Biosynthetic Modularity Rules in the Bisintercalator Family of Antitumor Compounds


Abstract
:
1. Antitumor Compounds from the Bisintercalators Family: The Origins

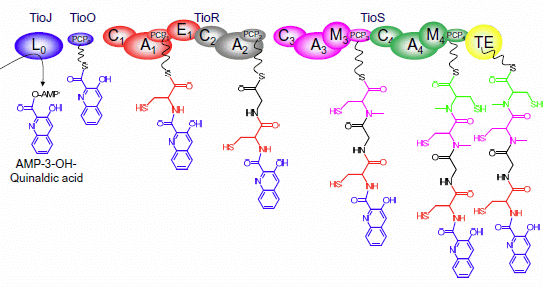
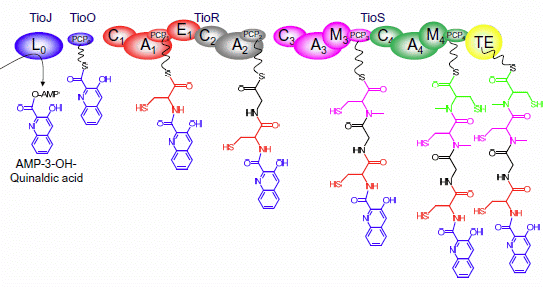
{kind=link}
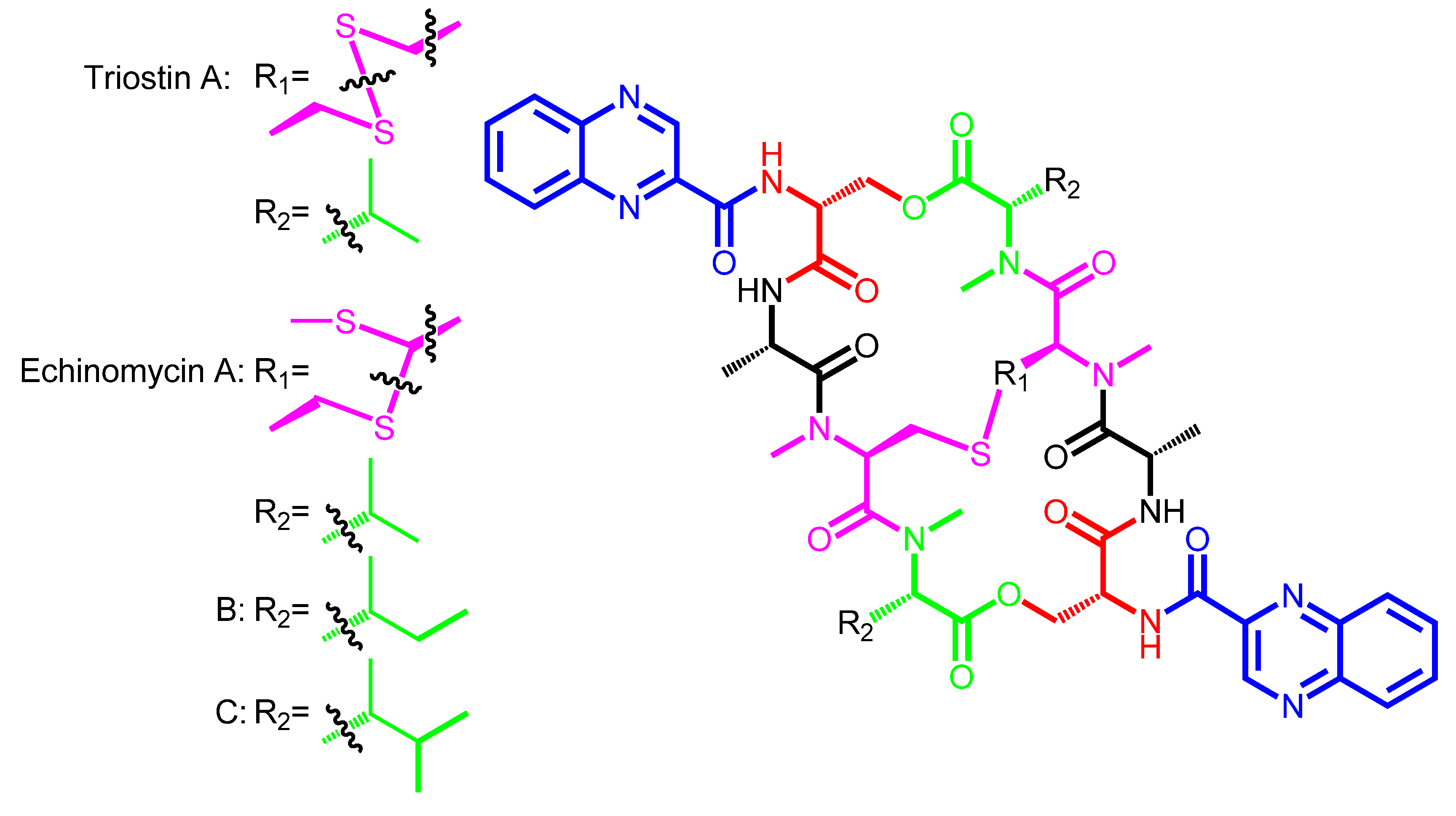
{kind=link}
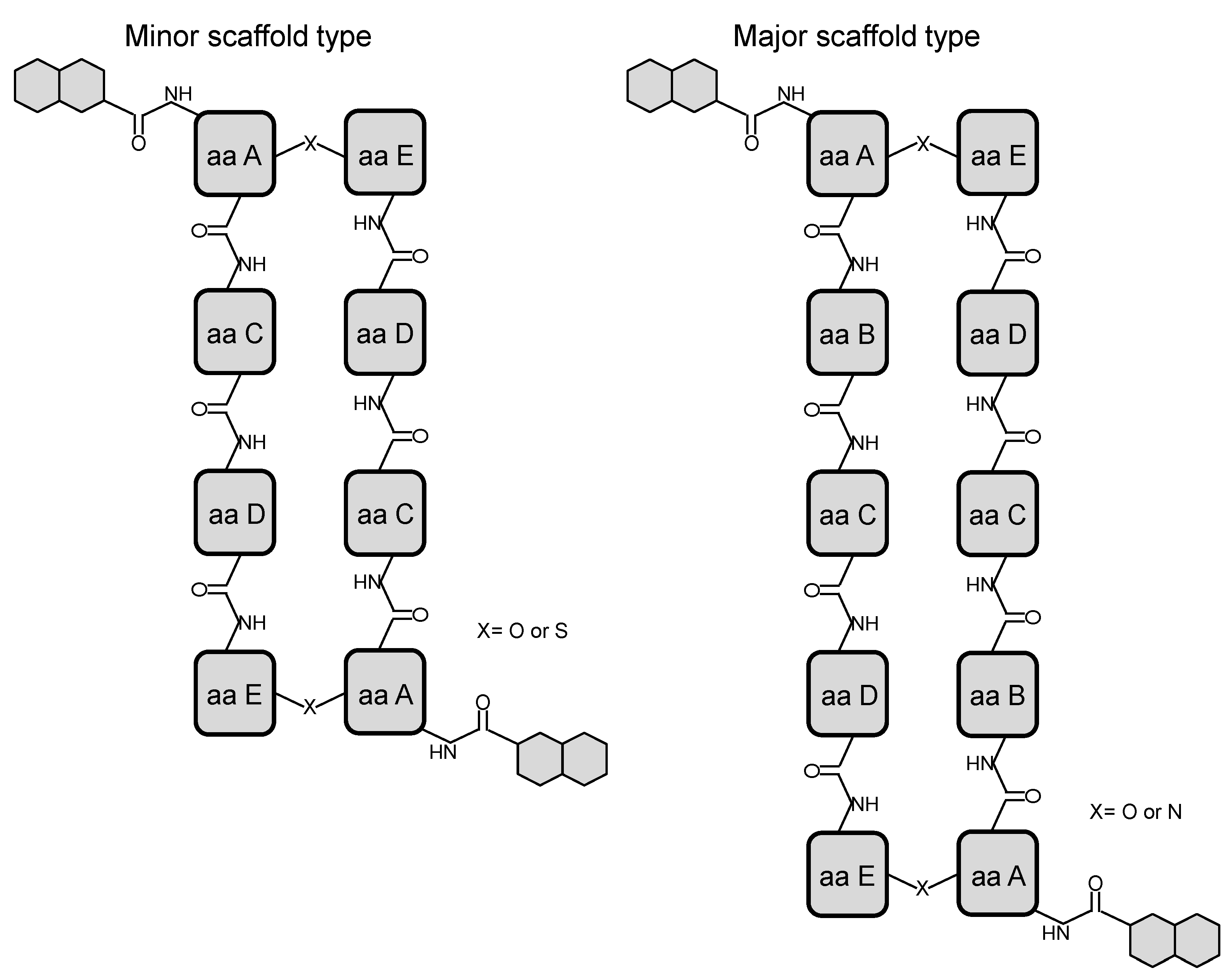{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Natural Derivatives | Producer | Origin of Strain | Synthetic Derivaties |
|---|---|---|---|---|
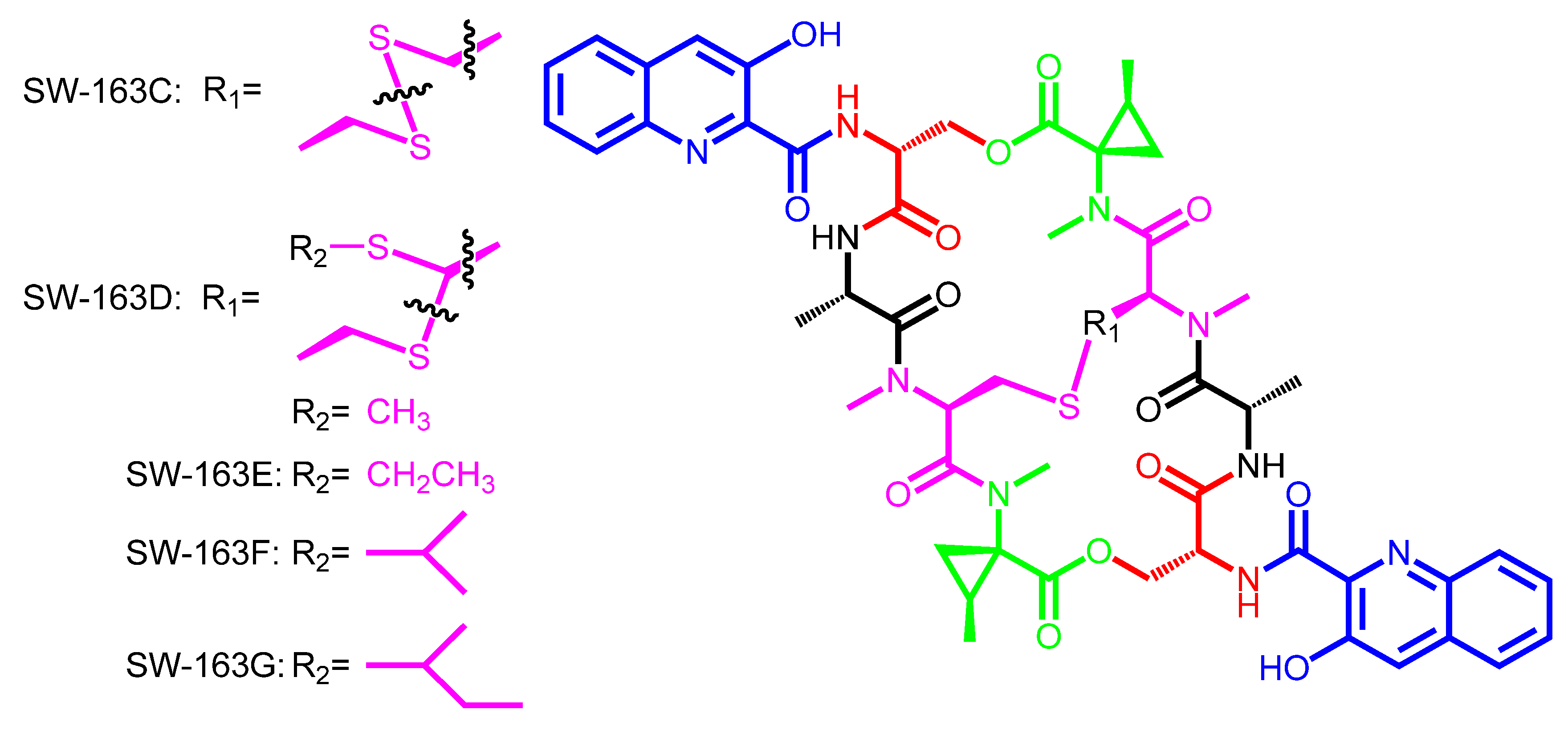
| SW-163C | SW-163D | Streptomyces sp. SNA15896 | Japan | − |
| SW-163E | ||||
| SW-163F | ||||
| SW-163G | ||||
| Triostin A | − | Streptomyces triostinicus | Japan | TANDEM |
| Streptomyces lasaliensis | SNAC-derivatives | |||
| Streptomyces aureus s-2-210-L | ||||
| Echinomycin A (Quinomycin A) | Quinomycin B | Streptomyces echinatus | Angola, Japan | Ecolymicin C |
| Quinomycin C | Streptomyces lasaliensis | 1QN | ||
| Quinomycin D | Streptomyces sp. KN-0647 | 2QN | ||
| Quinomycin E | Streptomyces sp. 732 | |||
| Streptomyces griseovariabilis subsp. bandungensis | ||||
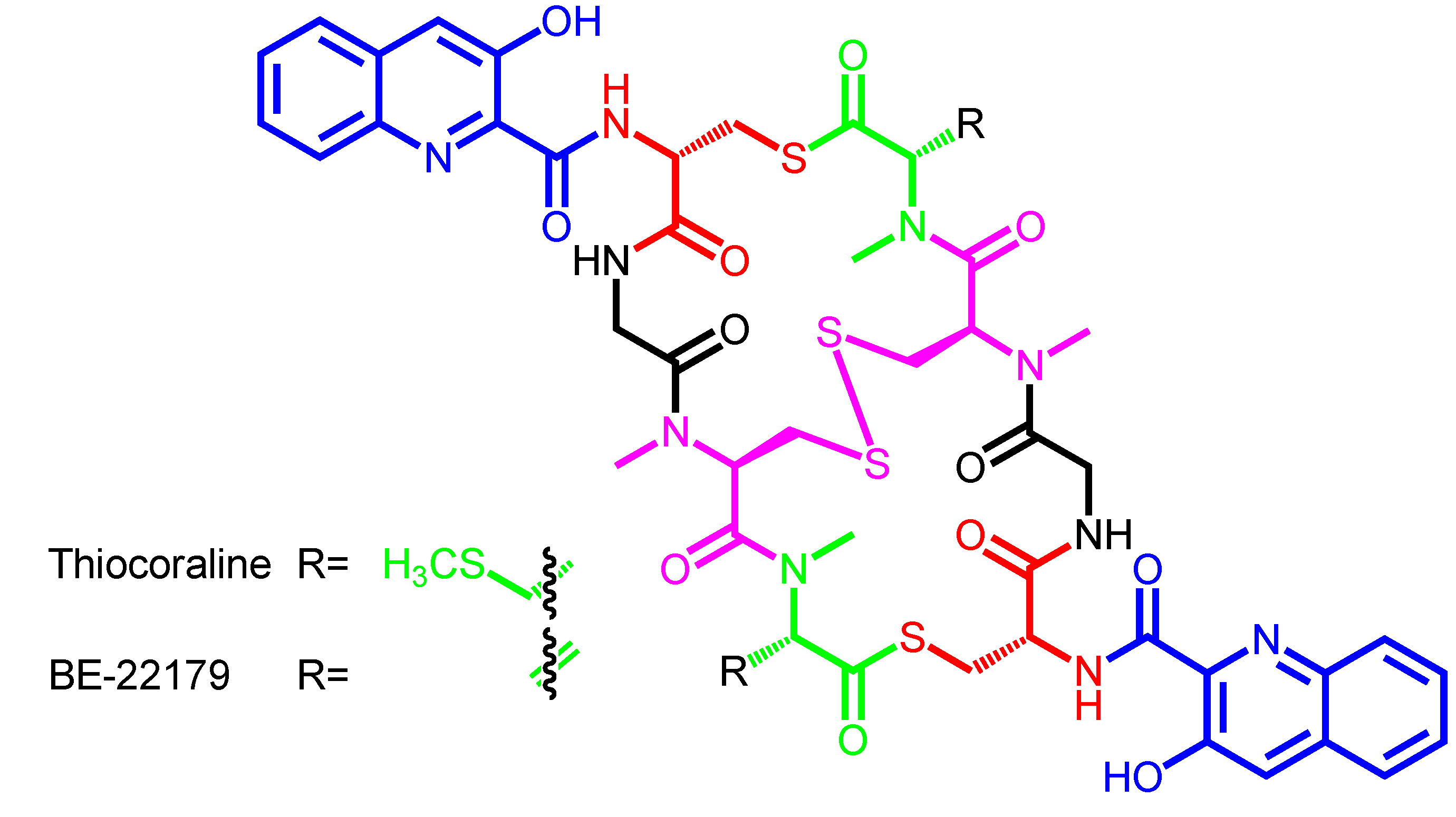
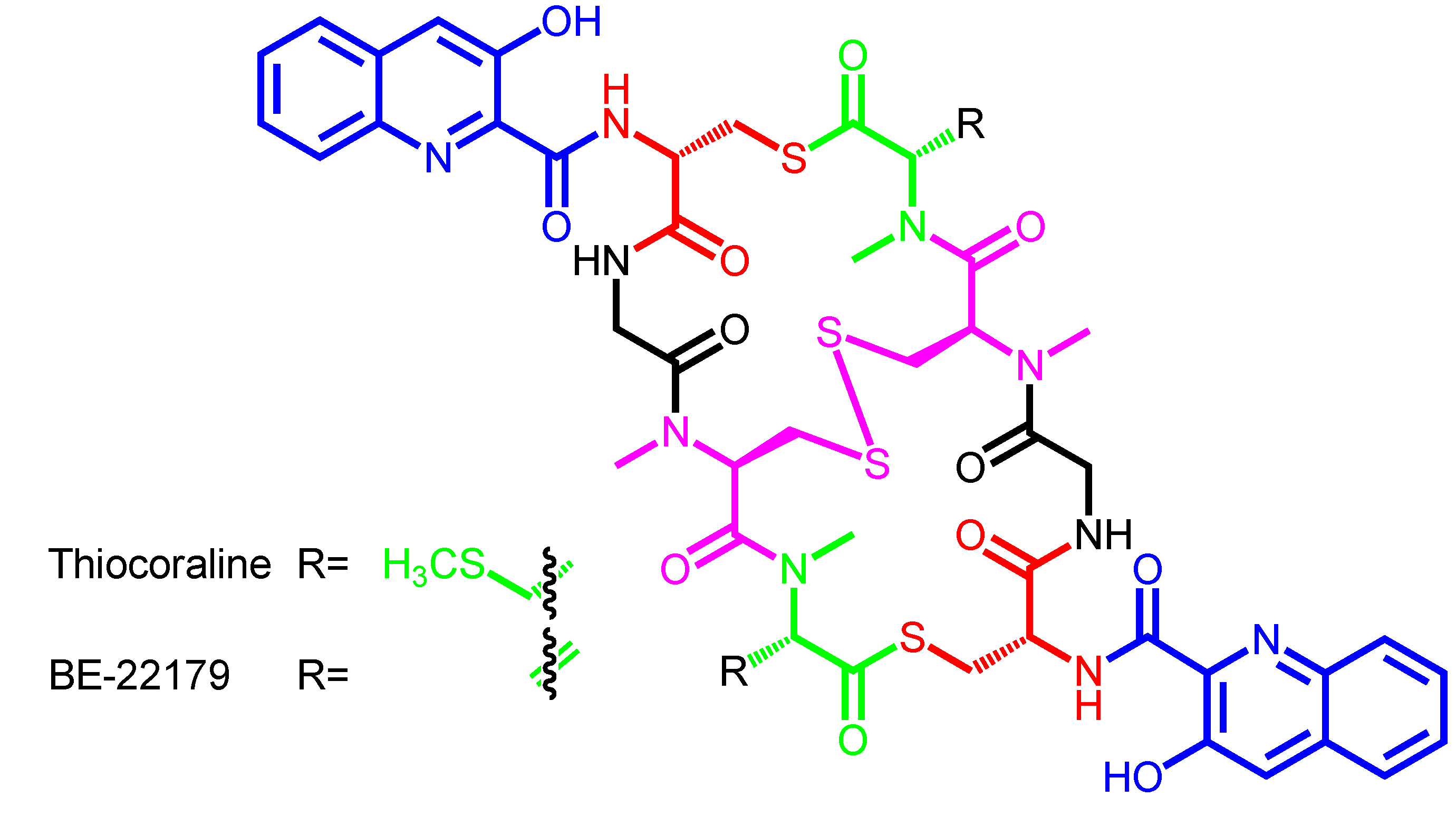
| Thiocoraline | − | Micromonospora marina ML1 | Mozambique Strait | Marburg-derivatives |
| Micromonospora marina ACM2-092 | Azathiocoraline | |||
| NMe-Azathiocoraline | ||||
| Oxathiocoraline | ||||
| BE-22179 | − | Streptomyces sp. A22179 | Japan | − |
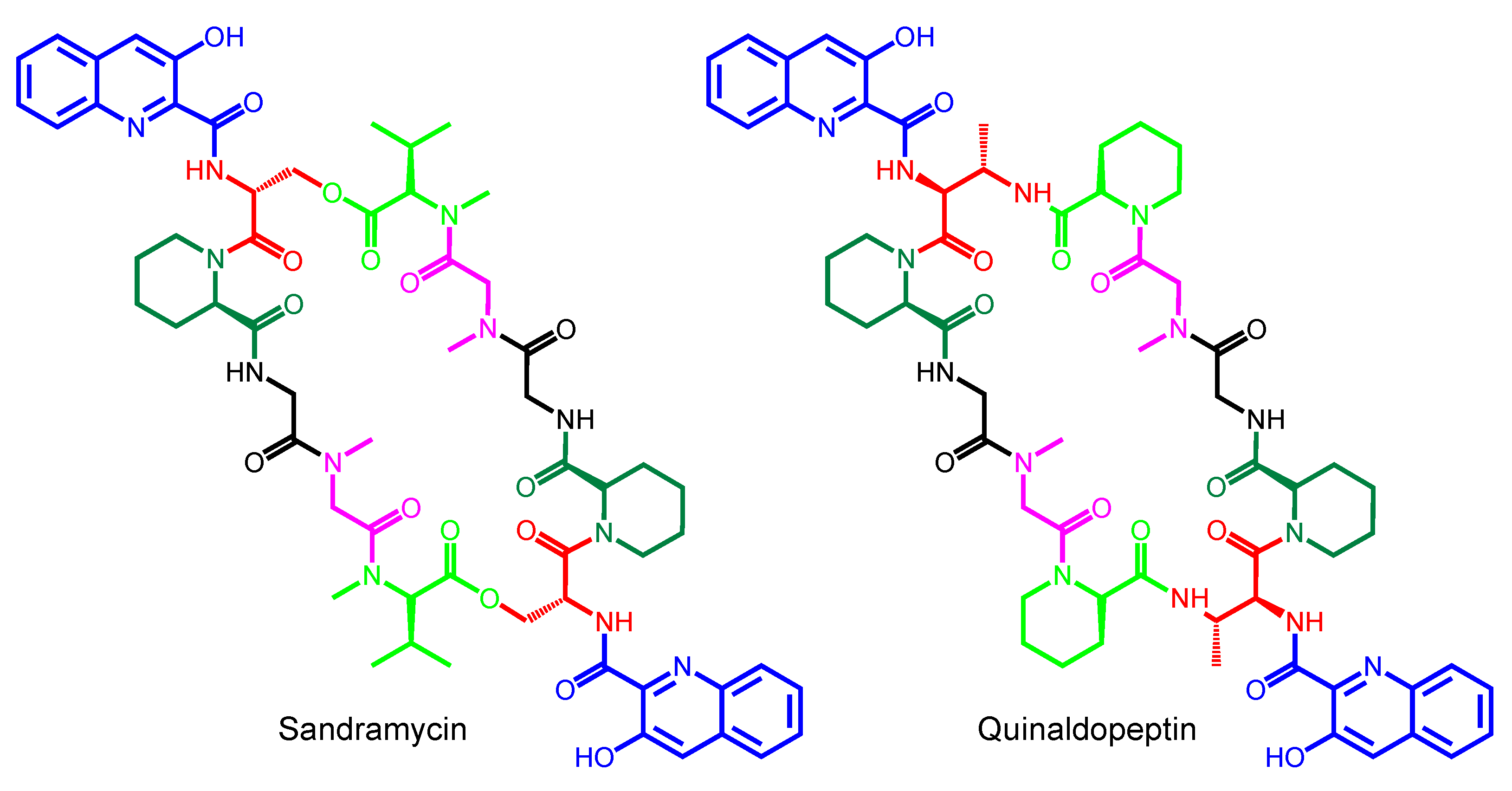
| Sandramycin | − | Nocardioides sp. ATCC 39419 | Mexico | − |
| Quinaldopeptin | − | Streptoverticillium album Q132-6 | India | − |
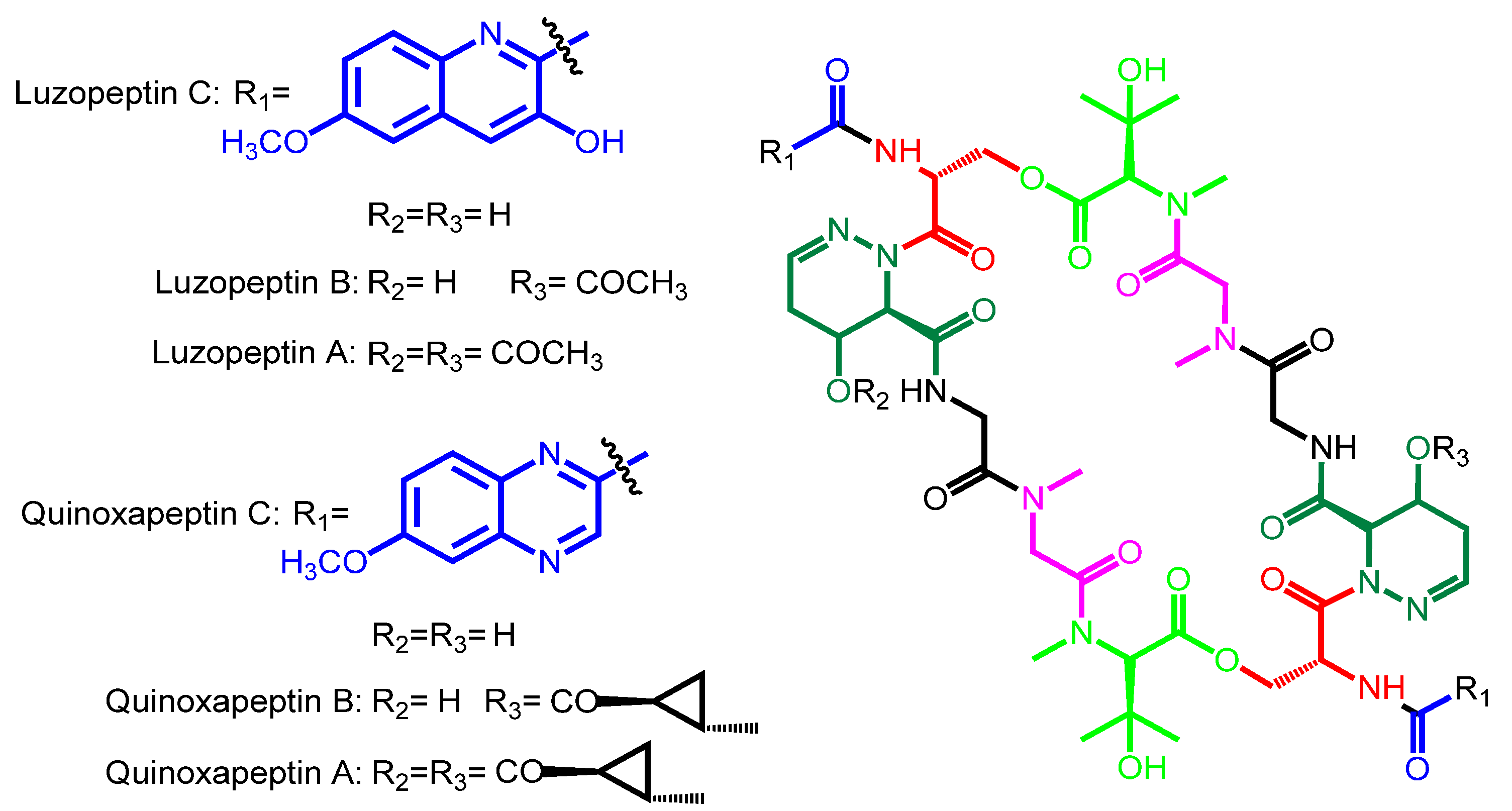
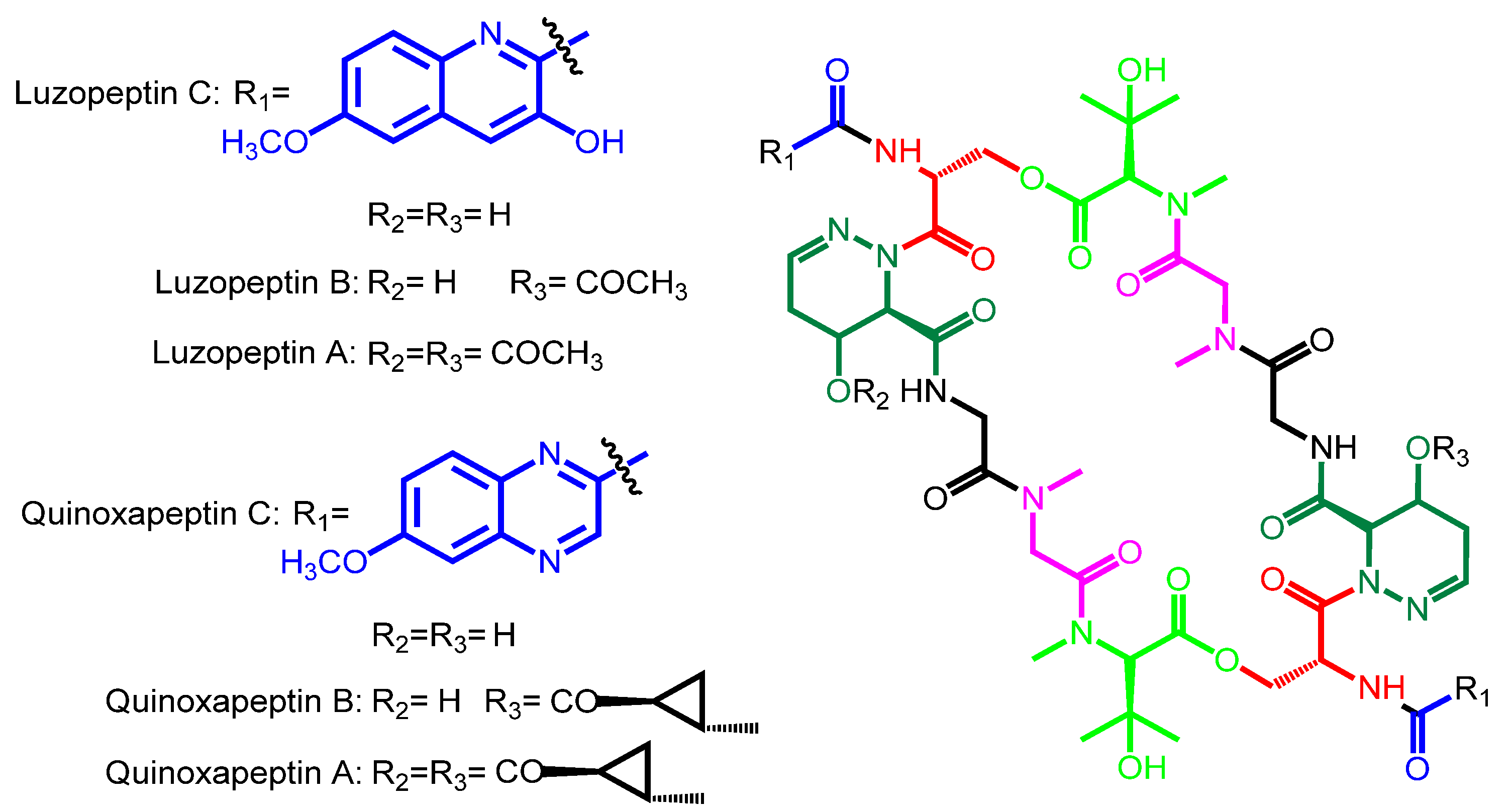
| Luzopeptin C | Luzopeptin A | Actinomadura luzonensis | Philippines | − |
| Luzopeptin B | ||||
| Quinoxapeptin C | Quinoxapeptin A Quinoxapeptin B | Unclassified nocardioform actinomycete | Alaska | − |
| Compound | Chromophore | Amino Acid A | Amino Acid B | Amino Acid C | Amino Acid D | Amino Acid E |
|---|---|---|---|---|---|---|
| (unknown ancestor X) | 3HQA | d-Ser | − | l-Ala | N-methyl-l-Cys | N-methyl-l-Val |
| (disulfide bridge) | ||||||
| SW-163C | 3HQA | d-Ser | − | l-Ala | N-methyl-l-Cys | N-methyl-Norcoronamic acid |
| (disulfide bridge) | ||||||
| SW-163D,E,F,G | 3HQA | d-Ser | − | l-Ala | N-methyl-l-Cys | N-methyl-Norcoronamic acid |
| (thioacetal bridge) | ||||||
| Triostin A | QXCA | d-Ser | − | l-Ala | N-methyl-l-Cys | N-methyl-l-Val |
| (disulfide bridge) | ||||||
| Echinomycin A | QXCA | d-Ser | − | l-Ala | N-methyl-l-Cys | N-methyl-l-Val |
| (thioacetal bridge) | ||||||
| Thiocoraline | 3HQA | d-Cys | − | Gly | N-methyl-l-Cys | N,S-dimethyl-l-Cys |
| (disulfide bridge) | ||||||
| BE-22179 | 3HQA | d-Cys | − | Gly | N-methyl-l-Cys | N-methyl-dehydro-Ala |
| (disulfide bridge) | ||||||
| Sandramycin | 3HQA | d-Ser | l-Pipecolic acid | Gly | Sarcosine | N-methyl-l-Val |
| Quinaldopeptin | 3HQA | d-DABA | l-Pipecolic acid | Gly | Sarcosine | l-Pipecolic acid |
| Luzopeptin C | 6-methoxy-3HQA | d-Ser | 4-OH-Δ-piperazic acid | Gly | Sarcosine | β-OH-N-methyl-l-Val |
| Quinoxapeptin C | 6-methoxy-QXCA | d-Ser | 4-OH-Δ-piperazic acid | Gly | Sarcosine | β-OH-N-methyl-l-Val |
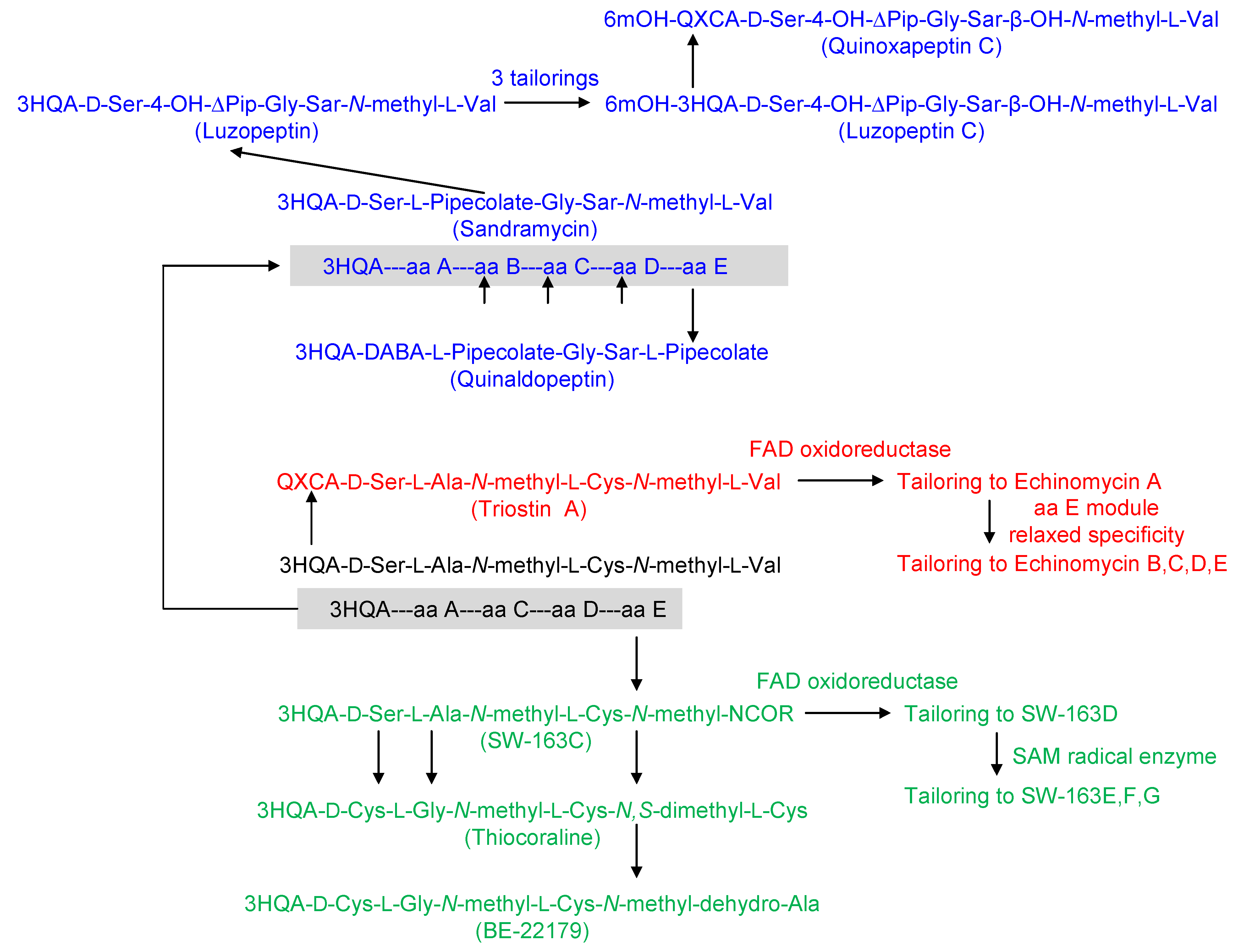
2. Structural Diversity and Modularity in the Bisintercalators Family: Suggestions of a Common Evolutionary Origin?

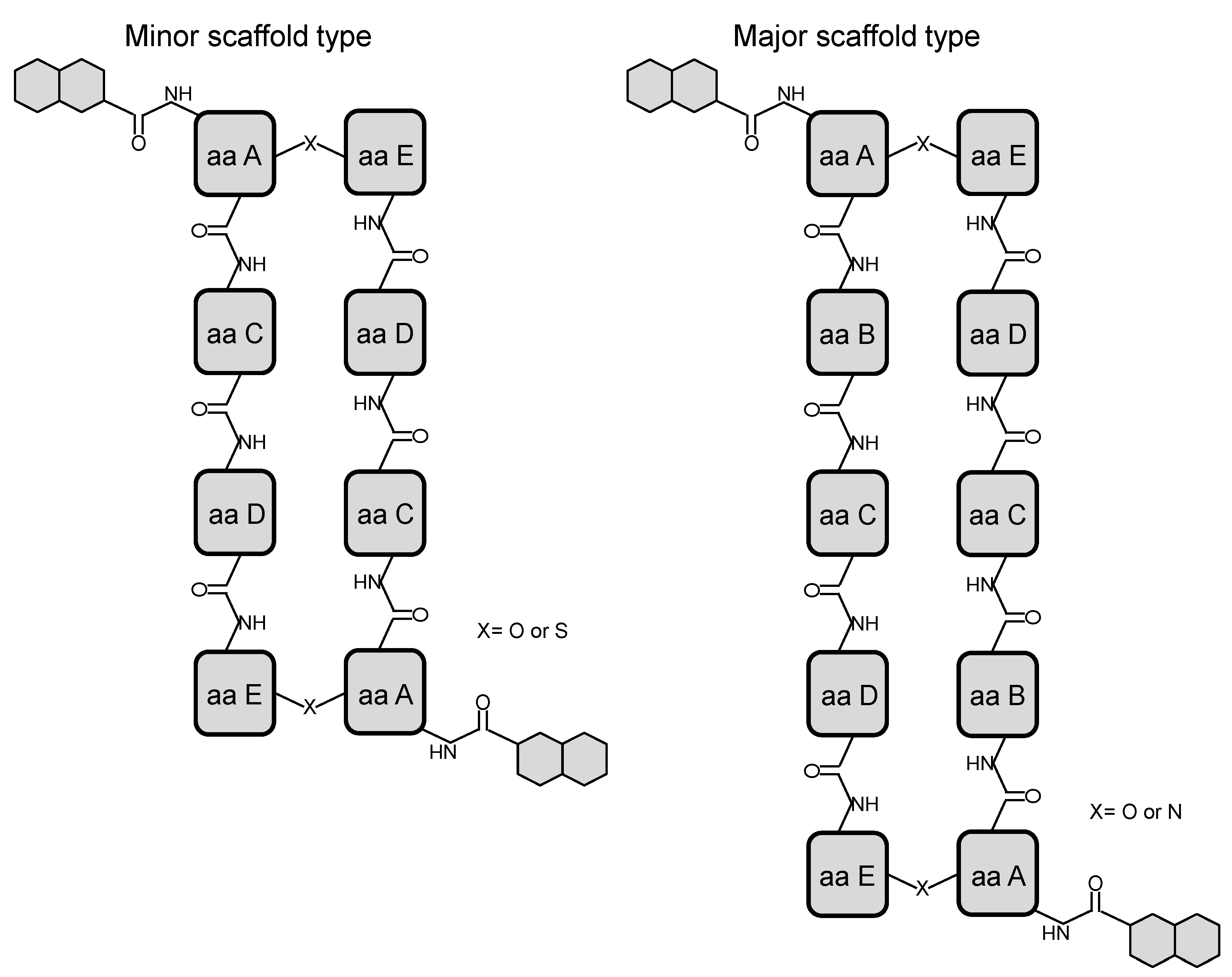
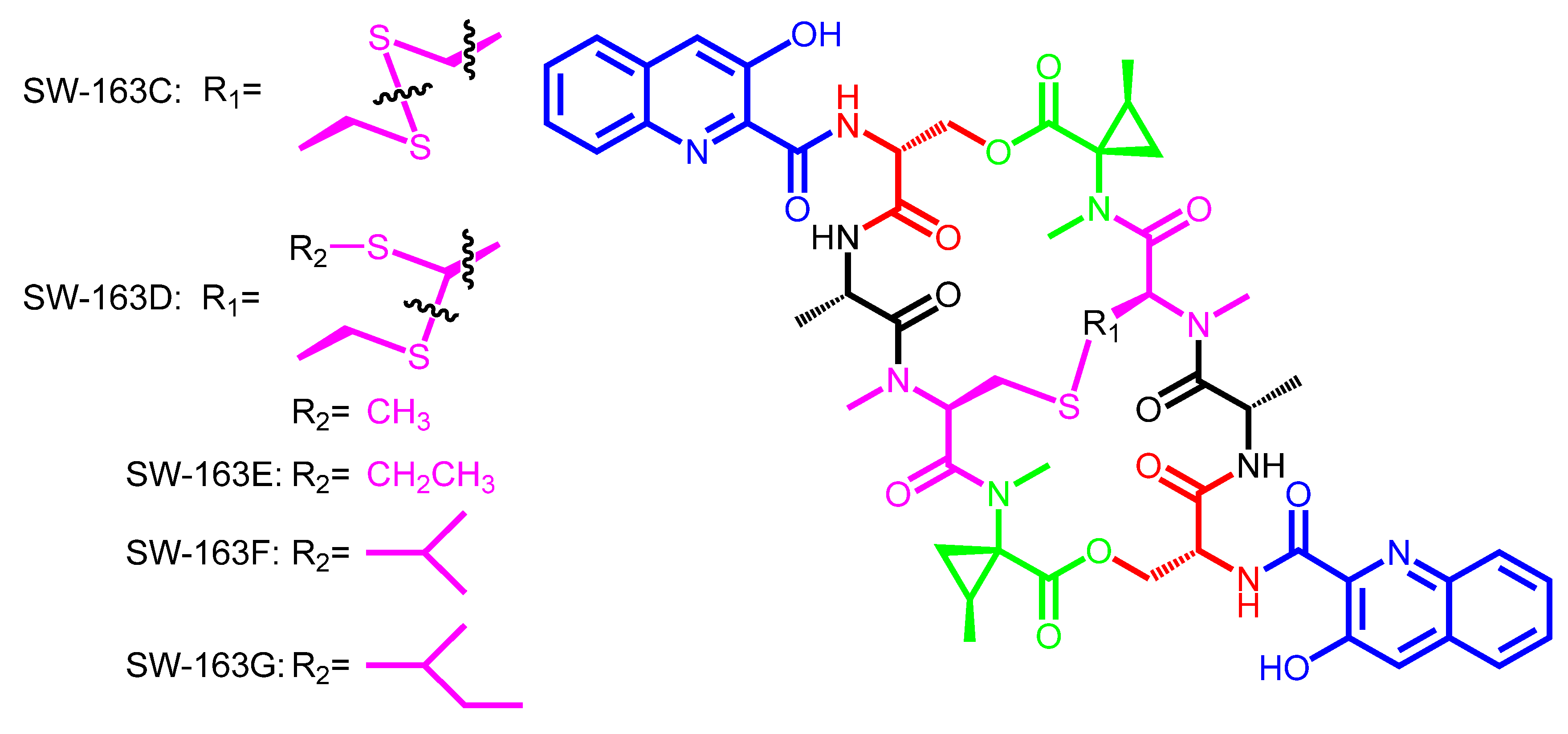
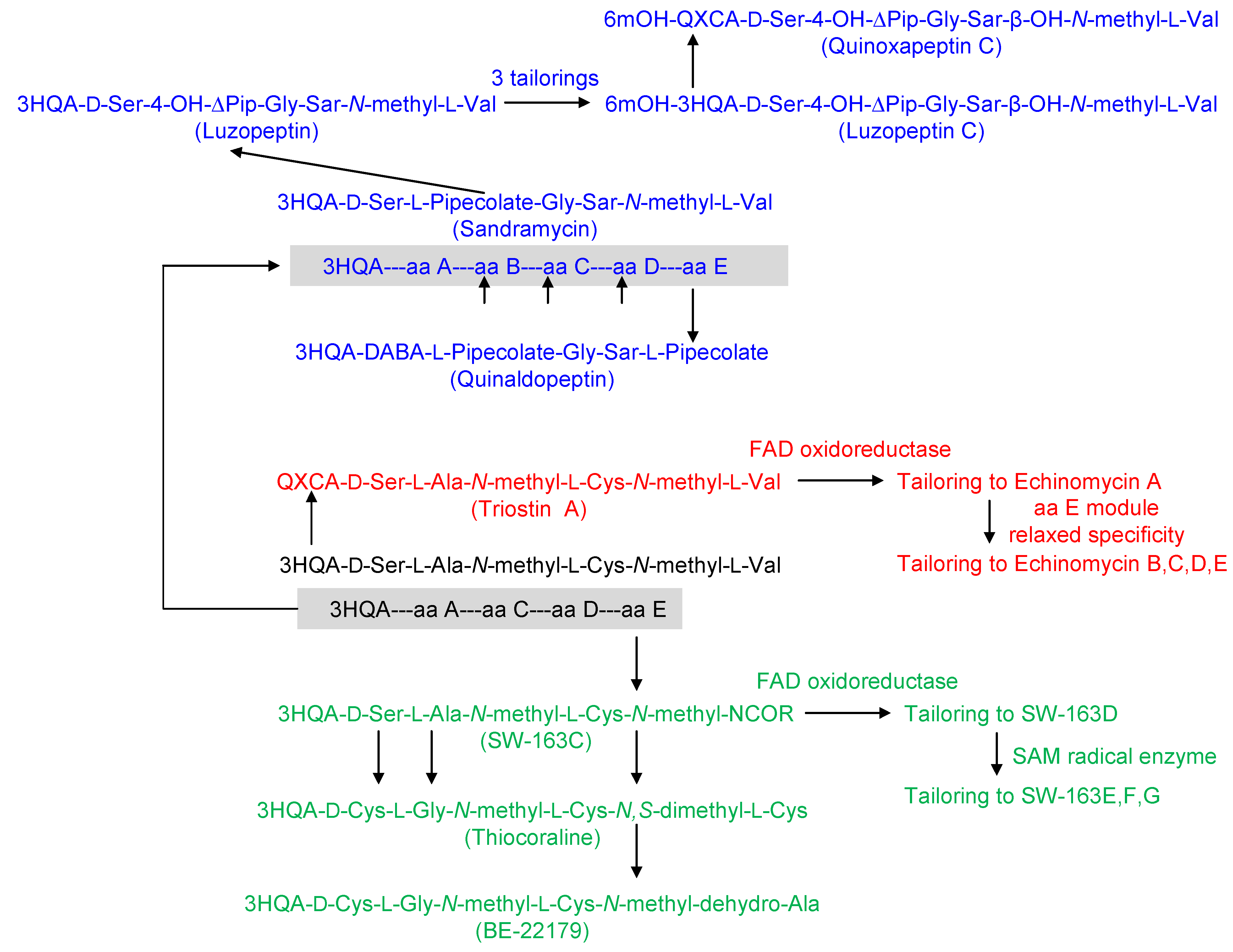
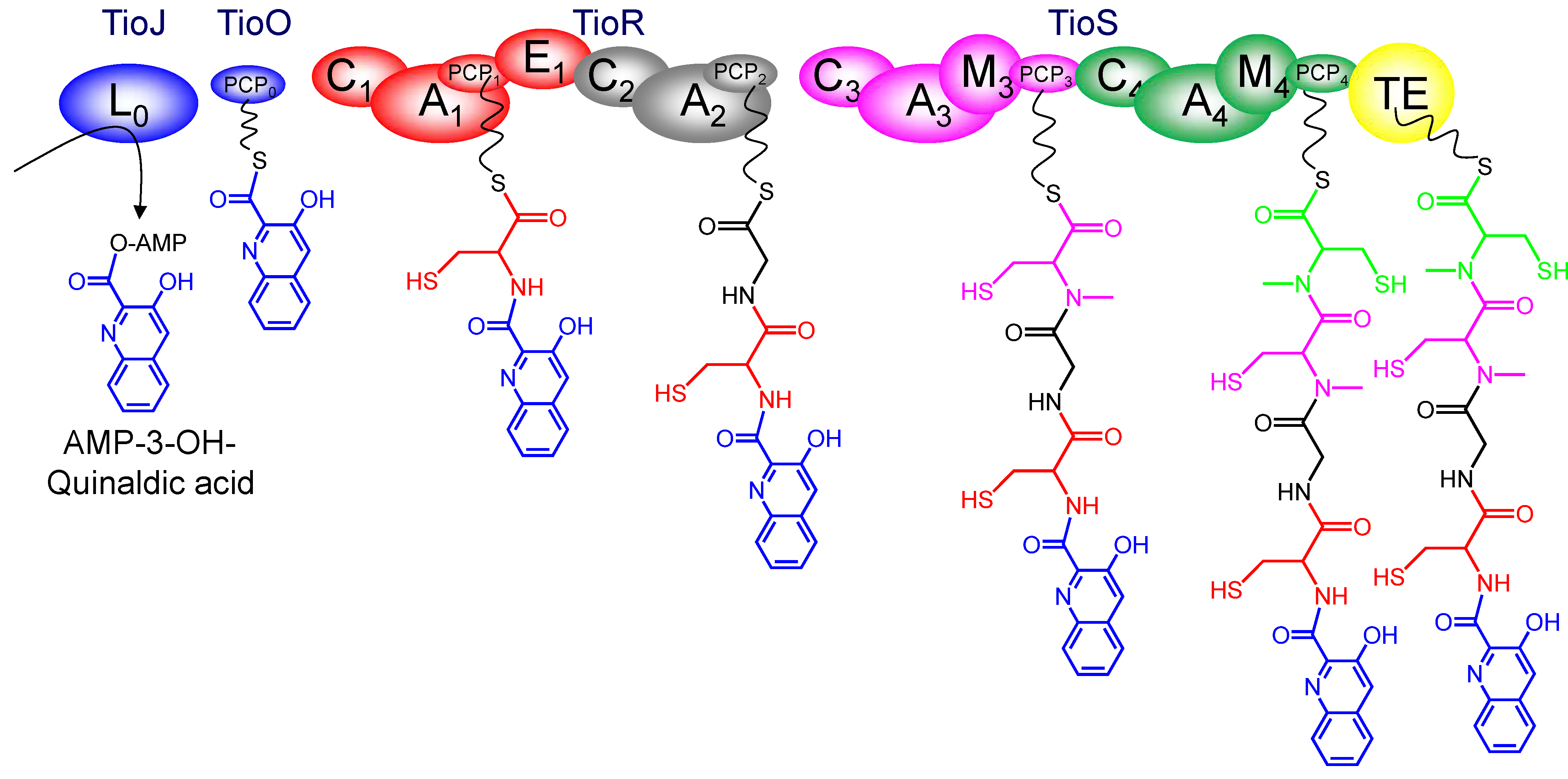
3. Modular Biosynthesis in the Bisintercalators Family
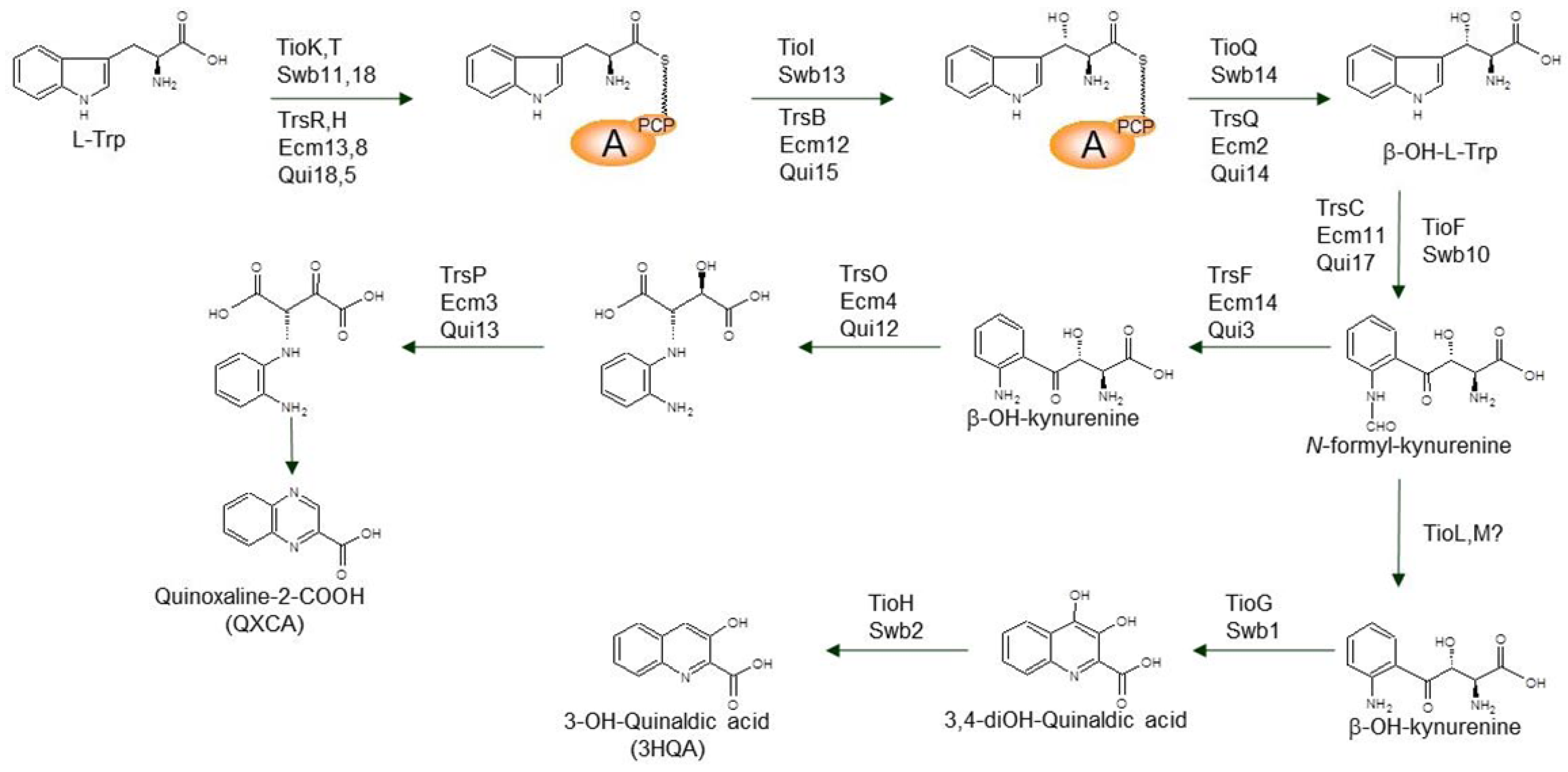
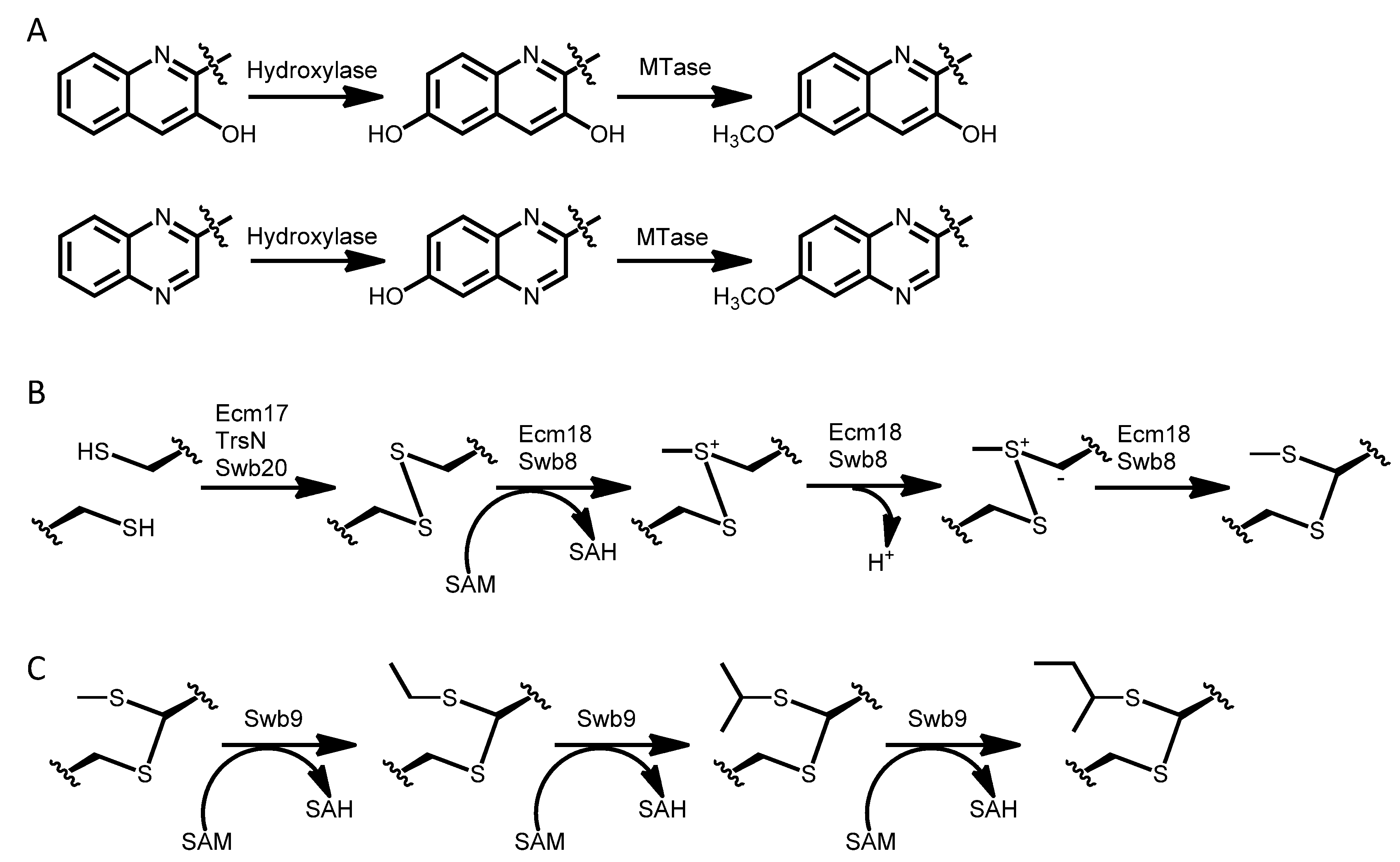
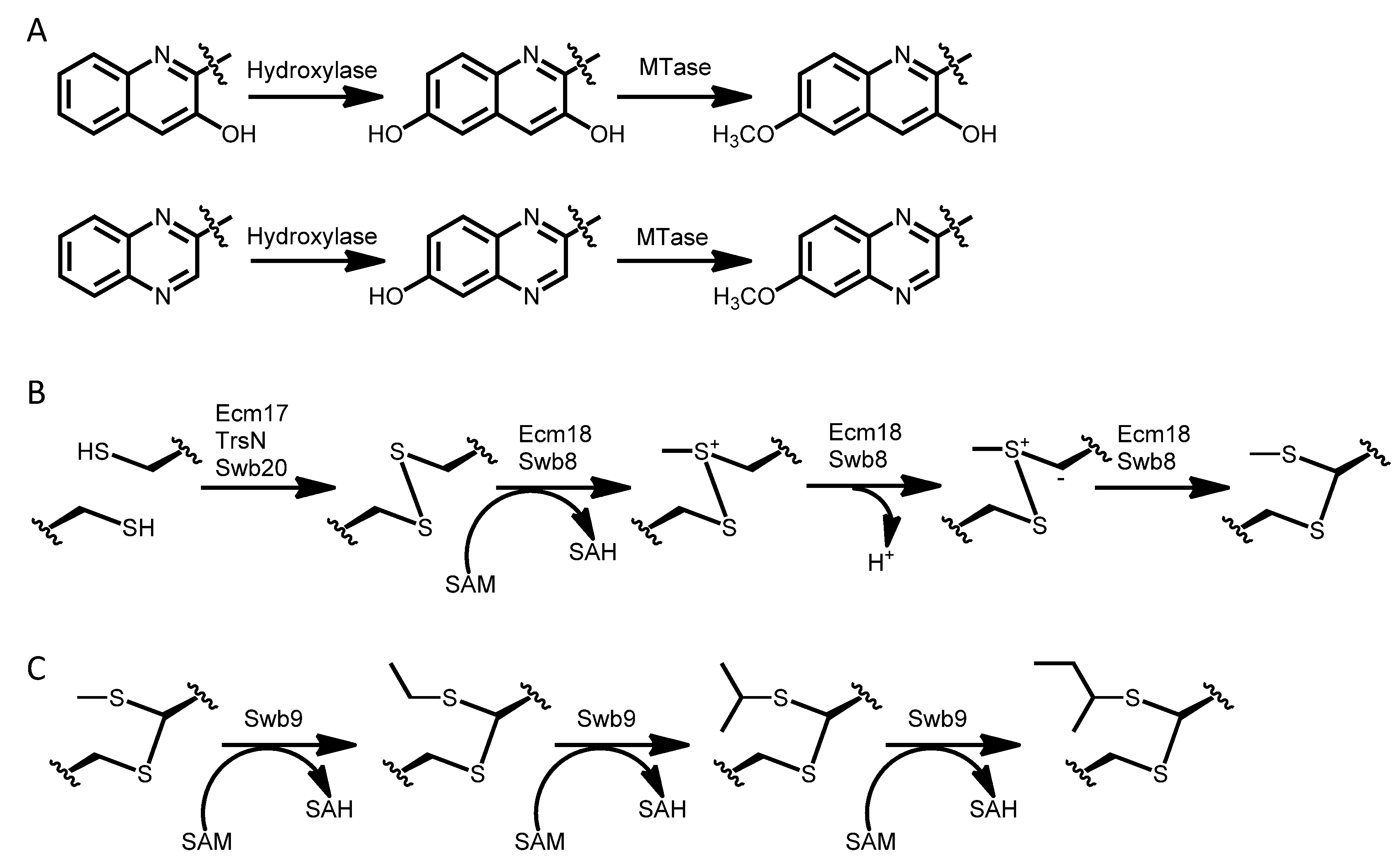
3.1. Biosynthesis of the Chromophore Moieties
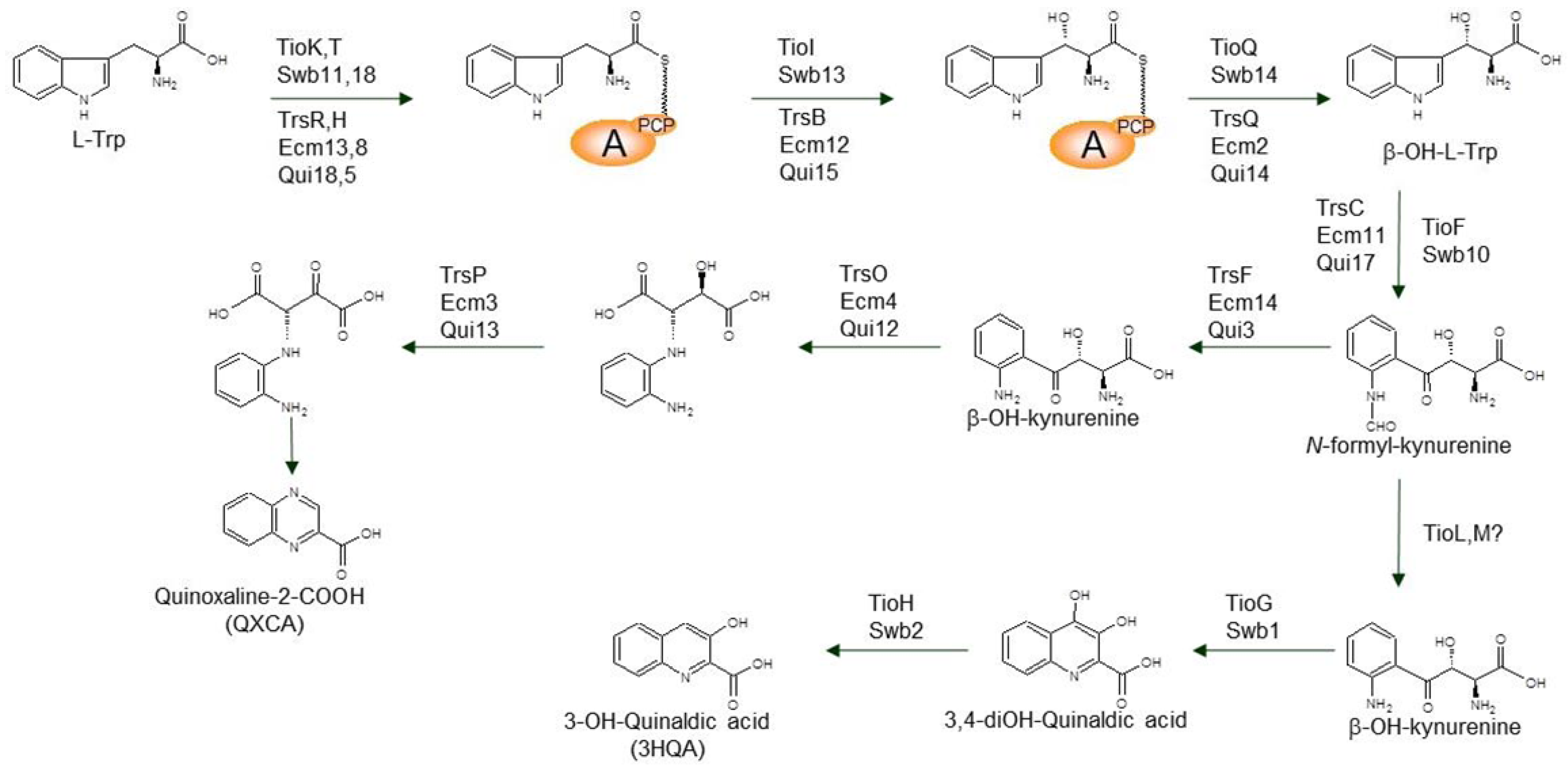
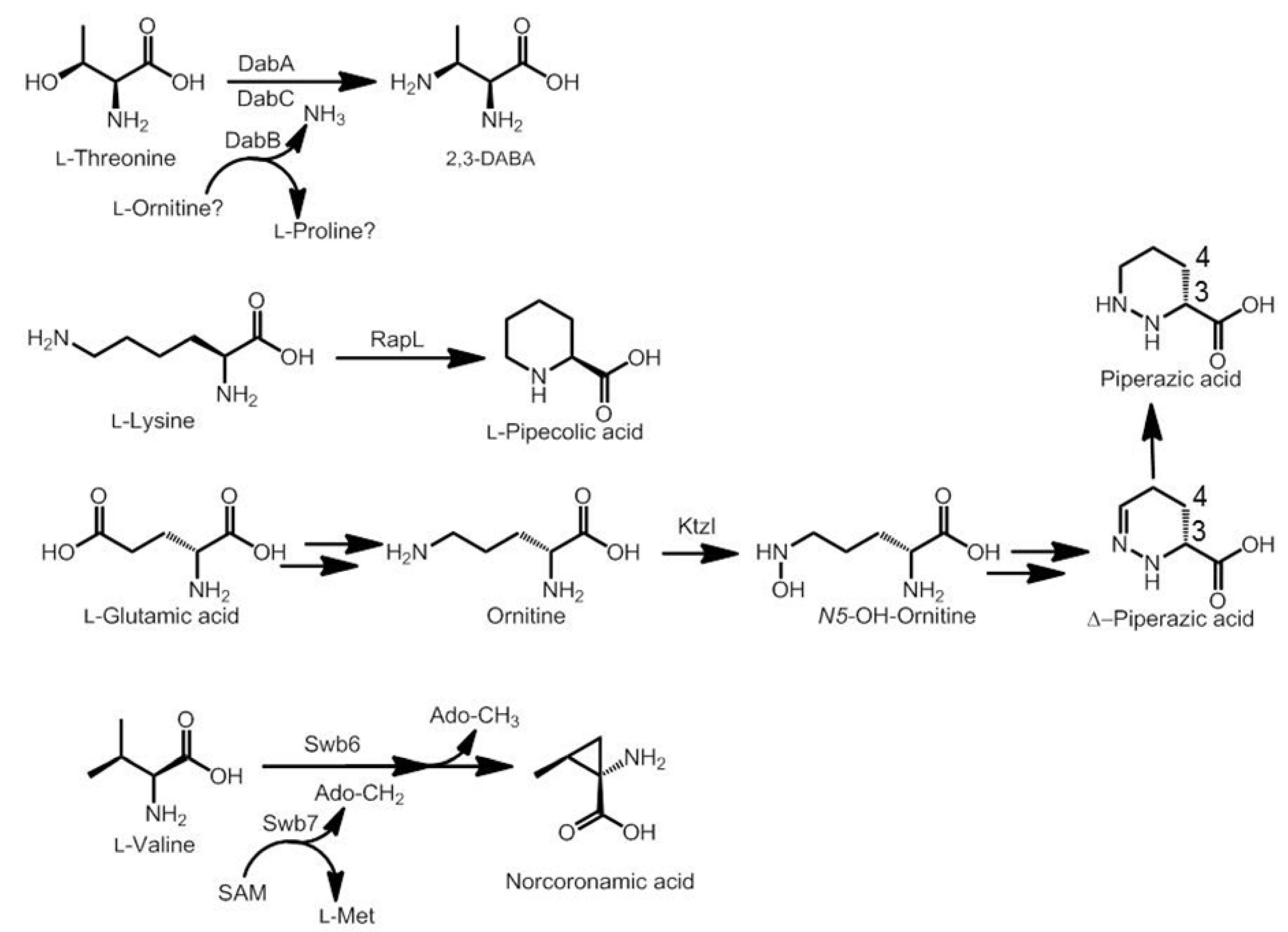
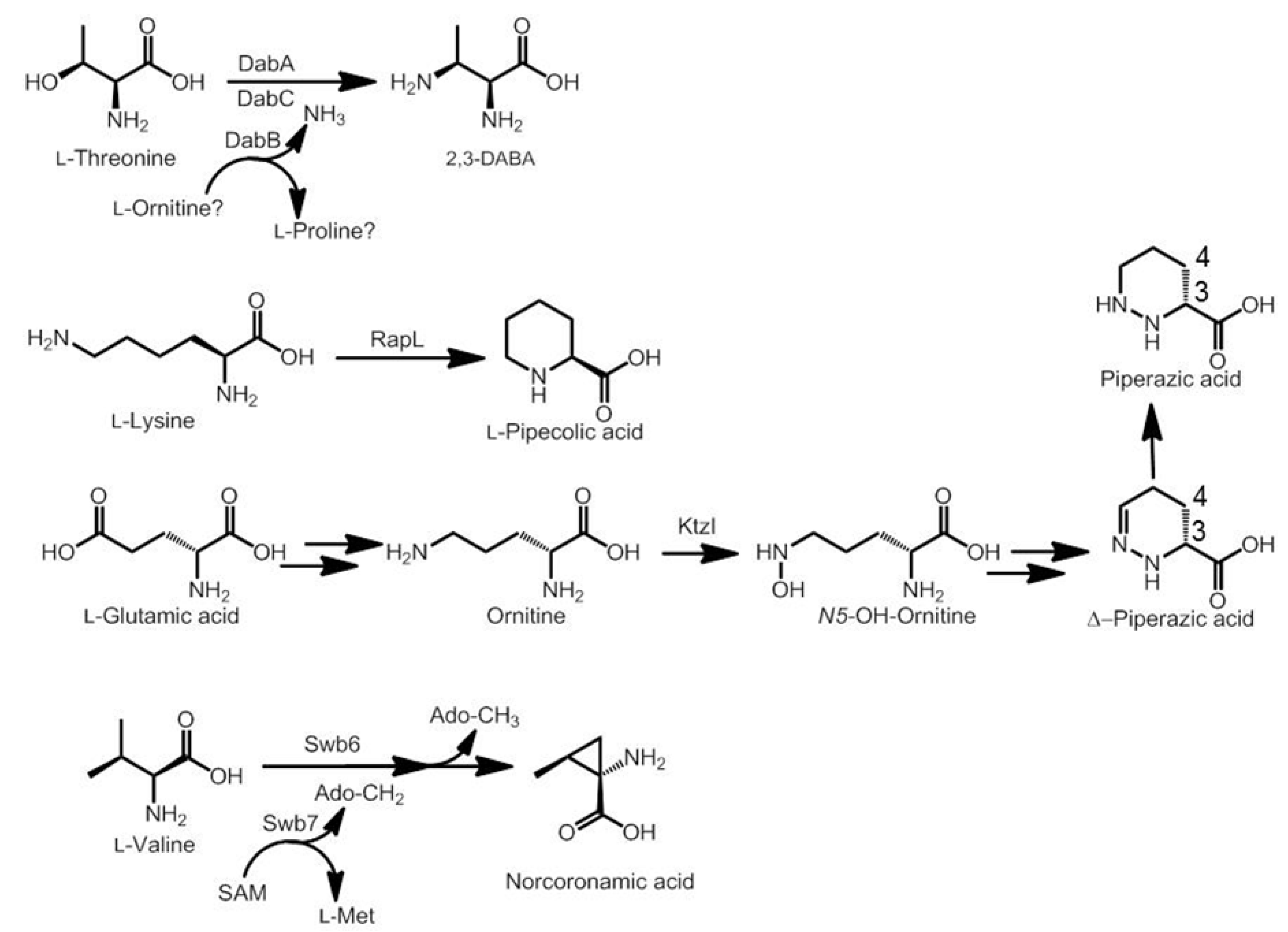
3.2. Biosynthesis of Distinctive Amino Acids
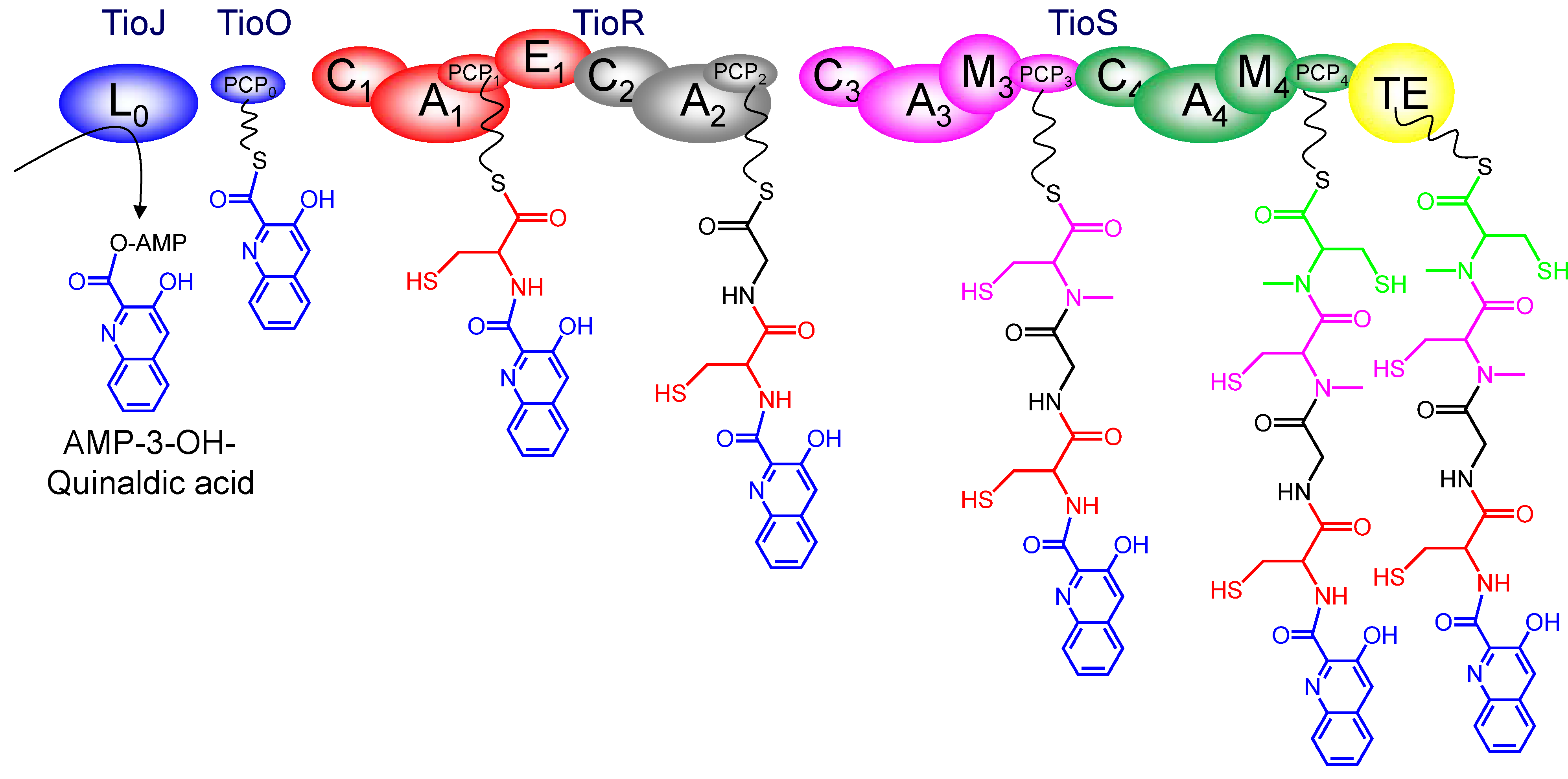
3.3. Use of the Chromophore Moiety as Starter in the NRPS Assembly Line
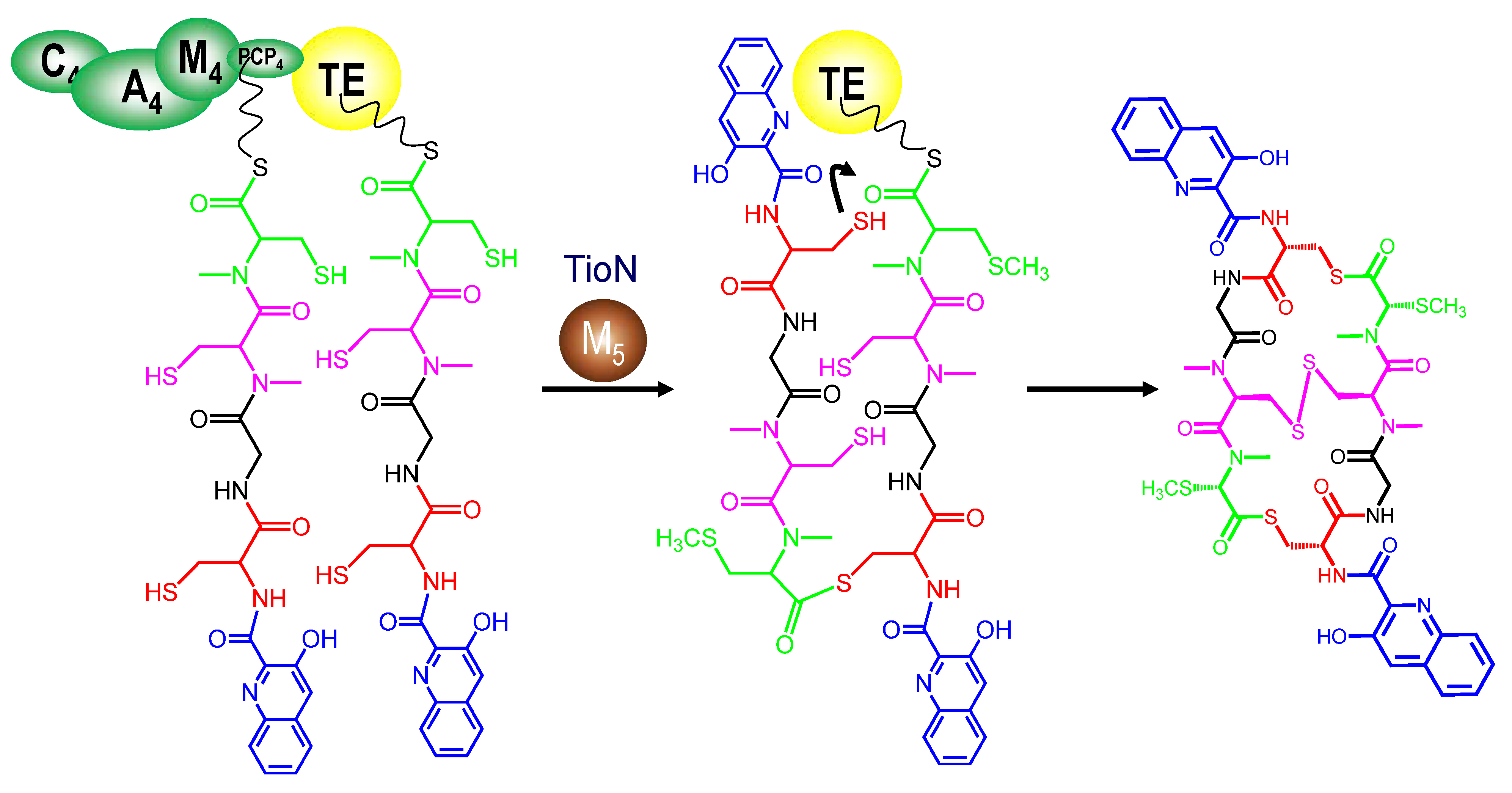
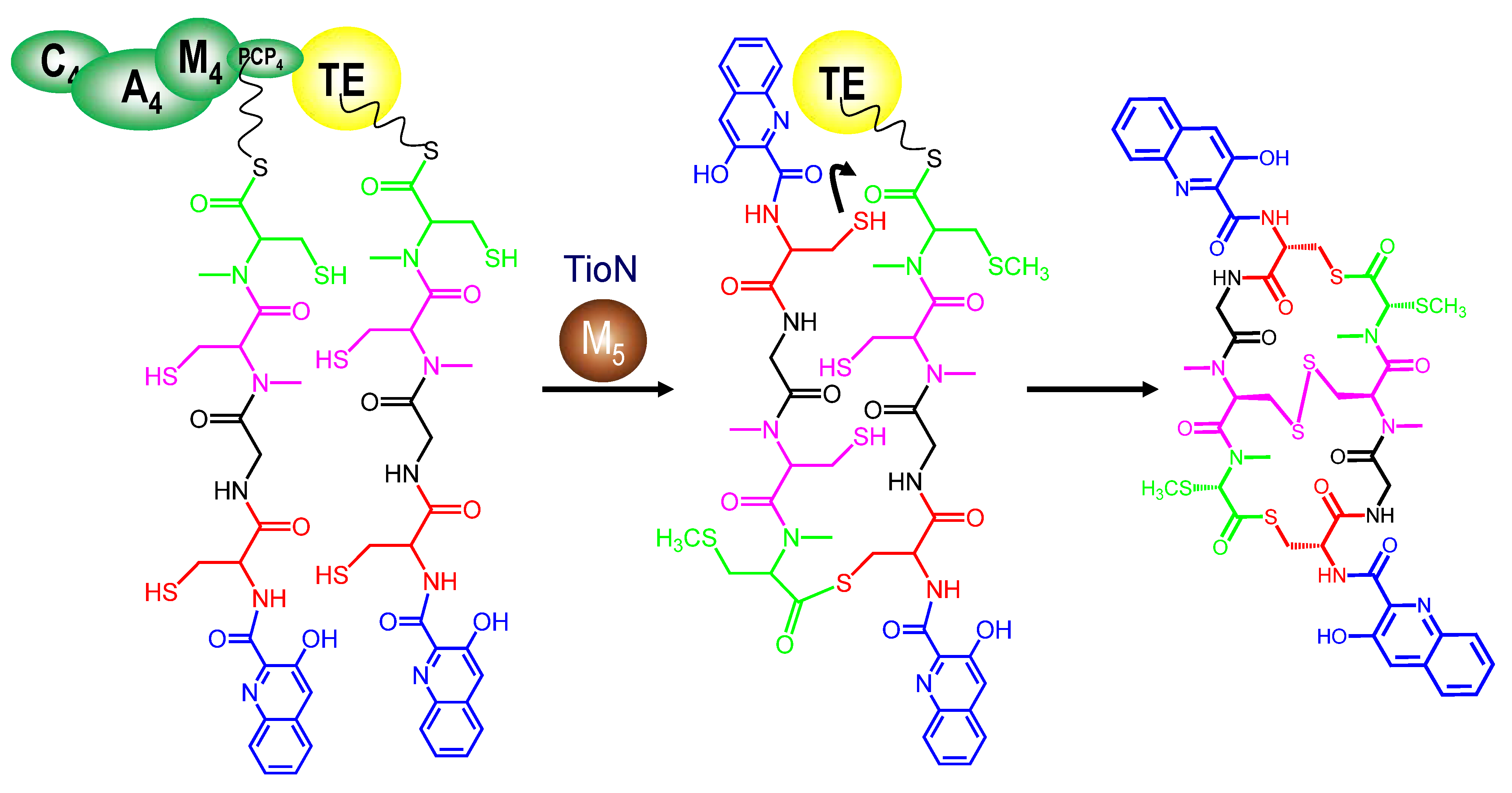
3.4. Dimerization, Cyclization and Scission of the NRPS Scaffold
3.5. Tailoring Enzymes
3.6. Resistance and Secretion Enzymes
4. Bioactivity of Bisintercalator Compounds
4.1. Binding to the DNA Minor Groove
4.2. Biological Activities
5. Unnatural Derivatives
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Keller, U.; Schauwecker, F. Nonribosomal biosynthesis of microbial chromopeptides. Prog. Nucleic Acid Res. Mol. Biol. 2001, 70, 233–289. [Google Scholar] [CrossRef]
- Quigley, G.J.; Ughetto, G.; Van der Marel, G.A.; Van Boom, J.H.; Wang, A.H.; Rich, A. Non-Watson-Crick G.C and A.T base pairs in a DNA-antibiotic complex. Science 1986, 232, 1255–1258. [Google Scholar]
