Advances in Mammalian Cell Line Development Technologies for Recombinant Protein Production

{kind=link}
{kind=link}
{kind=link}
Abstract
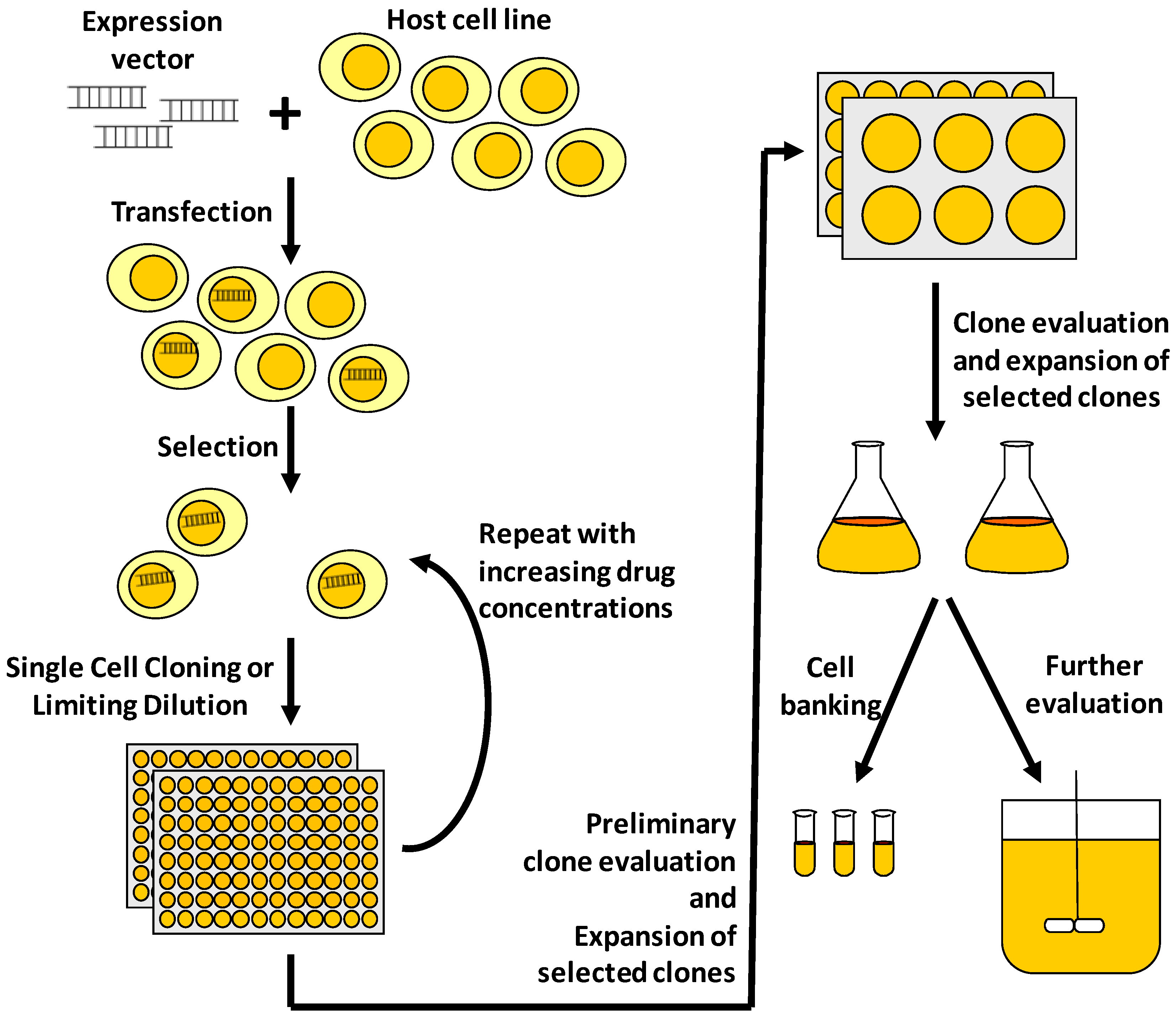
:1. Introduction

2. Protein Expression Technologies
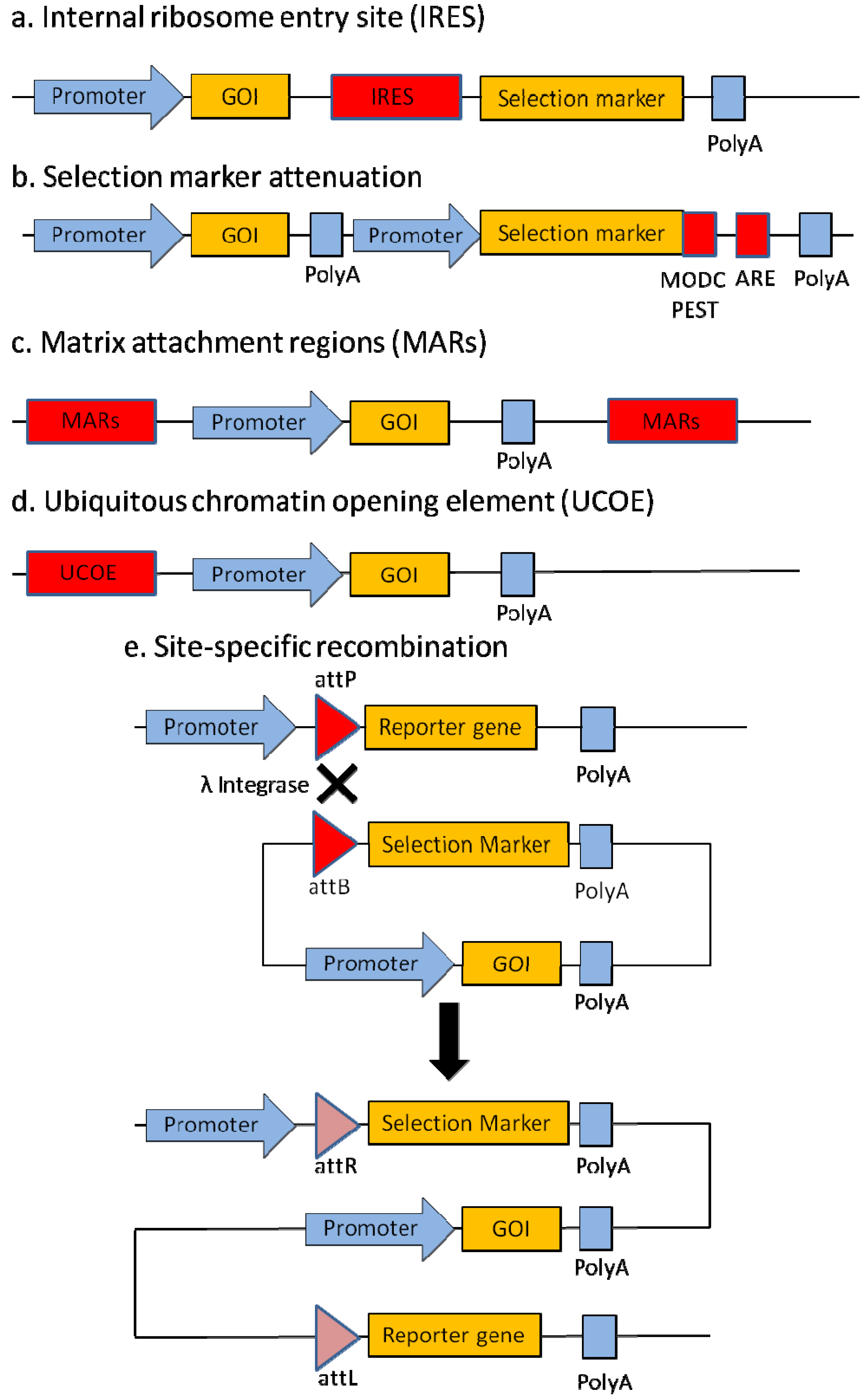
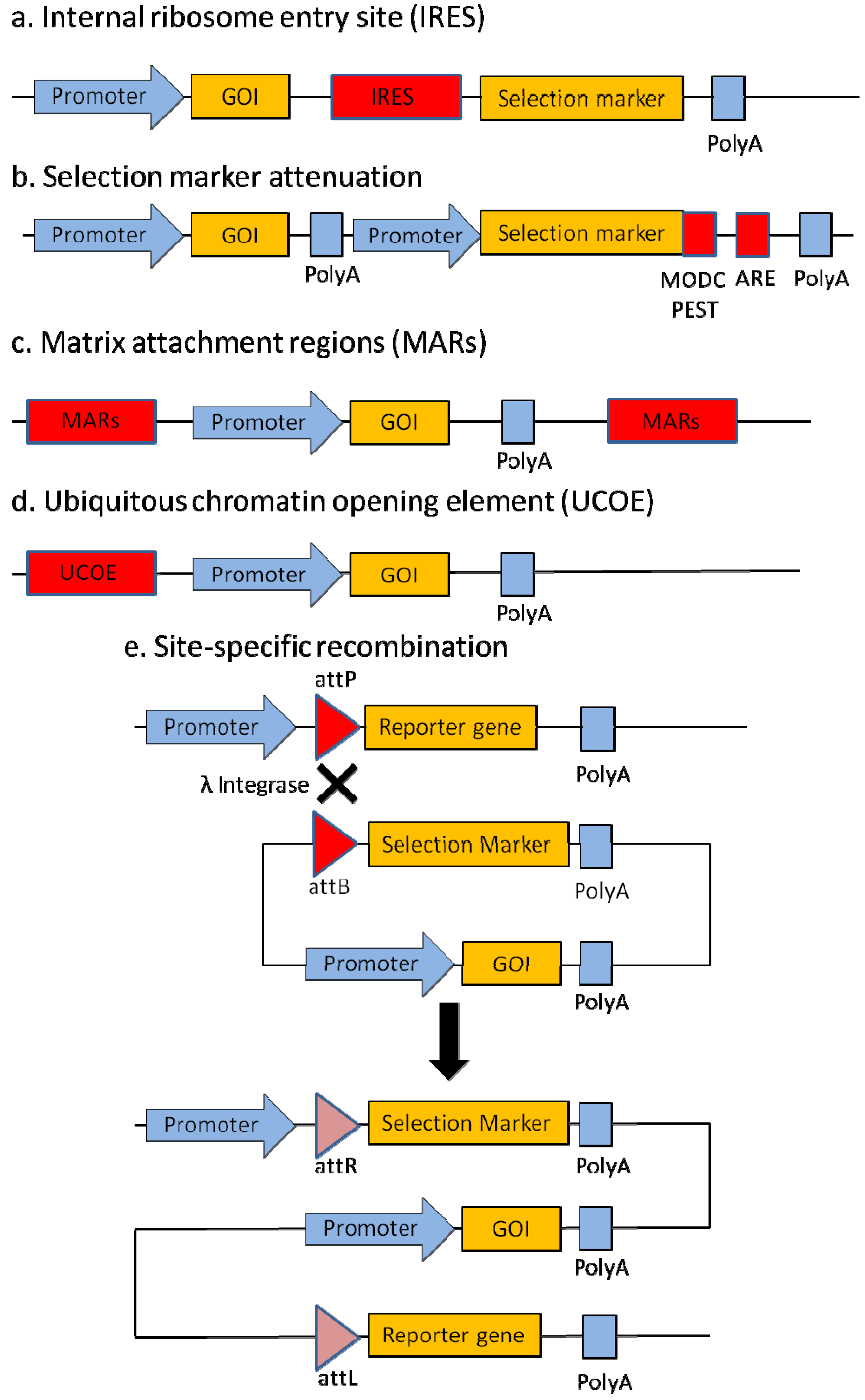
2.1. Internal Ribosome Entry Site (IRES)
2.2. Selection Marker Attenuation
2.3. Matrix Attachment Regions (MARs)
