Current Challenges and Future Directions in Recombinant AAV-Mediated Gene Therapy of Duchenne Muscular Dystrophy

Abstract
:1. Introduction
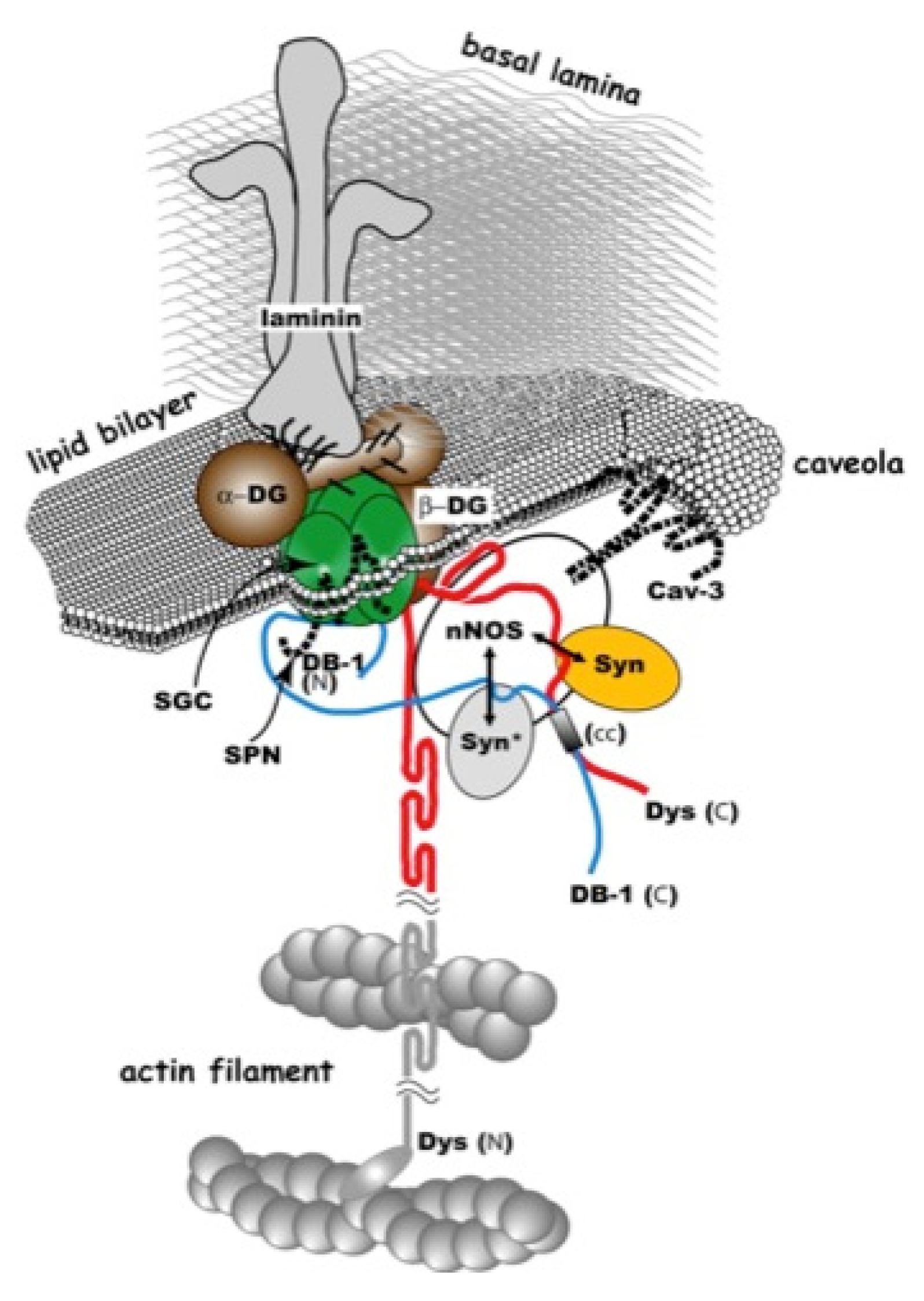
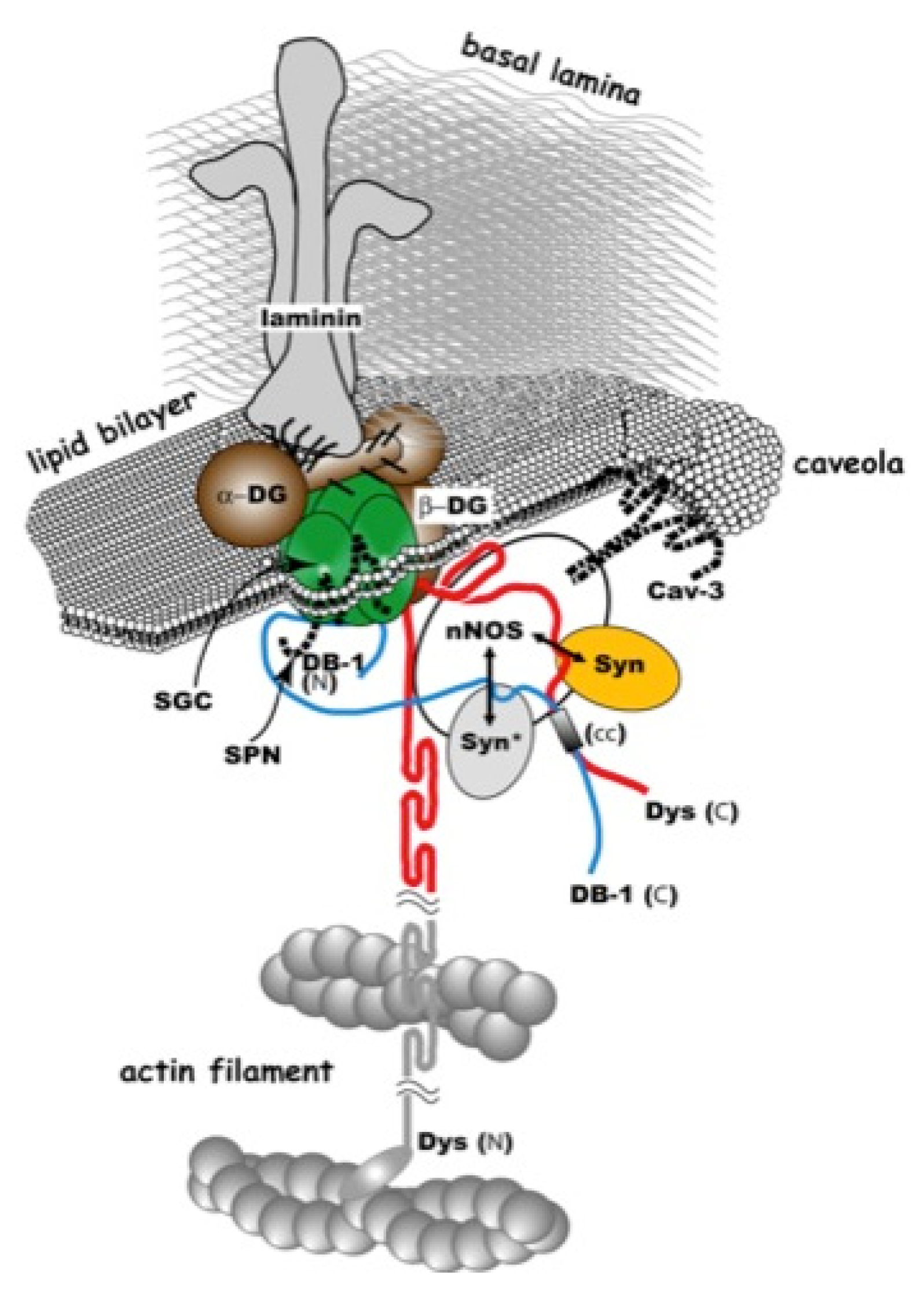
2. Gene-Replacement Strategies Using Virus Vectors
2.1. Choice of Vector

{kind=link}
{kind=link}
{kind=link}
| Tissue type | Effective serotype | Reference |
|---|---|---|
| Neurons and glial cells | AAV9, AAV7 > AAV8 > AAV5 > AAV2, AAV1 | [26,27,28] |
| Glioblastoma | AAV8, AAV7 > AAV6 > AAV2 > AAV5 | [28] |
| Cardiac tissue | AAV9 > AAV8 > AAV1, AAV6 > AAV2 | [29,30,31,32] |
| Muscle (systemic) | AAV8 | [33,34] |
| Muscle (local) | AAV1, AAV6 | [33,35,36,37] |
| Liver (hepatocytes) | AAV9, AAV8 | [38] |
| Pancreas | AAV8, AAV1 | [39,40] |
| Retina | AAV8, AAV5 > AAV4 > AAV1, AAV2 | [41,42,43] |
| Dendritic cells | AAV6 | [44] |
| Hematopoietic stem cells | AAV1 | [45] |
| Fibroblasts | AAV1, AAV6 > AAV2 | [46] |
2.2. Modification of the Dystrophin GENE
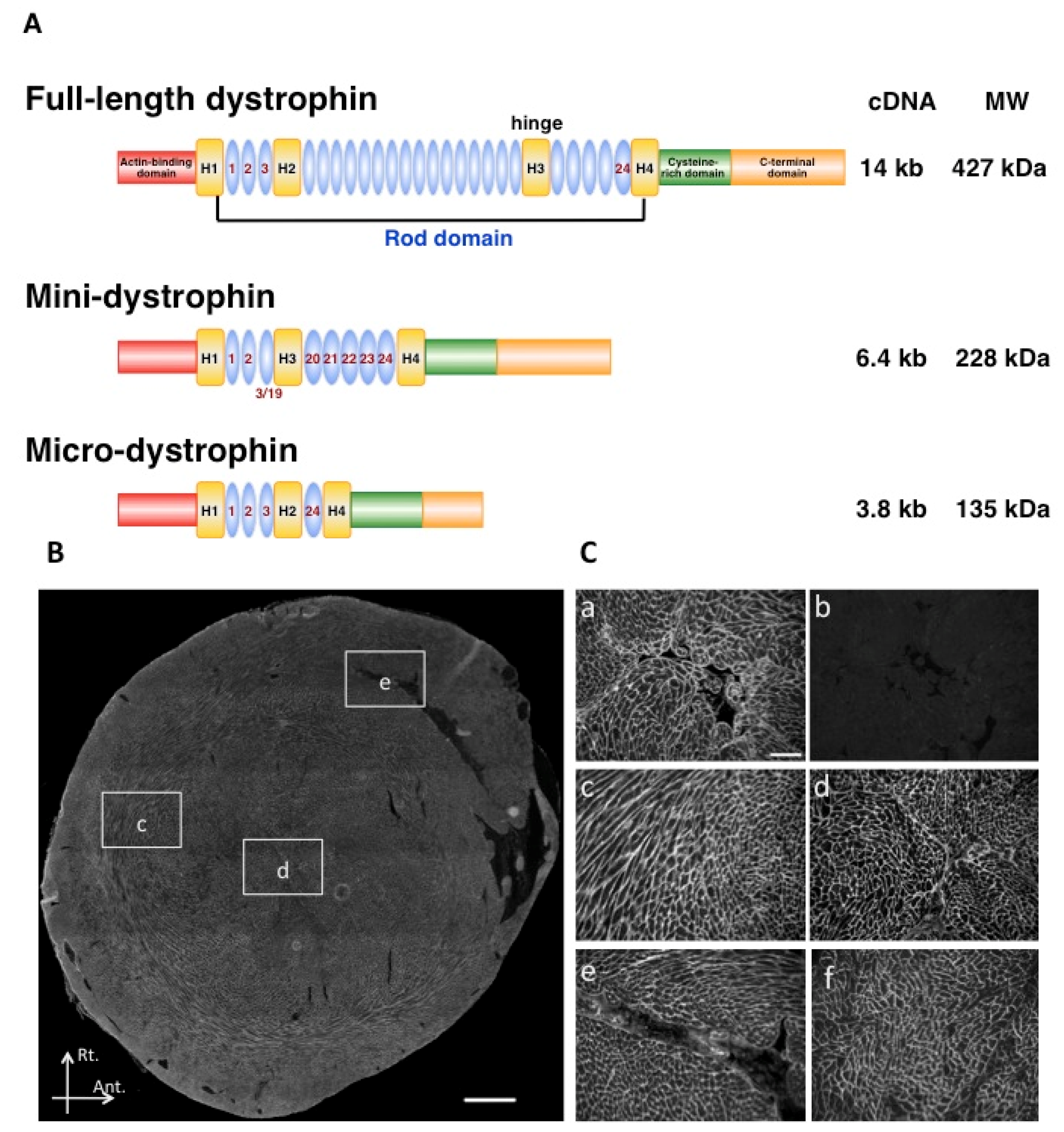
2.3. Use of Surrogate Genes
3. AAV-Mediated Transduction of Large Animal Models
3.1. Vector Production
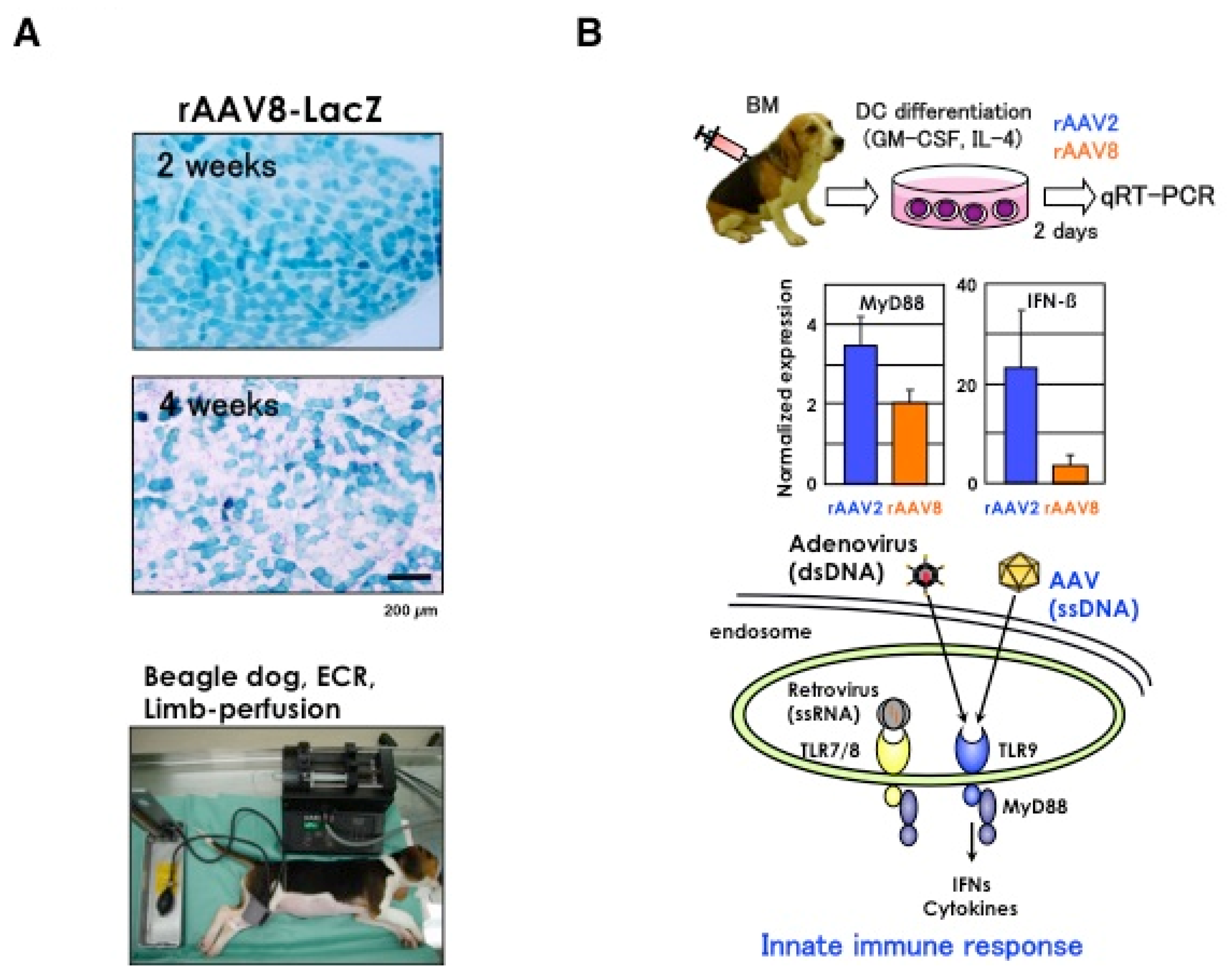
3.2. Canine Models for the Gene Transduction Study
3.3. Immunological Issues of rAAV
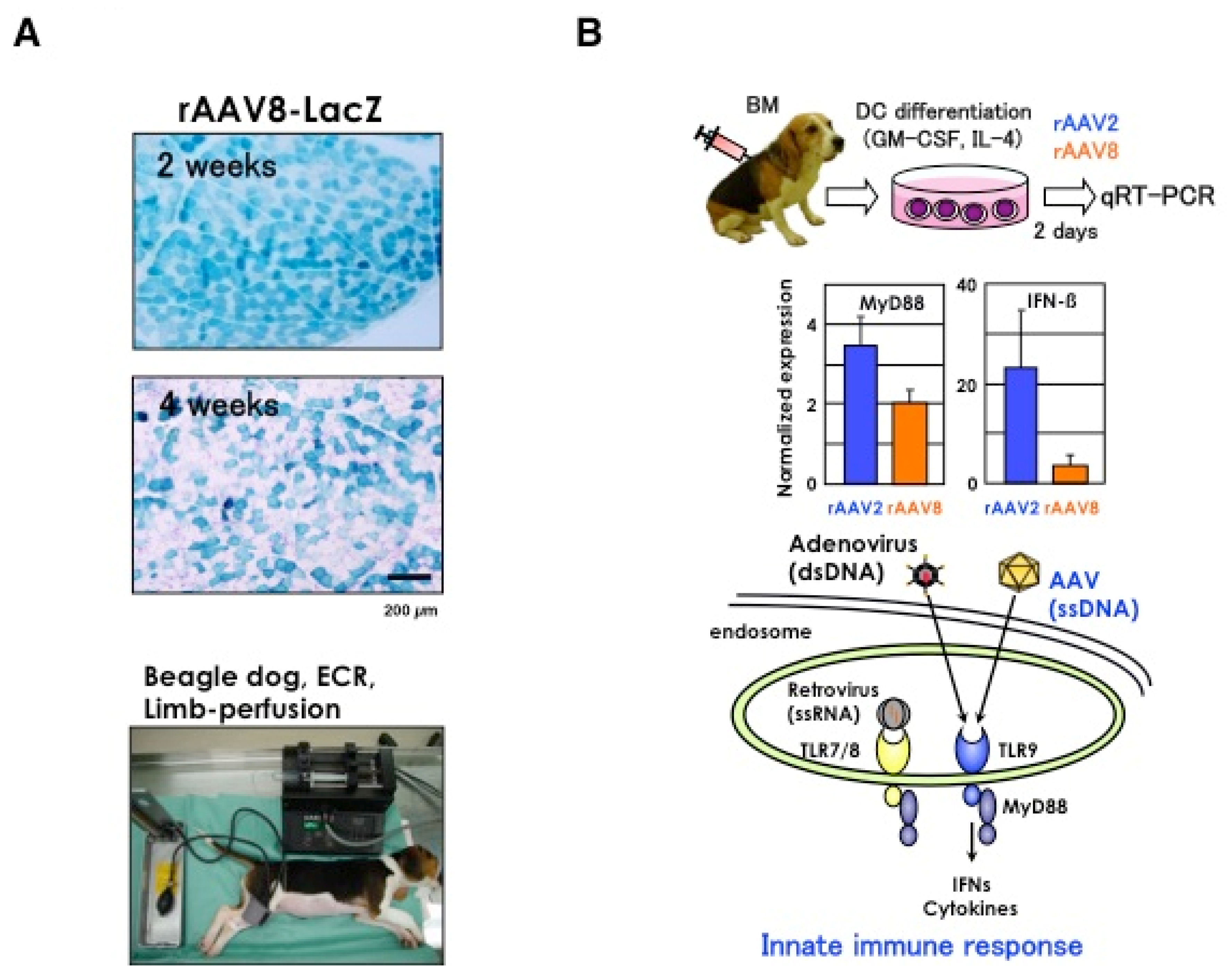
3.4. Intravascular Vector Administration by Limb Perfusion
3.5. Global Muscle Therapies
4. Safety and Potential Impact of Clinical Trials
4.1. Clinical Trials for Muscle Transduction
4.2. Gene Therapy Medicine
5. Future Perspectives
5.1. Modification of mRNA Splicing with rAAV-mediated Exon-Skipping
5.2. Pharmacological Intervention
5.3. Capsid Modification
5.4. AAV-Mediated Gene and Cell Therapy
5.5. Targeted Vector Integration
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Emery, A.E. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul. Disord. 1991, 1, 19–29. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Snow, W.M.; Anderson, J.E.; Jakobson, L.S. Neuropsychological and neurobehavioral functioning in Duchenne muscular dystrophy: A review. Neurosci. Biobehav. Rev. 2013, 37, 743–752. [Google Scholar] [CrossRef]
- Melacini, P.; Fanin, M.; Danieli, G.A.; Villanova, C.; Martinello, F.; Miorin, M.; Freda, M.P.; Miorelli, M.; Mostacciuolo, M.L.; Fasoli, G.; et al. Myocardial involvement is very frequent among patients affected with subclinical Becker’s muscular dystrophy. Circulation 1996, 94, 3168–3175. [Google Scholar] [CrossRef]
- Yoshida, M.; Hama, H.; Ishikawa-Sakurai, M.; Imamura, M.; Mizuno, Y.; Araishi, K.; Wakabayashi-Takai, E.; Noguchi, S.; Sasaoka, T.; Ozawa, E. Biochemical evidence for association of dystrobrevin with the sarcoglycan-sarcospan complex as a basis for understanding sarcoglycanopathy. Hum. Mol. Genet. 2000, 9, 1033–1040. [Google Scholar] [CrossRef]
- Beytia Mde, L.; Vry, J.; Kirschner, J. Drug treatment of Duchenne muscular dystrophy: Available evidence and perspectives. Acta Myol. 2012, 31, 4–8. [Google Scholar]
- Kirschner, J.; Schessl, J.; Schara, U.; Reitter, B.; Stettner, G.M.; Hobbiebrunken, E.; Wilichowski, E.; Bernert, G.; Weiss, S.; Stehling, F.; et al. Treatment of Duchenne muscular dystrophy with ciclosporin A: a randomised, double-blind, placebo-controlled multicentre trial. Lancet Neurol. 2010, 9, 1053–1059. [Google Scholar] [CrossRef]
- Pichavant, C.; Aartsma-Rus, A.; Clemens, P.R.; Davies, K.E.; Dickson, G.; Takeda, S.; Wilton, S.D.; Wolff, J.A.; Wooddell, C.I.; Xiao, X.; et al. Current status of pharmaceutical and genetic therapeutic approaches to treat DMD. Mol. Ther. 2011, 19, 830–840. [Google Scholar] [CrossRef]
- Takeshima, Y.; Nishio, H.; Sakamoto, H.; Nakamura, H.; Matsuo, M. Modulation of in vitro splicing of the upstream intron by modifying an intra-exon sequence which is deleted from the dystrophin gene in dystrophin Kobe. J. Clin. Invest. 1995, 95, 515–520. [Google Scholar] [CrossRef]
- Beroud, C.; Tuffery-Giraud, S.; Matsuo, M.; Hamroun, D.; Humbertclaude, V.; Monnier, N.; Moizard, M.P.; Voelckel, M.A.; Calemard, L.M.; Boisseau, P.; et al. Multiexon skipping leading to an artificial DMD protein lacking amino acids from exons 45 through 55 could rescue up to 63% of patients with Duchenne muscular dystrophy. Hum. Mutat. 2007, 28, 196–202. [Google Scholar] [CrossRef]
