Antioxidant and Anti-Inflammatory Activities of Barettin

 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Bioactivity Testing
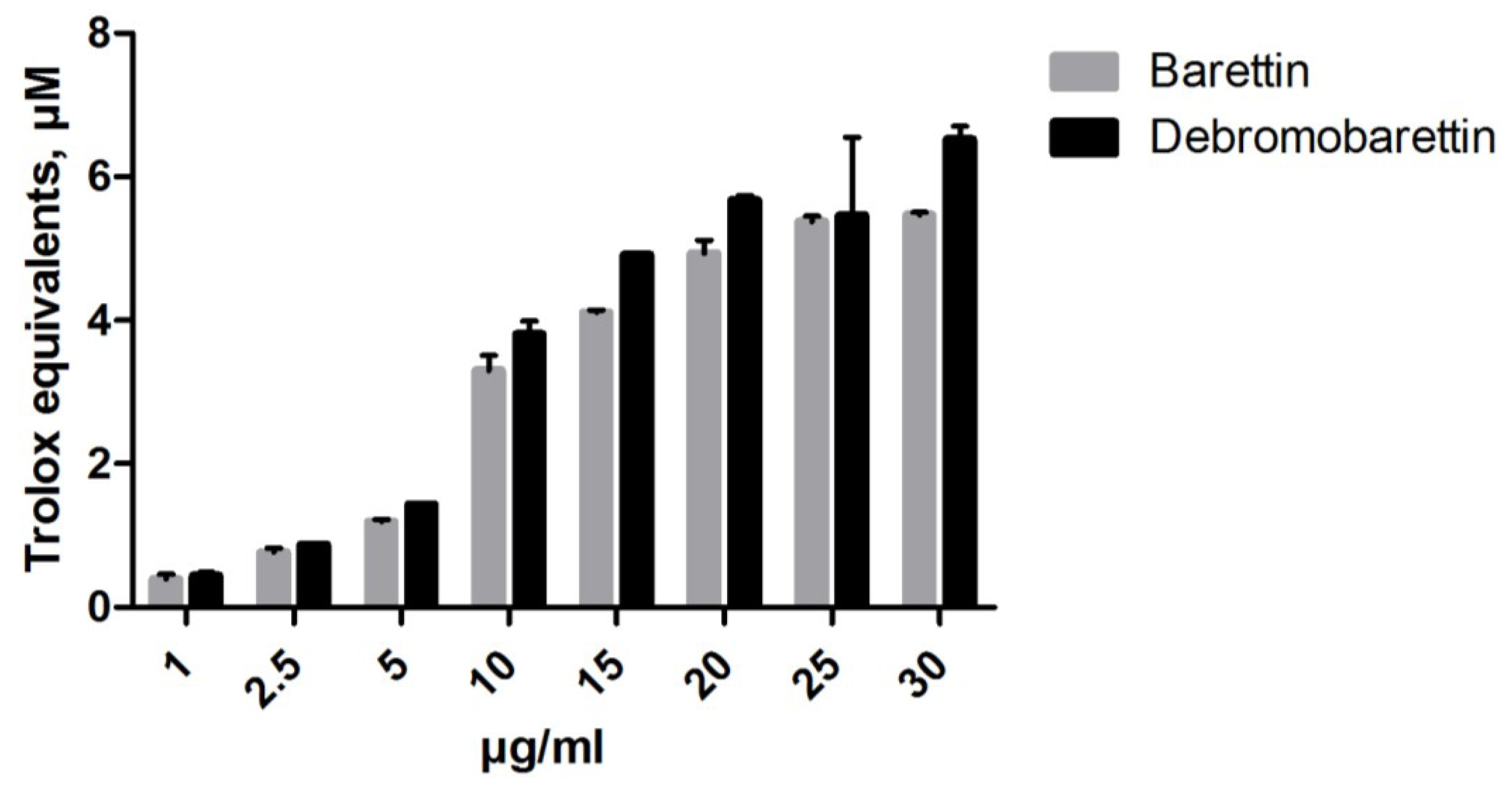
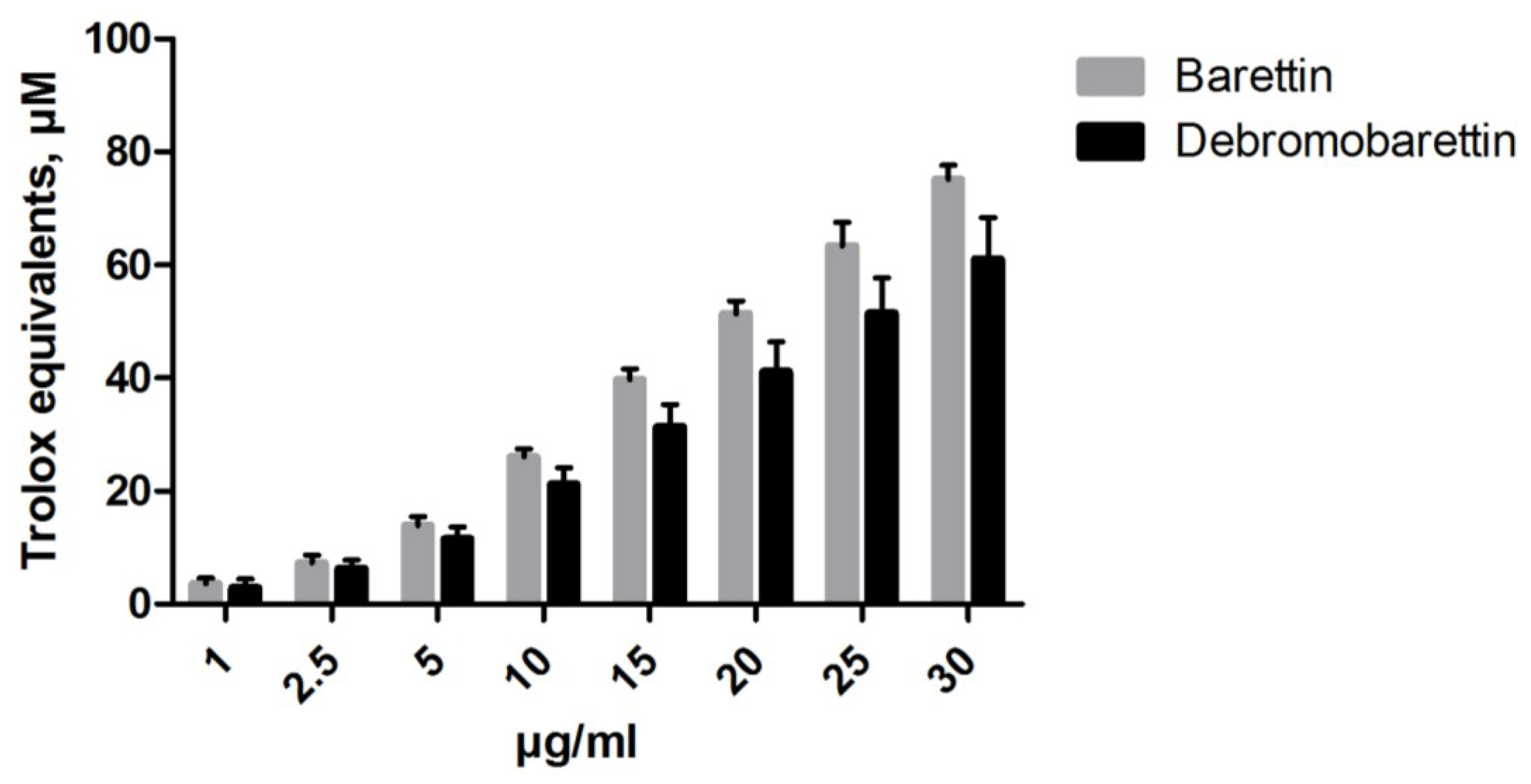
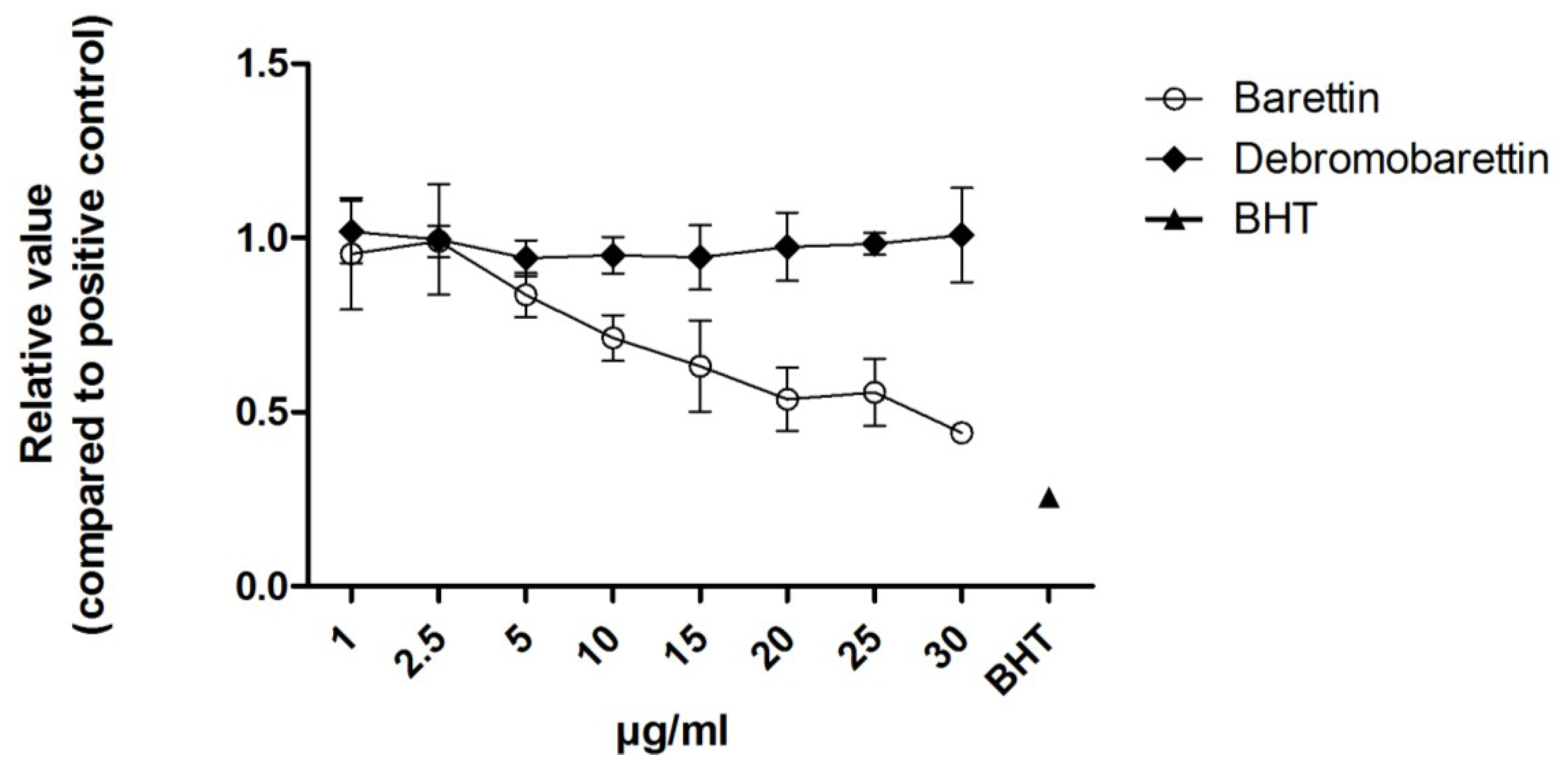
2.1.1. Antioxidant Activity
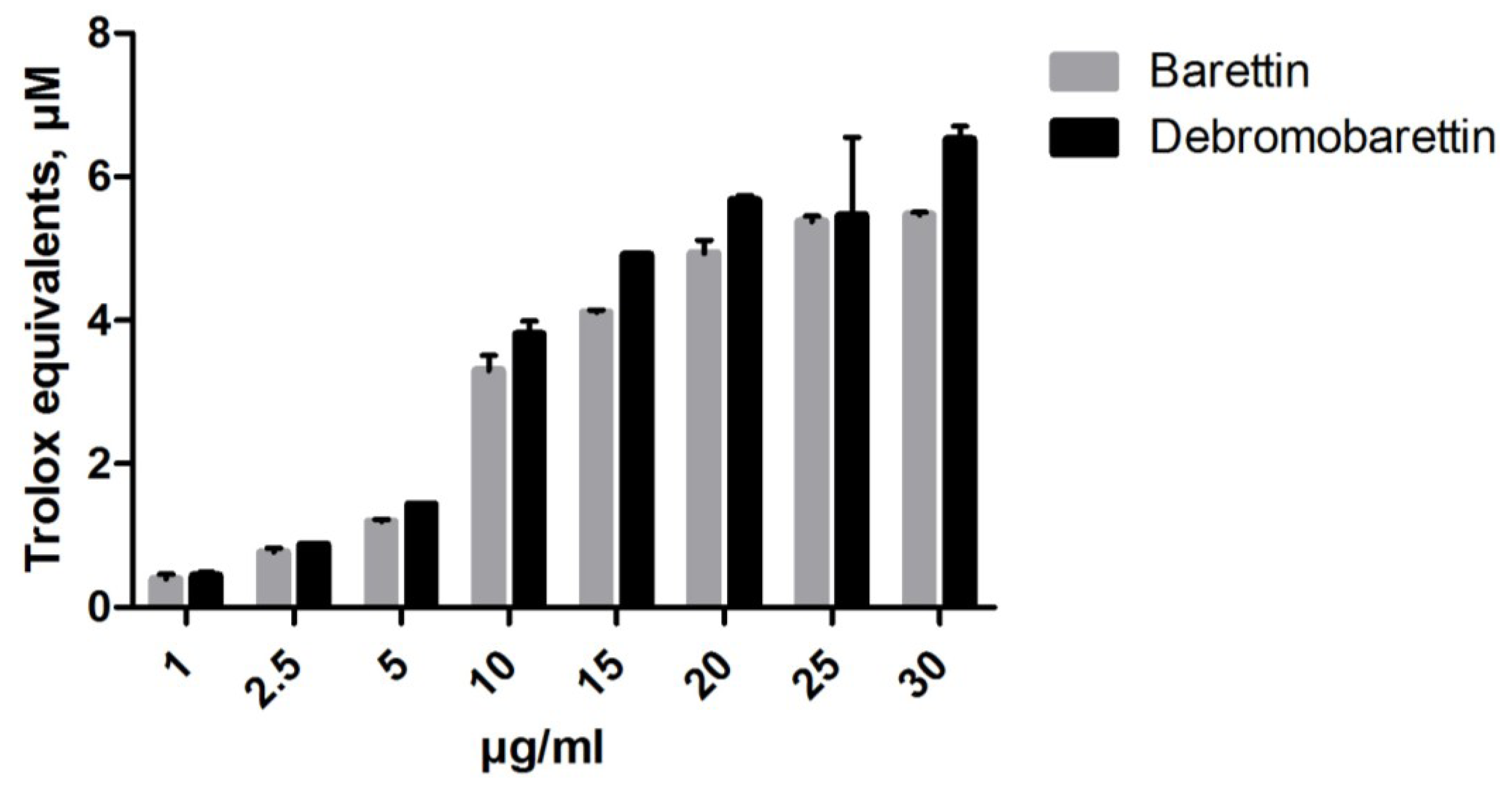
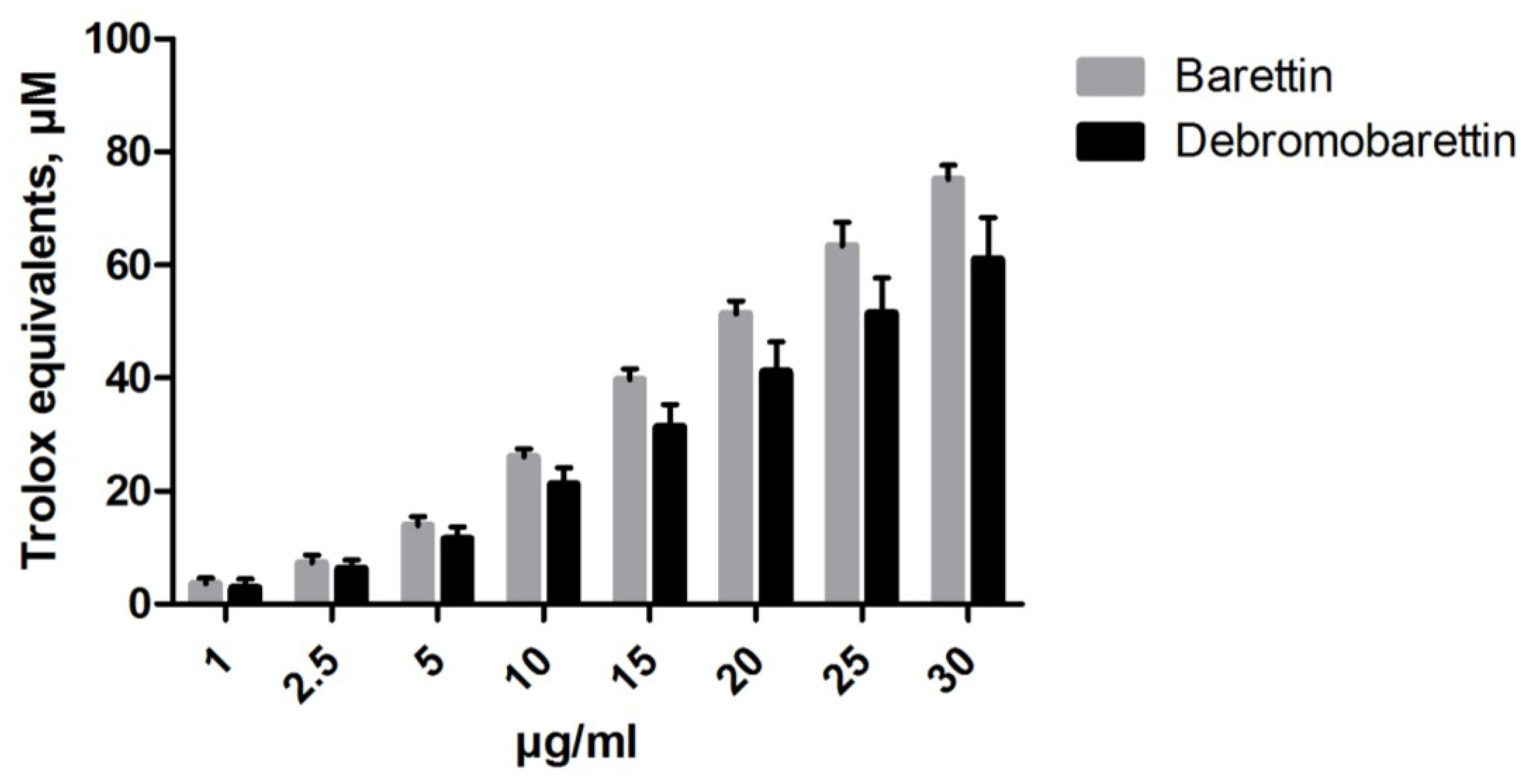
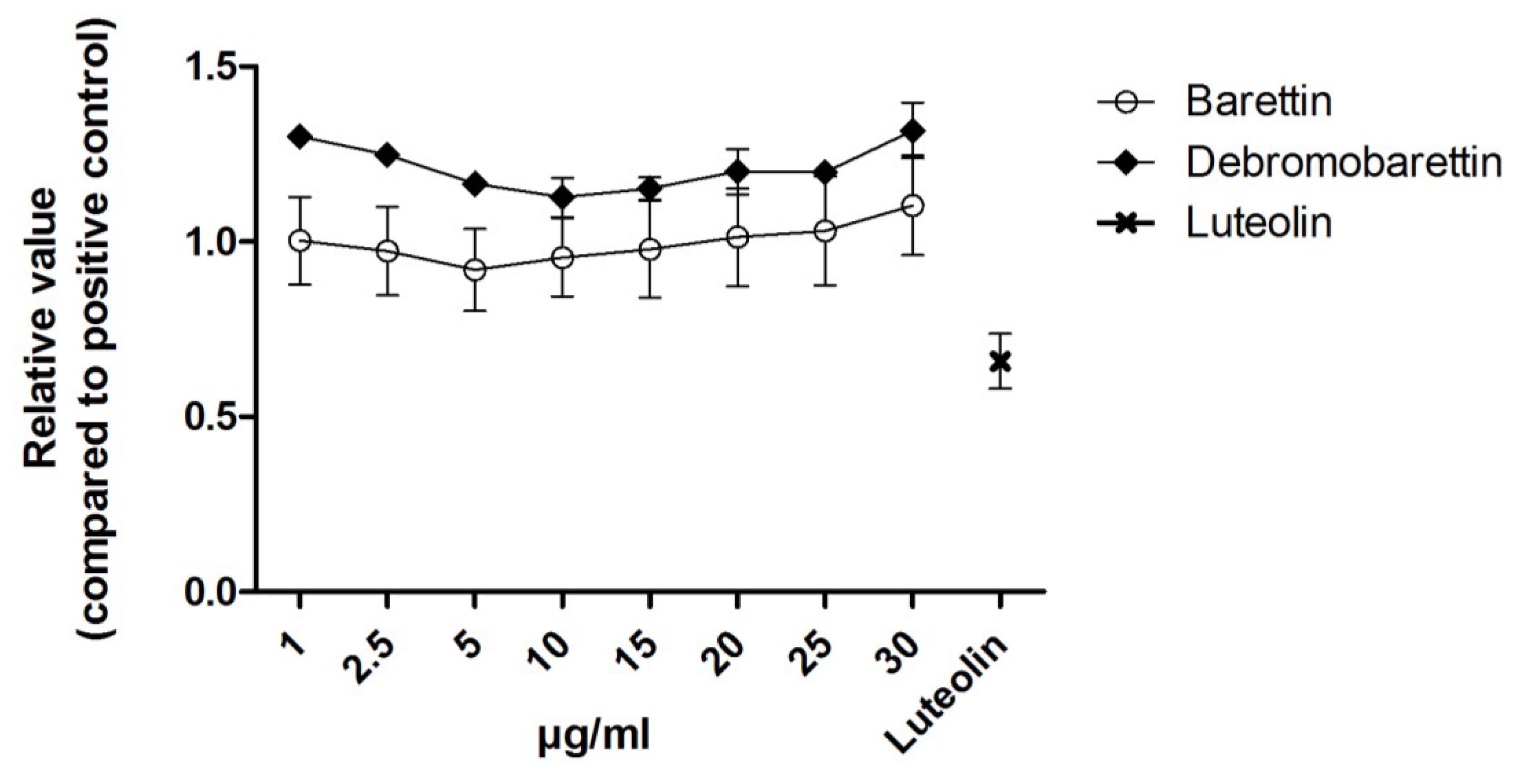
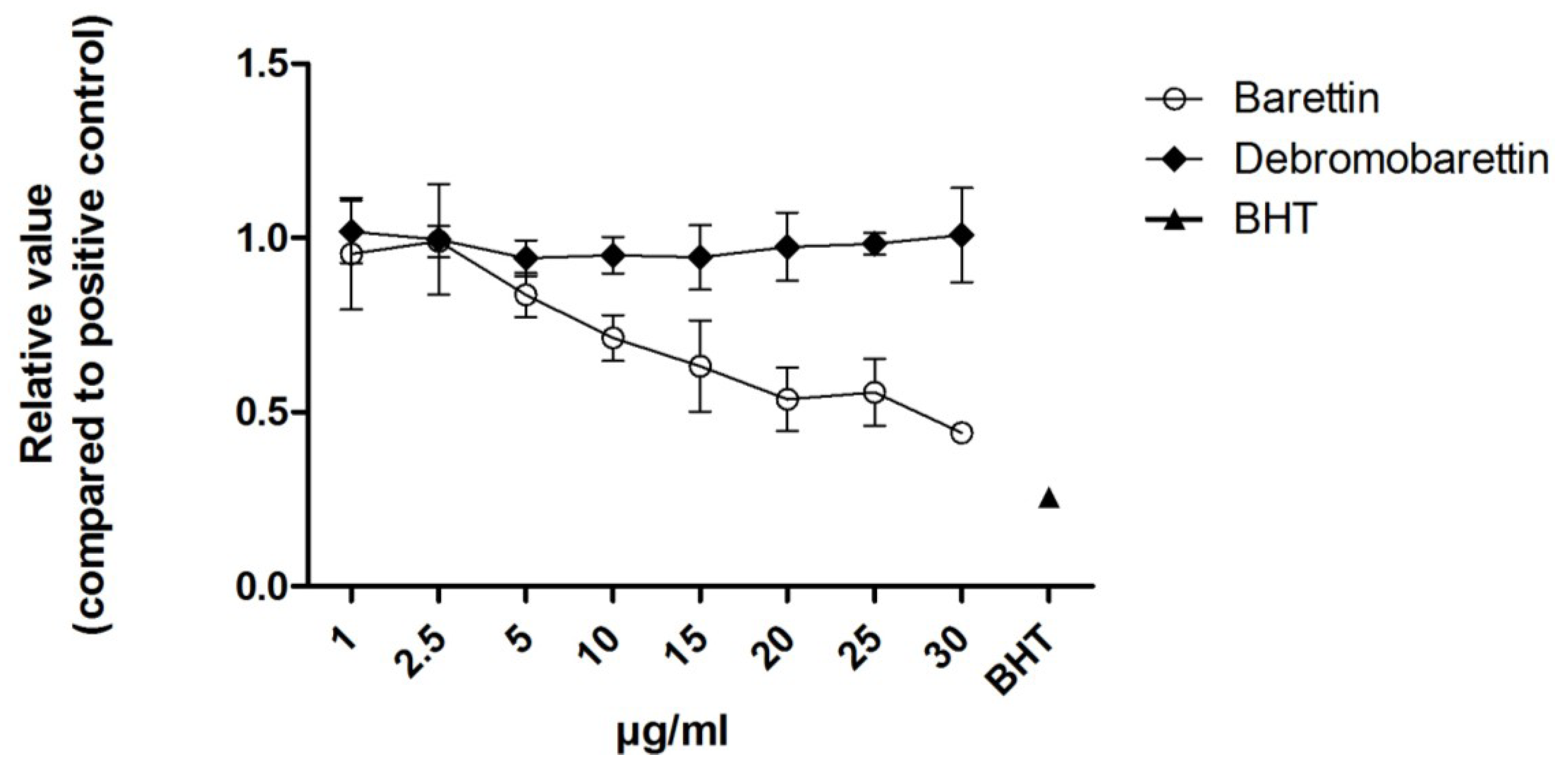
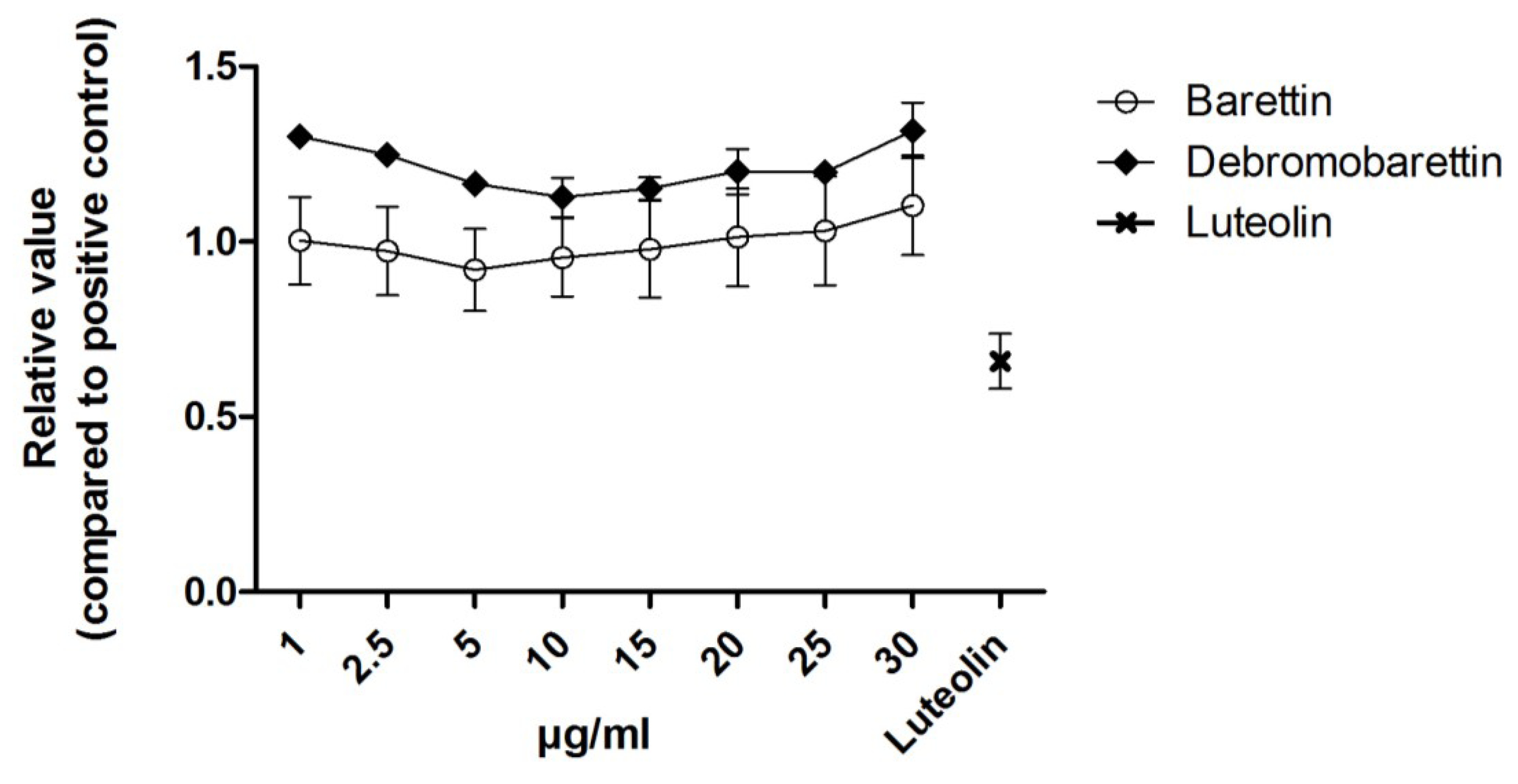
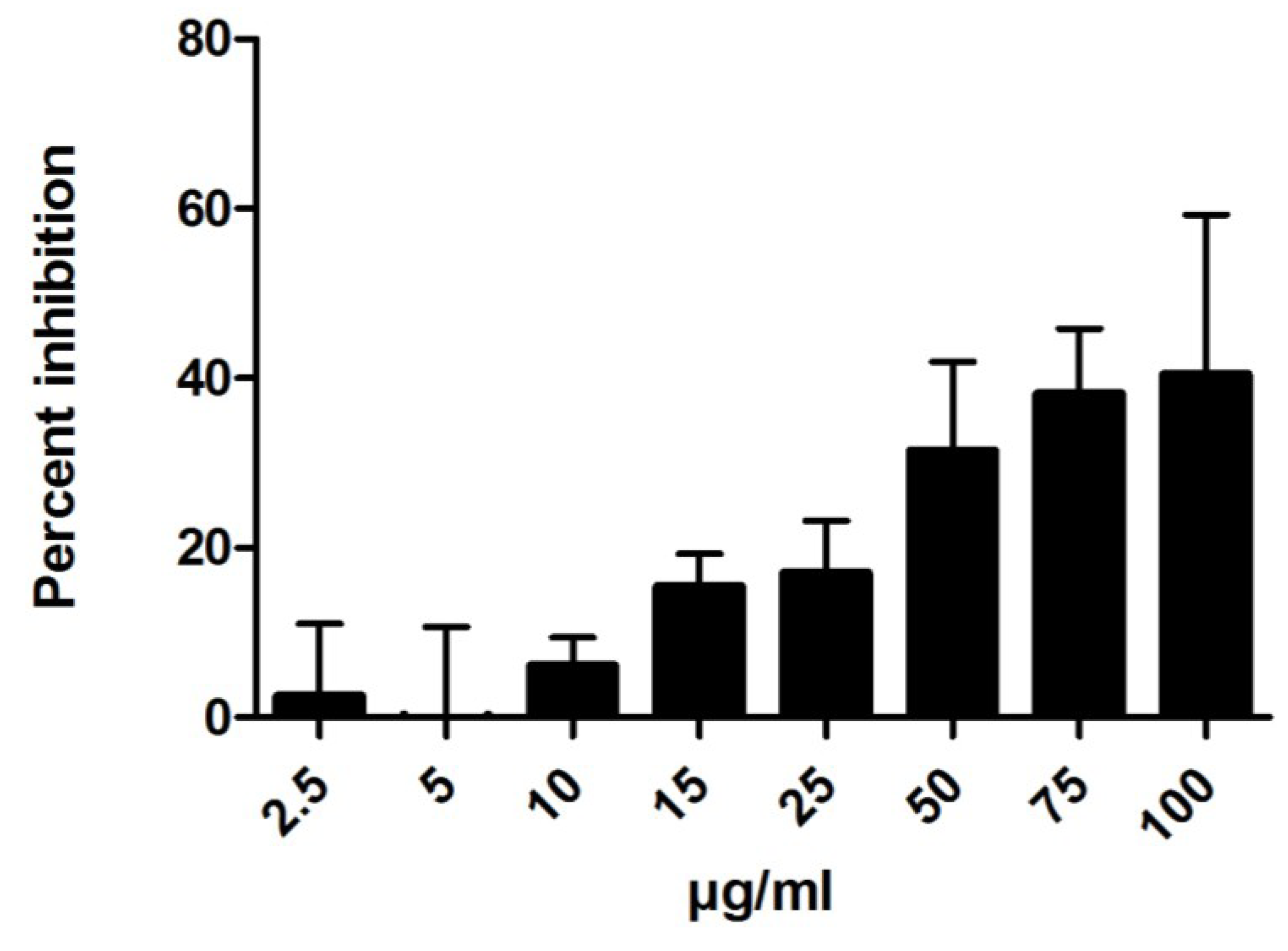
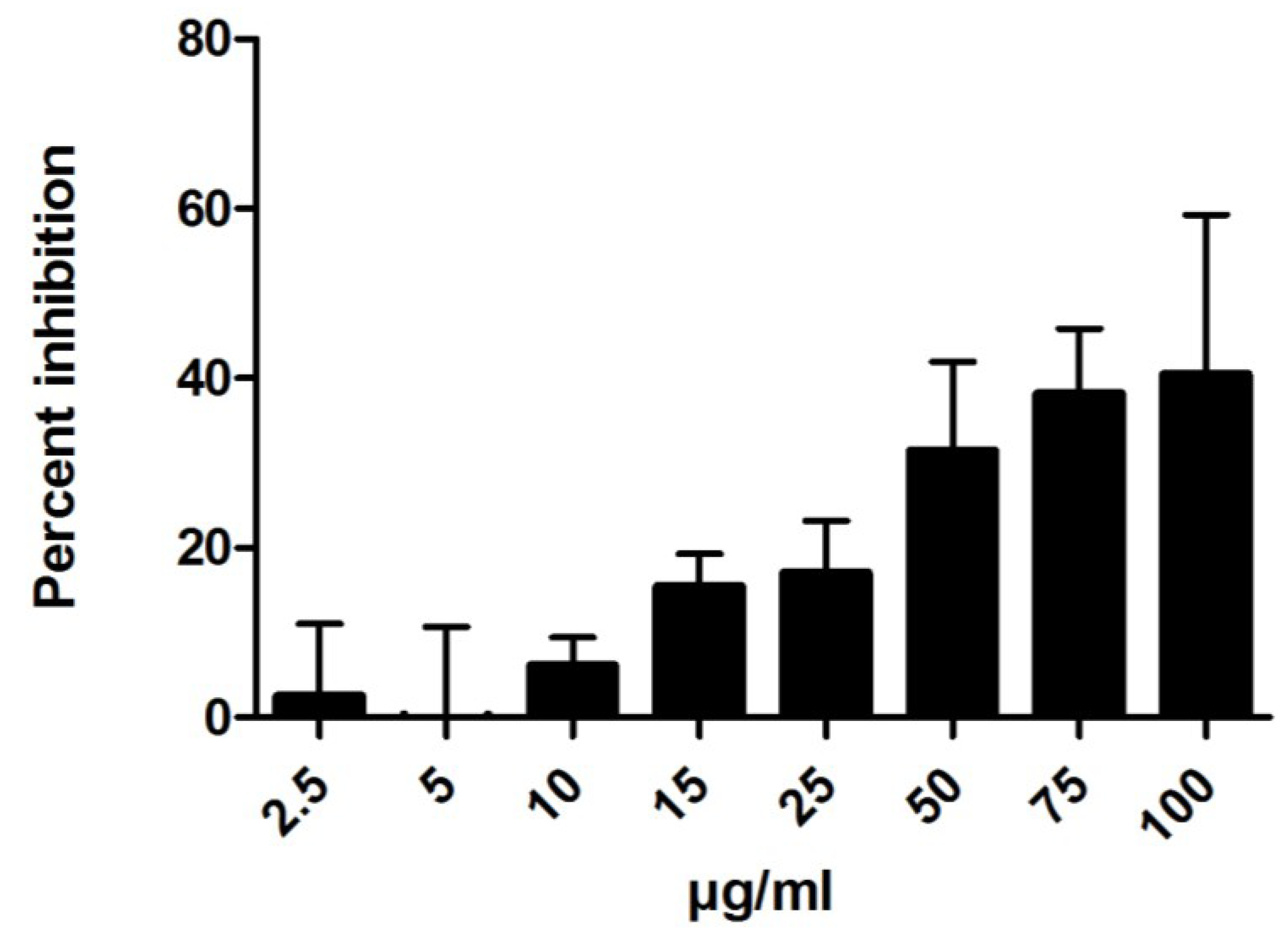
2.1.2. Anti-Inflammatory Activity
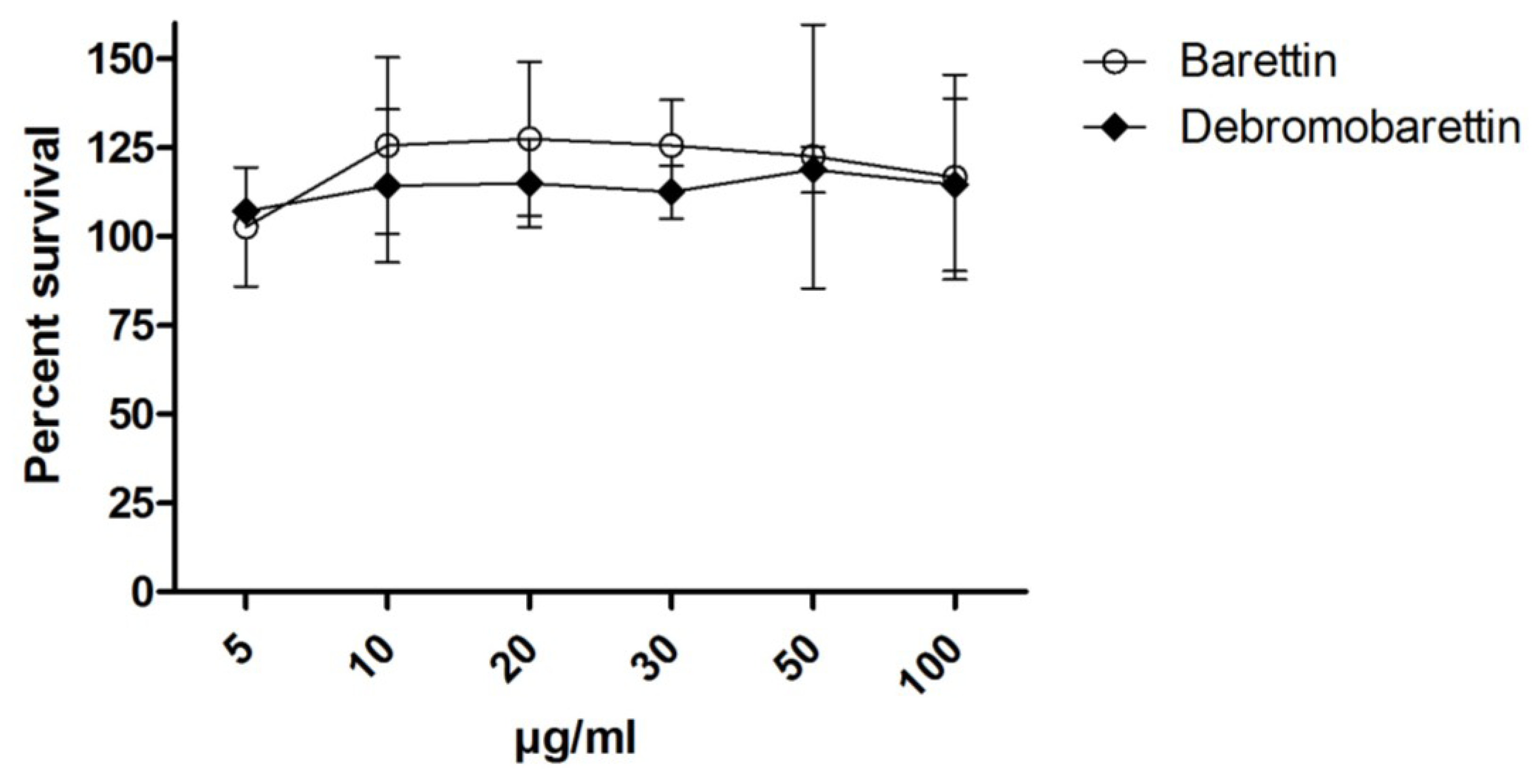
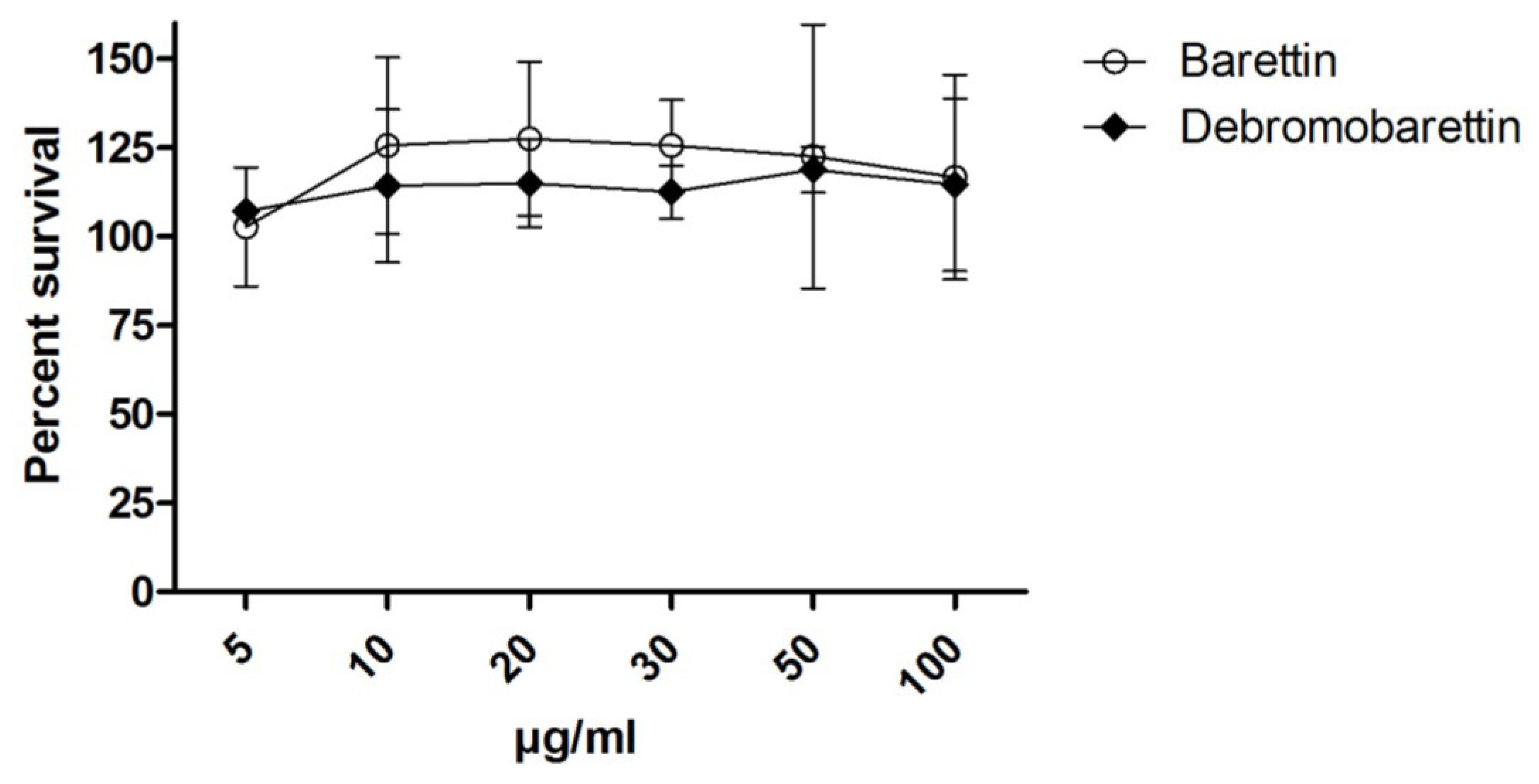
2.2. Cytotoxicity
3. Experimental Section
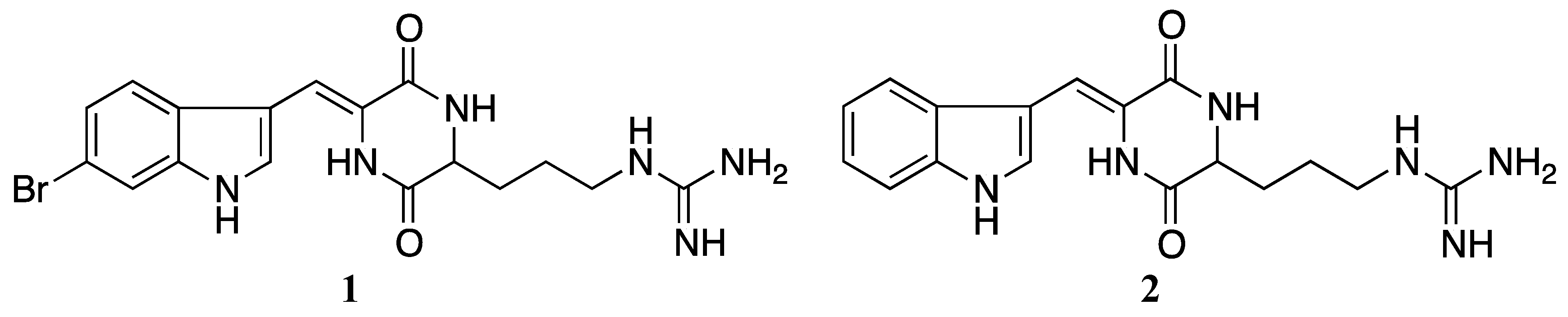
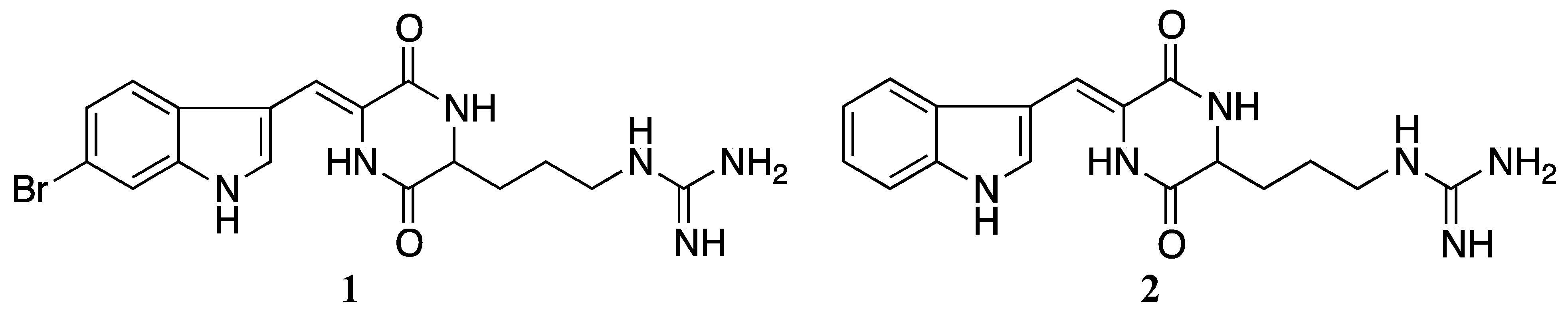
3.1. Purification, Isolation and Identification
3.2. Synthesis
3.3. Biochemical Assays
3.4. Cellular Assays
3.4.1. Cellular Lipid Peroxidation Antioxidant Activity (CLPAA) Assay
3.4.2. Cellular Antioxidant Activity (CAA) Assay
3.4.3. Cytotoxicity
3.4.4. Anti-Inflammatory
3.4.5. Endotoxin Removal
4. Conclusions
Acknowledgements
References
