Marinopyrrole Derivatives as Potential Antibiotic Agents against Methicillin-Resistant Staphylococcus aureus (I)

 ,
,
Abstract
:1. Introduction
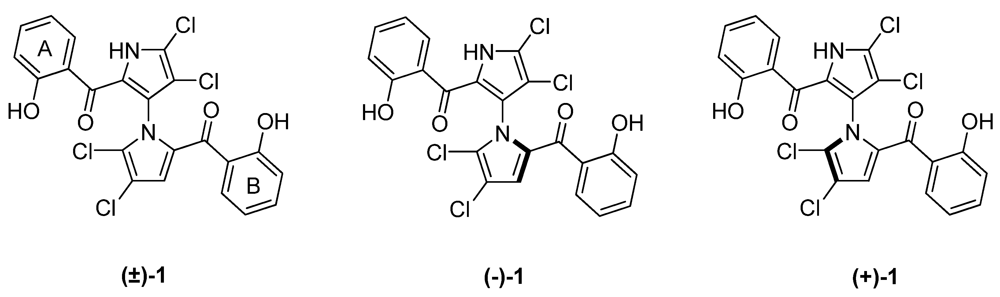
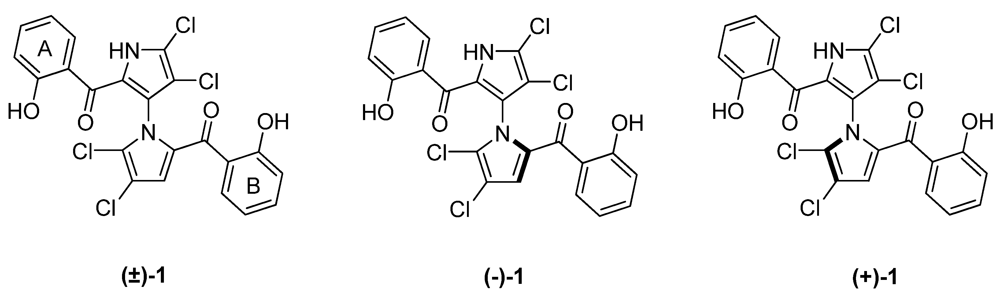
2. Results and Discussion
2.1. Synthesis and Structure Activity Relationships of Asymmetrical Marinopyrrole Derivatives
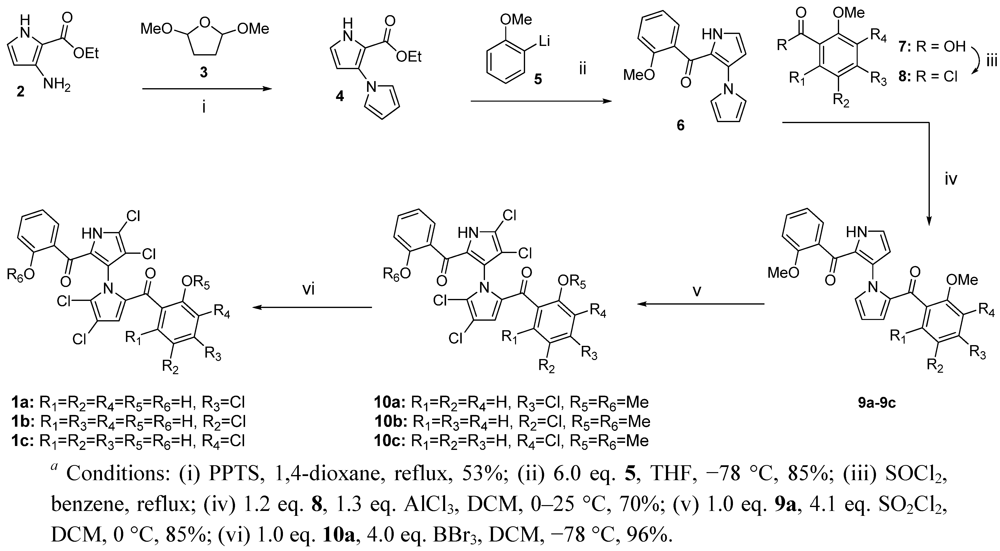

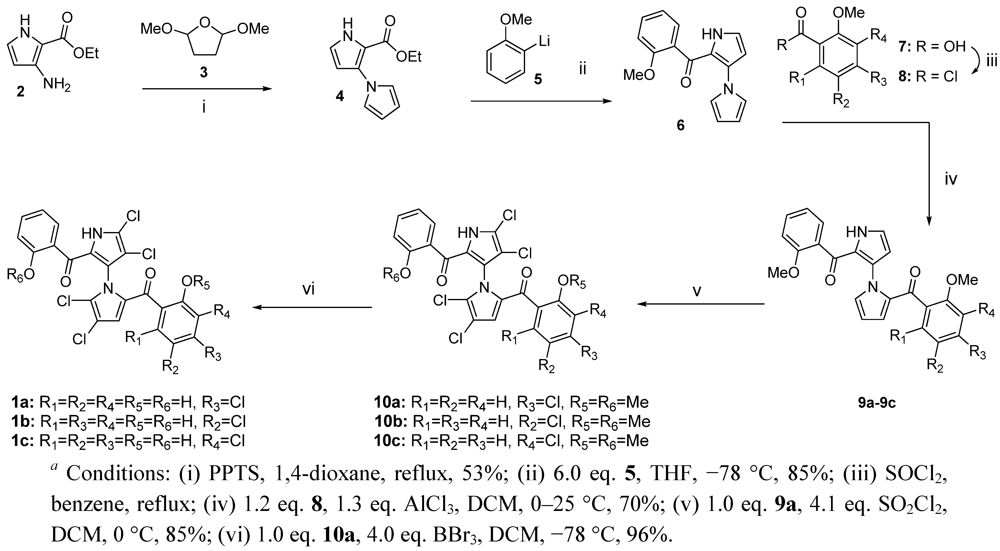
{kind=link}
{kind=link}
{kind=link}
| Compound | THB | THB + 20% Serum |
|---|---|---|
| 1 (parent) | 0.74 | 94–188 |
| 10a | >200 | >200 |
| 1a | 0.19–0.39 | 12.5–25 |
| 1b | 1.56 | >200 |
| 1c | 3.13 | >200 |
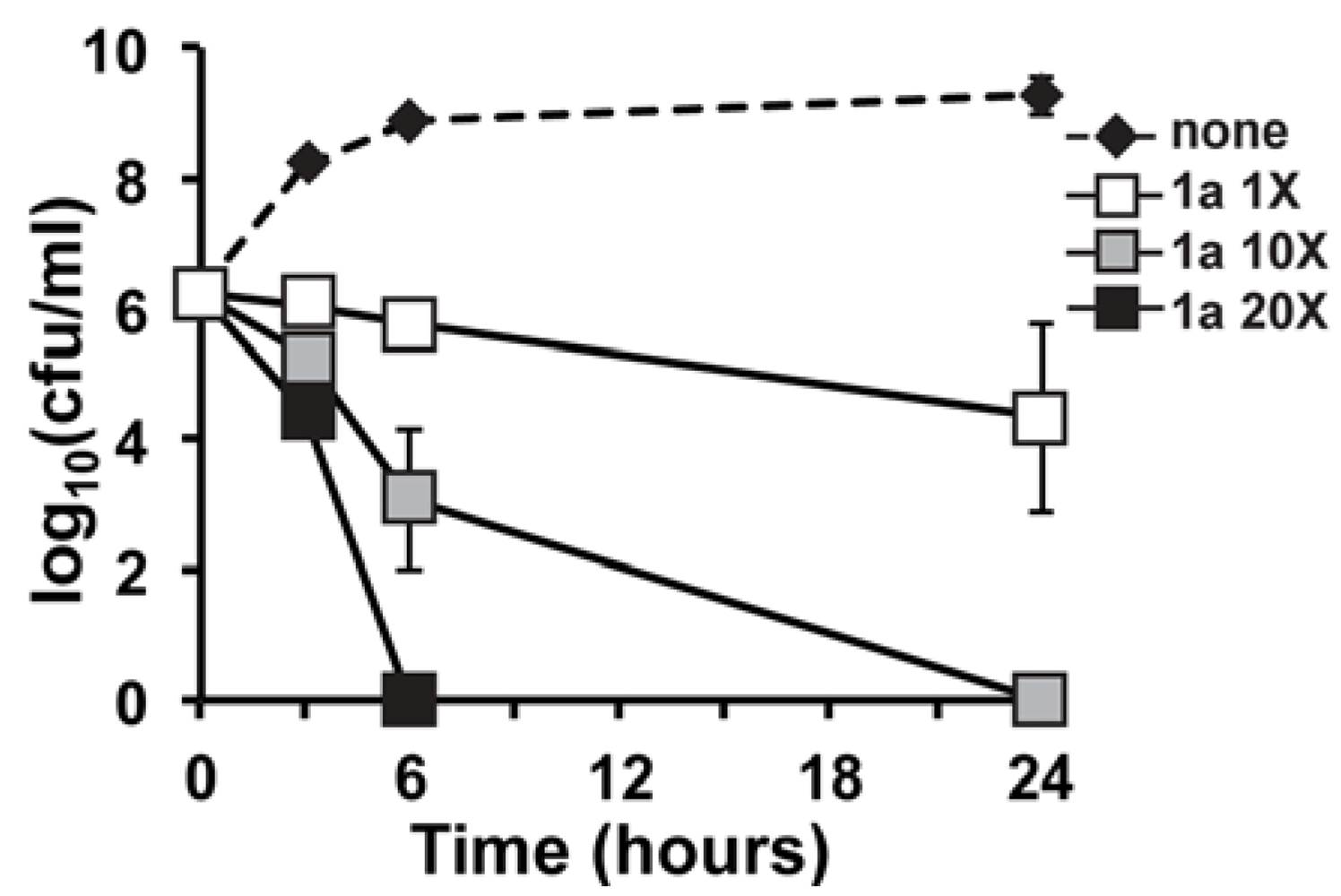
2.2. In Vitro Time-Kill of Marinopyrrole Derivative 1a

3. Experimental Section
3.1. Synthesis of Compounds 4–10c
3.2. Synthesis of Compounds 1a–1c
3.3. In Vitro Antibacterial Assays
3.4. In Vitro Time-Kill Analysis
4. Conclusions
Acknowledgments
References
- Hughes, C.C.; Prieto-Davo, A.; Jensen, P.R.; Fenical, W. The marinopyrroles, antibiotics of an unprecedented structure class from a marine Streptomyces sp. Org. Lett. 2008, 10, 629–631. [Google Scholar]