- Takeshima, Y.; Yagi, M.; Wada, H.; Ishibashi, K.; Nishiyama, A.; Kakumoto, M.; Sakaeda, T.; Saura, R.; Okumura, K.; Matsuo, M. Intravenous infusion of an antisense oligonucleotide results in exon skipping in muscle dystrophin mRNA of Duchenne muscular dystrophy. Pediatr. Res. 2006, 59, 690–694. [Google Scholar] [CrossRef]
- Van Deutekom, J.C.; Janson, A.A.; Ginjaar, I.B.; Frankhuizen, W.S.; Aartsma-Rus, A.; Bremmer-Bout, M.; den Dunnen, J.T.; Koop, K.; van der Kooi, A.J.; Goemans, N.M.; et al. Local dystrophin restoration with antisense oligonucleotide PRO051. N. Engl. J. Med. 2007, 357, 2677–2686. [Google Scholar] [CrossRef]
- Alter, J.; Lou, F.; Rabinowitz, A.; Yin, H.; Rosenfeld, J.; Wilton, S.D.; Partridge, T.A.; Lu, Q.L. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat. Med. 2006, 12, 175–177. [Google Scholar] [CrossRef]
- Yokota, T.; Lu, Q.; Partridge, T.; Kobayashi, M.; Nakamura, A.; Takeda, S.; Hoffman, E.P. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Annals Neurol. 2009, 65, 667–676. [Google Scholar]
- Wu, B.; Moulton, H.M.; Iversen, P.L.; Jiang, J.; Li, J.; Li, J.; Spurney, C.F.; Sali, A.; Guerron, A.D.; Nagaraju, K.; et al. Effective rescue of dystrophin improves cardiac function in dystrophin-deficient mice by a modified morpholino oligomer. Proc. Natl. Acad. Sci. USA 2008, 105, 14814–14819. [Google Scholar] [CrossRef]
- Malerba, A.; Sharp, P.S.; Graham, I.R.; Arechavala-Gomeza, V.; Foster, K.; Muntoni, F.; Wells, D.J.; Dickson, G. Chronic systemic therapy with low-dose morpholino oligomers ameliorates the pathology and normalizes locomotor behavior in mdx mice. Mol. Ther. 2011, 19, 345–354. [Google Scholar] [CrossRef]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef]
- Okada, T.; Ramsey, J.; Munir, J.; Wildner, O.; Blaese, M. Efficient directional cloning of recombinant adenovirus vectors using DNA-protein complex. Nucleic Acids Res. 1998, 26, 1947–1950. [Google Scholar] [CrossRef]
- Okada, T.; Caplen, N.J.; Ramsey, W.J.; Onodera, M.; Shimazaki, K.; Nomoto, T.; Ajalli, R.; Wildner, O.; Morris, J.; Kume, A.; et al. In situ generation of pseudotyped retroviral progeny by adenovirus-mediated transduction of tumor cells enhances the killing effect of HSV-tk suicide gene therapy in vitro and in vivo. J. Gene Med. 2004, 6, 288–299. [Google Scholar] [CrossRef]
- Hammerschmidt, D.E. Development of a gutless vector. J. Lab Clin. Med. 1999, 134, C3. [Google Scholar]
- Hoshiya, H.; Kazuki, Y.; Abe, S.; Takiguchi, M.; Kajitani, N.; Watanabe, Y.; Yoshino, T.; Shirayoshi, Y.; Higaki, K.; Messina, G.; et al. A highly Stable and Nonintegrated Human Artificial Chromosome (HAC) Containing the 2.4 Mb Entire Human Dystrophin Gene. Mol. Ther. 2008, 17, 309–317. [Google Scholar]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Okada, T.; Shimazaki, K.; Nomoto, T.; Matsushita, T.; Mizukami, H.; Urabe, M.; Hanazono, Y.; Kume, A.; Tobita, K.; Ozawa, K.; et al. Adeno-associated viral vector-mediated gene therapy of ischemia-induced neuronal death. Methods Enzymol. 2002, 346, 378–393. [Google Scholar]
- Ito, T.; Okada, T.; Miyashita, H.; Nomoto, T.; Nonaka-Sarukawa, M.; Uchibori, R.; Maeda, Y.; Urabe, M.; Mizukami, H.; Kume, A.; et al. Interleukin-10 expression mediated by an adeno-associated virus vector prevents monocrotaline-induced pulmonary arterial hypertension in rats. Circ. Res. 2007, 101, 734–741. [Google Scholar] [CrossRef]
- Yuasa, K.; Miyagoe, Y.; Yamamoto, K.; Nabeshima, Y.; Dickson, G.; Takeda, S. Effective restoration of dystrophin-associated proteins in vivo by adenovirus-mediated transfer of truncated dystrophin cDNAs. FEBS Lett. 1998, 425, 329–336. [Google Scholar] [CrossRef]
- Samaranch, L.; Salegio, E.A.; San Sebastian, W.; Kells, A.P.; Bringas, J.R.; Forsayeth, J.; Bankiewicz, K.S. Strong Cortical and Spinal Cord Transduction After AAV7 and AAV9 Delivery into the Cerebrospinal Fluid of Nonhuman Primates. Hum. Gene Ther. 2013, 24, 526–532. [Google Scholar] [CrossRef]
- Wu, K.; Meyer, E.M.; Bennett, J.A.; Meyers, C.A.; Hughes, J.A.; King, M.A. AAV2/5-mediated NGF gene delivery protects septal cholinergic neurons following axotomy. Brain Res. 2005, 1061, 107–113. [Google Scholar] [CrossRef]
