Atypical Reactive Center Kunitz-Type Inhibitor from the Sea Anemone Heteractis crispa

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Purification and Primary Structure Determination of InhVJ
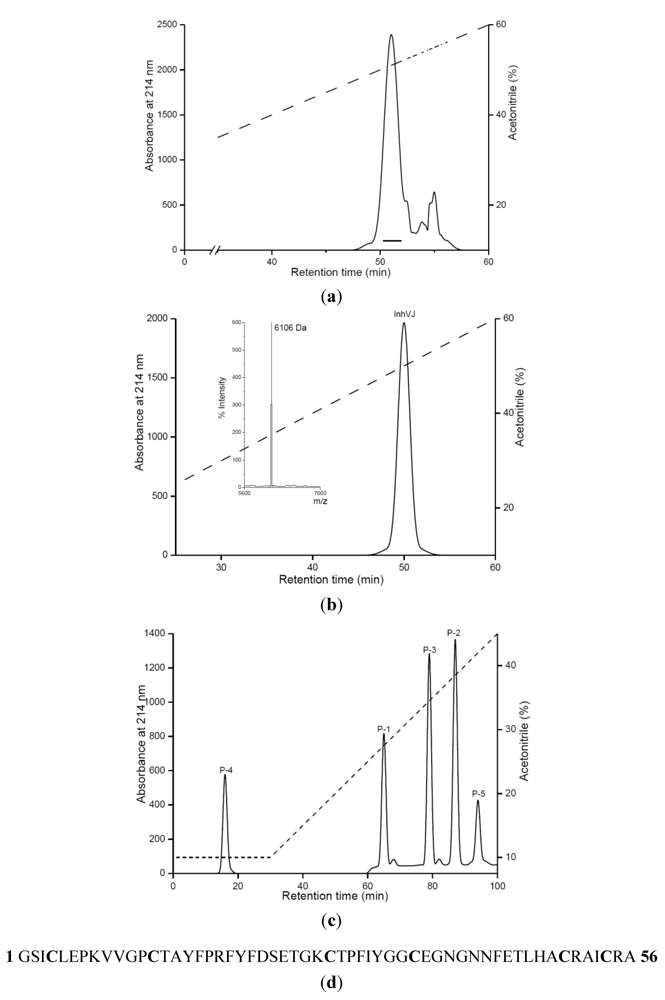

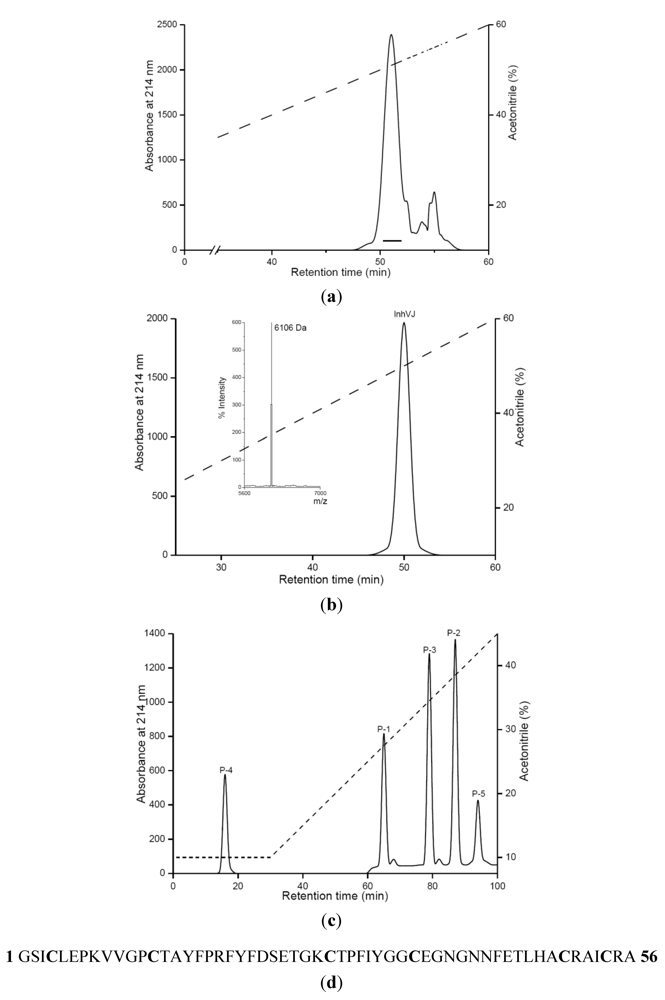{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Amino acid sequence | Molecular mass (MALDI)Dа | Molecular mass (theoretical)Da |
|---|---|---|---|
| P-1 | GSICLEPK | 951.31 | 951.01 |
| P-2 | VVGPCTAYFPR | 1314.91 | 1314.42 |
| P-3 | FYFDSETGK | 1094.54 | 1093.15 |
| P-4 | AICRA | 637.73 | 637.66 |
| P-5 | CTPFIYGGCEGNGNNFETLHACR | 2818.95 | 2818.75 |

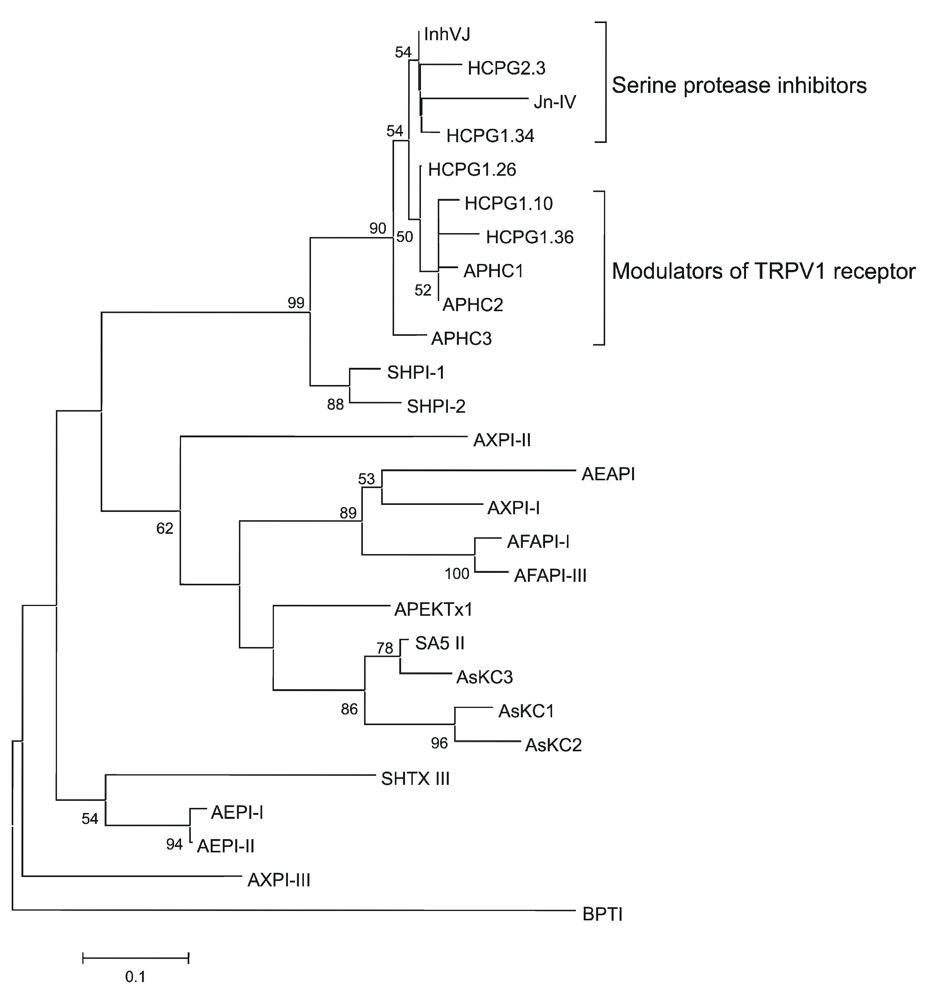
2.2. Amino Acid Sequences and Phylogenetic Analysis


2.3. Electrophysiological Experiments

2.4. Comparison of Inhibitor Protease Inhibiting Activity
| Sea anemone | Inhibitor | Кi (М) | |
|---|---|---|---|
| Trypsin | α-chymotrypsin | ||
| H. crispa | InhVJ | 7.38 × 10−8 [31] | 9.93 × 10−7 [31] |
| Jn-IV | 9.6 × 10−9 [15] | n.d. | |
| АРНС1 | 1.0 × 10−6 [21] | 5.0 × 10−6 [21] | |
| АРНС2 | 0.9 × 10−6 [22] | 4.5 × 10−6 [22] | |
| АРНС3 | 5.0 × 10−7 [22] | 7.0 × 10−6 [22] | |
| A. sulcata | SA5 II | 3.0 × 10−10 [5] | n.d. |
| AsKC1-AsKC3 | 30 × 10−9 [17] | n.d. | |
| R. rhodostoma | - | 9.5 × 10−10 [6] | 3.3 × 10−8 [6] |
| A. elegantissima | APEKTx1 | 124 × 10−9 [28] | n.d. |
| S. helianthus | SHPI-1 | 1.1 × 10−10 [11] | 2.3 × 10−9 [11] |
| B. taurus | BPTI | 6.0 × 10−14 [41] | 1.8 × 10−13 [42] |
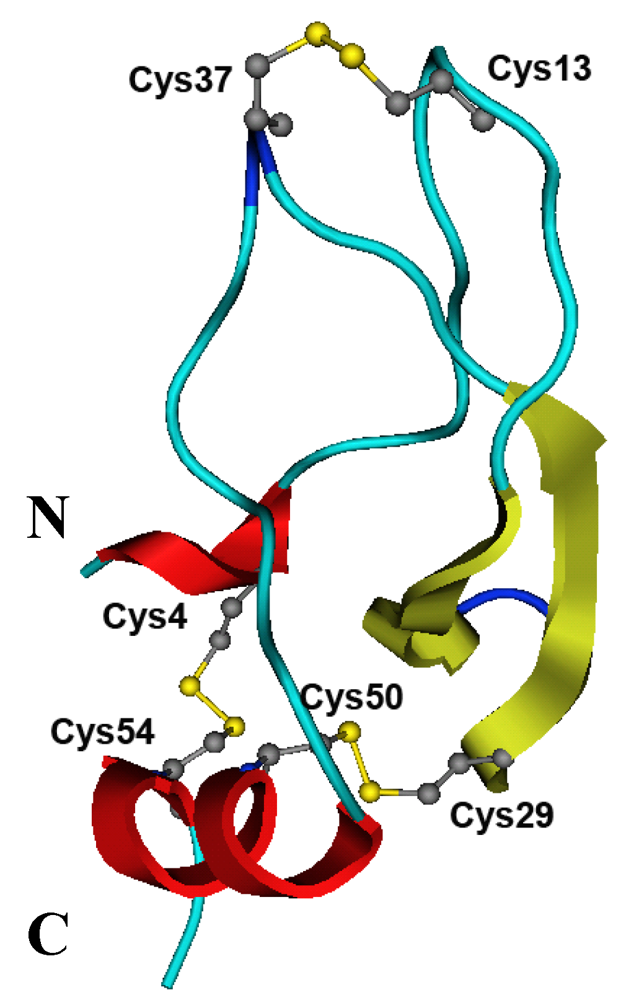
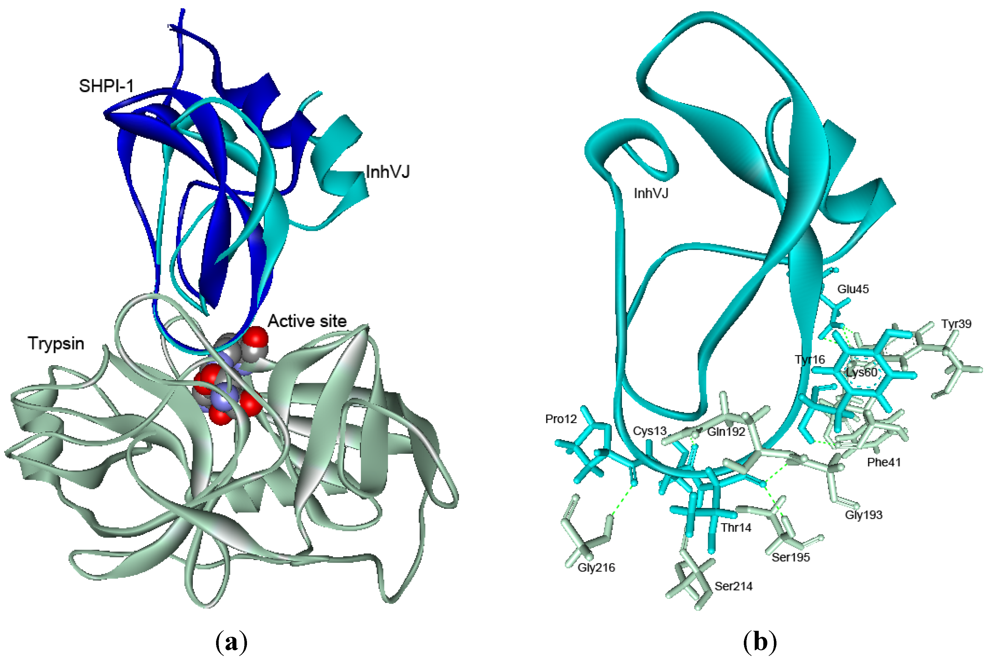
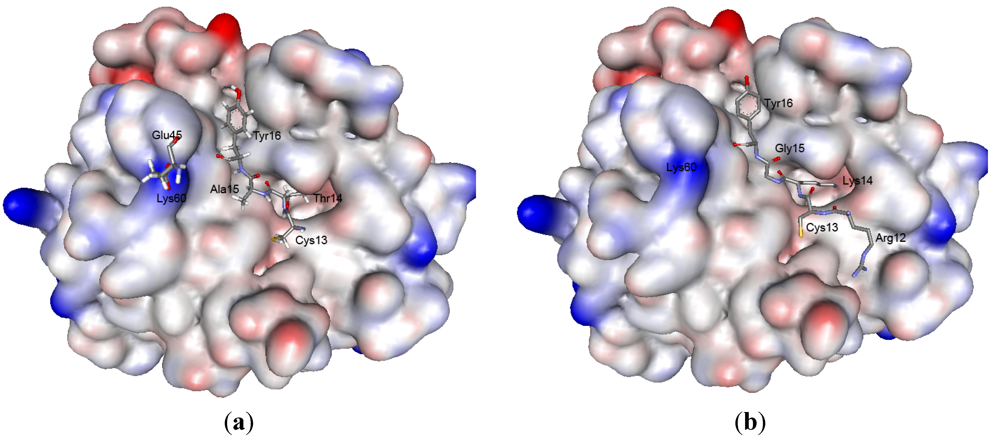
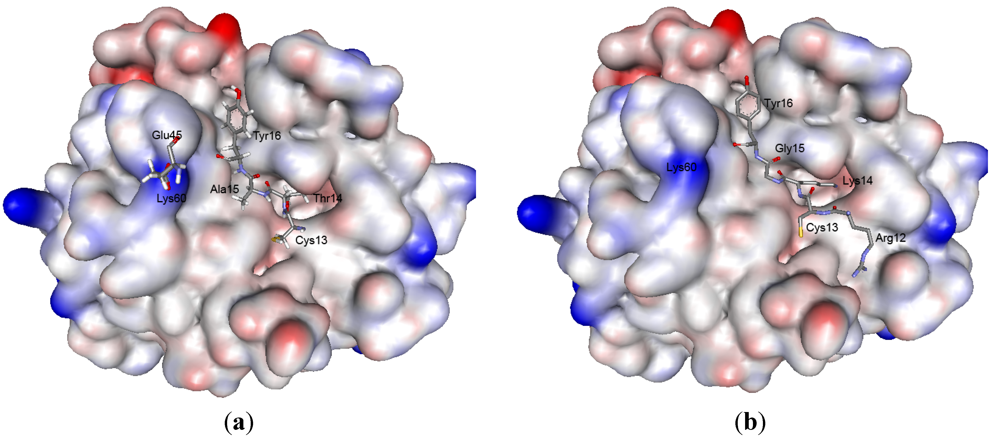
2.5. Structure Modeling

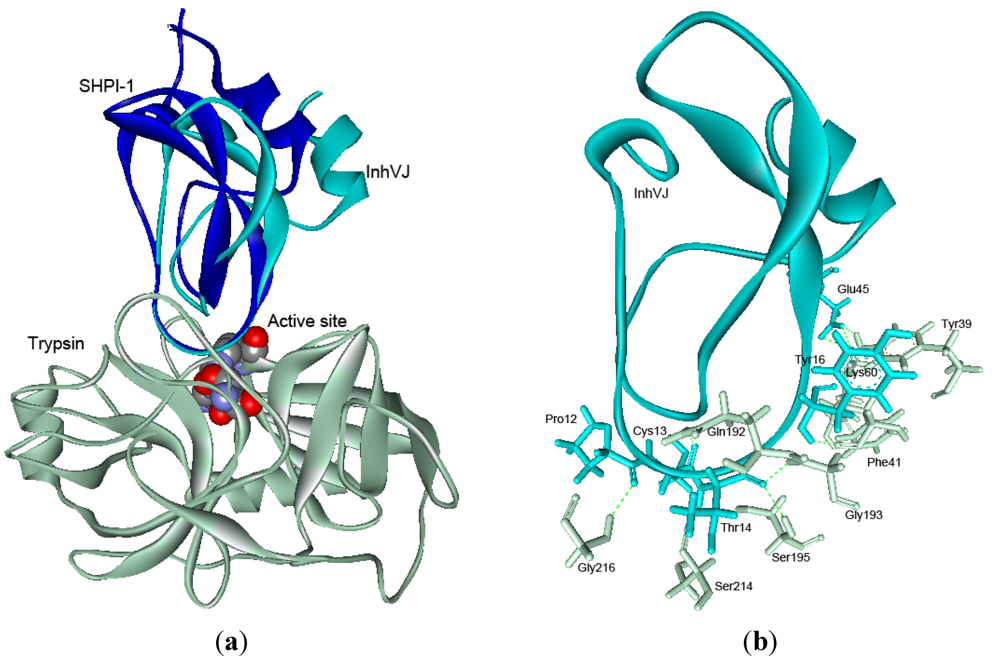