- Chen, H.; Patel, D.J. Solution structure of a quinomycin bisintercalator-DNA complex. J. Mol. Biol. 1995, 246, 164–179. [Google Scholar] [CrossRef]
- Park, J.Y.; Ryang, Y.S.; Shim, K.Y.; Lee, J.I.; Kim, H.S.; Kim, Y.H.; Kim, S.K. Molecular signaling cascade in DNA bisintercalator, echinomycin-induced apoptosis of HT-29 cells: Evidence of the apoptotic process via activation of the cytochrome c-ERK-caspase-3 pathway. Int. J. Biochem. Cell Biol. 2006, 38, 244–254. [Google Scholar] [CrossRef]
- Corbaz, R.; Ettlinger, L.; Gäumann, E.; Keller-Schierlein, W.; Kradolfer, F.; Neipp, L.; Prelog, V.; Reusser, P.; Zähner, H. Stoffwechselprodukte von Actinomyceten. 7. Echinomycin. Helv. Chim. Acta 1957, 40, 199–204. [Google Scholar] [CrossRef]
- Yoshida, T.; Katagiri, K.; Yokozawa, S. Studies on quinoxaline antibiotics. II. Isolation and properties of quinomycins A, B and C. J. Antibiot. 1961, 14, 330–334. [Google Scholar]
- Steinerová, N.; Lipavská, H.; Stajner, K.; Cáslavská, J.; Blumauerová, M.; Cudlín, J.; Vanĕk, Z. Production of quinomycin A in Streptomyces lasaliensis. Folia Microbiol. 1987, 32, 1–5. [Google Scholar] [CrossRef]
- Liu, H.; Qin, S.; Wang, Y.; Li, W.; Zhang, J. Insecticidal action of Quinomycin A from Streptomyces sp. KN-0647, isolated from a forest soil. World J. Microbiol. Biotechnol. 2008, 24, 2243–2248. [Google Scholar] [CrossRef]
- Zhang, C.; Kong, L.; Liu, Q.; Lei, X.; Zhu, T.; Yin, J.; Lin, B.; Deng, Z.; You, D. In vitro characterization of echinomycin biosynthesis: formation and hydroxylation of l-tryptophanyl-S-enzyme and oxidation of (2S,3S) β-hydroxytryptophan. PLoS One 2013, 8, e56772. [Google Scholar]
- Kinashi, H.; Otten, S.L.; Duncan, J.S.; Hutchinson, C.R. Frequent loss and restoration of antibiotic production by Streptomyces lasaliensis. J. Antibiot. 1988, 41, 624–637. [Google Scholar] [CrossRef]
- Kinashi, H. Giant linear plasmids in Streptomyces: A treasure trove of antibiotic biosynthetic clusters. J. Antibiot. 2011, 64, 19–25. [Google Scholar] [CrossRef]
- Dawson, S.; Malkinson, J.P.; Paumier, D.; Searcey, M. Bisintercalator natural products with potential therapeutic applications: Isolation, structure determination, synthetic and biological studies. Nat. Prod. Rep. 2007, 24, 109–126. [Google Scholar] [CrossRef]
- Zolova, O.E.; Mady, A.S.; Garneau-Tsodikova, S. Recent developments in bisintercalator natural products. Biopolymers 2010, 93, 777–790. [Google Scholar] [CrossRef]
- Cornish, A.; Waring, M.J.; Nolan, R.D. Conversion of triostins to quinomycins by protoplasts of Streptomyces echinatus. J. Antibiot. 1983, 36, 1664–1670. [Google Scholar] [CrossRef]
- Lombó, F.; Velasco, A.; Castro, A.; de la Calle, F.; Braña, A.F.; Sánchez-Puelles, J.M.; Méndez, C.; Salas, J.A. Deciphering the biosynthesis pathway of the antitumor thiocoraline from a marine actinomycete and its expression in two Streptomyces species. Chembiochem 2006, 7, 366–376. [Google Scholar] [CrossRef]
- Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 2013, 12, 371–387. [Google Scholar] [CrossRef]
- Ohkuma, H.; Sakai, F.; Nishiyama, Y.; Ohbayashi, M.; Imanishi, H.; Konishi, M.; Miyaki, T.; Koshiyama, H.; Kawaguchi, H. BBM-928, a new antitumor antibiotic complex. I. Production, isolation, characterization and antitumor activity. J. Antibiot. 1980, 33, 1087–1097. [Google Scholar] [CrossRef]
- Romero, F.; Espliego, F.; Pérez Baz, J.; García de Quesada, T.; Grávalos, D.; de la Calle, F.; Fernández-Puentes, J.L. Thiocoraline, a new depsipeptide with antitumor activity produced by a marine Micromonospora. I. Taxonomy, fermentation, isolation, and biological activities. J. Antibiot. 1997, 50, 734–737. [Google Scholar] [CrossRef]
- Matson, J.A.; Colson, K.L.; Belofsky, G.N.; Bleiberg, B.B. Sandramycin, a novel antitumor antibiotic produced by a Nocardioides sp. II. Structure determination. J. Antibiot. 1993, 46, 162–166. [Google Scholar] [CrossRef]
- Okada, H.; Suzuki, H.; Yoshinari, T.; Arakawa, H.; Okura, A.; Suda, H.; Yamada, A.; Uemura, D. A new topoisomerase II inhibitor, BE-22179, produced by a streptomycete. I. Producing strain, fermentation, isolation and biological activity. J. Antibiot. 1994, 47, 129–135. [Google Scholar] [CrossRef]
- Toda, S.; Sugawara, K.; Nishiyama, Y.; Ohbayashi, M.; Ohkusa, N.; Yamamoto, H.; Konishi, M.; Oki, T. Quinaldopeptin, a novel antibiotic of the quinomycin family. J. Antibiot. 1990, 43, 796–808. [Google Scholar] [CrossRef]
- Lingham, R.B.; Hsu, A.H.; O’Brien, J.A.; Sigmund, J.M.; Sanchez, M.; Gagliardi, M.M.; Heimbuch, B.K.; Genilloud, O.; Martin, I.; Diez, M.T.; et al. Quinoxapeptins: novel chromodepsipeptide inhibitors of HIV-1 and HIV-2 reverse transcriptase. I. The producing organism and biological activity. J. Antibiot. 1996, 49, 253–259. [Google Scholar] [CrossRef]