- Lidgren, G.; Bohlin, L.; Bergman, J. Studies of swedish marine organisms VII. A novel biologically active indole alkaloid from the sponge Geodia barretti. Tetrahedon Lett. 1986, 27, 3283–3284. [Google Scholar] [CrossRef]
- Sölter, S.; Dieckmann, R.; Blumenberg, M.; Francke, W. Barettin, revisited? Tetrahedon Lett. 2002, 43, 3385–3386. [Google Scholar] [CrossRef]
- Johnson, A.-L.; Bergman, J.; Sjögren, M.; Bohlin, L. Synthesis of barettin. Tetrahedon 2004, 60, 961–965. [Google Scholar]
- Sjögren, M.; Göransson, U.; Johnson, A.-L.; Dahlström, M.; Andersson, R.; Bergman, J.; Jonsson, P.R.; Bohlin, L. Antifouling activity of brominated cyclopeptides from the marine sponge Geodia barretti. J. Nat. Prod. 2004, 67, 368–372. [Google Scholar] [CrossRef]
- Sjögren, M.; Johnson, A.L.; Hedner, E.; Dahlström, M.; Göransson, U.; Shirani, H.; Bergman, J.; Jonsson, P.R.; Bohlin, L. Antifouling activity of synthesized peptide analogs of the sponge metabolite barettin. Peptides 2006, 27, 2058–2064. [Google Scholar] [CrossRef]
- Hedner, E.; Sjogren, M.; Hodzic, S.; Andersson, R.; Goransson, U.; Jonsson, P.R.; Bohlin, L. Antifouling activity of a dibrominated cyclopeptide from the marine sponge Geodia barretti. J. Nat. Prod. 2008, 71, 330–333. [Google Scholar] [CrossRef]
