A Sterol and Spiroditerpenoids from a Penicillium sp. Isolated from a Deep Sea Sediment Sample

Abstract
:1. Introduction
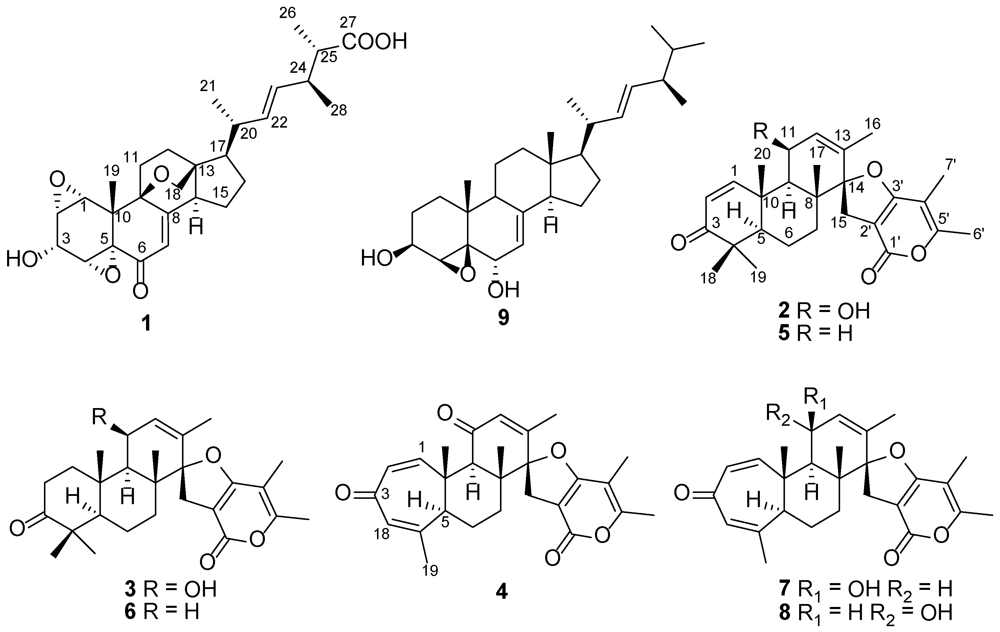
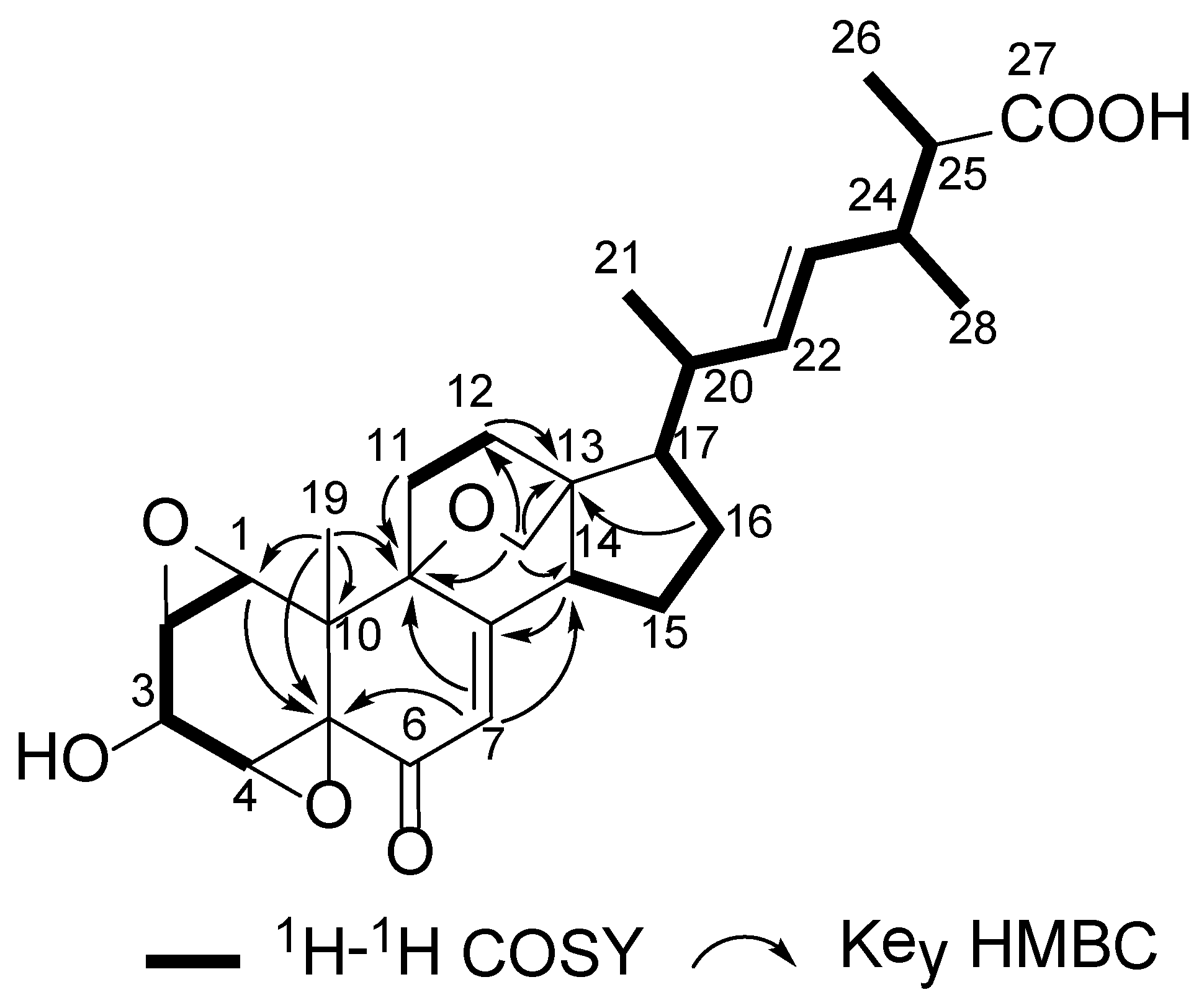
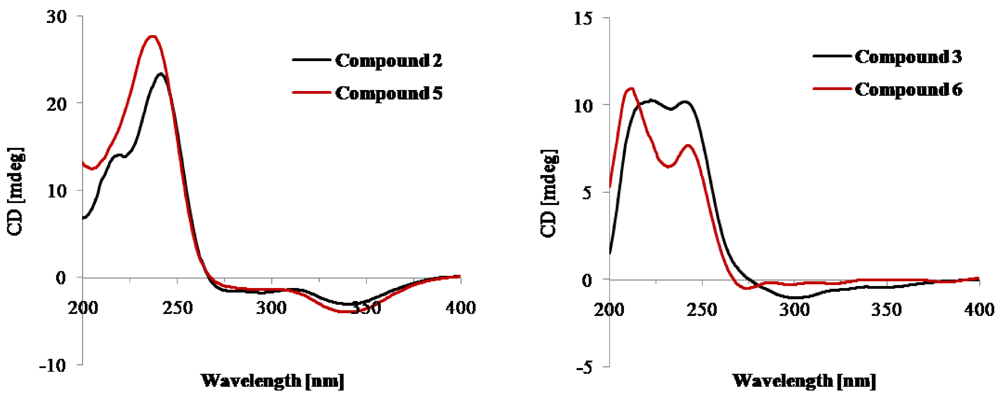
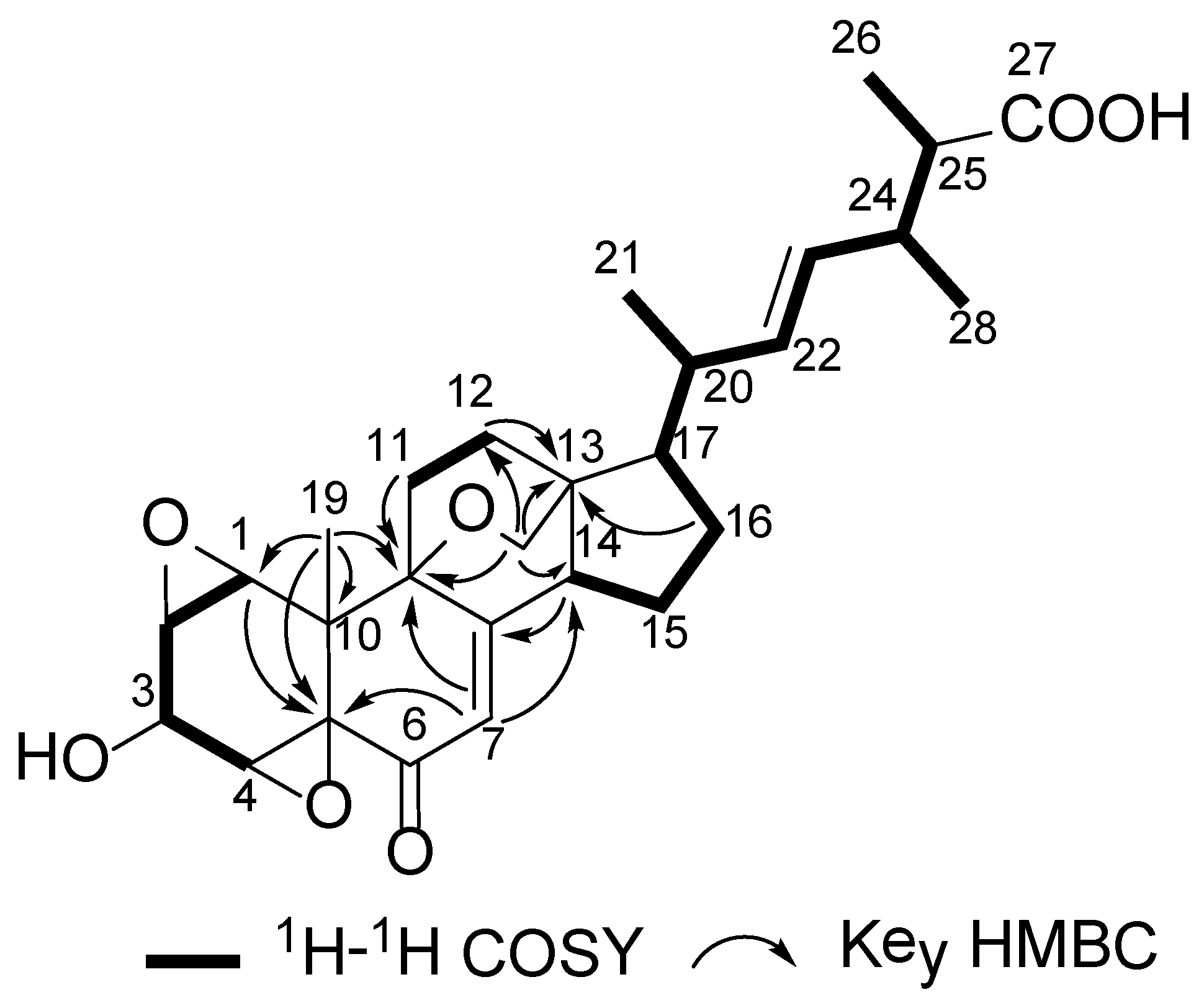
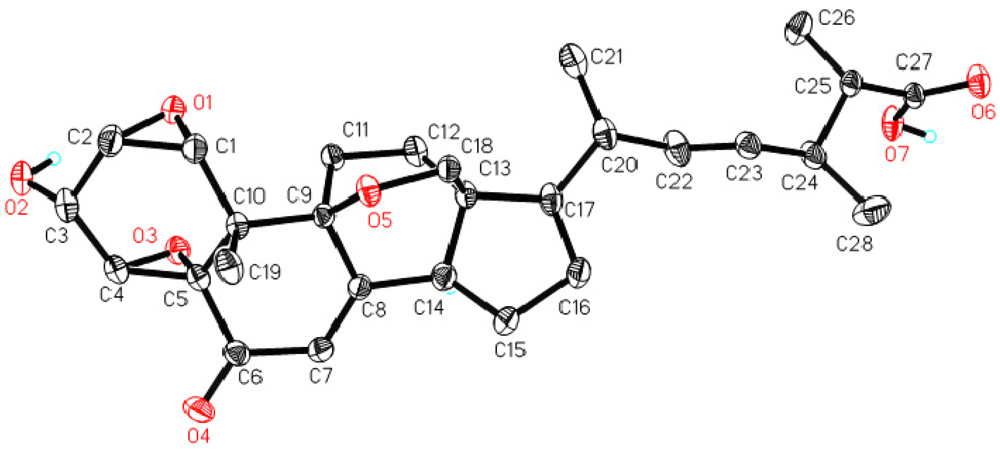
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | δCa, mult. | δHb (J in Hz) | HMBC (H→C#) |
|---|---|---|---|
| 1 | 58.9, CH | 3.45, s | 2, 5, 10, 19 |
| 2 | 52.6, CH | 3.35, s | 4 |
| 3 | 64.7, CH | 4.25, s | |
| 4 | 55.4, CH | 3.94, s | 2, 3 |
| 5 | 66.2, qC | ||
| 6 | 189.8, qC | ||
| 7 | 122.6, CH | 5.96, s | 5, 9, 14 |
| 8 | 167.8, qC | ||
| 9 | 74.1, qC | ||
| 10 | 38.9, qC | ||
| 11a | 30.6, CH2 | 1.43, m | |
| 11b | 2.51, m | 8, 9 | |
| 12a | 30.9, CH2 | 2.29, m | 11, 13 |
| 12b | 2.31, m | 13 | |
| 13 | 42.8, qC | ||
| 14 | 51.3, CH | 2.62, t (9.7) | 7, 8, 15, 18 |
| 15a | 25.1, CH2 | 1.69, m | 16 |
| 15b | 1.97, m | 16, 17 | |
| 16a | 29.6, CH2 | 1.54, m | 20, 17 |
| 16b | 2.00, m | 13, 15 | |
| 17 | 49.8, CH | 1.59, m | 16, 20 |
| 18a | 65.6, CH2 | 3.85, d (6.3) | 9, 12, 13, 14 |
| 18b | 3.92, d (6.3) | 12, 13, 14 | |
| 19 | 19.1, CH3 | 1.15, s | 1, 5, 9, 10 |