- Harding, T.C.; Dickinson, P.J.; Roberts, B.N.; Yendluri, S.; Gonzalez-Edick, M.; Lecouteur, R.A.; Jooss, K.U. Enhanced gene transfer efficiency in the murine striatum and an orthotopic glioblastoma tumor model, using AAV-7- and AAV-8-pseudotyped vectors. Hum. Gene Ther. 2006, 17, 807–820. [Google Scholar] [CrossRef]
- Inagaki, K.; Fuess, S.; Storm, T.A.; Gibson, G.A.; McTiernan, C.F.; Kay, M.A.; Nakai, H. Robust systemic transduction with AAV9 vectors in mice: Efficient global cardiac gene transfer superior to that of AAV8. Mol. Ther. 2006, 14, 45–53. [Google Scholar] [CrossRef]
- Du, L.; Kido, M.; Lee, D.V.; Rabinowitz, J.E.; Samulski, R.J.; Jamieson, S.W.; Weitzman, M.D.; Thistlethwaite, P.A. Differential myocardial gene delivery by recombinant serotype-specific adeno-associated viral vectors. Mol. Ther. 2004, 10, 604–608. [Google Scholar] [CrossRef]
- Pacak, C.A.; Mah, C.S.; Thattaliyath, B.D.; Conlon, T.J.; Lewis, M.A.; Cloutier, D.E.; Zolotukhin, I.; Tarantal, A.F.; Byrne, B.J. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ. Res. 2006, 99, e3–e9. [Google Scholar] [CrossRef]
- Kawamoto, S.; Shi, Q.; Nitta, Y.; Miyazaki, J.; Allen, M.D. Widespread and early myocardial gene expression by adeno-associated virus vector type 6 with a beta-actin hybrid promoter. Mol. Ther. 2005, 11, 980–985. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, T.; Qiao, C.; Zhou, L.; Wang, B.; Zhang, J.; Chen, C.; Li, J.; Xiao, X. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat. Biotechnol. 2005, 23, 321–328. [Google Scholar]
- Nishiyama, A.; Ampong, B.N.; Ohshima, S.; Shin, J.H.; Nakai, H.; Imamura, M.; Miyagoe-Suzuki, Y.; Okada, T.; Takeda, S. Recombinant adeno-associated virus type 8-mediated extensive therapeutic gene delivery into skeletal muscle of alpha-sarcoglycan-deficient mice. Hum. Gene Ther. 2008, 19, 719–730. [Google Scholar] [CrossRef]
- Mingozzi, F.; Meulenberg, J.J.; Hui, D.J.; Basner-Tschakarjan, E.; Hasbrouck, N.C.; Edmonson, S.A.; Hutnick, N.A.; Betts, M.R.; Kastelein, J.J.; Stroes, E.S.; et al. AAV-1-mediated gene transfer to skeletal muscle in humans results in dose-dependent activation of capsid-specific T cells. Blood 2009, 114, 2077–2086. [Google Scholar] [CrossRef]
- Brantly, M.L.; Chulay, J.D.; Wang, L.; Mueller, C.; Humphries, M.; Spencer, L.T.; Rouhani, F.; Conlon, T.J.; Calcedo, R.; Betts, M.R.; et al. Sustained transgene expression despite T lymphocyte responses in a clinical trial of rAAV1-AAT gene therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 16363–16368. [Google Scholar] [CrossRef]
- Wang, Z.; Storb, R.; Lee, D.; Kushmerick, M.J.; Chu, B.; Berger, C.; Arnett, A.; Allen, J.; Chamberlain, J.S.; Riddell, S.R.; et al. Immune responses to AAV in canine muscle monitored by cellular assays and noninvasive imaging. Mol. Ther. 2010, 18, 617–624. [Google Scholar] [CrossRef]
- Vandendriessche, T.; Thorrez, L.; Acosta-Sanchez, A.; Petrus, I.; Wang, L.; Ma, L.; de Waele, L.; Iwasaki, Y.; Gillijns, V.; Wilson, J.M.; et al. Efficacy and safety of adeno-associated viral vectors based on serotype 8 and 9 vs. lentiviral vectors for hemophilia B gene therapy. J. Thromb. Haemost. 2007, 5, 16–24. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.Y.; Peng, P.D.; Ehrhardt, A.; Storm, T.A.; Kay, M.A. Comparison of adenoviral and adeno-associated viral vectors for pancreatic gene delivery in vivo. Hum. Gene Ther. 2004, 15, 405–413. [Google Scholar] [CrossRef]
- Loiler, S.A.; Conlon, T.J.; Song, S.; Tang, Q.; Warrington, K.H.; Agarwal, A.; Kapturczak, M.; Li, C.; Ricordi, C.; Atkinson, M.A.; et al. Targeting recombinant adeno-associated virus vectors to enhance gene transfer to pancreatic islets and liver. Gene Ther. 2003, 10, 1551–1558. [Google Scholar] [CrossRef]
- Rabinowitz, J.E.; Rolling, F.; Li, C.; Conrath, H.; Xiao, W.; Xiao, X.; Samulski, R.J. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J. Virol. 2002, 76, 791–801. [Google Scholar] [CrossRef]
- Colella, P.; Auricchio, A. Gene therapy of inherited retinopathies: A long and successful road from viral vectors to patients. Hum. Gene Ther. 2012, 23, 796–807. [Google Scholar] [CrossRef]
