [Google Scholar] [CrossRef]
- Fuchser, J.; Thiericke, R.; Zeeck, A. Biosynthesis of aspinonene, a branched pnetaketide produced by Aspergillus ochraceus, related to aspyrone. J. Chem. Soc. Perkin Trans. I 1995, 1663–1666. [Google Scholar] [CrossRef]
- Balcells, M.; Canela, R.; Coll, J.; Sanchís, V.; Torres, M. Effect of fungal metabolites and some derivatives against Tribolium castaneum (Herbst) and Nezara viridula (L.). Pesitic. Sci. 1995, 45, 319–323. [Google Scholar] [CrossRef]
- Kimura, Y.; Nakahara, S.; Fujioka, S. Aspyrone, a nematicidal compound isolated from the fungus, Aspergillus melleus. Biosci. Biotech. Biochem. 1996, 60, 1375–1376. [Google Scholar] [CrossRef]
- Torres, M.; Balcells, M.; Sala, N.; Sanchís, V.; Canela, R. Bactericidal and fungicidal activity of Aspergillus ochraceus metabolites and some derivatives. Pesitic. Sci. 1998, 53, 9–14. [Google Scholar] [CrossRef]
- Zhang, D.; Yang, X.; Kang, J.S.; Choi, H.D.; Son, B.H. Chlorohydroaspyrones A and B, antibacterial aspyrone derivatives from the marine-derived fungus Exophiala sp. J. Nat. Prod. 2008, 71, 1458–1460. [Google Scholar] [CrossRef]
- Wu, C.-J.; Li, C.-W.; Cui, C.-B. Seven new and two known lipopeptides as well as five known polyketides: The activated production of silent metabolites in a marine-derived fungus by chemical mutagenesis strategy using diethyl sulphate. Mar. Drugs 2014, 12, 1815–1838. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, X.-W.; Li, C.-W.; Cui, C.-B.; Hua, W.; Zhu, T.-J.; Gu, Q.-Q. Nine New and Five Known Polyketides Derived from a Deep Sea-Sourced Aspergillus sp. 16-02-1. Mar. Drugs 2014, 12, 3116-3137. https://doi.org/10.3390/md12063116
Chen X-W, Li C-W, Cui C-B, Hua W, Zhu T-J, Gu Q-Q. Nine New and Five Known Polyketides Derived from a Deep Sea-Sourced Aspergillus sp. 16-02-1. Marine Drugs. 2014; 12(6):3116-3137. https://doi.org/10.3390/md12063116
Chicago/Turabian StyleChen, Xiu-Wen, Chang-Wei Li, Cheng-Bin Cui, Wei Hua, Tian-Jiao Zhu, and Qian-Qun Gu. 2014. "Nine New and Five Known Polyketides Derived from a Deep Sea-Sourced Aspergillus sp. 16-02-1" Marine Drugs 12, no. 6: 3116-3137. https://doi.org/10.3390/md12063116