| 20 | 38.9, CH | 2.02, m | 16 |
| 21 | 21.4, CH3 | 1.06, d (6.7) | 17, 20, 22 |
| 22 | 136.3, CH | 5.28, dd (15.0, 8.0) | 20, 24 |
| 23 | 130.6, CH | 5.19, dd (15.0, 8.0) | 20, 24 |
| 24 | 39.6, CH | 2.42, q (6.9) | 22, 23, 25, 26, 27, 28 |
| 25 | 44.8, CH | 2.34, q (6.8) | 23, 24, 26, 27, 28 |
| 26 | 14.0, CH3 | 1.10, d (6.8) | 24, 25, 27 |
| 27 | 180.1, qC | ||
| 28 | 18.8, CH3 | 1.04, d (6.9) | 23, 24, 25 |
| Breviones I (2) | Breviones J (3) | Breviones K (4) | ||||||
|---|---|---|---|---|---|---|---|---|
| Position | δC a, mult. | δH b (J in Hz) | δC c, mult. | δH b (J in Hz) | δC c, mult. | δH b (J in Hz) | ||
| 1a | 158.4, CH | 7.50, d (11) | 38.4, CH2 | 2.29, ddd (16, 6.0, 3.2) | 155.2, CH | 6.83, d (13) | ||
| 1b | 2.34, ddd (16, 6.0, 3.2) | |||||||
| 2a | 125.6, CH | 5.80, d (11) | 33.8, CH2 | 2.69, ddd (16, 6.0, 3.2) | 128.4, CH | 5.70, dd (13, 2.1) | ||
| 2b | 2.74, ddd (16, 6.0, 3.2) | |||||||
| 3 | 203.9, qC | 215.1, qC | 192.5, qC | |||||
| 4 | 44.8, qC | 50.8, qC | 154.0, qC | |||||
| 5 | 54.6, CH | 1.63, m | 56.3, CH | 1.75, m | 48.6, CH | 2.93, d (13) | ||
| 6a | 19.3, CH2 | 1.64, m | 18.8, CH2 | 1.63, m | 21.7, CH2 | 1.83, dt (13, 3.5) | ||
| 6b | 1.80, m | 1.82, m | 1.93, td (13, 3.5) | |||||
