New Dimeric Members of the Phomoxanthone Family: Phomolactonexanthones A, B and Deacetylphomoxanthone C Isolated from the Fungus Phomopsis sp.

Abstract
:1. Introduction
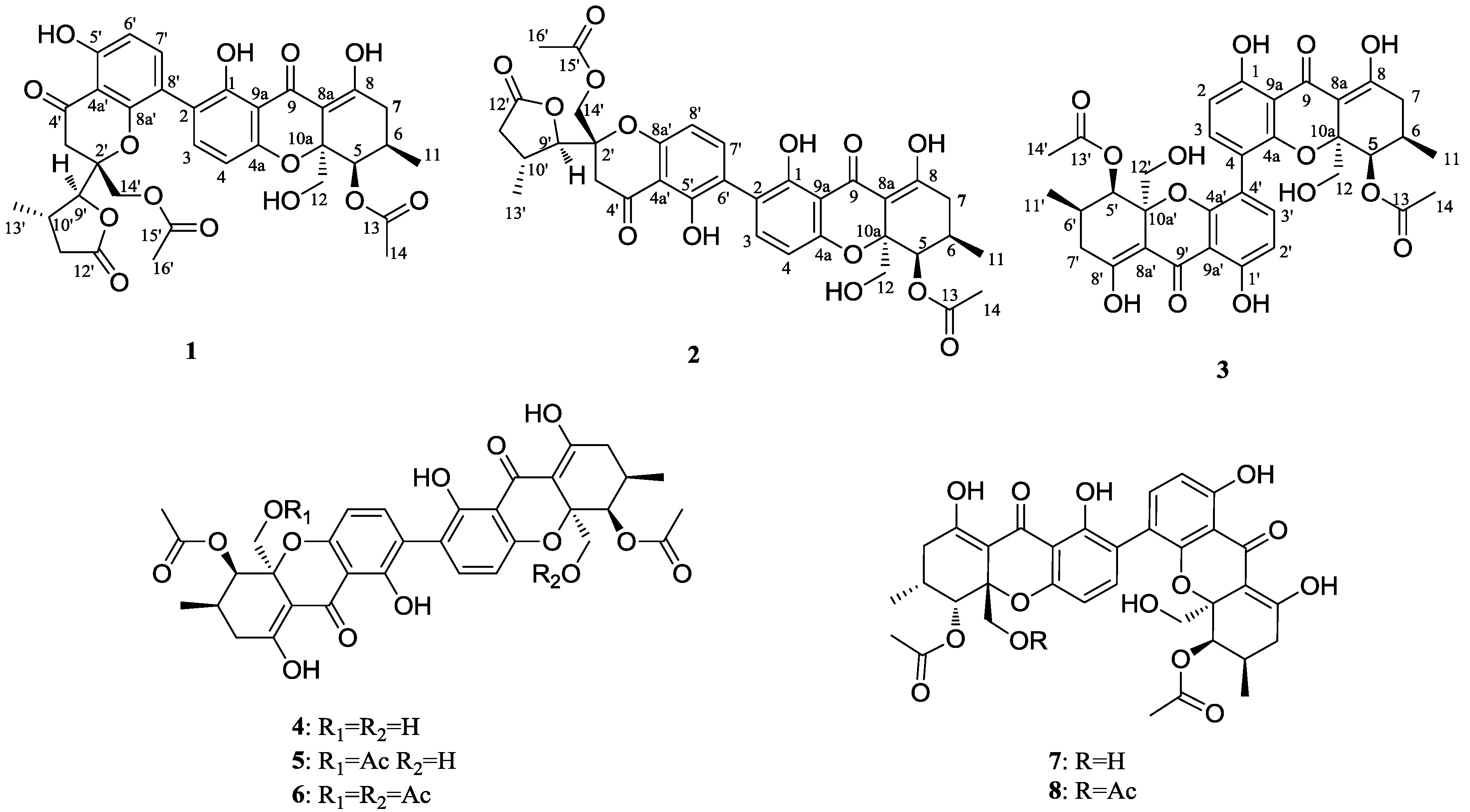
2. Results and Discussion
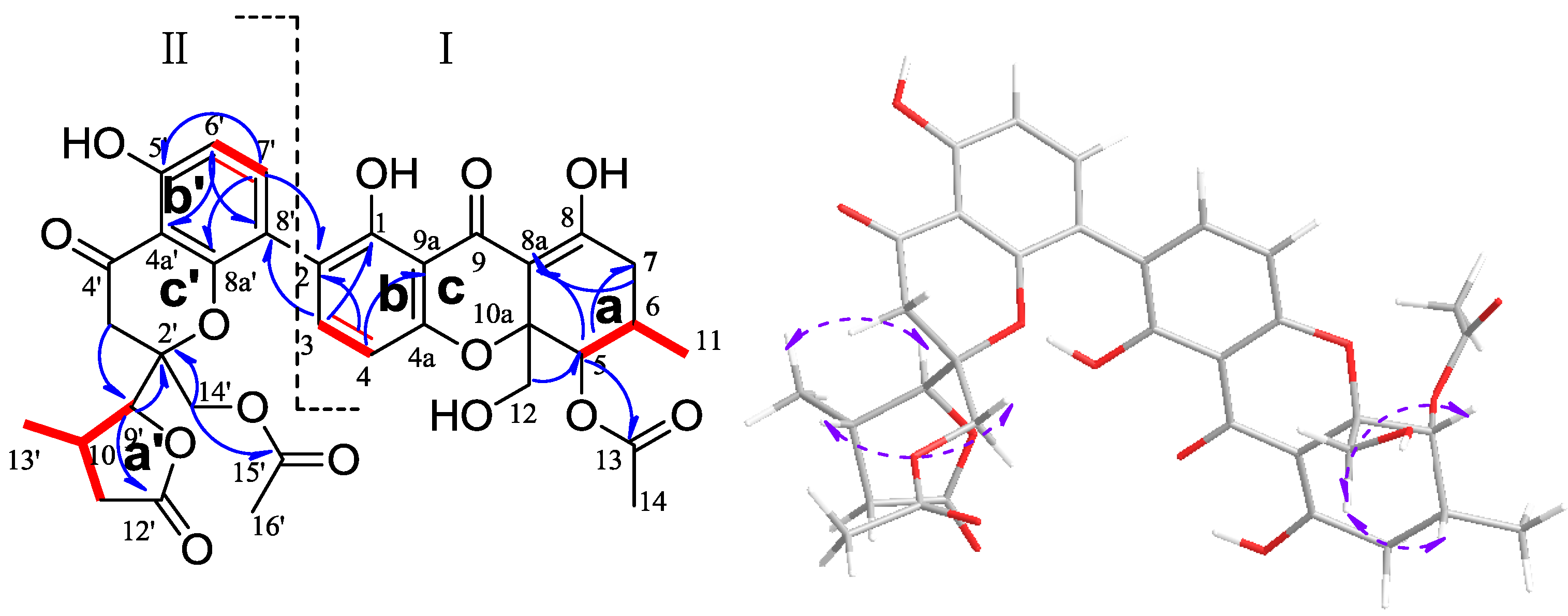
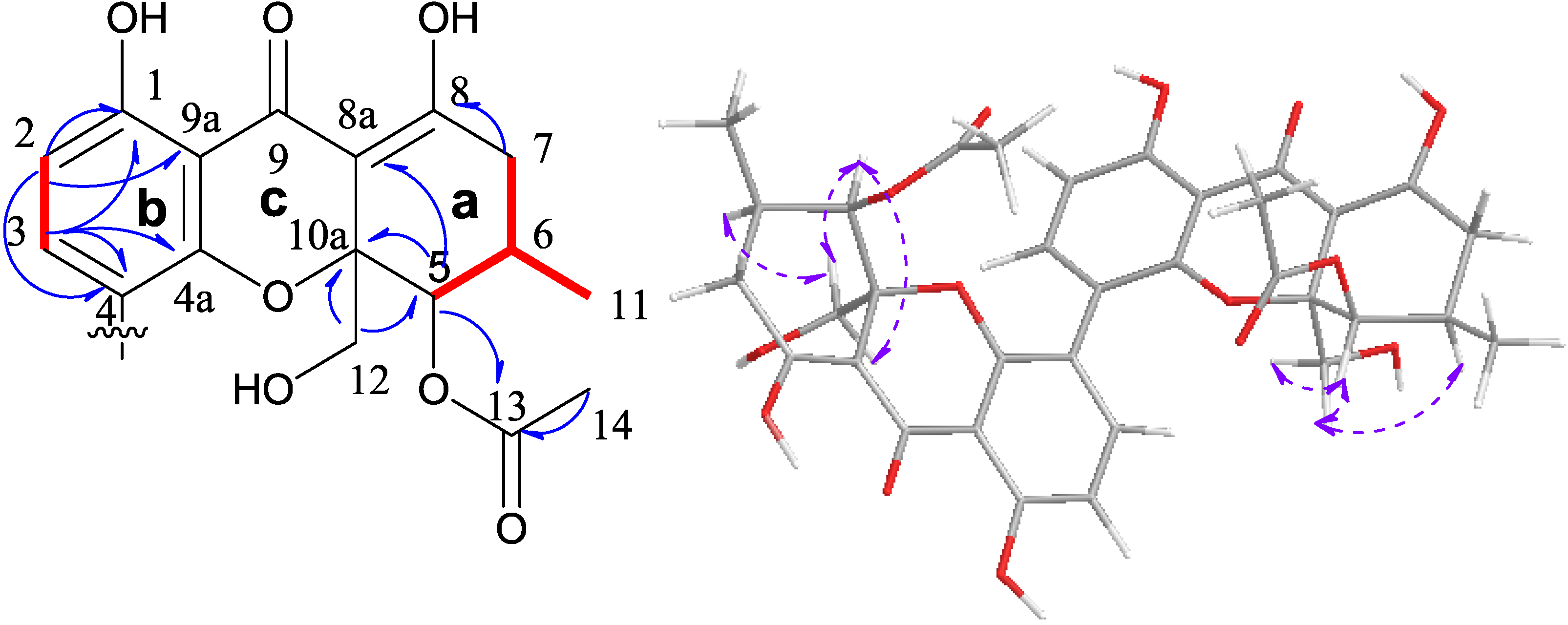
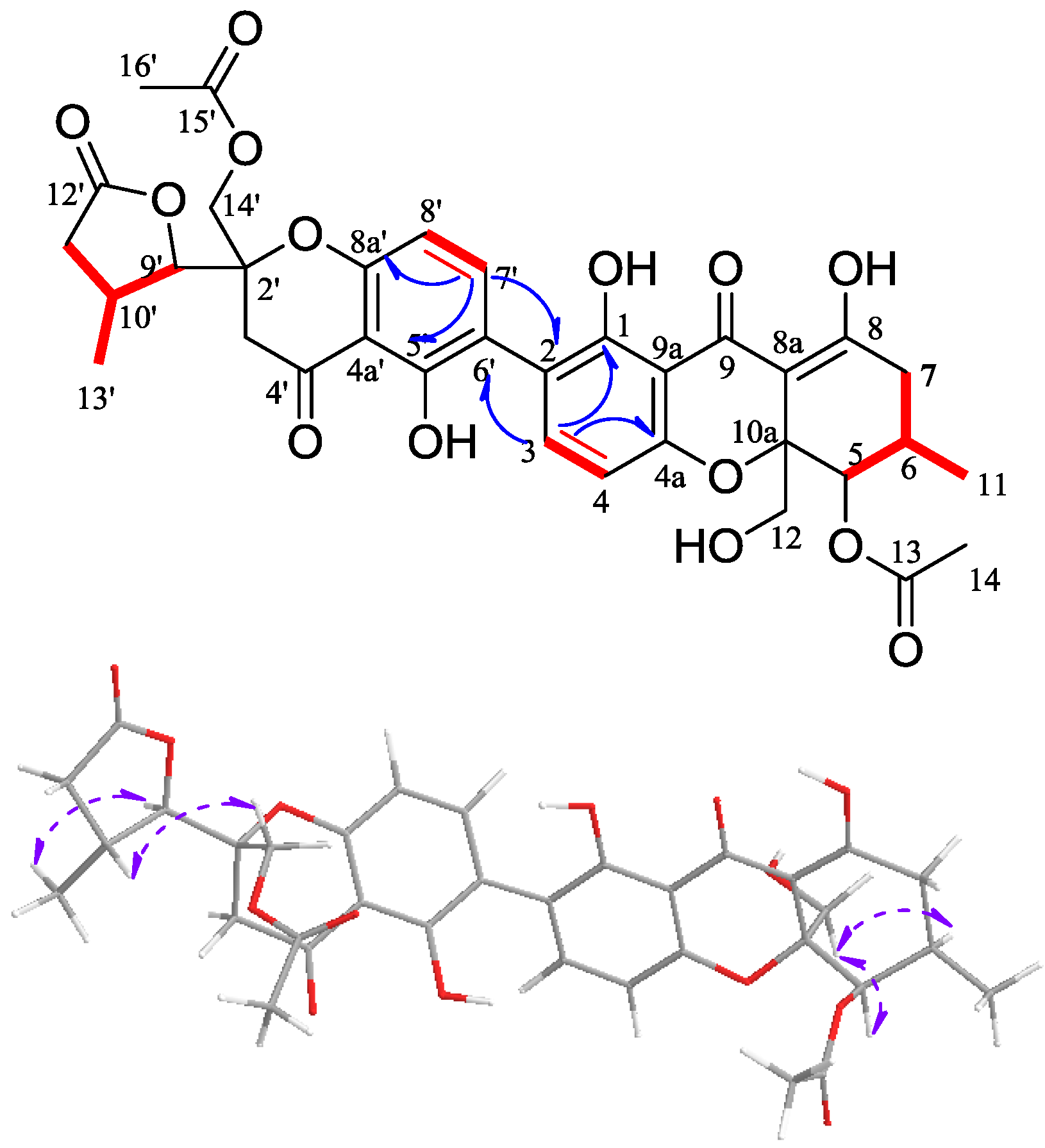
2.1. Structural Elucidation of New Compounds

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC, Type | δH, Mult. (J in Hz) | δC, Type | δH, Mult. (J in Hz) | |
| 1 | 159.4, C | 159.6, C | ||
| 2 | 118.6, C | 117.7, C | ||
| 3 | 139.6, CH | 7.24, d (8.4) | 140.2, CH | 7.41, d (8.5) |
| 4 | 108.2, CH | 6.51, d (8.4) | 108.0, CH | 6.51, d (8.5) |
| 4a | 157.4, C | 157.3, C | ||
| 5 | 70.2, CH | 5.57, br s | 70.3, CH | 5.75, br s |