- Baba, Y.; Satoh, S.; Otsu, M.; Sasaki, E.; Okada, T.; Watanabe, S. In vitro cell subtype-specific transduction of adeno-associated virus in mouse and marmoset retinal explant culture. Biochimie 2012, 94, 2716–2722. [Google Scholar] [CrossRef]
- Aldrich, W.A.; Ren, C.; White, A.F.; Zhou, S.Z.; Kumar, S.; Jenkins, C.B.; Shaw, D.R.; Strong, T.V.; Triozzi, P.L.; Ponnazhagan, S. Enhanced transduction of mouse bone marrow-derived dendritic cells by repetitive infection with self-complementary adeno-associated virus 6 combined with immunostimulatory ligands. Gene Ther. 2006, 13, 29–39. [Google Scholar] [CrossRef]
- Zhong, L.; Li, W.; Li, Y.; Zhao, W.; Wu, J.; Li, B.; Maina, N.; Bischof, D.; Qing, K.; Weigel-Kelley, K.A.; et al. Evaluation of primitive murine hematopoietic stem and progenitor cell transduction in vitro and in vivo by recombinant adeno-associated virus vector serotypes 1 through 5. Hum. Gene Ther. 2006, 17, 321–333. [Google Scholar]
- Ellis, B.L.; Hirsch, M.L.; Barker, J.C.; Connelly, J.P.; Steininger, R.J., 3rd; Porteus, M.H. A survey of ex vivo/in vitro transduction efficiency of mammalian primary cells and cell lines with Nine natural adeno-associated virus (AAV1–9) and one engineered adeno-associated virus serotype. Virol. J. 2013, 10, 74. [Google Scholar]
- Chiorini, J.A.; Kim, F.; Yang, L.; Kotin, R.M. Cloning and characterization of adeno-associated virus type 5. J. Virol. 1999, 73, 1309–1319. [Google Scholar]
- Yoshioka, T.; Okada, T.; Maeda, Y.; Ikeda, U.; Shimpo, M.; Nomoto, T.; Takeuchi, K.; Nonaka-Sarukawa, M.; Ito, T.; Takahashi, M.; et al. Adeno-associated virus vector-mediated interleukin-10 gene transfer inhibits atherosclerosis in apolipoprotein E-deficient mice. Gene Ther. 2004, 11, 1772–1779. [Google Scholar] [CrossRef]
- Nonaka-Sarukawa, M.; Okada, T.; Ito, T.; Yamamoto, K.; Yoshioka, T.; Nomoto, T.; Hojo, Y.; Shimpo, M.; Urabe, M.; Mizukami, H.; Kume, A.; et al. Adeno-associated virus vector-mediated systemic interleukin-10 expression ameliorates hypertensive organ damage in Dahl salt-sensitive rats. J. Gene Med. 2008, 10, 368–374. [Google Scholar]
- Nomoto, T.; Okada, T.; Shimazaki, K.; Yoshioka, T.; Nonaka-Sarukawa, M.; Ito, T.; Takeuchi, K.; Katsura, K.I.; Mizukami, H.; Kume, A.; et al. Systemic delivery of IL-10 by an AAV vector prevents vascular remodeling and end-organ damage in stroke-prone spontaneously hypertensive rat. Gene Ther. 2009, 16, 383–391. [Google Scholar] [CrossRef]
- Ohshima, S.; Shin, J.H.; Yuasa, K.; Nishiyama, A.; Kira, J.; Okada, T.; Takeda, S. Transduction Efficiency and Immune Response Associated With the Administration of AAV8 Vector Into Dog Skeletal Muscle. Mol. Ther. 2009, 17, 73–80. [Google Scholar] [CrossRef]
- Shin, J.H.; Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Ohshima-Hosoyama, S.; Kinoshita, K.; Chiyo, T.; Okada, H.; Okada, T.; Takeda, S. Improvement of cardiac fibrosis in dystrophic mice by rAAV9-mediated microdystrophin transduction. Gene Ther. 2011, 18, 910–919. [Google Scholar] [CrossRef]
- Kawano, R.; Ishizaki, M.; Maeda, Y.; Uchida, Y.; Kimura, E.; Uchino, M. Transduction of full-length dystrophin to multiple skeletal muscles improves motor performance and life span in utrophin/dystrophin double knockout mice. Mol. Ther. 2008, 16, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Li, J.; Xiao, X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl. Acad. Sci. USA 2000, 97, 13714–13719. [Google Scholar] [CrossRef]
- Sakamoto, M.; Yuasa, K.; Yoshimura, M.; Yokota, T.; Ikemoto, T.; Suzuki, M.; Dickson, G.; Miyagoe-Suzuki, Y.; Takeda, S. Micro-dystrophin cDNA ameliorates dystrophic phenotypes when introduced into mdx mice as a transgene. Biochem. Biophys. Res. Commun. 2002, 293, 1265–1272. [Google Scholar] [CrossRef]
- Yoshimura, M.; Sakamoto, M.; Ikemoto, M.; Mochizuki, Y.; Yuasa, K.; Miyagoe-Suzuki, Y.; Takeda, S. AAV vector-mediated microdystrophin expression in a relatively small percentage of mdx myofibers improved the mdx phenotype. Mol. Ther. 2004, 10, 821–828. [Google Scholar]