| 7a | 33.0, CH2 | 1.50, m | 32.3, CH2 | 1.48, m | 30.3, CH2 | 1.65, td (13, 3.5) | ||
| 7b | 1.62, m | 1.56, m | 1.74, dt (13, 3.5) | |||||
| 8 | 41.9, qC | 40.8, qC | 45.4, qC | |||||
| 9 | 47.1, CH | 1.95, d (4.5) | 47.2, CH | 1.91, br | 54.8, CH | 3.15, s | ||
| 10 | 41.3, qC | 40.2, qC | 42.0, qC | |||||
| 11 | 64.4, CH | 4.78, s | 64.1, CH | 4.51, s | 198.2, qC | |||
| 12 | 131.3, CH | 5.79, s | 131.0, CH | 5.72, d (5.0) | 130.2, CH | 5.91, s | ||
| 13 | 133.1, qC | 132.2, qC | 151.8, qC | |||||
| 14 | 99.8, qC | 99.1, qC | 97.7, qC | |||||
| 15a | 29.5, CH2 | 2.96, d (16) | 29.7, CH2 | 2.94, d (16) | 30.1, CH2 | 3.15, s | ||
| 15b | 3.01, d (16) | 2.97, d (16) | ||||||
| 16 | 18.8, CH3 | 1.74, s | 18.2, CH3 | 1.71, s | 18.5, CH3 | 1.97, s | ||
| 17 | 20.0, CH3 | 1.32, s | 16.7, CH3 | 1.29, s | 18.3, CH3 | 1.18, s | ||
| 18 | 27.8, CH3 | 1.08, s | 26.0, CH3 | 1.05, s | 131.7, CH | 5.99, s | ||
| 19 | 22.2, CH3 | 1.06, s | 21.4, CH3 | 1.01, s | 24.1, CH3 | 1.99, s | ||
| 20 | 21.5, CH3 | 1.67, s | 19.4, CH3 | 1.59, s | 14.3, CH3 | 1.41, s | ||
| 1' | 171.3, qC | 170.6, qC | 170.8, qC | |||||
| 2' | 99.8, qC | 99.3, qC | 100.1, qC | |||||
| 3' | 161.2, qC | 159.4, qC | 161.8, qC | |||||