| 6 | 27.9, CH | 2.41, m | 27.8, CH | 2.40, m |
| 7 | 33.5, CH2 | 2.42-2.50 m | 33.5, CH2 | 2.41–2.48, m |
| 8 | 178.7, C | 178.1, C | ||
| 8a | 101.1, C | 101.0, C | ||
| 9 | 187.7, C | 187.8, C | ||
| 9a | 106.2, C | 106.5, C | ||
| 10a | 82.7, C | 82.6, C | ||
| 11 | 17.6, CH3 | 1.08, d (6.0) | 17.7, CH3 | 1.07, d (5.8) |
| 12α | 65.7, CH2 | 3.97, d (12.9) | 65.7, CH2 | 4.09, d (13.0) |
| 12β | 3.58, d (12.9) | 3.54, d (13.0) | ||
| 13 | 170.6, C | 170.7, C | ||
| 14 | 21.4, CH3 | 2.11, s | 21.0, CH3 | 2.10, s |
| 1-OH | 11.84, s | 11.86, s | ||
| 2′ | 82.2, C | 81.8, C | ||
| 3′α | 38.9, CH2 | 3.37, d (17.3) | 38.8, CH2 | 3.34, d (17.4) |
| 3′β | 2.76, d (17.4) | 2.81, d (17.4) | ||
| 4′ | 196.6, C | 196.1, C | ||
| 4a′ | 107.2, C | 107.2, C | ||