- Huang, C.H.; Mirabelli, C.K.; Mong, S.; Crooke, S.T. Intermolecular cross-linking of DNA through bifunctional intercalation of an antitumor antibiotic, luzopeptin A (BBM-928A). Cancer Res. 1983, 43, 2718–2724. [Google Scholar]
- Sheldrick, G.M.; Heine, A.; Schmidt-Bäse, K.; Pohl, E.; Jones, P.G.; Paulus, E.; Waring, M.J. Structures of quinoxaline antibiotics. Acta Crystallogr. B 1995, 51, 987–999. [Google Scholar] [CrossRef]
- Perez Baz, J.; Cañedo, L.M.; Fernández Puentes, J.L.; Silva Elipe, M.V. Thiocoraline, a novel depsipeptide with antitumor activity produced by a marine Micromonospora. II. Physico-chemical properties and structure determination. J. Antibiot. 1997, 50, 738–741. [Google Scholar] [CrossRef]
- Praseuth, A.P.; Wang, C.C.; Watanabe, K.; Hotta, K.; Oguri, H.; Oikawa, H. Complete sequence of biosynthetic gene cluster responsible for producing triostin A and evaluation of quinomycin-type antibiotics from Streptomyces triostinicus. Biotechnol. Prog. 2008, 24, 1226–1231. [Google Scholar] [CrossRef]
- Watanabe, K.; Hotta, K.; Nakaya, M.; Praseuth, A.P.; Wang, C.C.; Inada, D.; Takahashi, K.; Fukushi, E.; Oguri, H.; Oikawa, H. Escherichia coli allows efficient modular incorporation of newly isolated quinomycin biosynthetic enzyme into echinomycin biosynthetic pathway for rational design and synthesis of potent antibiotic unnatural natural product. J. Am. Chem. Soc. 2009, 131, 9347–9353. [Google Scholar] [CrossRef]
- Takahashi, K.; Koshino, H.; Esumi, Y.; Tsuda, E.; Kurosawa, K. SW-163C and E, novel antitumor depsipeptides produced by Streptomyces sp. II. Structure elucidation. J. Antibiot. 2001, 54, 622–627. [Google Scholar] [CrossRef]
- Nakaya, M.; Oguri, H.; Takahashi, K.; Fukushi, E.; Watanabe, K.; Oikawa, H. Relative and absolute configuration of antitumor agent SW-163D. Biosci. Biotechnol. Biochem. 2007, 71, 2969–2976. [Google Scholar] [CrossRef]
- Shoji, J.; Konaka, R.; Kawano, K.; Higuchi, N.; Kyogoku, Y. Presence of isomers in quinomycin E. J. Antibiot. 1976, 29, 1246–1248. [Google Scholar]
- Yoshida, T.; Katagiri, K. Influence of isoleucine upon quinomycin biosynthesis by Streptomyces sp. 732. J. Bacteriol. 1967, 93, 1327–1331. [Google Scholar]
- Boger, D.L.; Schüle, G. Synthesis of Acyclic Precursors to (3S,4S)-4-Hydroxy-2,3,4,5-tetrahydropyridazine-3-carboxylic Acid and Incorporation into a Luzopeptin/Quinoxapeptin Dipeptide. J. Org. Chem. 1998, 63, 6421–6424. [Google Scholar] [CrossRef]
- Boger, D.L.; Ledeboer, M.W.; Kume, M.; Jin, Q. Total Synthesis of Quinoxapeptin A–C: Establishment of Absolute Stereochemistry. Angew. Chem. Int. Ed. Engl. 1999, 38, 2424–2426. [Google Scholar] [CrossRef]
- Sieber, S.A.; Marahiel, M.A. Molecular mechanisms underlying nonribosomal peptide synthesis: Approaches to new antibiotics. Chem. Rev. 2005, 105, 715–738. [Google Scholar] [CrossRef]
- von Döhren, H.; Dieckmann, R.; Pavela-Vrancic, M. The nonribosomal code. Chem. Biol. 1999, 6, R273–R279. [Google Scholar] [CrossRef]
- Süssmuth, R.D.; Wohlleben, W. The biosynthesis of glycopeptide antibiotics—A model for complex, non-ribosomally synthesized, peptidic secondary metabolites. Appl. Microbiol. Biotechnol. 2004, 63, 344–350. [Google Scholar] [CrossRef]
- Lambalot, R.H.; Gehring, A.M.; Flugel, R.S.; Zuber, P.; LaCelle, M.; Marahiel, M.A.; Reid, R.; Khosla, C.; Walsh, C.T. A new enzyme superfamily—The phosphopantetheinyl transferases. Chem. Biol. 1996, 3, 923–936. [Google Scholar] [CrossRef]
- Quadri, L.E.; Weinreb, P.H.; Lei, M.; Nakano, M.M.; Zuber, P.; Walsh, C.T. Characterization of Sfp, a Bacillus subtilis phosphopantetheinyl transferase for peptidyl carrier protein domains in peptide synthetases. Biochemistry 1998, 37, 1585–1595. [Google Scholar]
- Stachelhaus, T.; Mootz, H.D.; Marahiel, M.A. The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol. 1999, 6, 493–505. [Google Scholar] [CrossRef]
- Challis, G.L.; Ravel, J.; Townsend, C.A. Predictive, structure-based model of amino acid recognition by nonribosomal peptide synthetase adenylation domains. Chem. Biol. 2000, 7, 211–224. [Google Scholar] [CrossRef]
- Yoshida, T.; Kimura, Y.; Katagiri, K. The biosynthesis of the quinoxaline antibiotic, triostin, by Streptomyces S-2-2 1. On the role of quinoxaline-2-carboxylic acid. In Progress in Antimicrobial and Anticancer Chemotherapy; University Park Press: Baltimore, MD, USA, 1970; pp. 1160–1165. [Google Scholar]
- Cornish, A.; Fox, K.R.; Santikarn, S.; Waring, M.J.; Williams, D.H. Incorporation of fluorotryptophan into triostin antibiotics by Streptomyces triostinicus. J. Gen. Microbiol. 1985, 131, 561–570. [Google Scholar]