| Reactive site | Weak contact site | |||||||
|---|---|---|---|---|---|---|---|---|
| BPTI (3OTJ) | Pro12 | Cys13 | Lys14 | Ala15 | Arg16 | Cys37 | Arg38 | Lys45 |
| ΔGbind, Kcal/Mol | −2.1 | −4.8 | −8.1 | −3.3 | −3.2 | −1.8 | −2 | 1.1 |
| ΔSASA, Ǻ2 | 33 | 51.6 | 165.2 | 36 | 110.8 | 27.2 | 97.2 | 0 |
| BPTI mutant (3BTT) | Pro12 | Cys13 | Thr14 | Ala15 | Arg16 | Cys37 | Arg38 | Lys45 |
| ΔGbind, Kcal/Mol | −2.1 | −4,6 | −4.4 | −3.5 | −3.5 | −1.7 | −1.6 | 0.3 |
| ΔSASA, Ǻ2 | 37.9 | 55.5 | 101.4 | 36.5 | 118.3 | 28.6 | 94.6 | 0 |
| SHPI-1 (3M7Q) | Arg12 | Cys13 | Lys14 | Gly15 | Tyr16 | Cys37 | Gly38 | Glu45 |
| ΔGbind, Kcal/Mol | −3.4 | −2.9 | −8.4 | −2.2 | −4.5 | −1.4 | −0.1 | −0.5 |
| ΔSASA, Ǻ2 | 143.8 | 52 | 163.6 | 19.9 | 88.7 | 30 | 0.9 | 0 |
| InhVJ | Pro12 | Cys13 | Thr14 | Ala15 | Tyr16 | Cys37 | Glu38 | Glu45 |
| ΔGbind, Kcal/Mol | −2.1 | −4.4 | −4.7 | −2.6 | −5.9 | −1.1 | −0.2 | −8.4 |
| ΔSASA, Ǻ2 | 50.5 | 53 | 106.1 | 30.5 | 102.2 | 37.2 | 0 | 37.9 |
3. Experimental Section
3.1. Purification of InhVJ
3.2. Chemical and Enzymatic Treatment
3.3. Physical-Chemical Characterization
3.4. RACE Techniques, Cloning Experiments and Sequencing
3.5. Phylogenetic Analysis
3.6. Expression of Voltage-Gated Ion Channels in Xenopus Laevis Oocytes
3.7. Electrophysiological Recordings
3.8. Homology Modeling and Docking
4. Conclusions
Acknowledgments
References
- Kristeller, J.L.; Roslund, B.P.; Stahl, R.F. Benefits and risks of aprotinin use during cardiac surgery. Pharmacotherapy 2008, 28, 112–124. [Google Scholar] [CrossRef]
- Zhou, L.W.; Wang, Y.L.; Yan, X.T.; He, X.H. Urinary trypsin inhibitor treatment ameliorates acute lung and liver injury resulting from sepsis in a rat model. Saudi Med. J. 2008, 29, 368–373. [Google Scholar]