| 5′ | 161.5, C | 159.1, C | ||
| 6′ | 110.3, CH | 6.61, d (8.6) | 117.9, C | |
| 7′ | 140.3, CH | 7.31, d (8.6) | 140.8, CH | 7.45, d (8.5) |
| 8′ | 116.0, C | 107.1, CH | 6.45,d (8.5) | |
| 8a′ | 155.3, C | 157.9, C | ||
| 9′ | 87.3, CH | 4.10, d (2.7) | 86.5, CH | 4.23, d (3.7) |
| 10′ | 29.3, CH | 2.62, m | 29.5, CH | 2.92, m |
| 11′ α | 35.9, CH2 | 2.18, dd ( 17.8, 9.2) | 36.6, CH2 | 2.94, m |
| 11′ β | 1.85, dd (17.8, 9.2) | 2.24, m | ||
| 12′ | 176.0, C | 175.5, C | ||
| 13′ | 21.0, CH3 | 1.12, d (7.1) | 20.6, CH3 | 1.28, d (6.7) |
| 14′α | 63.2, CH2 | 4.45, d (12.2) | 63.8 CH2 | 4.39, d (12.1) |
| 14′β | 4.32, d (12.3) | 4.31, d (12.1) | ||
| 15′ | 169.9, C | 170.0, C | ||
| 16′ | 20.6, CH3 | 2.03, s | 20.9, CH3 | 2.03, s |
| 5′-OH | 11.63, s | 11.98, s | ||
| Position | δC, Type | δH, Mult. (J in Hz) |