- Sato, M.; Nakazawa, T.; Tsunematsu, Y.; Hotta, K.; Watanabe, K. Echinomycin biosynthesis. Curr. Opin. Chem. Biol. 2013, 17, 537–545. [Google Scholar] [CrossRef]
- Mady, A.S.; Zolova, O.E.; Millán, M.Á.; Villamizar, G.; de la Calle, F.; Lombó, F.; Garneau-Tsodikova, S. Characterization of TioQ, a type II thioesterase from the thiocoraline biosynthetic cluster. Mol. Biosyst. 2011, 7, 1999–2011. [Google Scholar] [CrossRef]
- Koketsu, K.; Oguri, H.; Watanabe, K.; Oikawa, H. Identification and stereochemical assignment of the beta-hydroxytryptophan intermediate in the echinomycin biosynthetic pathway. Org. Lett. 2006, 8, 4719–4722. [Google Scholar] [CrossRef]
- Sheoran, A.; King, A.; Velasco, A.; Pero, J.M.; Garneau-Tsodikova, S. Characterization of TioF, a tryptophan 2,3-dioxygenase involved in 3-hydroxyquinaldic acid formation during thiocoraline biosynthesis. Mol. Biosyst. 2008, 4, 622–628. [Google Scholar] [CrossRef]
- Hirose, Y.; Watanabe, K.; Minami, A.; Nakamura, T.; Oguri, H.; Oikawa, H. Involvement of common intermediate 3-hydroxy-l-kynurenine in chromophore biosynthesis of quinomycin family antibiotics. J. Antibiot. 2011, 64, 117–122. [Google Scholar] [CrossRef]
- van der Goot, A.T.; Nollen, E.A. Tryptophan metabolism: Entering the field of aging and age-related pathologies. Trends Mol. Med. 2013, 19, 336–344. [Google Scholar] [CrossRef]
- Gauvreau, D.; Waring, M.J. Directed biosynthesis of novel derivatives of echinomycin by Streptomyces echinatus. I. Effect of exogenous analogues of quinoxaline-2-carboxylic acid on the fermentation. Can. J. Microbiol. 1984, 30, 439–450. [Google Scholar] [CrossRef]
- Glund, K.; Schlumbohm, W.; Bapat, M.; Keller, U. Biosynthesis of quinoxaline antibiotics: Purification and characterization of the quinoxaline-2-carboxylic acid activating enzyme from Streptomyces triostinicus. Biochemistry 1990, 29, 3522–3527. [Google Scholar] [CrossRef]
- Müller, C.; Nolden, S.; Gebhardt, P.; Heinzelmann, E.; Lange, C.; Puk, O.; Welzel, K.; Wohlleben, W.; Schwartz, D. Sequencing and analysis of the biosynthetic gene cluster of the lipopeptide antibiotic Friulimicin in Actinoplanes friuliensis. Antimicrob. Agents Chemother. 2007, 51, 1028–1037. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, Y.; Shen, Q.; Yin, X. Molecular cloning and identification of the laspartomycin biosynthetic gene cluster from Streptomyces viridochromogenes. Gene 2011, 483, 11–21. [Google Scholar] [CrossRef]
- Beasley, F.C.; Cheung, J.; Heinrichs, D.E. Mutation of l-2,3-diaminopropionic acid synthase genes blocks staphyloferrin B synthesis in Staphylococcus aureus. BMC Microbiol. 2011, 11. [Google Scholar] [CrossRef]
- Lam, W.H.; Rychli, K.; Bugg, T.D. Identification of a novel beta-replacement reaction in the biosynthesis of 2,3-diaminobutyric acid in peptidylnucleoside mureidomycin A. Org. Biomol. Chem. 2008, 6, 1912–1917. [Google Scholar] [CrossRef]
- Gatto, G.J., Jr.; Boyne, M.T., II; Kelleher, N.L.; Walsh, C.T. Biosynthesis of pipecolic acid by RapL, a lysine cyclodeaminase encoded in the rapamycin gene cluster. J. Am. Chem. Soc. 2006, 128, 3838–3847. [Google Scholar] [CrossRef]
- Huang, D.; Xia, M.; Li, S.; Wen, J.; Jia, X. Enhancement of FK506 production by engineering secondary pathways of Streptomyces tsukubaensis and exogenous feeding strategies. J. Ind. Microbiol. Biotechnol. 2013, 40, 1023–1037. [Google Scholar] [CrossRef]
- Namwat, W.; Kamioka, Y.; Kinoshita, H.; Yamada, Y.; Nihira, T. Characterization of virginiamycin S biosynthetic genes from Streptomyces virginiae. Gene 2002, 286, 283–290. [Google Scholar] [CrossRef]
- Chai, Y.; Pistorius, D.; Ullrich, A.; Weissman, K.J.; Kazmaier, U.; Müller, R. Discovery of 23 natural tubulysins from Angiococcus disciformis An d48 and Cystobacter SBCb004. Chem. Biol. 2010, 17, 296–309. [Google Scholar] [CrossRef]
- Oelke, A.J.; France, D.J.; Hofmann, T.; Wuitschik, G.; Ley, S.V. Piperazic acid-containing natural products: Isolation, biological relevance and total synthesis. Nat. Prod. Rep. 2011, 28, 1445–1471. [Google Scholar] [CrossRef]
- Qu, X.; Jiang, N.; Xu, F.; Shao, L.; Tang, G.; Wilkinson, B.; Liu, W. Cloning, sequencing and characterization of the biosynthetic gene cluster of sanglifehrin A, a potent cyclophilin inhibitor. Mol. Biosyst. 2011, 7, 852–861. [Google Scholar] [CrossRef]
- Neumann, C.S.; Jiang, W.; Heemstra, J.R., Jr.; Gontang, E.A.; Kolter, R.; Walsh, C.T. Biosynthesis of piperazic acid via N5-hydroxy-ornithine in Kutzneria spp. 744. Chembiochem 2012, 13, 972–976. [Google Scholar] [CrossRef]
- Schmoock, G.; Pfennig, F.; Jewiarz, J.; Schlumbohm, W.; Laubinger, W.; Schauwecker, F.; Keller, U. Functional cross-talk between fatty acid synthesis and nonribosomal peptide synthesis in quinoxaline antibiotic-producing streptomycetes. J. Biol. Chem. 2005, 280, 4339–4349. [Google Scholar]