- Lin, Y.F.; Zhang, N.; Guo, H.S.; Kong, D.S.; Jiang, T.; Liang, W.; Zhao, Z.H.; Tang, Q.Q.; Ma, D. Recombinant tissue factor pathway inhibitor induces apoptosis in cultured rat mesangial cells via its Kunitz-3 domain and C-terminal through inhibiting PI3-kinase/Akt pathway. Apoptosis 2007, 12, 2163–2173. [Google Scholar] [CrossRef]
- Fritz, H.; Brey, B.; Beress, L. Polyvalent isoinhibitors of trypsin, chymotrypsin, plasmin and kallikrein from sea anemone (Anemonia sulcata), isolation, inhibition and amino acid composition. Hoppe Seyler Z. Physiol. Chem. 1972, 353, 19–30. [Google Scholar] [CrossRef]
- Wunderer, G.; Beress, L.; Machleidt, W.; Fritz, H. Broad Specificity. Inhibitors from Sea Anemones. In Methods in Enzymology; Lorand, L., Ed.; Academic Press: San Francisco, CA, USA, 1976. [Google Scholar]
- Mebs, D.; Liebrich, M.; Reul, A.; Samejima, Y. Hemolysins and proteinase inhibitors from sea anemones of the Gulf of Aqaba. Toxicon 1983, 21, 257–264. [Google Scholar]
- Shiomi, K.; Ishikawa, M.; Yamanaka, H.; Kikuchi, T. Isolation and properties of four serine protease inhibitors from water extracts of sea anemone Actinia equine. Nippon Suisan Gakkaishi 1989, 55, 1235–1241. [Google Scholar] [CrossRef]
- Mebs, D.; Gebauer, E. Isolation of proteinase inhibitory, toxic and hemolytic polypeptides from the sea anemone Stichodactyla sp. Toxicon 1980, 18, 257–264. [Google Scholar]
- Minagawa, S.; Ishida, M.; Shimakura, K.; Nagashima, Y.; Shiomi, K. Isolation and amino acid sequences of two Kunitz-type protease inhibitors from the sea anemone Anthopleura aff. xanthogrammica. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1997, 118, 381–386. [Google Scholar] [CrossRef]