2.4. Ubiquitous Chromatin Opening Element (UCOE)
2.5. Site-Specific Recombination
2.6. Artificial Chromosome Expression (ACE) System
2.7. Cell Line Engineering
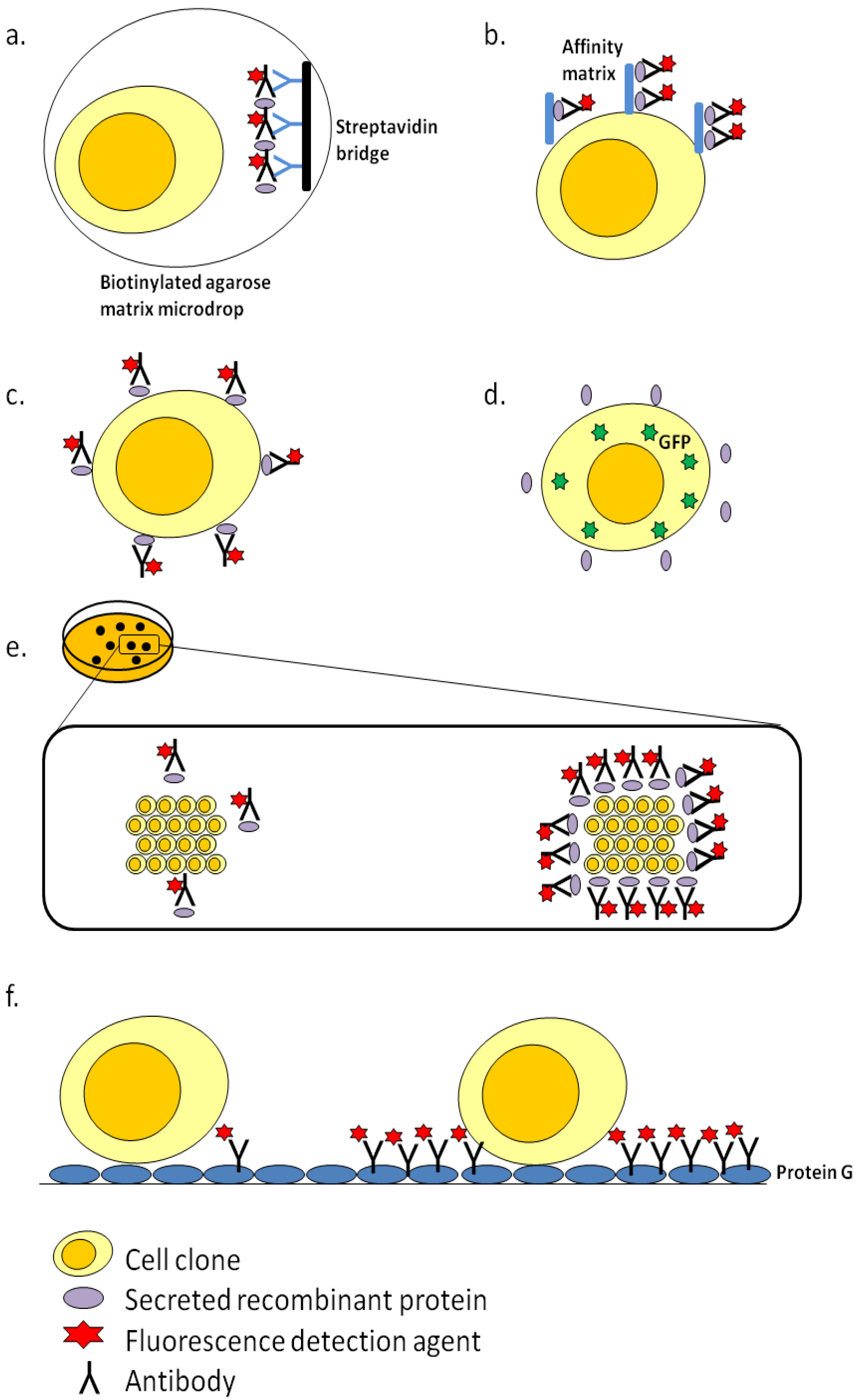
3. Clone Screening Technologies
3.1. Fluorescence-Activated Cell Sorting (FACS)-Based Screening
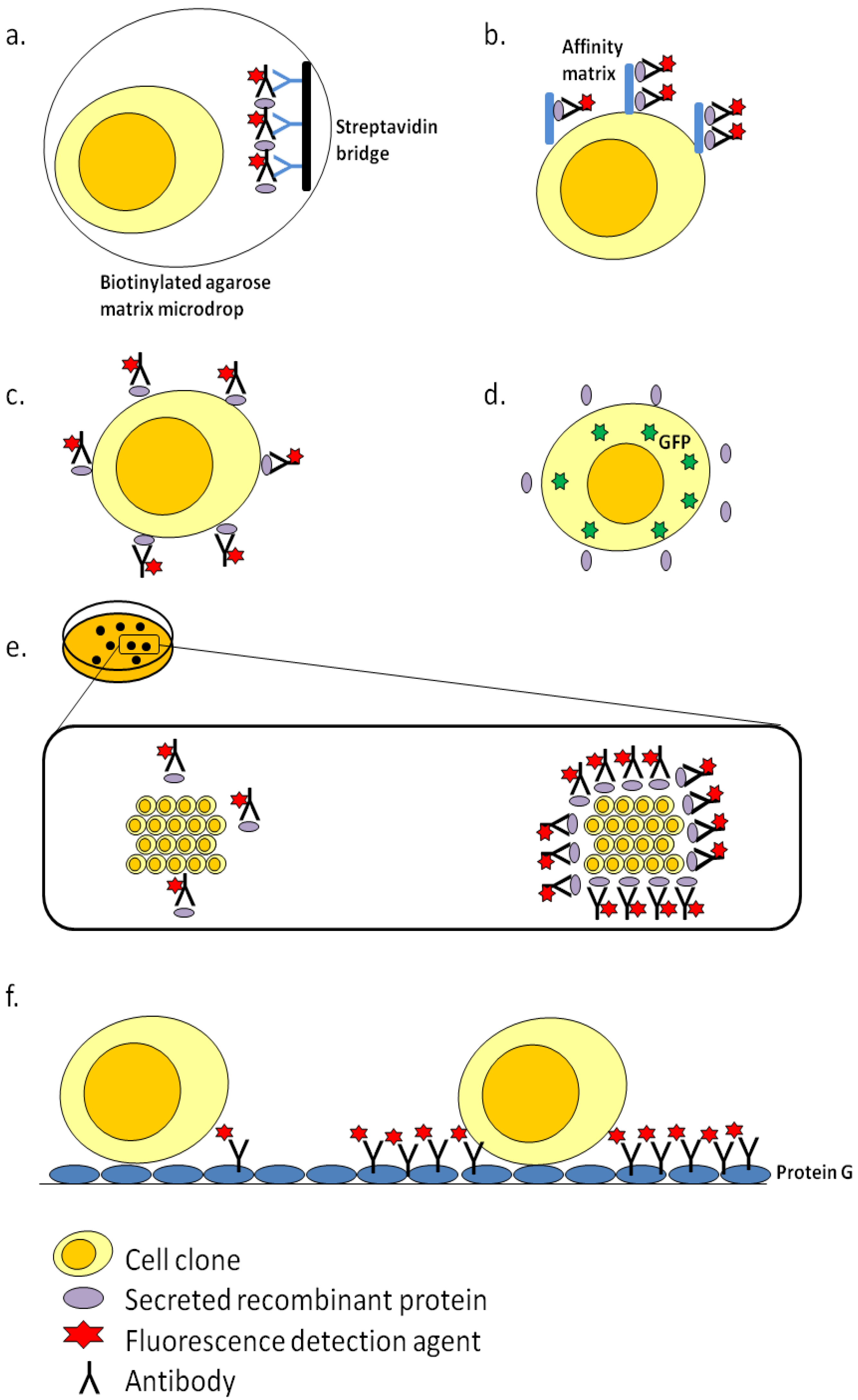
3.2. ClonePix
3.3. Cell XpressTM
4. Conclusions
Acknowledgments