- Robbel, L.; Hoyer, K.M.; Marahiel, M.A. TioS T-TE—A prototypical thioesterase responsible for cyclodimerization of the quinoline- and quinoxaline-type class of chromodepsipeptides. FEBS J. 2009, 276, 1641–1653. [Google Scholar] [CrossRef]
- Koketsu, K.; Oguri, H.; Watanabe, K.; Oikawa, H. Enzymatic macrolactonization in the presence of DNA leading to triostin A analogs. Chem. Biol. 2008, 15, 818–828. [Google Scholar] [CrossRef]
- Hotta, K.; Keegan, R.M.; Ranganathan, S.; Fang, M.; Bibby, J.; Winn, M.D.; Sato, M.; Lian, M.; Watanabe, K.; Rigden, D.J.; et al. Conversion of a Disulfide Bond into a Thioacetal Group during Echinomycin Biosynthesis. Angew. Chem. Int. Ed. Engl. 2014, 53, 824–828. [Google Scholar]
- Scharf, D.H.; Remme, N.; Heinekamp, T.; Hortschansky, P.; Brakhage, A.A.; Hertweck, C. Transannular disulfide formation in gliotoxin biosynthesis and its role in self-resistance of the human pathogen Aspergillus fumigatus. J. Am. Chem. Soc. 2010, 132, 10136–10141. [Google Scholar] [CrossRef]
- Scharf, D.H.; Remme, N.; Habel, A.; Chankhamjon, P.; Scherlach, K.; Heinekamp, T.; Hortschansky, P.; Brakhage, A.A.; Hertweck, C. A dedicated glutathione S-transferase mediates carbon-sulfur bond formation in gliotoxin biosynthesis. J. Am. Chem. Soc. 2011, 133, 12322–12325. [Google Scholar] [CrossRef]
- Biswas, T.; Zolova, O.E.; Lombó, F.; de la Calle, F.; Salas, J.A.; Tsodikov, O.V.; Garneau-Tsodikova, S. A new scaffold of an old protein fold ensures binding to the bisintercalator thiocoraline. J. Mol. Biol. 2010, 397, 495–507. [Google Scholar] [CrossRef]
- Sugiyama, M.; Kumagai, T.; Hayashida, M.; Maruyama, M.; Matoba, Y. The 1.6-A crystal structure of the copper(II)-bound bleomycin complexed with the bleomycin-binding protein from bleomycin-producing Streptomyces verticillus. J. Biol. Chem. 2002, 277, 2311–2320. [Google Scholar]
- Waring, M.J.; Wakelin, L.P. Echinomycin: A bifunctional intercalating antibiotic. Nature 1974, 252, 653–657. [Google Scholar] [CrossRef]
- Takusagawa, F. The role of the cyclic depsipeptide rings in antibiotics. J. Antibiot. 1985, 38, 1596–1604. [Google Scholar] [CrossRef]
- Low, C.M.; Olsen, R.K.; Waring, M.J. Sequence preferences in the binding to DNA of triostin A and TANDEM as reported by DNase I footprinting. FEBS Lett. 1984, 176, 414–420. [Google Scholar] [CrossRef]
- Robinson, H.; Priebe, W.; Chaires, J.B.; Wang, A.H. Binding of two novel bisdaunorubicins to DNA studied by NMR spectroscopy. Biochemistry 1997, 36, 8663–8670. [Google Scholar]
- Guéron, M.; Leroy, J.L. Studies of base pair kinetics by NMR measurement of proton exchange. Methods Enzymol. 1995, 261, 383–413. [Google Scholar] [CrossRef]
- Negri, A.; Marco, E.; García-Hernández, V.; Domingo, A.; Llamas-Saiz, A.L.; Porto-Sandá, S.; Riguera, R.; Laine, W.; David-Cordonnier, M.H.; Bailly, C.; et al. Antitumor activity, X-ray crystal structure, and DNA binding properties of thiocoraline A, a natural bisintercalating thiodepsipeptide. J. Med. Chem. 2007, 50, 3322–3333. [Google Scholar] [CrossRef]
- Wang, A.H.; Ughetto, G.; Quigley, G.J.; Hakoshima, T.; van der Marel, G.A.; van Boom, J.H.; Rich, A. The molecular structure of a DNA-triostin A complex. Science 1984, 225, 1115–1121. [Google Scholar]
- Mazzitelli, C.L.; Brodbelt, J.S. Probing ligand binding to duplex DNA using KMnO4 reactions and electrospray ionization tandem mass spectrometry. Anal. Chem. 2007, 79, 4636–4647. [Google Scholar] [CrossRef]
- Boger, D.L.; Chen, J.H.; Saionz, K.W.; Jin, Q. Synthesis of key sandramycin analogs: Systematic examination of the intercalation chromophore. Bioorg. Med. Chem. 1998, 6, 85–102. [Google Scholar] [CrossRef]
- Mazzitelli, C.L.; Chu, Y.; Reczek, J.J.; Iverson, B.L.; Brodbelt, J.S. Screening of threading bis-intercalators binding to duplex DNA by electrospray ionization tandem mass spectrometry. J. Am. Soc. Mass Spectrom. 2007, 18, 311–321. [Google Scholar] [CrossRef]
- Rackham, B.D.; Howell, L.A.; Round, A.N.; Searcey, M. Non-covalent duplex to duplex crosslinking of DNA in solution revealed by single molecule force spectroscopy. Org. Biomol. Chem. 2013, 11, 8340–8347. [Google Scholar] [CrossRef]
- Ughetto, G.; Wang, A.H.; Quigley, G.J.; van der Marel, G.A.; van Boom, J.H.; Rich, A. A comparison of the structure of echinomycin and triostin A complexed to a DNA fragment. Nucleic Acids Res. 1985, 13, 2305–2323. [Google Scholar] [CrossRef]
- Lee, J.S.; Waring, M.J. Interaction between synthetic analogues of quinoxaline antibiotics and nucleic acids. Changes in mechanism and specificity related to structural alterations. Biochem. J. 1978, 173, 129–144. [Google Scholar]