| 4' | 103.0, qC | 102.4, qC | 103.2, qC | |||||
| 5' | 161.0, qC | 160.4, qC | 160.7, qC | |||||
| 6' | 9.5, CH3 | 1.85, s | 8.7, CH3 | 1.86, s | 9.5, CH3 | 1.92, s | ||
| 7' | 17.1, CH3 | 2.17, s | 16.4, CH3 | 2.17, s | 17.2, CH3 | 2.20, s | ||
| OH-11 | 4.01, br | 3.72, d (5.0) | ||||||

3. Experimental Section
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Extraction and Isolation
3.4. X-ray Crystallographic Analysis of 1
3.5. MTS Assay
4. Conclusions
Acknowledgments
Supplementary Files
References and Notes
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2004, 21, 1–49. [Google Scholar]
- Blunt, J.W.; Copp, B.R.; Munro, M.H.G.; Northcote, P.T.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2011, 28, 196–268. [Google Scholar]
- Gautschi, J.T.; Amagata, T.; Amagata, A.; Valeriote, F.A.; Mooberry, S.L.; Crews, P. Expanding the strategies in natural product studies of marine-derived fungi: A chemical investigation of Penicillium obtained from deep water sediment. J. Nat. Prod. 2004, 67, 1638–1638. [Google Scholar]
- Lebar, M.D.; Heimbegnier, J.L.; Baker, B.J. Cold-water marine natural products. Nat. Prod. Rep. 2007, 24, 774–797. [Google Scholar]