References
- Boeger, H.; Bushnell, D.A.; Davis, R.; Griesenbeck, J.; Lorch, Y.; Strattan, J.S.; Westover, K.D.; Kornberg, R.D. Structural basis of eukaryotic gene transcription. FEBS Lett. 2005, 579, 899–903. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kim, Y.G.; Lee, G.M. CHO cells in biotechnology for production of recombinant proteins: Current state and further potential. Appl. Microbiol. Biotechnol. 2012, 93, 917–930. [Google Scholar] [CrossRef]
- Ghaderi, D.; Zhang, M.; Hurtado-Ziola, N.; Varki, A. Production platforms for biotherapeutic glycoproteins. Occurrence, impact, and challenges of non-human sialylation. Biotechnol. Genet. Eng. Rev. 2012, 28, 147–175. [Google Scholar]
- Wiberg, F.C.; Rasmussen, S.K.; Frandsen, T.P.; Rasmussen, L.K.; Tengbjerg, K.; Coljee, V.W.; Sharon, J.; Yang, C.Y.; Bregenholt, S.; Nielsen, L.S.; et al. Production of target-specific recombinant human polyclonal antibodies in mammalian cells. Biotechnol. Bioeng. 2006, 94, 396–405. [Google Scholar] [CrossRef]
- TOP 30 Biologics 2011. Available online: http://www.pipelinereview.com/index.php/archive/view/listid-1-la-merie-daily/mailid-35-La-Merie-Daily-TOP-30-Biologics-2011-new-free-report/tmpl-component (Accessed on 16 April 2013).