- Sjögren, M.; Dahlström, M.; Göransson, U.; Jonsson, P.R.; Bohlin, L. Recruitment in the field of Balanus improvisus and Mytilus edulis in response to the antifouling cyclopeptides barettin and 8,9-dihydrobarettin from the marine sponge Geodia barretti. Biofouling 2004, 20, 291–297. [Google Scholar] [CrossRef]
- Hedner, E.; Sjögren, M.; Frändberg, P.-A.; Johansson, T.; Göransson, U.; Dahlström, M.; Jonsson, P.; Nyberg, F.; Bohlin, L. Brominated cyclodipeptides from the marine sponge Geodia barretti as selective 5-HT ligands. J. Nat. Prod. 2006, 69, 1421–1424. [Google Scholar] [CrossRef]
- Sjögren, M.; Jonsson, P.R.; Dahlström, M.; Lundälv, T.; Burman, R.; Göransson, U.; Bohlin, L. Two brominated cyclic dipeptides released by the coldwater marine sponge Geodia barretti act in synergy as chemical defense. J. Nat. Prod. 2011, 74, 449–454. [Google Scholar] [CrossRef]
- Svenson, J. MabCent: Arctic marine bioprospecting in Norway. Phytochem. Rev. 2012. [Google Scholar] [CrossRef] [Green Version]
- Kang, J.; Xie, C.; Li, Z.; Nagarajan, S.; Schauss, A.G.; Wu, T.; Wu, X. Flavonoids from acai (Euterpe oleracea Mart.) pulp and their antioxidant and anti-inflammatory activities. Food Chem. 2011, 128, 152–157. [Google Scholar]