- Li, D.H.; Wang, F.P.; Xiao, X.; Fang, Y.C.; Zhu, T.J.; Gu, Q.Q.; Zhu, W.M. Trisorbicillinone A, a novel sorbicillin trimer, from a deep sea fungus, Phialocephala sp. FL30r. Tetrahedron Lett. 2007, 48, 5235–5238. [Google Scholar]
- Skropeta, D. Deep-sea natural products. Nat. Prod. Rep. 2008, 25, 1131–1166. [Google Scholar]
- Wilson, Z.E.; Brimble, M.A. Molecule derived from the extremes of life. Nat. Prod. Rep. 2009, 26, 44–71. [Google Scholar]
- Li, Y.; Ye, D.Z.; Chen, X.L.; Lu, X.H.; Shao, Z.Z.; Zhang, H.; Che, Y.S. Breviane spiroditerpenoids from an extreme-tolerant Penicillium sp. isolated from a deep sea sediment sample. J. Nat. Prod. 2009, 72, 912–916. [Google Scholar] [CrossRef]
- Li, D.H.; Cai, S.X.; Zhu, T.J.; Wang, F.P.; Xiao, X.; Gu, Q.Q. Three new sorbicillin trimers, trisorbicillinones B, C, and D, from a deep ocean sediment derived fungus, Phialocephala sp. FL30r. Tetrahedron 2010, 66, 5101–5106. [Google Scholar]
- Macias, F.A.; Varela, R.M.; Simonet, A.M.; Cutler, H.G.; Cutler, S.J.; Ross, S.A.; Dunbar, D.C.; Dugan, F.M.; Hill, R.A. (+)-Brevione A. The first member of a novel family of bioactive spiroditerpenoids isolated from Penicillium brevicompactum Dierckx. Tetrahedron Lett. 2000, 41, 2683–2686. [Google Scholar]