- Lanthier, M.; Behrman, R.; Nardinelli, C. Economic issues with follow-on protein products. Nat. Rev. Drug Discov. 2008, 7, 733–737. [Google Scholar] [CrossRef]
- Kaufman, R.J.; Sharp, P.A. Amplification and expression of sequences cotransfected with a modular dihydrofolate reductase complementary dna gene. J. Mol. Biol. 1982, 159, 601–621. [Google Scholar] [CrossRef]
- Chu, L.; Robinson, D.K. Industrial choices for protein production by large-scale cell culture. Curr. Opin. Biotechnol. 2001, 12, 180–187. [Google Scholar] [CrossRef]
- Bebbington, C.R.; Renner, G.; Thomson, S.; King, D.; Abrams, D.; Yarranton, G.T. High-level expression of a recombinant antibody from myeloma cells using a glutamine synthetase gene as an amplifiable selectable marker. Biotechnology (NY) 1992, 10, 169–175. [Google Scholar]
- Browne, S.M.; Al-Rubeai, M. Selection methods for high-producing mammalian cell lines. Trends Biotechnol. 2007, 25, 425–432. [Google Scholar] [CrossRef]
- Wigler, M.; Perucho, M.; Kurtz, D.; Dana, S.; Pellicer, A.; Axel, R.; Silverstein, S. Transformation of mammalian cells with an amplifiable dominant-acting gene. Proc. Natl. Acad. Sci. USA 1980, 77, 3567–3570. [Google Scholar] [CrossRef]
- Urlaub, G.; Chasin, L.A. Isolation of Chinese hamster cell mutants deficient in dihydrofolate reductase activity. Proc. Natl. Acad. Sci. USA 1980, 77, 4216–4220. [Google Scholar] [CrossRef]
- Liu, P.Q.; Chan, E.M.; Cost, G.J.; Zhang, L.; Wang, J.; Miller, J.C.; Guschin, D.Y.; Reik, A.; Holmes, M.C.; Mott, J.E.; et al. Generation of a triple-gene knockout mammalian cell line using engineered zinc-finger nucleases. Biotechnol. Bioeng. 2010, 106, 97–105. [Google Scholar]
- Lonza launches next generation GS gene expression system. Available online: http://www.lonza.com/about-lonza/media-center/news/2012/120710-GS-System-e.aspx (Accessed on 15 February 2013).