- Gregorevic, P.; Allen, J.M.; Minami, E.; Blankinship, M.J.; Haraguchi, M.; Meuse, L.; Finn, E.; Adams, M.E.; Froehner, S.C.; Murry, C.E.; et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 2006, 12, 787–789. [Google Scholar] [CrossRef]
- Gregorevic, P.; Blankinship, M.J.; Allen, J.M.; Chamberlain, J.S. Systemic microdystrophin gene delivery improves skeletal muscle structure and function in old dystrophic mdx mice. Mol. Ther. 2008, 16, 657–664. [Google Scholar] [CrossRef]
- Bostick, B.; Shin, J.H.; Yue, Y.; Wasala, N.B.; Lai, Y.; Duan, D. AAV micro-dystrophin gene therapy alleviates stress-induced cardiac death but not myocardial fibrosis in >21-m-old mdx mice, an end-stage model of Duchenne muscular dystrophy cardiomyopathy. J. Mol. Cell Cardiol. 2012, 53, 217–222. [Google Scholar]
- Foster, H.; Sharp, P.S.; Athanasopoulos, T.; Trollet, C.; Graham, I.R.; Foster, K.; Wells, D.J.; Dickson, G. Codon and mRNA sequence optimization of microdystrophin transgenes improves expression and physiological outcome in dystrophic mdx mice following AAV2/8 gene transfer. Mol. Ther. 2008, 16, 1825–1832. [Google Scholar] [CrossRef]
- Li, X.; Eastman, E.M.; Schwartz, R.J.; Draghia-Akli, R. Synthetic muscle promoters: Activities exceeding naturally occurring regulatory sequences. Nat. Biotechnol. 1999, 17, 241–245. [Google Scholar]
- Tinsley, J.M.; Fairclough, R.J.; Storer, R.; Wilkes, F.J.; Potter, A.C.; Squire, S.E.; Powell, D.S.; Cozzoli, A.; Capogrosso, R.F.; Lambert, A.; et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One 2011, 6, e19189. [Google Scholar]
- Odom, G.L.; Gregorevic, P.; Allen, J.M.; Finn, E.; Chamberlain, J.S. Microutrophin delivery through rAAV6 increases lifespan and improves muscle function in dystrophic dystrophin/utrophin-deficient mice. Mol. Ther. 2008, 16, 1539–1545. [Google Scholar] [CrossRef]
- Li, D.; Bareja, A.; Judge, L.; Yue, Y.; Lai, Y.; Fairclough, R.; Davies, K.E.; Chamberlain, J.S.; Duan, D. Sarcolemmal nNOS anchoring reveals a qualitative difference between dystrophin and utrophin. J. Cell Sci. 2010, 123, 2008–2013. [Google Scholar] [CrossRef]
- Ramachandran, J.; Schneider, J.S.; Crassous, P.A.; Zheng, R.; Gonzalez, J.P.; Xie, L.H.; Beuve, A.; Fraidenraich, D.; Peluffo, R.D. Nitric oxide signalling pathway in Duchenne muscular dystrophy mice: Up-regulation of L-arginine transporters. Biochem. J. 2013, 449, 133–142. [Google Scholar] [CrossRef]
- Qiao, C.; Li, J.; Jiang, J.; Zhu, X.; Wang, B.; Li, J.; Xiao, X. Myostatin propeptide gene delivery by adeno-associated virus serotype 8 vectors enhances muscle growth and ameliorates dystrophic phenotypes in mdx mice. Hum. Gene Ther. 2008, 19, 241–254. [Google Scholar]
- Schertzer, J.D.; Lynch, G.S. Comparative evaluation of IGF-I gene transfer and IGF-I protein administration for enhancing skeletal muscle regeneration after injury. Gene Ther. 2006, 13, 1657–1664. [Google Scholar] [CrossRef]
- Fukushima, K.; Nakamura, A.; Ueda, H.; Yuasa, K.; Yoshida, K.; Takeda, S.; Ikeda, S. Activation and localization of matrix metalloproteinase-2 and -9 in the skeletal muscle of the muscular dystrophy dog (CXMDJ). BMC Musculoskelet. Disord. 2007, 8, 54. [Google Scholar] [CrossRef]
- Gargioli, C.; Coletta, M.; De Grandis, F.; Cannata, S.M.; Cossu, G. PlGF-MMP-9-expressing cells restore microcirculation and efficacy of cell therapy in aged dystrophic muscle. Nat. Med. 2008, 14, 973–978. [Google Scholar] [CrossRef]
- Matsushita, T.; Elliger, S.; Elliger, C.; Podsakoff, G.; Villarreal, L.; Kurtzman, G.J.; Iwaki, Y.; Colosi, P. Adeno-associated virus vectors can be efficiently produced without helper virus. Gene Ther. 1998, 5, 938–945. [Google Scholar]
- Yamaguchi, T.; Okada, T.; Takeuchi, K.; Tonda, T.; Ohtaki, M.; Shinoda, S.; Masuzawa, T.; Ozawa, K.; Inaba, T. Enhancement of thymidine kinase-mediated killing of malignant glioma by BimS, a BH3-only cell death activator. Gene Ther. 2003, 10, 375–385. [Google Scholar] [CrossRef]
- Okada, T.; Mizukami, H.; Urabe, M.; Nomoto, T.; Matsushita, T.; Hanazono, Y.; Kume, A.; Tobita, K.; Ozawa, K. Development and characterization of an antisense-mediated prepackaging cell line for adeno-associated virus vector production. Biochem. Biophys. Res. Commun. 2001, 288, 62–68. [Google Scholar] [CrossRef]