- Minagawa, S.; Ishida, M.; Shimakura, K.; Nagashima, Y.; Shiomi, K. Amino acid sequence and biological activities of another Kunitz-type protease inhibitor from the sea anemone Anthopleura aff. xanthogrammica. Fish Sci. 1998, 64, 157–161. [Google Scholar]
- Delfín, J.; Martínez, I.; Antuch, W.; Morera, V.; González, Y.; Rodríguez, R.; Márquez, M.; Saroyán, A.; Larionova, N.; Díaz, J.; et al. Purification, characterization and immobilization of proteinase inhibitors from Stichodactyla helianthus. Toxicon 1996, 34, 1367–1376. [Google Scholar]
- Díaz, J.; Morera, V.; Delfín, J.; Huerta, V.; Lima, G.; Rodriguex de la Vega, M.; Garcia, B.; Padrón, G.; Assfalg-Machleidt, I.; Machleidt, W.; et al. Purification and partial characterization of a novel proteinase inhibitor from the sea anemone Stichodactyla helianthus. Toxicon 1998, 36, 1275–1276. [Google Scholar]
- Kolkenbrock, H.; Tschesche, H.A. New inhibitor of elastase from the sea anemone Anemonia sulcata. Biol. Chem. Hoppe Seyler 1987, 368, 93–99. [Google Scholar] [CrossRef]
- Ishida, M.; Minagawa, S.; Miyauchi, K.; Shimakura, K.; Nagashima, Y.; Shiomi, K. Amino acid sequences of Kunitz-type protease inhibitors from the sea anemone Actinia equine. Fish Sci. 1997, 63, 794–798. [Google Scholar]
- Zykova, T.A.; Vinokurov, L.M.; Markova, L.F.; Kozlovskaya, E.P.; Elyakov, G.B. Amino-acid sequence of trypsin inhibitor IV from Radianthus macrodactylus. Bioorg. Khim. 1985, 11, 293–301. [Google Scholar]
- Wunderer, G.; Machleidt, W.; Fritz, H. The Broad-Specificity Proteinase Inhibitor 5 II from the Sea Anemone Anemonia sulcata. In Methods in Enzymology; Lorand, L., Ed.; Academic Press: San Francisco , CA, USA, 1981; pp. 816–820. [Google Scholar]