- Nicolaou, K.C.; Simmons, N.L.; Chen, J.S.; Haste, N.M.; Nizet, V. Total synthesis and biological evaluation of marinopyrrole A and analogues. Tetrahedron Lett. 2011, 52, 2041–2043. [Google Scholar]
- Hughes, C.C.; Yang, Y.L.; Liu, W.T.; Dorrestein, P.C.; la Clair, J.J.; Fenical, W. Marinopyrrole A target elucidation by acyl dye transfer. J. Am. Chem. Soc. 2009, 131, 12094–12096. [Google Scholar]
- Hughes, C.C.; Kauffman, C.A.; Jensen, P.R.; Fenical, W. Structures, reactivities, and antibiotic properties of the marinopyrroles A–F. J. Org. Chem. 2010, 75, 3240–3250. [Google Scholar]
- Kanakis, A.A.; Sarli, V. Total synthesis of (±)-marinopyrrole A via copper-mediated N-arylation. Org. Lett. 2010, 12, 4872–4875. [Google Scholar]
- Cheng, C.; Pan, L.; Chen, Y.; Song, H.; Qin, Y.; Li, R. Total synthesis of (±)-marinopyrrole a and its library as potential antibiotic and anticancer agents. J. Comb. Chem. 2010, 12, 541–547. [Google Scholar]
- Grundmann, H.; Aires-de-Sousa, M.; Boyce, J.; Tiemersma, E. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet 2006, 368, 874–885. [Google Scholar]