- Zimmer, A.R.; Leonardi, B.; Miron, D.; Schapoval, E.; Oliveira, J.R.; Gosmann, G. Antioxidant and anti-inflammatory properties of Capsicum baccatum: from traditional use to scientific approach. J. Ethnopharmacol. 2012, 139, 228–233. [Google Scholar] [CrossRef]
- Peng, J.; Yuan, J.-P.; Wu, C.-F.; Wang, J.-H. Fucoxanthin, a marine carotenoid present in brown seaweeds and diatoms: Metabolism and bioactivities relevant to human health. Mar. Drugs 2011, 9, 1806–1828. [Google Scholar] [CrossRef]
- Li, H.; Xia, N.; Förstermann, U. Cardiovascular effects and molecular targets of resveratrol. Nitric Oxide 2012, 26, 102–110. [Google Scholar] [CrossRef]
- Wolfe, K.L.; Liu, R.H. Structure-activity relationships of flavonoids in the cellular antioxidant activity assay. J. Agric. Food Chem. 2008, 56, 8404–8411. [Google Scholar] [CrossRef]
- Lü, J.-M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med. 2010, 14, 840–860. [Google Scholar] [CrossRef]
- Goya, L.; Mateos, R.; Bravo, L. Effect of the olive oil phenol hydroxytyrosol on human hepatoma HepG2 cells. Eur. J. Nutr. 2007, 46, 70–78. [Google Scholar] [CrossRef]