- Okada, T.; Nomoto, T.; Yoshioka, T.; Nonaka-Sarukawa, M.; Ito, T.; Ogura, T.; Iwata-Okada, M.; Uchibori, R.; Shimazaki, K.; Mizukami, H.; et al. Large-scale production of recombinant viruses by use of a large culture vessel with active gassing. Hum. Gene Ther. 2005, 16, 1212–1218. [Google Scholar] [CrossRef]
- Okada, T.; Nonaka-Sarukawa, M.; Uchibori, R.; Kinoshita, K.; Hayashita-Kinoh, H.; Nitahara-Kasahara, Y.; Takeda, S.; Ozawa, K. Scalable purification of adeno-associated virus serotype 1 (AAV1) and AAV8 vectors, using dual ion-exchange adsorptive membranes. Hum. Gene Ther. 2009, 20, 1013–1021. [Google Scholar] [CrossRef]
- Valentine, B.A.; Cooper, B.J.; de Lahunta, A.; O'Quinn, R.; Blue, J.T. Canine X-linked muscular dystrophy. An animal model of Duchenne muscular dystrophy: Clinical studies. J. Neurol. Sci. 1988, 88, 69–81. [Google Scholar]
- Shimatsu, Y.; Yoshimura, M.; Yuasa, K.; Urasawa, N.; Tomohiro, M.; Nakura, M.; Tanigawa, M.; Nakamura, A.; Takeda, S. Major clinical and histopathological characteristics of canine X-linked muscular dystrophy in Japan, CXMDJ. Acta Myol. 2005, 24, 145–154. [Google Scholar]
- Yuasa, K.; Yoshimura, M.; Urasawa, N.; Ohshima, S.; Howell, J.M.; Nakamura, A.; Hijikata, T.; Miyagoe-Suzuki, Y.; Takeda, S. Injection of a recombinant AAV serotype 2 into canine skeletal muscles evokes strong immune responses against transgene products. Gene Ther. 2007, 14, 1249–1260. [Google Scholar]
- Urasawa, N.; Wada, M.R.; Machida, N.; Yuasa, K.; Shimatsu, Y.; Wakao, Y.; Yuasa, S.; Sano, T.; Nonaka, I.; Nakamura, A.; et al. Selective vacuolar degeneration in dystrophin-deficient canine Purkinje fibers despite preservation of dystrophin-associated proteins with overexpression of Dp71. Circulation 2008, 117, 2437–2448. [Google Scholar] [CrossRef]
- Yuasa, K.; Sakamoto, M.; Miyagoe-Suzuki, Y.; Tanouchi, A.; Yamamoto, H.; Li, J.; Chamberlain, J.S.; Xiao, X.; Takeda, S. Adeno-associated virus vector-mediated gene transfer into dystrophin-deficient skeletal muscles evokes enhanced immune response against the transgene product. Gene Ther. 2002, 9, 1576–1588. [Google Scholar] [CrossRef]
- Li, C.; Hirsch, M.; Asokan, A.; Zeithaml, B.; Ma, H.; Kafri, T.; Samulski, R.J. Adeno-associated virus type 2 (AAV2) capsid-specific cytotoxic T lymphocytes eliminate only vector-transduced cells coexpressing the AAV2 capsid in vivo. J. Virol. 2007, 81, 7540–7547. [Google Scholar] [CrossRef]
- Zhang, Y.; Chirmule, N.; Gao, G.; Wilson, J. CD40 ligand-dependent activation of cytotoxic T lymphocytes by adeno-associated virus vectors in vivo: Role of immature dendritic cells. J. Virol. 2000, 74, 8003–8010. [Google Scholar] [CrossRef]
- Wang, Z.; Allen, J.M.; Riddell, S.R.; Gregorevic, P.; Storb, R.; Tapscott, S.J.; Chamberlain, J.S.; Kuhr, C.S. Immunity to adeno-associated virus-mediated gene transfer in a random-bred canine model of Duchenne muscular dystrophy. Hum. Gene Ther. 2007, 18, 18–26. [Google Scholar]
- Manno, C.S.; Pierce, G.F.; Arruda, V.R.; Glader, B.; Ragni, M.; Rasko, J.J.; Ozelo, M.C.; Hoots, K.; Blatt, P.; Konkle, B.; et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006, 12, 342–347. [Google Scholar] [CrossRef]
- Scallan, C.D.; Jiang, H.; Liu, T.; Patarroyo-White, S.; Sommer, J.M.; Zhou, S.; Couto, L.B.; Pierce, G.F. Human immunoglobulin inhibits liver transduction by AAV vectors at low AAV2 neutralizing titers in SCID mice. Blood 2006, 107, 1810–1817. [Google Scholar] [CrossRef]
- Vandenberghe, L.H.; Wang, L.; Somanathan, S.; Zhi, Y.; Figueredo, J.; Calcedo, R.; Sanmiguel, J.; Desai, R.A.; Chen, C.S.; Johnston, J.; et al. Heparin binding directs activation of T cells against adeno-associated virus serotype 2 capsid. Nat Med 2006, 12, 967–971. [Google Scholar]
- Lorain, S.; Gross, D.A.; Goyenvalle, A.; Danos, O.; Davoust, J.; Garcia, L. Transient immunomodulation allows repeated injections of AAV1 and correction of muscular dystrophy in multiple muscles. Mol. Ther. 2008, 16, 541–547. [Google Scholar]
- Wang, Z.; Kuhr, C.S.; Allen, J.M.; Blankinship, M.; Gregorevic, P.; Chamberlain, J.S.; Tapscott, S.J.; Storb, R. Sustained AAV-mediated Dystrophin Expression in a Canine Model of Duchenne Muscular Dystrophy with a Brief Course of Immunosuppression. Mol. Ther. 2007, 15, 1160–1166. [Google Scholar]