- Schweitz, H.; Bruhn, T.; Guillemar, E.; Moinier, D.; Lancelin, J.M.; Béress, L.; Lazdunski, M. Kalicludines and Kaliseptine: Two different classes of sea anemone toxins for voltage sensitive K+ cannels. J. Biol. Chem. 1995, 270, 25121–25126. [Google Scholar]
- Sokotun, I.N.; Il’ina, A.P.; Monastyrnaya, M.M.; Leychenko, E.V.; Es’kov, A.A.; Anastuk, S.D.; Kozlovskaya, E.P. Proteinase inhibitors from the tropical sea anemone Radianthus macrodactylus: Isolation and characteristic. Biochemistry 2007, 72, 301–306. [Google Scholar]
- Sokotun, I.N.; Leichenko, E.V.; Vakorina, T.I.; Es’kov, A.A.; Il’ina, A.P.; Monastyrnaia, M.M.; Kozlovskaia, E.P. A serine protease inhibitor from the anemone Radianthus macrodactylus: Isolation and physicochemical characteristics. Bioorg. Khim. 2007, 33, 448–455. [Google Scholar]
- Honma, T.; Kawahata, S.; Ishida, M.; Nagai, H.; Nagashima, Y.; Shiomi, K. Novel peptide toxins from the sea anemone Stichodactyla haddoni. Peptides 2008, 29, 536–544. [Google Scholar] [CrossRef]
- Andreev, Y.A.; Kozlov, S.A.; Koshelev, S.G.; Ivanova, E.A.; Monastyrnaya, M.M.; Kozlovskaya, E.P.; Grishin, E.V. Analgesic compound from sea anemone Heteractis crispa is the first polypeptide inhibitor of vanilloid receptor 1 (TRPV1). J. Biol. Chem. 2008, 283, 23914–23921. [Google Scholar]
- Kozlov, S.A.; Andreev, Y.A.; Murashev, A.N.; Skobtsov, D.I.; D’yachenko, I.A.; Grishin, E.V. New polypeptide components from the Heteractis crispa sea anemone with analgesic activity. Bioorg. Khim. 2009, 35, 789–798. [Google Scholar]
- Minagawa, S.; Sugiyama, M.; Ishida, M.; Nagashima, Y.; Shiomi, K. Kunitz-type protease inhibitors from acrorhagi of three species of sea anemones. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2008, 150, 240–245. [Google Scholar] [CrossRef]