- Macias, F.A.; Varela, R.M.; Simonet, A.M.; Cutler, H.G.; Cutler, S.J.; Dugan, F.M.; Hill, R.A. Novel bioactive breviane spiroditerpenoids from Penicillium brevicompactum Dierckx. J. Org. Chem. 2000, 65, 9039–9046. [Google Scholar]
- Vanderah, D.J.; Djerassi, C. Marine natural products. Synthesis of four naturally occurring 20.beta.-H cholanic acid derivatives. J. Org. Chem. 1978, 43, 1442–1448. [Google Scholar] [CrossRef]
- Iorizzi, M.; Minale, L.; Riccio, R.; Debray, M.; Menou, J.L. Starfish saponins, part 23. Steroidal glycosides from the starfish Halityle regularis. J. Nat. Prod. 1986, 49, 67–78. [Google Scholar] [CrossRef]
- Mansoor, T.A.; Hong, J.; Lee, C.O.; Bae, S.J.; Im, K.S.; Jung, J.H. Cytotoxic sterol derivatives from a marine sponge Homaxinella sp. J. Nat. Prod. 2005, 68, 331–336. [Google Scholar]
- Krohn, K.; Kouam, S.F.; Kuigoua, G.M.; Hussain, H.; Flörke, S.C.-B.U.; Kurtán, T.; Pescitelli, G.; Di Bari, L.; Draeger, S.; Schulz, B. Xanthones and oxepino[2, 3-b]chromones from three endophytic fungi. Chem. Eur. J. 2009, 15, 12121–12132. [Google Scholar]
- SHELXL-97, program for x-ray crystal structure solution and refinement. University of Göttingen: Göttingen, Germany, 1997.