- Como-Sabetti, K.; Harriman, K.H.; Buck, J.M.; Glennen, A.; Boxrud, D.J.; Lynfield, R. Community-associated methicillin-resistant Staphylococcus aureus: Trends in case and isolate characteristics from six years of prospective surveillance. Public Health Rep. 2009, 124, 427–435. [Google Scholar]
- Chambers, H.F.; Deleo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef]
- Deleo, F.R.; Otto, M.; Kreiswirth, B.N.; Chambers, H.F. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 2010, 375, 1557–1568. [Google Scholar]
- Lowy, F.D. Antimicrobial resistance: The example of Staphylococcus aureus. J. Clin. Invest. 2003, 111, 1265–1273. [Google Scholar]
- Zhao, K.; Reiner, J.; Xie, W. FDA new drug approvals in 2000. Front. Biotechnol. Pharm. 2001, 2, 329–349. [Google Scholar]
- Eisenstein, B.I. Lipopeptides, focusing on daptomycin, for the treatment of Gram-positive infections. Expert. Opin. Investig. Drugs 2004, 13, 1159–1169. [Google Scholar] [CrossRef]
- FDA Approves Teflaro for Bacterial Infections. FDA News Events 29 October 2010. Available online: http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm231594.htm (accessed on 1 March 2012).