|---|---|---|
| 1,1′ | 161.8, C | |
| 2,2′ | 110.2, CH | 6.58, d (8.6) |
| 3,3′ | 141.1, CH | 7.12, d (8.6) |
| 4,4′ | 115.4, C | |
| 4a,4a′ | 153.7, C | |
| 5,5′ | 69.7, CH | 5.48, br s |
| 6,6′ | 27.9, CH | 2.27, m |
| 7,7′ | 33.3, CH2 | 2.37-2.41, m |
| 8,8′ | 177.7, C | |
| 8a,8a′ | 101.5, C | |
| 9,9′ | 188.1, C | |
| 9a,9a′ | 107.1, C | |
| 10a,10a′ | 82.2, C | |
| 11,11′ | 17.6, CH3 | 1.01, d (6.4) |
| 12α,12′α | 64.6 CH2 | 3.87, d (13.0) |
| 12β,12′β | 3.42, d (13.0) | |
| 13,13′ | 169.9, C | |
| 14,14′ | 21.0, CH3 | 2.14, s |
| 1,1′-OH | 11.44, s | |
| 8,8′-OH | 13.97, s |
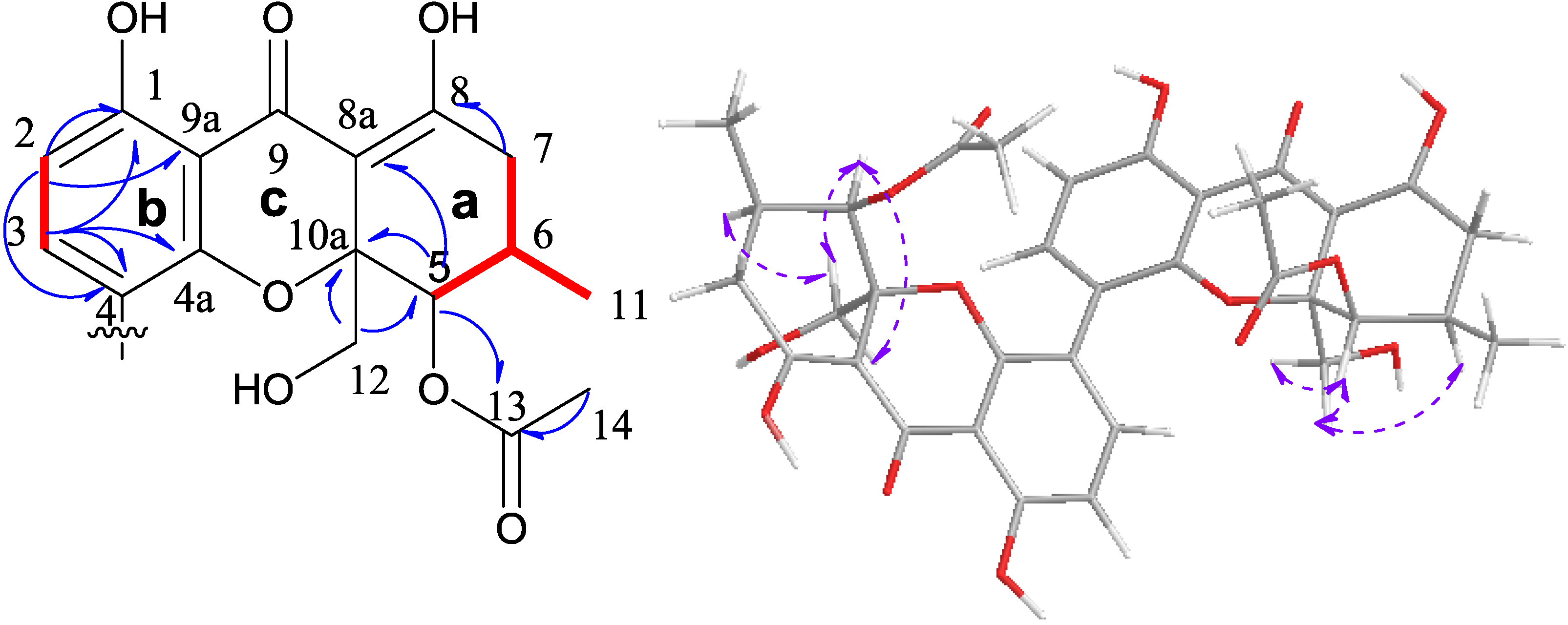
2.2. Cytotoxic Results
| Coumpound | IC50 (μM) | ||||
|---|---|---|---|---|---|
| MDA-MB-435 c | HCT-116 c | Calu-3 c | Huh7 c | MCF-10A c | |