- Murakami, C.; Hirakawa, Y.; Inui, H.; Nakano, Y.; Yoshida, H. Effect of tea catechins on cellular lipid peroxidation and cytotoxicity in HepG2 cells. Biosci. Biotechnol. Biochem. 2002, 66, 1559–1562. [Google Scholar] [CrossRef]
- Alía, M.; Ramos, S.; Mateos, R.; Granado-Serrano, A.B.; Bravo, L.; Goya, L. Quercetin protects human hepatoma HepG2 against oxidative stress induced by tert-butyl hydroperoxide. Toxicol. Appl. Pharmacol. 2006, 212, 110–118. [Google Scholar] [CrossRef]
- Wolfe, K.L.; Liu, R.H. Cellular antioxidant activity (CAA) assay for assessing antioxidants, foods, and dietary supplements. J. Agric. Food Chem. 2007, 55, 8896–8907. [Google Scholar] [CrossRef]
- Hofer, T.; Eriksen, T.E.; Hansen, E.; Varmedal, I.; Jensen, I.-J.; Hammer Andersen, J.; Olsen, R.L. Cellular and chemical assays for discovery of novel antioxidants in marine organisms. In Studies on Experimental Models; Basu, S., Wiklund, L., Eds.; Humana Press: New York, NY, USA, 2011; pp. 637–657. [Google Scholar]
- Takamatsu, S.; Hodges, T.W.; Rajbhandari, I.; Gerwick, W.H.; Hamann, M.T.; Nagle, D.G. Marine natural products as novel antioxidant prototypes. J. Nat. Prod. 2003, 66, 605–608. [Google Scholar] [CrossRef]