- Kleimann, C.; Sischka, A.; Spiering, A.; Tönsing, K.; Sewald, N.; Diederichsen, U.; Anselmetti, D. Binding kinetics of bisintercalator Triostin a with optical tweezers force mechanics. Biophys. J. 2009, 97, 2780–2784. [Google Scholar] [CrossRef]
- Gilbert, D.E.; Feigon, J. The DNA sequence at echinomycin binding sites determines the structural changes induced by drug binding: NMR studies of echinomycin binding to [d(ACGTACGT)]2 and [d(TCGATCGA)]2. Biochemistry 1991, 30, 2483–2494. [Google Scholar] [CrossRef]
- Van Dyke, M.M.; Dervan, P.B. Echinomycin binding sites on DNA. Science 1984, 225, 1122–1127. [Google Scholar]
- Dell, A.; Williams, D.H.; Morris, H.R.; Smith, G.A.; Feeney, J.; Roberts, G.C. Structure revision of the antibiotic echinomycin. J. Am. Chem. Soc. 1975, 97, 2497–2502. [Google Scholar] [CrossRef]
- Gause, G.G., Jr.; Loshkareva, N.P.; Zbarsky, I.B. Effect of olivomycin and echinomycin on initiation and growth of RNA chains catalyzed by RNA polymerase. Biochim. Biophys. Acta 1968, 166, 752–754. [Google Scholar] [CrossRef]
- Takusagawa, H.L.; Takusagawa, F. Crystallization and preliminary X-ray diffraction studies of d(ACGTAGCTACGT)2:[actinomycin D, (echinomycin)2] and d(ACGTAGCTACGT)2:[actinomycin D, (triostin A)2] complexes. Acta Crystallogr. D Biol. Crystallogr. 2000, 56, 344–347. [Google Scholar] [CrossRef]
- Boger, D.L.; Ichikawa, S.; Tse, W.C.; Hedrick, M.P.; Jin, Q. Total syntheses of thiocoraline and BE-22179 and assessment of their DNA binding and biological properties. J. Am. Chem. Soc. 2001, 123, 561–568. [Google Scholar] [CrossRef]
- Verma, R.P.; Hansch, C. Investigation of DNA-binding properties of organic molecules using quantitative structure-activity relationship (QSAR) models. J. Pharm. Sci. 2008, 97, 88–110. [Google Scholar] [CrossRef]
- Boger, D.L.; Saionz, K.W. DNA binding properties of key sandramycin analogues: Systematic examination of the intercalation chromophore. Bioorg. Med. Chem. 1999, 7, 315–321. [Google Scholar] [CrossRef]
- Leroy, J.L.; Gao, X.L.; Misra, V.; Guéron, M.; Patel, D.J. Proton exchange in DNA-luzopeptin and DNA-echinomycin bisintercalation complexes: Rates and processes of base-pair opening. Biochemistry 1992, 31, 1407–1415. [Google Scholar] [CrossRef]
- Berge, T.; Haken, E.L.; Waring, M.J.; Henderson, R.M. The binding mode of the DNA bisintercalator luzopeptin investigated using atomic force microscopy. J. Struct. Biol. 2003, 142, 241–246. [Google Scholar] [CrossRef]
- Bailly, C.; Crow, S.; Minnock, A.; Waring, M.J. DNA recognition by quinoline antibiotics: Use of base-modified DNA molecules to investigate determinants of sequence-specific binding of luzopeptin. Nucleosides Nucleotides Nucleic Acids 2000, 19, 1337–1353. [Google Scholar] [CrossRef]
- Zhang, X.L.; Patel, D.J. Solution structure of the luzopeptin-DNA complex. Biochemistry 1991, 30, 4026–4041. [Google Scholar] [CrossRef]
- Huang, C.H.; Crooke, S.T. Effects of structural modifications of antitumor antibiotics (luzopeptins) on the interactions with deoxyribonucleic acid. Cancer Res. 1985, 45, 3768–3773. [Google Scholar]
- Bergamaschi, D.; Faretta, M.; Ronzoni, S.; Taverna, S.; De Feudis, P.; Bonfanti, M.; Guidi, G.; Faircloth, M.; Jimeno, J.; D’Incalci, M.; et al. Flow cytometric analysis of cell cycle phase perturbations induced by Thiocoraline, a new marine-derived anticancer compound. Eur. J. Histochem. 1997, 41, 63–64. [Google Scholar]
- Muss, H.B.; Blessing, J.A.; Hanjani, P.; Malfetano, J.H.; Kemp, G.M.; Webster, K. Echinomycin (NSC 526417) in recurrent and metastatic nonsquamous cell carcinoma of the cervix. A phase II trial of the Gynecologic Oncology Group. Am. J. Clin. Oncol. 1992, 15, 363–364. [Google Scholar] [CrossRef]
- Rance, M.J.; Ruddock, J.C.; Pacey, M.S.; Cullen, W.P.; Huang, L.H.; Jefferson, M.T.; Whipple, E.B.; Maeda, H.; Tone, J. UK-63,052 complex, new quinomycin antibiotics from Streptomyces braegensis subsp. japonicus; taxonomy, fermentation, isolation, characterisation and antimicrobial activity. J. Antibiot. 1989, 42, 206–217. [Google Scholar] [CrossRef]
- Castillo, U.; Harper, J.K.; Strobel, G.A.; Sears, J.; Alesi, K.; Ford, E.; Lin, J.; Hunter, M.; Maranta, M.; Ge, H.; et al. Kakadumycins, novel antibiotics from Streptomyces sp NRRL 30566, an endophyte of Grevillea pteridifolia. FEMS Microbiol. Lett. 2003, 224, 183–190. [Google Scholar] [CrossRef]
- Socha, A.M.; Laplante, K.L.; Russell, D.J.; Rowley, D.C. Structure-activity studies of echinomycin antibiotics against drug-resistant and biofilm-forming Staphylococcus aureus and Enterococcus faecalis. Bioorg. Med. Chem. Lett. 2009, 19, 1504–1507. [Google Scholar] [CrossRef]
- Park, Y.S.; Shin, W.S.; Kim, S.K. In vitro and in vivo activities of echinomycin against clinical isolates of Staphylococcus aureus. J. Antimicrob. Chemother. 2008, 61, 163–168. [Google Scholar]