- Schimke, R.T. Gene amplification in cultured animal cells. Cell 1984, 37, 705–713. [Google Scholar] [CrossRef]
- Niwa, H.; Yamamura, K.; Miyazaki, J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 1991, 108, 193–199. [Google Scholar] [CrossRef]
- Sautter, K.; Enenkel, B. Selection of high-producing CHO cells using NPT selection marker with reduced enzyme activity. Biotechnol. Bioeng. 2005, 89, 530–538. [Google Scholar] [CrossRef]
- Wurtele, H.; Little, K.C.; Chartrand, P. Illegitimate DNA integration in mammalian cells. Gene Ther. 2003, 10, 1791–1799. [Google Scholar] [CrossRef]
- West, A.G.; Fraser, P. Remote control of gene transcription. Hum. Mol. Genet. 2005, 14, R101–R111. [Google Scholar] [CrossRef]
- Wurm, F.M. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotechnol. 2004, 22, 1393–1398. [Google Scholar] [CrossRef]
- Davies, S.L.; Lovelady, C.S.; Grainger, R.K.; Racher, A.J.; Young, R.J.; James, D.C. Functional heterogeneity and heritability in CHO cell populations. Biotechnol. Bioeng. 2013, 110, 260–274. [Google Scholar] [CrossRef]
- Pilbrough, W.; Munro, T.P.; Gray, P. Intraclonal protein expression heterogeneity in recombinant CHO cells. PLoS One 2009, 4, e8432. [Google Scholar] [CrossRef]
- Derouazi, M.; Martinet, D.; Besuchet Schmutz, N.; Flaction, R.; Wicht, M.; Bertschinger, M.; Hacker, D.L.; Beckmann, J.S.; Wurm, F.M. Genetic characterization of CHO production host DG44 and derivative recombinant cell lines. Biochem. Biophys. Res. Commun. 2006, 340, 1069–1077. [Google Scholar] [CrossRef]
- Lattenmayer, C.; Loeschel, M.; Schriebl, K.; Steinfellner, W.; Sterovsky, T.; Trummer, E.; Vorauer-Uhl, K.; Muller, D.; Katinger, H.; Kunert, R. Protein-free transfection of CHO host cells with an IgG-fusion protein: Selection and characterization of stable high producers and comparison to conventionally transfected clones. Biotechnol. Bioeng. 2007, 96, 1118–1126. [Google Scholar] [CrossRef]
- Kim, S.J.; Kim, N.S.; Ryu, C.J.; Hong, H.J.; Lee, G.M. Characterization of chimeric antibody producing CHO cells in the course of dihydrofolate reductase-mediated gene amplification and their stability in the absence of selective pressure. Biotechnol. Bioeng. 1998, 58, 73–84. [Google Scholar]
- Kim, N.S.; Byun, T.H.; Lee, G.M. Key determinants in the occurrence of clonal variation in humanized antibody expression of cho cells during dihydrofolate reductase mediated gene amplification. Biotechnol. Prog. 2001, 17, 69–75. [Google Scholar]
- Kaufman, R.J.; Wasley, L.C.; Spiliotes, A.J.; Gossels, S.D.; Latt, S.A.; Larsen, G.R.; Kay, R.M. Coamplification and coexpression of human tissue-type plasminogen activator and murine dihydrofolate reductase sequences in Chinese hamster ovary cells. Mol. Cell. Biol. 1985, 5, 1750–1759. [Google Scholar]
- Chusainow, J.; Yang, Y.S.; Yeo, J.H.; Toh, P.C.; Asvadi, P.; Wong, N.S.; Yap, M.G. A study of monoclonal antibody-producing CHO cell lines: What makes a stable high producer? Biotechnol. Bioeng. 2009, 102, 1182–1196. [Google Scholar] [CrossRef]
- Fussenegger, M.; Bailey, J.E.; Hauser, H.; Mueller, P.P. Genetic optimization of recombinant glycoprotein production by mammalian cells. Trends Biotechnol. 1999, 17, 35–42. [Google Scholar] [CrossRef]
- Baird, S.D.; Turcotte, M.; Korneluk, R.G.; Holcik, M. Searching for IRES. RNA 2006, 12, 1755–1785. [Google Scholar] [CrossRef]
- Ho, S.C.; Bardor, M.; Feng, H.; Mariati; Tong, Y.W.; Song, Z.; Yap, M.G.; Yang, Y. IRES-mediated Tricistronic vectors for enhancing generation of high monoclonal antibody expressing CHO cell lines. J. Biotechnol. 2012, 157, 130–139. [Google Scholar]