- Gentry, C.L.; Egleton, R.D.; Gillespie, T.; Abbruscato, T.J.; Bechowski, H.B.; Hruby, V.J.; Davis, T.P. The effect of halogenation on blood-brain barrier permeability of a novel peptide drug. Peptides 1999, 20, 1229–1238. [Google Scholar] [CrossRef]
- Gerebtzoff, G.; Li-Blatter, X.; Fischer, H.; Frentzel, A.; Seelig, A. Halogenation of drugs enhances membrane binding and permeation. ChemBioChem 2004, 5, 676–684. [Google Scholar] [CrossRef]
- Osterud, B.; Bjorklid, E. Role of monocytes in atherogenesis. Physiol. Rev. 2003, 83, 1069–1112. [Google Scholar]
- Patel, R.P.; Moellering, D.; Murphy-Ullrich, J.; Jo, H.; Beckman, J.S.; Darley-Usmar, V.M. Cell signaling by reactive nitrogen and oxygen species in atherosclerosis. Free Radic. Biol. Med. 2000, 28, 1780–1794. [Google Scholar] [CrossRef]
- Greenberg, M.E.; Li, X.-M.; Gugiu, B.G.; Gu, X.; Qin, J.; Salomon, R.G.; Hazen, S.L. The lipid whisker model of the structure of oxidized cell membranes. J. Biol. Chem. 2008, 283, 2385–2396. [Google Scholar]
- Stocker, R.; Keaney, J.F. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef]
- Xie, C.; Kang, J.; Burris, R.; Ferguson, M.E.; Schauss, A.G.; Nagarajan, S.; Wu, X. Açaí juice attenuates atherosclerosis in ApoE deficient mice through antioxidant and anti-inflammatory activities. Atherosclerosis 2011, 216, 327–333. [Google Scholar] [CrossRef]