| phomolactonexanthone A (1) | >50 | >50 | 43.45 ± 2.51 | >50 | >50 |
| phomolactonexanthone B (2) | >50 | >50 | >50 | >50 | >50 |
| phomolactonexanthone C (3) | >50 | 44.06 ± 3.29 | 43.35 ± 2.09 | >50 | >50 |
| dicerandrol A (4) | 3.03 ± 0.12 | 2.64 ± 0.03 | 1.76 ± 0.02 | 4.19 ± 0.08 | 28.32 ± 3.57 |
| dicerandrol B (5) | 8.65 ± 0.66 | 3.94 ± 0.39 | 4.10 ± 0.08 | 30.37 ± 1.10 | 8.14 ± 1.27 |
| dicerandrol C (6) | 44.10 ± 2.45 | 42.63 ± 2.90 | 36.52 ± 3.32 | >50 | 33.05 ± 2.74 |
| deacetylphomoxanthone B (7) | 14.40 ± 1.18 | 7.12 ± 0.70 | 4.14 ± 0.02 | 29.20 ± 1.19 | >50 |
| penexanthone A (8) | 7.90 ± 0.58 | 6.92 ± 0.38 | 6.44 ± 0.86 | 42.82 ± 3.58 | 16.13 ± 1.57 |
| epirubicin b | 0.56 ± 0.06 | 0.48 ± 0.029 | 1.03 ± 0.10 | 0.96 ± 0.01 | 0.48 ± 0.08 |
3. Experimental Section
3.1. General
3.2. Fungal Material
3.3. Extraction and Isolation
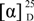 −75 (c, 0.2, MeOH), IR νmax (KBr): 3428, 2917, 2521, 2138, 1798, 1635, 1436, 1388, 1061 cm−1, UV (PDA) λmax 241.6, 340.3 nm; for 1H and 13C NMR data see Table 1; HRESIMS m/z 665.18707 [M − H]− (calcd for C34H33O14, 665.18758). −21.7 (c, 0.6, MeOH), IR νmax (KBr): 3429, 2919, 2516, 1796, 1628, 1434, 1057 cm−1, UV (PDA) λmax 245.2, 340.3 nm; for 1H and 13C NMR data see Table 1; HRESIMS m/z 665.18731 [M − H]− (calcd for C34H33O14, 665.18758). +101.6 (c, 0.44, MeOH), IR νmax (KBr): 3402, 2925, 1743, 1614, 1465, 1366, 1224, 1045 cm−1, UV (PDA) λmax 216.9, 248.7, 337.9 nm; for 1H and 13C NMR data see Table 2; HRESIMS m/z 665.18739 [M − H]− (calcd for C34H33O14, 665.18758).