- Kunitz, M.; Northrop, J. Isolation from beef pancreas of crystalline trypsinogen, trypsin, a trypsin inhibitor and intibular trypsin compound. J. Gen. Physiol. 1936, 19, 991–1007. [Google Scholar] [CrossRef]
- Bode, W.; Huger, R. Structural basis of the endoproteinase-protein inhibitor interaction. Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000, 1477, 241–252. [Google Scholar] [CrossRef]
- Krowarsch, D.; Cierpicki, T.; Jelen, F.; Otlewski, J. Canonical protein inhibitors of serine proteases. Cell Mol. Life Sci. 2003, 60, 2427–2444. [Google Scholar] [CrossRef]
- Gil, D.F.; García-Fernández, R.; Alonso-del-Rivero, M.; Lamazares, E.; Pérez, M.; Varas, L.; Díaz, J.; Chávez, M.A.; González-González, Y.; Mansur, M. Recombinant expression of ShPI-1A, a non-specific BPTI-Kunitz-type inhibitor, and its protection effect on proteolytic degradation of recombinant human miniproinsulin expressed in Pichia pastoris. FEMS Yeast Res. 2011, 11, 575–586. [Google Scholar] [CrossRef]
- Peigneur, S.; Billen, B.; Derua, R.; Waelkens, E.; Debaveye, S.; Béress, L.; Tytgat, J. A bifunctional sea anemone peptide with Kunitz type protease and potassium channel inhibiting properties. Biochem. Pharmacol. 2011, 82, 81–90. [Google Scholar]
- Zelepuga, E.; Tabakmakher, V.; Monastyrnaya, M.; Lukyanov, P.; Kozlovskaya, E. In Silico Investigation of Interaction between Human Neutrophil Elastase and Sea Anemone Heteractis crispa Kunitz Polypeptide. In Proceedings of the 5th International Conference on Bioinformatics and Biomedical Engineering (ICBBE 2011), Wuhan, China, 10-12 May 2011; pp. 1–4.