- Gorbet, M.B.; Sefton, M.V. Endotoxin: The uninvited guest. Biomaterials 2005, 26, 6811–6817. [Google Scholar] [CrossRef]
- Lieder, R.; Gaware, V.S.; Thormodsson, F.; Einarsson, J.M.; Ng, C.-H.; Gislason, J.; Masson, M.; Petersen, P.H.; Sigurjonsson, O.E. Endotoxins affect bioactivity of chitosan derivatives in cultures of bone marrow-derived human mesenchymal stem cells. Acta Biomater. 2013, 9, 4771–4778. [Google Scholar] [CrossRef]
- Gómez-Lechón, M.J.; Castell, J.V.; Donato, M.T. Hepatocytes-the choice to investigate drug metabolism and toxicity in man: In vitro variability as a reflection of in vivo. Chem. Biol. Interact. 2007, 168, 30–50. [Google Scholar] [CrossRef]
- Nakamura, K.; Mizutani, R.; Sanbe, A.; Enosawa, S.; Kasahara, M.; Nakagawa, A.; Ejiri, Y.; Murayama, N.; Miyamoto, Y.; Torii, T.; et al. Evaluation of drug toxicity with hepatocytes cultured in a micro-space cell culture system. J. Biosci. Bioeng. 2011, 111, 78–84. [Google Scholar] [CrossRef]
- Benzie, I.F.F.; Strain, J.J. The ferric reducing ability of plasma (FRAP) as a measure of “antioxidant power”: The FRAP assay. Anal. Biochem. 1996, 239, 70–76. [Google Scholar] [CrossRef]