−75 (c, 0.2, MeOH), IR νmax (KBr): 3428, 2917, 2521, 2138, 1798, 1635, 1436, 1388, 1061 cm−1, UV (PDA) λmax 241.6, 340.3 nm; for 1H and 13C NMR data see Table 1; HRESIMS m/z 665.18707 [M − H]− (calcd for C34H33O14, 665.18758). −21.7 (c, 0.6, MeOH), IR νmax (KBr): 3429, 2919, 2516, 1796, 1628, 1434, 1057 cm−1, UV (PDA) λmax 245.2, 340.3 nm; for 1H and 13C NMR data see Table 1; HRESIMS m/z 665.18731 [M − H]− (calcd for C34H33O14, 665.18758). +101.6 (c, 0.44, MeOH), IR νmax (KBr): 3402, 2925, 1743, 1614, 1465, 1366, 1224, 1045 cm−1, UV (PDA) λmax 216.9, 248.7, 337.9 nm; for 1H and 13C NMR data see Table 2; HRESIMS m/z 665.18739 [M − H]− (calcd for C34H33O14, 665.18758).3.4. Antitumor Activity in Vitro
3.4.1. Cell Culture
3.4.2. Assessment of Antitumor Activity by MTT Assay
4. Conclusions
Acknowledgements
Conflicts of Interest
References
- Hostettmann, K.; Wagner, H. Xanthone glycosides. Phytochemistry 1977, 16, 821–829. [Google Scholar] [CrossRef]
- Na, Y. Recent cancer drug development with xanthone structures. J. Pharm. Pharmacol. 2009, 61, 707–712. [Google Scholar] [CrossRef]
- Pinto, M.M.M.; Sousa, M.E.; Nascimento, M.S.J. Xanthone Derivatives: New Insights in Biological Activities. Curr. Med. Chem. 2005, 12, 2517–2538. [Google Scholar]
- Yang, C.-H.; Ma, L.; Wei, Z.-P.; Han, F.; Gao, J. Advances in Isolation and Synthesis of Xanthone Derivatives. Chin. Herb. Med. 2012, 4, 87–102. [Google Scholar]
- Masters, K.-S.; Bräse, S. Xanthones from Fungi, Lichens, and Bacteria: The Natural Products and Their Synthesis. Chem. Rev. 2012, 112, 3717–3776. [Google Scholar] [CrossRef]
- Lim, C.; Kim, J.; Choi, J.N.; Ponnusamy, K.; Jeon, Y.; Kim, S.U.; Kim, J.G.; Lee, C. Identification, fermentation, and bioactivity against Xanthomonas oryzae of antimicrobial metabolites isolated from Phomopsis longicolla S1B4. J. Microbiol. Biotechnol. 2010, 20, 494–500. [Google Scholar]
- Elsässer, B.; Krohn, K.; Flörke, U.; Root, N.; Aust, H.-J.; Draeger, S.; Schulz, B.; Antus, S.; Kurtán, T. X-ray Structure Determination, Absolute Configuration and Biological Activity of Phomoxanthone A. Eur. J. Org. Chem. 2005, 2005, 4563–4570. [Google Scholar] [CrossRef]
- Isaka, M.; Jaturapat, A.; Rukseree, K.; Danwisetkanjana, K.; Tanticharoen, M.; Thebtaranonth, Y. Phomoxanthones A and B, Novel Xanthone Dimers from the Endophytic Fungus Phomopsis Species. J. Nat. Prod. 2001, 64, 1015–1018. [Google Scholar] [CrossRef]
- Wagenaar, M.M.; Clardy, J. Dicerandrols, New Antibiotic and Cytotoxic Dimers Produced by the Fungus Phomopsis longicolla Isolated from an Endangered Mint. J. Nat. Prod. 2001, 64, 1006–1009. [Google Scholar] [CrossRef]
- Rukachaisirikul, V.; Sommart, U.; Phongpaichit, S.; Sakayaroj, J.; Kirtikara, K. Metabolites from the endophytic fungus Phomopsis sp. PSU-D15. Phytochemistry 2008, 69, 783–787. [Google Scholar] [CrossRef]