- Wang, Z.; Storb, R.; Halbert, C.L.; Banks, G.B.; Butts, T.M.; Finn, E.E.; Allen, J.M.; Miller, A.D.; Chamberlain, J.S.; Tapscott, S.J. Successful regional delivery and long-term expression of a dystrophin gene in canine muscular dystrophy: A preclinical model for human therapies. Mol. Ther. 2012, 20, 1501–1507. [Google Scholar]
- Monteilhet, V.; Saheb, S.; Boutin, S.; Leborgne, C.; Veron, P.; Montus, M.F.; Moullier, P.; Benveniste, O.; Masurier, C. A 10 patient case report on the impact of plasmapheresis upon neutralizing factors against adeno-associated virus (AAV) types 1, 2, 6, and 8. Mol. Ther. 2011, 19, 2084–2091. [Google Scholar] [CrossRef]
- Hagstrom, J.E.; Hegge, J.; Zhang, G.; Noble, M.; Budker, V.; Lewis, D.L.; Herweijer, H.; Wolff, J.A. A facile nonviral method for delivering genes and siRNAs to skeletal muscle of mammalian limbs. Mol. Ther. 2004, 10, 386–398. [Google Scholar] [CrossRef]
- Sun, B.; Li, S.; Bird, A.; Koeberl, D.D. Hydrostatic isolated limb perfusion with adeno-associated virus vectors enhances correction of skeletal muscle in Pompe disease. Gene Ther. 2010, 17, 1500–1505. [Google Scholar] [CrossRef]
- Rodino-Klapac, L.R.; Montgomery, C.L.; Mendell, J.R.; Chicoine, L.G. AAV-mediated gene therapy to the isolated limb in rhesus macaques. Methods Mol. Biol. 2011, 709, 287–298. [Google Scholar] [CrossRef]
- Townsend, D.; Yasuda, S.; Li, S.; Chamberlain, J.S.; Metzger, J.M. Emergent dilated cardiomyopathy caused by targeted repair of dystrophic skeletal muscle. Mol. Ther. 2008, 16, 832–835. [Google Scholar] [CrossRef]
- Bostick, B.; Yue, Y.; Lai, Y.; Long, C.; Li, D.; Duan, D. Adeno-associated virus serotype-9 microdystrophin gene therapy ameliorates electrocardiographic abnormalities in mdx mice. Hum Gene Ther. 2008, 19, 851–856. [Google Scholar] [CrossRef]
- Yue, Y.; Ghosh, A.; Long, C.; Bostick, B.; Smith, B.F.; Kornegay, J.N.; Duan, D. A single intravenous injection of adeno-associated virus serotype-9 leads to whole body skeletal muscle transduction in dogs. Mol. Ther. 2008, 16, 1944–1952. [Google Scholar] [CrossRef]
- Reay, D.P.; Bilbao, R.; Koppanati, B.M.; Cai, L.; O'Day, T.L.; Jiang, Z.; Zheng, H.; Watchko, J.F.; Clemens, P.R. Full-length dystrophin gene transfer to the mdx mouse in utero. Gene Ther. 2008, 15, 531–536. [Google Scholar] [CrossRef]
- Rodino-Klapac, L.R.; Chicoine, L.G.; Kaspar, B.K.; Mendell, J.R. Gene therapy for duchenne muscular dystrophy: Expectations and challenges. Arch. Neurol. 2007, 64, 1236–1241. [Google Scholar]
- Mendell, J.R.; Campbell, K.; Rodino-Klapac, L.; Sahenk, Z.; Shilling, C.; Lewis, S.; Bowles, D.; Gray, S.; Li, C.; Galloway, G.; et al. Dystrophin immunity in Duchenne's muscular dystrophy. N. Engl. J. Med. 2010, 363, 1429–1437. [Google Scholar] [CrossRef]
- Nathwani, A.C.; Tuddenham, E.G.; Rangarajan, S.; Rosales, C.; McIntosh, J.; Linch, D.C.; Chowdary, P.; Riddell, A.; Pie, A.J.; Harrington, C.; et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011, 365, 2357–2365. [Google Scholar] [CrossRef]
- Buchlis, G.; Podsakoff, G.M.; Radu, A.; Hawk, S.M.; Flake, A.W.; Mingozzi, F.; High, K.A. Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood 2012, 119, 3038–3041. [Google Scholar] [CrossRef]
- Goyenvalle, A.; Vulin, A.; Fougerousse, F.; Leturcq, F.; Kaplan, J.C.; Garcia, L.; Danos, O. Rescue of dystrophic muscle through U7 snRNA-mediated exon skipping. Science 2004, 306, 1796–1799. [Google Scholar]
- Denti, M.A.; Incitti, T.; Sthandier, O.; Nicoletti, C.; De Angelis, F.G.; Rizzuto, E.; Auricchio, A.; Musaro, A.; Bozzoni, I. Long-term benefit of adeno-associated virus/antisense-mediated exon skipping in dystrophic mice. Hum. Gene Ther. 2008, 19, 601–608. [Google Scholar] [CrossRef]
- Okada, T.; Uchibori, R.; Iwata-Okada, M.; Takahashi, M.; Nomoto, T.; Nonaka-Sarukawa, M.; Ito, T.; Liu, Y.; Mizukami, H.; Kume, A.; et al. A histone deacetylase inhibitor enhances recombinant adeno-associated virus-mediated gene expression in tumor cells. Mol. Ther. 2006, 13, 738–746. [Google Scholar] [CrossRef]