- Huang, D.; Ou, B.; Hampsch-Woodill, M.; Flanagan, J.A.; Prior, R.L. High-throughput assay of oxygen radical absorbance capacity (ORAC) using a multichannel liquid handling system coupled with a microplate fluorescence reader in 96-well format. J. Agric. Food Chem. 2002, 50, 4437–4444. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lind, K.F.; Hansen, E.; Østerud, B.; Eilertsen, K.-E.; Bayer, A.; Engqvist, M.; Leszczak, K.; Jørgensen, T.Ø.; Andersen, J.H. Antioxidant and Anti-Inflammatory Activities of Barettin. Mar. Drugs 2013, 11, 2655-2666. https://doi.org/10.3390/md11072655
Lind KF, Hansen E, Østerud B, Eilertsen K-E, Bayer A, Engqvist M, Leszczak K, Jørgensen TØ, Andersen JH. Antioxidant and Anti-Inflammatory Activities of Barettin. Marine Drugs. 2013; 11(7):2655-2666. https://doi.org/10.3390/md11072655
Chicago/Turabian StyleLind, Karianne F., Espen Hansen, Bjarne Østerud, Karl-Erik Eilertsen, Annette Bayer, Magnus Engqvist, Kinga Leszczak, Trond Ø. Jørgensen, and Jeanette H. Andersen. 2013. "Antioxidant and Anti-Inflammatory Activities of Barettin" Marine Drugs 11, no. 7: 2655-2666. https://doi.org/10.3390/md11072655