- Kim, J.B.; Lee, G.S.; Kim, Y.B.; Kim, S.K.; Kim, Y.H. In vitro antibacterial activity of echinomycin and a novel analogue, YK2000, against vancomycin-resistant enterococci. Int. J. Antimicrob. Agents 2004, 24, 613–615. [Google Scholar] [CrossRef]
- Minor, P.D.; Dimmock, N.J. Selective inhibition of influenza virus protein synthesis by inhibitors of DNA function. Virology 1977, 78, 393–406. [Google Scholar] [CrossRef]
- Jayasuriya, H.; Zink, D.L.; Polishook, J.D.; Bills, G.F.; Dombrowski, A.W.; Genilloud, O.; Pelaez, F.F.; Herranz, L.; Quamina, D.; Lingham, R.B.; et al. Identification of diverse microbial metabolites as potent inhibitors of HIV-1 Tat transactivation. Chem. Biodivers. 2005, 2, 112–122. [Google Scholar] [CrossRef]
- Espinosa, A.; Socha, A.M.; Ryke, E.; Rowley, D.C. Antiamoebic properties of the actinomycete metabolites echinomycin A and tirandamycin A. Parasitol. Res. 2012, 111, 2473–2477. [Google Scholar] [CrossRef]
- Kong, D.; Park, E.J.; Stephen, A.G.; Calvani, M.; Cardellina, J.H.; Monks, A.; Fisher, R.J.; Shoemaker, R.H.; Melillo, G. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res. 2005, 65, 9047–9055. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer. 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Wang, R.; Zhou, S.; Li, S. Cancer therapeutic agents targeting hypoxia-inducible factor-1. Curr. Med. Chem. 2011, 18, 3168–3189. [Google Scholar] [CrossRef]
- Yonekura, S.; Itoh, M.; Okuhashi, Y.; Takahashi, Y.; Ono, A.; Nara, N.; Tohda, S. Effects of the HIF1 inhibitor, echinomycin, on growth and NOTCH signalling in leukaemia cells. Anticancer Res. 2013, 33, 3099–3103. [Google Scholar]
- Zimmermann, S.M.; Würgler-Hauri, C.C.; Wanner, G.A.; Simmen, H.P.; Werner, C.M. Echinomycin in the prevention of heterotopic ossification—An experimental antibiotic agent shows promising results in a murine model. Injury 2013, 44, 570–575. [Google Scholar] [CrossRef]
- Lee, Y.K.; Hyung Park, J.; Tae Moon, H.; Yun Lee, D.; Han Yun, J.; Byun, Y. The short-term effects on restenosis and thrombosis of echinomycin-eluting stents topcoated with a hydrophobic heparin-containing polymer. Biomaterials 2007, 28, 1523–1530. [Google Scholar] [CrossRef]
- Tesfazghi, S.; Eide, J.; Dammalapati, A.; Korlesky, C.; Wyche, T.P.; Bugni, T.S.; Chen, H.; Jaskula-Sztul, R. Thiocoraline alters neuroendocrine phenotype and activates the Notch pathway in MTC-TT cell line. Cancer Med. 2013, 2, 734–743. [Google Scholar]
- Erba, E.; Bergamaschi, D.; Ronzoni, S.; Faretta, M.; Taverna, S.; Bonfanti, M.; Catapano, C.V.; Faircloth, G.; Jimeno, J.; D’Incalci, M. Mode of action of thiocoraline, a natural marine compound with anti-tumour activity. Br. J. Cancer 1999, 80, 971–980. [Google Scholar] [CrossRef]
- Yoshinari, T.; Okada, H.; Yamada, A.; Uemura, D.; Oka, H.; Suda, H.; Okura, A. Inhibition of topoisomerase II by a novel antitumor cyclic depsipeptide, BE-22179. Jpn. J. Cancer Res. 1994, 85, 550–555. [Google Scholar] [CrossRef]
- Rose, W.C.; Huftalen, J.B.; Bradner, W.T.; Schurig, J.E. In vivo characterization of P388 leukemia resistant to mitomycin C. In Vivo 1987, 1, 47–52. [Google Scholar]
- Yung, B.Y.; Busch, H.; Chan, P.K. Effects of luzopeptins on protein B23 translocation and ribosomal RNA synthesis in HeLa cells. Cancer Res. 1986, 46, 922–925. [Google Scholar]
- Lee, S.; Inselburg, J. In vitro sensitivity of Plasmodium falciparum to drugs that bind DNA or inhibit its synthesis. J. Parasitol. 1993, 79, 780–782. [Google Scholar] [CrossRef]
- Inouye, Y.; Take, Y.; Nakamura, S.; Nakashima, H.; Yamamoto, N.; Kawaguchi, H. Screening for inhibitors of avian myeloblastosis virus reverse transcriptase and effect on the replication of AIDS-virus. J. Antibiot. 1987, 40, 100–104. [Google Scholar] [CrossRef]
- Ciufolini, M.A.; Valognes, D.; Xi, N. Total Synthesis of Luzopeptin E2. Angew. Chem. Int. Ed. Engl. 2000, 39, 2493–2495. [Google Scholar] [CrossRef]
- Katayama, K.; Nakagawa, K.; Takeda, H.; Matsuda, A.; Ichikawa, S. Total Synthesis of Sandramycin and Its Analogues via a Multicomponent Assemblage. Org. Lett. 2014, 16, 428–431. [Google Scholar] [CrossRef]
- Ichikawa, S.; Okamura, T.; Matsuda, A. Total synthesis of quinaldopeptin and its analogues. J. Org. Chem. 2013, 78, 12662–12670. [Google Scholar] [CrossRef]
- Malkinson, J.P.; Anim, M.K.; Zloh, M.; Searcey, M.; Hampshire, A.J.; Fox, K.R. Efficient solid-phase-based total synthesis of the bisintercalator TANDEM. J. Org. Chem. 2005, 70, 7654–7661. [Google Scholar] [CrossRef]
- Bayó-Puxan, N.; Fernández, A.; Tulla-Puche, J.; Riego, E.; Cuevas, C.; Alvarez, M.; Albericio, F. Total solid-phase synthesis of the azathiocoraline class of symmetric bicyclic peptides. Chemistry 2006, 12, 9001–9009. [Google Scholar] [CrossRef]