- Butler, M.S.; Buss, A.D. Natural products—The future scaffolds for novel antibiotics? Biochem. Pharmacol. 2006, 71, 919–929. [Google Scholar] [CrossRef]
- Haste, N.M.; Hughes, C.C.; Tran, D.N.; Fenical, W.; Jensen, P.R.; Nizet, V.; Hensler, M.E. Pharmacological properties of the marine natural product marinopyrrole A against methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2011, 55, 3305–3312. [Google Scholar] [CrossRef]
- Furneaux, R.H.; Tyler, P.C. Improved Syntheses of 3H,5H-Pyrrolo[3,2-d]pyrimidines. J. Org. Chem. 1999, 64, 8411–8412. [Google Scholar] [CrossRef]
- Rochais, C.; Lisowski, V.; Dallemagne, P.; Rault, S. Synthesis and biological evaluation of novel pyrrolopyrrolizinones as anticancer agents. Bioorg. Med. Chem. 2006, 14, 8162–8175. [Google Scholar]
- Chin, J.N.; Rybak, M.J.; Cheung, C.M.; Savage, P.B. Antimicrobial activities of ceragenins against clinical isolates of resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2007, 51, 1268–1273. [Google Scholar] [CrossRef]
- Credito, K.; Lin, G.; Appelbaum, P.C. Activity of daptomycin alone and in combination with rifampin and gentamicin against Staphylococcus aureus assessed by time-kill methodology. Antimicrob. Agents Chemother. 2007, 51, 1504–1507. [Google Scholar] [CrossRef]
- Ueda, Y.; Kanazawa, K.; Eguchi, K.; Takemoto, K.; Eriguchi, Y.; Sunagawa, M. In vitro and in vivo antibacterial activities of SM-216601, a new broad-spectrum parenteral carbapenem. Antimicrob. Agents Chemother. 2005, 49, 4185–4196. [Google Scholar] [CrossRef]
- Samples Availability: Available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, Y.; Haste, N.M.; Thienphrapa, W.; Nizet, V.; Hensler, M.; Li, R. Marinopyrrole Derivatives as Potential Antibiotic Agents against Methicillin-Resistant Staphylococcus aureus (I). Mar. Drugs 2012, 10, 953-962. https://doi.org/10.3390/md10040953
Liu Y, Haste NM, Thienphrapa W, Nizet V, Hensler M, Li R. Marinopyrrole Derivatives as Potential Antibiotic Agents against Methicillin-Resistant Staphylococcus aureus (I). Marine Drugs. 2012; 10(4):953-962. https://doi.org/10.3390/md10040953
Chicago/Turabian StyleLiu, Yan, Nina M. Haste, Wdee Thienphrapa, Victor Nizet, Mary Hensler, and Rongshi Li. 2012. "Marinopyrrole Derivatives as Potential Antibiotic Agents against Methicillin-Resistant Staphylococcus aureus (I)" Marine Drugs 10, no. 4: 953-962. https://doi.org/10.3390/md10040953