- Isaeva, M.P.; Chausova, V.E.; Zelepuga, E.A.; Guzev, K.V.; Tabakmakher, V.M.; Monastyrnaya, M.M.; Kozlovskaya, E.P. A new multigene superfamily of Kunitz-type protease inhibitors from sea anemone Heteractis crispa. Peptides 2012, 34, 88–97. [Google Scholar] [CrossRef]
- Sokotun, I.N.; Gnedenko, O.V.; Leychenko, E.V.; Monastyrnaya, M.M.; Kozlovskaya, E.P.; Molnar, A.A.; Ivanov, A.S. Study of the interaction of trypsin inhibitor from the sea anemone Radianthus macrodactylus with proteases. Biochem. (Mosc.) Suppl. Ser. B Biomed. Chem. 2007, 1, 139–142. [Google Scholar]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar]
- Kassell, B. Trypsin-Kallikrein Inhibitor (Kunitz Inhibitor, Basic Pancreatic Trypsin Inhibitor, Polyvalent Inhibitor from Bovine Organs). In Methods in Enzymology; Lorand, L., Ed.; Academic Press: San Francisco, CA, USA, 1970; Volume 19, pp. 844–852. [Google Scholar]
- Olivera, B.M.; Hillyard, D.R.; Marsh, M.; Yoshikami, D. Combinatorial peptide libraries in drug design: Lessons from venomous cone snails. Trends Biotechnol. 1995, 13, 422–426. [Google Scholar] [CrossRef]
- Sollod, B.L.; Wilson, D.; Zhaxybayeva, O.; Gogarten, J.P.; Drinkwater, R.; King, G.F. Were arachnids the first to use combinatorial peptide libraries? Peptides 2005, 26, 131–139. [Google Scholar] [CrossRef]
- Kozminsky-Atias, A.; Bar-Shalom, A.; Mishmar, D.; Zilberberg, N. Assembling an arsenal, the scorpion way. BMC Evol. Biol. 2008, 8, 333–346. [Google Scholar]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Lancelin, J.M.; Foray, M.F.; Poncin, M.; Hollecker, M.; Marion, D. Proteinase inhibitor homologues as potassium channel blockers. Nat. Struct. Biol. 1994, 4, 246–250. [Google Scholar]
- Zelepuga, E.A.; Tabakmakher, V.M.; Chausova, V.E.; Monastyrnaya, M.M.; Isaeva, M.P.; Kozlovskaya, E.P. Interaction of sea anemone Heteractis crispa Kunitz type polypeptides with pain vanilloid receptor TRPV1: In silico investigation. Russ. J. Bioorg. Chem. 2012, 38, 159–170. [Google Scholar] [CrossRef]
- Helland, R.; Otlewski, J.; Sundheim, O.; Dadlez, M.; Smalås, A.O. The crystal structures of the complexes between bovine beta-trypsin and ten P1 variants of BPTI. J. Mol. Biol. 1999, 287, 923–942. [Google Scholar] [CrossRef]
- Vincent, J.-P.; Lazdunski, M. Trypsin-pancreatic trypsin inhibitor association. Dynamics of the interaction and role of disulfide bridges. Biochemistry 1972, 11, 2967–2977. [Google Scholar] [CrossRef]
- Delaria, K.A.; Muller, D.K.; Marlor, C.W.; Brown, J.E.; Das, R.C.; Roczniak, S.O.; Tamburini, P.P. Characterization of placental bikunin, a novel human serine protease inhibitor. J. Biol. Chem. 1997, 272, 12209–12214. [Google Scholar]
- Czapinska, H.; Helland, R.; Smalås, A.O.; Otlewski, J. Crystal structures of five bovine chymotrypsin complexes with P1 BPTI variants. J. Mol. Biol. 2004, 344, 1005–1020. [Google Scholar] [CrossRef]
- Krowarsch, D.; Dadlez, M.; Buczek, O.; Krokoszynska, I.; Smalås, A.O.; Otlewski, J. Interscaffolding additivity: Binding of P1 variants of bovine pancreatic trypsin inhibitor to four serine proteases. J. Mol. Biol. 1999, 289, 175–186. [Google Scholar] [CrossRef]
- Wang, G.J.; Gao, C.F.; Wei, D.; Wang, C.; Ding, S.Q. Acute pancreatitis: Etiology and common pathogenesis. World J. Gastroenterol. 2009, 15, 1427–1430. [Google Scholar] [CrossRef]
- Averianov, A.V. Role of neutrophil elastase in pathogenesis of chronic obstructive pulmonary disease. Cytokines Inflamm. 2007, 6, 3–8. [Google Scholar]
- Antuch, W.; Berndt, D.K.; Chávez, A.M.; Delfín, J.; Wüthrich, K. The NMR solution structure of a Kunitz-type proteinase inhibitor from the sea anemone Stichodactyla helianthus. Eur. J. Biochem. 1993, 212, 675–684. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL Workspace: A web-based environment for protein structure homology modeling. Bioinformatics 2006, 22, 195–201. [Google Scholar] [CrossRef]
- Rizzo, R.C.; Jorgensen, W.L. OPLS all-atom model for amines: Resolution of the amine hydration problem. J. Am. Chem. Soc. 1999, 121, 4827–4836. [Google Scholar] [CrossRef]