- Kim, Y.B.; Kim, Y.H.; Park, J.Y.; Kim, S.K. Synthesis and biological activity of new quinoxaline antibiotics of echinomycin analogues. Bioorg. Med. Chem. Lett. 2004, 14, 541–544. [Google Scholar] [CrossRef]
- Jarikote, D.V.; Li, W.; Jiang, T.; Eriksson, L.A.; Murphy, P.V. Towards echinomycin mimetics by grafting quinoxaline residues on glycophane scaffolds. Bioorg. Med. Chem. 2011, 19, 826–835. [Google Scholar] [CrossRef]
- Fox, K.R.; Gauvreau, D.; Goodwin, D.C.; Waring, M.J. Binding of quinoline analogues of echinomycin to deoxyribonucleic acid. Role of the chromophores. Biochem. J. 1980, 191, 729–742. [Google Scholar]
- Cornish, A.; Fox, K.R.; Waring, M.J. Preparation and DNA-binding properties of substituted triostin antibiotics. Antimicrob. Agents Chemother. 1983, 23, 221–231. [Google Scholar] [CrossRef]
- Hamphsire, A.J.; Rusling, D.A.; Bryan, S.; Paumier, D.; Dawson, S.J.; Malkinson, J.P.; Searcey, M.; Fox, K.R. DNA binding by analogues of the bifunctional intercalator TANDEM. Biochemistry 2008, 47, 7900–7906. [Google Scholar] [CrossRef]
- Tulla-Puche, J.; Góngora-Benítez, M.; Bayó-Puxan, N.; Francesch, A.M.; Cuevas, C.; Albericio, F. Enzyme-labile protecting groups for the synthesis of natural products: Solid-phase synthesis of thiocoraline. Angew. Chem. Int. Ed. Engl. 2013, 52, 5726–5730. [Google Scholar] [CrossRef]
- Tulla-Puche, J.; Marcucci, E.; Fermin, M.; Bayó-Puxan, N.; Albericio, F. Protection by conformationally restricted mobility: First solid-phase synthesis of triostin A. Chemistry 2008, 14, 4475–4478. [Google Scholar] [CrossRef]
- Tulla-Puche, J.; Marcucci, E.; Prats-Alfonso, E.; Bayó-Puxan, N.; Albericio, F. NMe amide as a synthetic surrogate for the thioester moiety in thiocoraline. J. Med. Chem. 2009, 52, 834–839. [Google Scholar] [CrossRef]
- Zamudio-Vázquez, R.; Albericio, F.; Tulla-Puche, J.; Fox, K.R. Thioester bonds in thiocoraline can be replaced with NMe-amide bridges without affecting its DNA-binding properties. Med. Chem. Lett. 2014, 5, 45–50. [Google Scholar] [CrossRef]
- Garcia-Martin, F.; Cruz, L.J.; Rodriguez-Mias, R.A.; Giralt, E.; Albericio, F. Design and synthesis of FAJANU: A de novo C(2) symmetric cyclopeptide family. J. Med. Chem. 2008, 51, 3194–3202. [Google Scholar] [CrossRef]
- Tulla-Puche, J.; Auriemma, S.; Falciani, C.; Albericio, F. Orthogonal Chemistry for the Synthesis of Thiocoraline-Triostin Hybrids. Exploring their Structure-Activity Relationship. J. Med. Chem. 2013, 56, 5587–5323. [Google Scholar] [CrossRef]
- Tulla-Puche, J.; Bayó-Puxan, N.; Moreno, J.A.; Francesch, A.M.; Cuevas, C.; Alvarez, M.; Albericio, F. Solid-phase synthesis of oxathiocoraline by a key intermolecular disulfide dimer. J. Am. Chem. Soc. 2007, 129, 5322–5323. [Google Scholar] [CrossRef]
- Watanabe, K.; Hotta, K.; Praseuth, A.P.; Koketsu, K.; Migita, A.; Boddy, C.N.; Wang, C.C.; Oguri, H.; Oikawa, H. Total biosynthesis of antitumor nonribosomal peptides in Escherichia coli. Nat. Chem. Biol. 2006, 2, 423–428. [Google Scholar] [CrossRef]
- Watanabe, K.; Oguri, H.; Oikawa, H. Diversification of echinomycin molecular structure by way of chemoenzymatic synthesis and heterologous expression of the engineered echinomycin biosynthetic pathway. Curr. Opin. Chem. Biol. 2009, 13, 189–196. [Google Scholar] [CrossRef]
- Watanabe, K.; Oikawa, H. Robust platform for de novo production of heterologous polyketides and nonribosomal peptides in Escherichia coli. Org. Biomol. Chem. 2007, 5, 593–602. [Google Scholar] [CrossRef]
- Praseuth, A.P.; Praseuth, M.B.; Oguri, H.; Oikawa, H.; Watanabe, K.; Wang, C.C. Improved production of triostin A in engineered Escherichia coli with furnished quinoxaline chromophore by design of experiments in small-scale culture. Biotechnol. Prog. 2008, 24, 134–139. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fernández, J.; Marín, L.; Álvarez-Alonso, R.; Redondo, S.; Carvajal, J.; Villamizar, G.; Villar, C.J.; Lombó, F. Biosynthetic Modularity Rules in the Bisintercalator Family of Antitumor Compounds. Mar. Drugs 2014, 12, 2668-2699. https://doi.org/10.3390/md12052668
Fernández J, Marín L, Álvarez-Alonso R, Redondo S, Carvajal J, Villamizar G, Villar CJ, Lombó F. Biosynthetic Modularity Rules in the Bisintercalator Family of Antitumor Compounds. Marine Drugs. 2014; 12(5):2668-2699. https://doi.org/10.3390/md12052668
Chicago/Turabian StyleFernández, Javier, Laura Marín, Raquel Álvarez-Alonso, Saúl Redondo, Juan Carvajal, Germán Villamizar, Claudio J. Villar, and Felipe Lombó. 2014. "Biosynthetic Modularity Rules in the Bisintercalator Family of Antitumor Compounds" Marine Drugs 12, no. 5: 2668-2699. https://doi.org/10.3390/md12052668