- SADABS, version 2.03; program for empirical absorption correction of area detector data. University of Göttingen: Göttingen, Germany, 1999.
- Cambridge Crystallographic Data Centre. Fax: +44-1223-336033; E-Mail: [email protected].
- Zhang, N.; Chen, Y.L.; Jiang, R.X.; Li, E.W.; Chen, X.L.; Xi, Z.J.; Guo, Y.L.; Liu, X.Z.; Zhou, Y.G.; Che, Y.S.; et al. PARP and RIP1 are required for autophagy induced by 11'-deoxyverticillin A, which precedes caspase-dependent apoptosis. Autophagy 2011, 7, 598–612. [Google Scholar] [CrossRef]
- Mansoor, T.A.; Lee, Y.M.; Hong, J.; Lee, C.O.; Im, K.S.; Jung, J.H. 5,6:8,9-Diepoxy and other cytotoxic sterols from the marine sponge Homaxinella sp. J. Nat. Prod. 2006, 69, 131–134. [Google Scholar] [CrossRef]
- Li, J.Z.; Qing, C.; Chen, C.X.; Hao, X.J.; Liu, H.Y. Cytotoxicity of cardenolides and cardenolide glycosides from Asclepias curassavica. Bioorg. Med. Chem. Lett. 2009, 19, 1956–1959. [Google Scholar]
- Wu, J.; Choi, J.H.; Yoshida, M.; Hirai, H.; Harada, E.; Masuda, K.; Koyama, T.; Yazawa, K.; Noguchi, K.; Nagasawa, K.; et al. Osteoclast-forming suppressing compounds, gargalols A, B, and C, from the edible mushroom Grifola gargal. Tetrahedron 2011, 67, 6576–6581. [Google Scholar]
- Takikawa, H.; Hirooka, M.; Sasaki, M. Synthetic studies on breviones: Construction of the CDE ring system. Tetrahedron Lett. 2002, 43, 1713–1716. [Google Scholar]
- Takikawa, H.; Hirooka, M.; Sasaki, M. The first synthesis of (±)-brevione B, an allelopathic agent isolated from Penicillium sp. Tetrahedron Lett. 2003, 44, 5235–5238. [Google Scholar]
- Takikawa, H.; Imamura, Y.; Sasaki, M. Synthesis and absolute configuration of brevione B, an allelochemical isolated from Penicillium sp. Tetrahedron 2006, 62, 39–48. [Google Scholar] [CrossRef]
- Macías, F.A.; Carrera, C.; Chinchilla, N.; Fronczek, F.R.; Galindo, J.C. Synthesis of the western half of breviones C, D, F and G. Tetrahedron 2010, 66, 4125–4132. [Google Scholar] [CrossRef]
- Yokoe, H.; Mitsuhashi, C.; Matsuoka, Y.; Yoshimura, T.; Yoshida, M.; Shishido, K. Enantiocontrolled total syntheses of breviones A, B, and C. J. Am. Chem. Soc. 2011, 133, 8854–8857. [Google Scholar]
- Samples Availability: Available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, Y.; Ye, D.; Shao, Z.; Cui, C.; Che, Y. A Sterol and Spiroditerpenoids from a Penicillium sp. Isolated from a Deep Sea Sediment Sample. Mar. Drugs 2012, 10, 497-508. https://doi.org/10.3390/md10020497
Li Y, Ye D, Shao Z, Cui C, Che Y. A Sterol and Spiroditerpenoids from a Penicillium sp. Isolated from a Deep Sea Sediment Sample. Marine Drugs. 2012; 10(2):497-508. https://doi.org/10.3390/md10020497
Chicago/Turabian StyleLi, Yan, Dezan Ye, Zongze Shao, Chengbin Cui, and Yongsheng Che. 2012. "A Sterol and Spiroditerpenoids from a Penicillium sp. Isolated from a Deep Sea Sediment Sample" Marine Drugs 10, no. 2: 497-508. https://doi.org/10.3390/md10020497