- Choi, J.N.; Kim, J.; Ponnusamy, K.; Lim, C.; Kim, J.G.; Muthaiya, M.J.; Lee, C.H. Identification of a new phomoxanthone antibiotic from Phomopsis longicolla and its antimicrobial correlation with other metabolites during fermentation. J. Antibiot. 2013, 66, 231–233. [Google Scholar] [CrossRef]
- Cao, S.; McMillin, D.W.; Tamayo, G.; Delmore, J.; Mitsiades, C.S.; Clardy, J. Inhibition of Tumor Cells Interacting with Stromal Cells by Xanthones Isolated from a Costa Rican Penicillium sp. J. Nat. Prod. 2012, 75, 793–797. [Google Scholar] [CrossRef]
- Wen, L.; Cai, X.; Xu, F.; She, Z.; Chan, W.L.; Vrijmoed, L.L.P.; Jones, E.B.G.; Lin, Y. Three Metabolites from the Mangrove Endophytic Fungus Sporothrix sp. (#4335) from the South China Sea. J. Org. Chem. 2009, 74, 1093–1098. [Google Scholar] [CrossRef]
- Huang, X.; Huang, H.; Li, H.; Sun, X.; Huang, H.; Lu, Y.; Lin, Y.; Long, Y.; She, Z. Asperterpenoid A, a New Sesterterpenoid as an Inhibitor of Mycobacterium tuberculosis Protein Tyrosine Phosphatase B from the Culture of Aspergillus sp. 16-5c. Org. Lett. 2013, 15, 721–723. [Google Scholar] [CrossRef]
- Xiao, Z.E.; Huang, H.R.; Shao, C.L.; Xia, X.K.; Ma, L.; Huang, X.S.; Lu, Y.J.; Lin, Y.C.; Long, Y.H.; She, Z.G. Asperterpenols A and B, New Sesterterpenoids Isolated from a Mangrove Endophytic Fungus Aspergillus sp 085242. Org. Lett. 2013, 15, 2522–2525. [Google Scholar] [CrossRef]
- Zhang, W.; Krohn, K.; Zia, U.; Flörke, U.; Pescitelli, G.; Di Bari, L.; Antus, S.; Kurtán, T.; Rheinheimer, J.; Draeger, S.; et al. New Mono- and Dimeric Members of the Secalonic Acid Family: Blennolides A–G Isolated from the Fungus Blennoria sp. Chem. Eur. J. 2008, 14, 4913–4923. [Google Scholar] [CrossRef]
- Chen, H.; Zhong, L.; Long, Y.; Li, J.; Wu, J.; Liu, L.; Chen, S.; Lin, Y.; Li, M.; Zhu, X.; et al. Studies on the Synthesis of Derivatives of Marine-Derived Bostrycin and Their Structure-Activity Relationship against Tumor Cells. Mar. Drugs 2012, 10, 932–952. [Google Scholar] [CrossRef]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ding, B.; Yuan, J.; Huang, X.; Wen, W.; Zhu, X.; Liu, Y.; Li, H.; Lu, Y.; He, L.; Tan, H.; et al. New Dimeric Members of the Phomoxanthone Family: Phomolactonexanthones A, B and Deacetylphomoxanthone C Isolated from the Fungus Phomopsis sp. Mar. Drugs 2013, 11, 4961-4972. https://doi.org/10.3390/md11124961
Ding B, Yuan J, Huang X, Wen W, Zhu X, Liu Y, Li H, Lu Y, He L, Tan H, et al. New Dimeric Members of the Phomoxanthone Family: Phomolactonexanthones A, B and Deacetylphomoxanthone C Isolated from the Fungus Phomopsis sp. Marine Drugs. 2013; 11(12):4961-4972. https://doi.org/10.3390/md11124961
Chicago/Turabian StyleDing, Bo, Jie Yuan, Xishan Huang, Weitao Wen, Xu Zhu, Yayue Liu, Hanxiang Li, Yongjun Lu, Lei He, Hongmei Tan, and et al. 2013. "New Dimeric Members of the Phomoxanthone Family: Phomolactonexanthones A, B and Deacetylphomoxanthone C Isolated from the Fungus Phomopsis sp." Marine Drugs 11, no. 12: 4961-4972. https://doi.org/10.3390/md11124961