- Li, W.; Asokan, A.; Wu, Z.; van Dyke, T.; DiPrimio, N.; Johnson, J.S.; Govindaswamy, L.; Agbandje-McKenna, M.; Leichtle, S.; Redmond, D.E., Jr.; et al. Engineering and selection of shuffled AAV genomes: A new strategy for producing targeted biological nanoparticles. Mol. Ther. 2008, 16, 1252–1260. [Google Scholar] [CrossRef]
- Muzyczka, N.; Warrington, K.H., Jr. Custom adeno-associated virus capsids: The next generation of recombinant vectors with novel tropism. Hum. Gene Ther. 2005, 16, 408–416. [Google Scholar] [CrossRef]
- Arnold, G.S.; Sasser, A.K.; Stachler, M.D.; Bartlett, J.S. Metabolic biotinylation provides a unique platform for the purification and targeting of multiple AAV vector serotypes. Mol. Ther. 2006, 14, 97–106. [Google Scholar]
- Nitahara-Kasahara, Y.; Hayashita-Kinoh, H.; Ohshima-Hosoyama, S.; Okada, H.; Wada-Maeda, M.; Nakamura, A.; Okada, T.; Takeda, S. Long-term engraftment of multipotent mesenchymal stromal cells that differentiate to form myogenic cells in dogs with Duchenne muscular dystrophy. Mol. Ther. 2012, 20, 168–177. [Google Scholar] [CrossRef]
- Chiu, R.C. MSC immune tolerance in cellular cardiomyoplasty. Semin. Thorac. Cardiovasc. Surg. 2008, 20, 115–118. [Google Scholar] [CrossRef]
- Millard, S.M.; Fisk, N.M. Mesenchymal stem cells for systemic therapy: Shotgun approach or magic bullets? Bioessays 2013, 35, 173–182. [Google Scholar]
- Casiraghi, F.; Azzollini, N.; Todeschini, M.; Cavinato, R.A.; Cassis, P.; Solini, S.; Rota, C.; Morigi, M.; Introna, M.; Maranta, R.; et al. Localization of Mesenchymal Stromal Cells Dictates Their Immune or Proinflammatory Effects in Kidney Transplantation. Am. J. Transplant. 2012.
- Studeny, M.; Marini, F.C.; Dembinski, J.L.; Zompetta, C.; Cabreira-Hansen, M.; Bekele, B.N.; Champlin, R.E.; Andreeff, M. Mesenchymal stem cells: Potential precursors for tumor stroma and targeted-delivery vehicles for anticancer agents. J. Natl. Cancer Inst. 2004, 96, 1593–1603. [Google Scholar] [CrossRef]
- Okada, T.; Ozawa, K. Vector-producing tumor-tracking multipotent mesenchymal stromal cells for suicide cancer gene therapy. Front. Biosci. 2008, 13, 1887–1891. [Google Scholar] [CrossRef]
- Uchibori, R.; Okada, T.; Ito, T.; Urabe, M.; Mizukami, H.; Kume, A.; Ozawa, K. Retroviral vector-producing mesenchymal stem cells for targeted suicide cancer gene therapy. J. Gene Med. 2009, 11, 373–381. [Google Scholar] [CrossRef]
- Sampaolesi, M.; Blot, S.; D'Antona, G.; Granger, N.; Tonlorenzi, R.; Innocenzi, A.; Mognol, P.; Thibaud, J.L.; Galvez, B.G.; Barthelemy, I.; et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 2006, 444, 574–579. [Google Scholar] [CrossRef] [Green Version]
- Chun, J.L.; O'Brien, R.; Song, M.H.; Wondrasch, B.F.; Berry, S.E. Injection of vessel-derived stem cells prevents dilated cardiomyopathy and promotes angiogenesis and endogenous cardiac stem cell proliferation in mdx/utrn-/- but not aged mdx mouse models for duchenne muscular dystrophy. Stem Cells Transl. Med. 2013, 2, 68–80. [Google Scholar]
- Kotin, R.M.; Linden, R.M.; Berns, K.I. Characterization of a preferred site on human chromosome 19q for integration of adeno-associated virus DNA by non-homologous recombination. Embo J. 1992, 11, 5071–5078. [Google Scholar]
- Goncalves, M.A.; van Nierop, G.P.; Tijssen, M.R.; Lefesvre, P.; Knaan-Shanzer, S.; van der Velde, I.; van Bekkum, D.W.; Valerio, D.; de Vries, A.A. Transfer of the full-length dystrophin-coding sequence into muscle cells by a dual high-capacity hybrid viral vector with site-specific integration ability. J. Virol. 2005, 79, 3146–3162. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Okada, T.; Takeda, S. Current Challenges and Future Directions in Recombinant AAV-Mediated Gene Therapy of Duchenne Muscular Dystrophy. Pharmaceuticals 2013, 6, 813-836. https://doi.org/10.3390/ph6070813
Okada T, Takeda S. Current Challenges and Future Directions in Recombinant AAV-Mediated Gene Therapy of Duchenne Muscular Dystrophy. Pharmaceuticals. 2013; 6(7):813-836. https://doi.org/10.3390/ph6070813
Chicago/Turabian StyleOkada, Takashi, and Shin'ichi Takeda. 2013. "Current Challenges and Future Directions in Recombinant AAV-Mediated Gene Therapy of Duchenne Muscular Dystrophy" Pharmaceuticals 6, no. 7: 813-836. https://doi.org/10.3390/ph6070813