- Ramachandran, G.N.; Sasisekharan, V. Conformations of polypeptides and proteins. Adv. Protein Chem. 1968, 23, 283–437. [Google Scholar] [CrossRef]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK—a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Koradi, R.; Billeter, M.; Wüthrich, K. MOLMOL: A program for display and analysis of macromolecular structures. J. Mol. Graph. 1996, 14, 51–55. [Google Scholar] [CrossRef]
- Kawamura, K.; Yamada, T.; Kurihara, K.; Tamada, T.; Kuroki, R.; Tanaka, I.; Takahashi, H.; Niimura, N. X-ray and neutron protein crystallographic analysis of the trypsin-BPTI complex. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 140–148. [Google Scholar] [CrossRef]
- Huber, R.; Kukla, D.; Bode, W.; Schwager, P.; Bartels, K.; Deisenhofer, J.; Steigemann, W. Structure of the complex formed by bovine trypsin and bovine pancreatic trypsin inhibitor: II. Crystallographic refinement at 1.9 Å resolution. J. Mol. Biol. 1974, 89, 73–101. [Google Scholar] [CrossRef]
- Il’ina, A.; Lipkin, A.; Barsova, E.; Issaeva, M.; Leychenko, E.; Guzev, K.; Monastyrnaya, M.; Lukyanov, S.; Kozlovskaya, E. Amino acid sequence of RTX-A’s isoform actinoporin from the sea anemone, Radianthus macrodactylus. Toxicon 2006, 47, 517–520. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Zuckerkandl, E.; Pauling, L. Evolutionary Divergence and Convergence in Proteins. In Evolving Genes and Proteins; Bryson, V., Vogel, H.J., Eds.; Academic Press: San Francisco, CA, USA, 1965; pp. 97–166. [Google Scholar]
- Liman, E.R.; Tytgat, J.; Hess, P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron 1992, 9, 861–871. [Google Scholar] [CrossRef]
- Caterina, M.J.; Schumacher, M.A.; Tominaga, M.; Rosen, T.A.; Levine, J.D.; Julius, D. The capsaicin receptor: A heat-activated ion channel in the pain pathway. Nature 1997, 389, 816–824. [Google Scholar]
- Kozakov, D.; Brenke, R.; Comeau, S.R.; Vajda, S. PIPER: An FFT-based protein docking program with pairwise potentials. Protein 2006, 65, 392–406. [Google Scholar] [CrossRef]
- Marquart, M.; Walter, J.; Deisenhofer, J.; Bode, W.; Huber, R. The Geometry of the reactive site and of the peptide groups in trypsin, trypsinogen and its complexes with inhibitors. Acta Cryst. Sect. B 1983, 39, 480–490. [Google Scholar] [CrossRef]
- Scheidig, A.J.; Hynes, T.R.; Pelletier, L.A.; Wells, J.A.; Kossiakoff, A.A. Crystal structures of bovine chymotrypsin and trypsin complexed to the inhibitor domain of Alzheimer's amyloid beta-protein precursor (APPI) and basic pancreatic trypsin inhibitor (BPTI): Engineering of inhibitors with altered specificities. Protein Sci. 1997, 6, 1806–1824. [Google Scholar] [CrossRef]
- Meireles, L.M.C.; Dömling, A.S.; Camacho, C.J. ANCHOR: A web server and database for analysis of protein-protein interaction binding pockets for drug discovery. Nucleic Acids Res. 2010, 38, 407–411. [Google Scholar]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar]
- Krissinel, E. Crystal contacts as nature’s docking solutions. J. Comput. Chem. 2010, 31, 133–143. [Google Scholar] [CrossRef]
- Camacho, C.J.; Zhang, C. FastContact: Rapid estimate of contact and binding free energies. Bioinformatics 2005, 21, 2534–2536. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gladkikh, I.; Monastyrnaya, M.; Leychenko, E.; Zelepuga, E.; Chausova, V.; Isaeva, M.; Anastyuk, S.; Andreev, Y.; Peigneur, S.; Tytgat, J.; et al. Atypical Reactive Center Kunitz-Type Inhibitor from the Sea Anemone Heteractis crispa. Mar. Drugs 2012, 10, 1545-1565. https://doi.org/10.3390/md10071545
Gladkikh I, Monastyrnaya M, Leychenko E, Zelepuga E, Chausova V, Isaeva M, Anastyuk S, Andreev Y, Peigneur S, Tytgat J, et al. Atypical Reactive Center Kunitz-Type Inhibitor from the Sea Anemone Heteractis crispa. Marine Drugs. 2012; 10(7):1545-1565. https://doi.org/10.3390/md10071545
Chicago/Turabian StyleGladkikh, Irina, Margarita Monastyrnaya, Elena Leychenko, Elena Zelepuga, Victoria Chausova, Marina Isaeva, Stanislav Anastyuk, Yaroslav Andreev, Steve Peigneur, Jan Tytgat, and et al. 2012. "Atypical Reactive Center Kunitz-Type Inhibitor from the Sea Anemone Heteractis crispa" Marine Drugs 10, no. 7: 1545-1565. https://doi.org/10.3390/md10071545