- Trill, J.J.; Shatzman, A.R.; Ganguly, S. Production of monoclonal antibodies in COS and CHO cells. Curr. Opin. Biotechnol. 1995, 6, 553–560. [Google Scholar] [CrossRef]
- Ng, S.K.; Lin, W.; Sachdeva, R.; Wang, D.I.; Yap, M.G. Vector fragmentation: Characterizing vector integrity in transfected clones by Southern blotting. Biotechnol. Prog. 2010, 26, 11–20. [Google Scholar]
- Kaufman, R.J.; Davies, M.V.; Wasley, L.C.; Michnick, D. Improved vectors for stable expression of foreign genes in mammalian cells by use of the untranslated leader sequence from EMC virus. Nucleic Acids Res. 1991, 19, 4485–4490. [Google Scholar] [CrossRef]
- Rees, S.; Coote, J.; Stables, J.; Goodson, S.; Harris, S.; Lee, M.G. Bicistronic vector for the creation of stable mammalian cell lines that predisposes all antibiotic-resistant cells to express recombinant protein. Biotechniques 1996, 20, 102–104, 106, 108–110. [Google Scholar]
- Gurtu, V.; Yan, G.; Zhang, G. IRES bicistronic expression vectors for efficient creation of stable mammalian cell lines. Biochem. Biophys. Res. Commun. 1996, 229, 295–298. [Google Scholar] [CrossRef]
- Kolb, A.F.; Siddell, S.G. Expression of a recombinant monoclonal antibody from a bicistronic mRNA. Hybridoma 1997, 16, 421–426. [Google Scholar] [CrossRef]
- Novo, J.B.; Morganti, L.; Moro, A.M.; Paes Leme, A.F.; Serrano, S.M.; Raw, I.; Ho, P.L. Generation of a Chinese hamster ovary cell line producing recombinant human glucocerebrosidase. J. Biomed. Biotechnol. 2012, 2012, 875383. [Google Scholar]
- Ng, S.K.; Tan, T.R.; Wang, Y.; Ng, D.; Goh, L.T.; Bardor, M.; Wong, V.V.; Lam, K.P. Production of Functional Soluble Dectin-1 Glycoprotein Using an IRES-Linked Destabilized-Dihydrofolate Reductase Expression Vector. PLoS One 2012, 7, e52785. [Google Scholar]
- Gross, G.; Hauser, H. Heterologous expression as a tool for gene identification and analysis. J. Biotechnol. 1995, 41, 91–110. [Google Scholar] [CrossRef]
- Westwood, A.D.; Rowe, D.A.; Clarke, H.R. Improved recombinant protein yield using a codon deoptimized DHFR selectable marker in a CHEF1 expression plasmid. Biotechnol. Prog. 2010, 26, 1558–1566. [Google Scholar] [CrossRef]
- Ng, S.K.; Wang, D.I.; Yap, M.G. Application of destabilizing sequences on selection marker for improved recombinant protein productivity in CHO-DG44. Metab. Eng. 2007, 9, 304–316. [Google Scholar] [CrossRef]
- Mirkovitch, J.; Mirault, M.E.; Laemmli, U.K. Organization of the higher-order chromatin loop: Specific DNA attachment sites on nuclear scaffold. Cell 1984, 39, 223–232. [Google Scholar] [CrossRef]
- Jost, J.P.; Oakeley, E.J.; Zhu, B.; Benjamin, D.; Thiry, S.; Siegmann, M.; Jost, Y.C. 5-Methylcytosine DNA glycosylase participates in the genome-wide loss of DNA methylation occurring during mouse myoblast differentiation. Nucleic Acids Res. 2001, 29, 4452–4461. [Google Scholar] [CrossRef]
- Zhu, B.; Benjamin, D.; Zheng, Y.; Angliker, H.; Thiry, S.; Siegmann, M.; Jost, J.P. Overexpression of 5-methylcytosine DNA glycosylase in human embryonic kidney cells EcR293 demethylates the promoter of a hormone-regulated reporter gene. Proc. Natl. Acad. Sci. USA 2001, 98, 5031–5036. [Google Scholar]
- Girod, P.A.; Nguyen, D.Q.; Calabrese, D.; Puttini, S.; Grandjean, M.; Martinet, D.; Regamey, A.; Saugy, D.; Beckmann, J.S.; Bucher, P.; et al. Genome-wide prediction of matrix attachment regions that increase gene expression in mammalian cells. Nat. Methods 2007, 4, 747–753. [Google Scholar] [CrossRef]
- Bell, A.C.; West, A.G.; Felsenfeld, G. The protein CTCF is required for the enhancer blocking activity of vertebrate insulators. Cell 1999, 98, 387–396. [Google Scholar] [CrossRef]
- Bidwell, J.P.; Torrungruang, K.; Alvarez, M.; Rhodes, S.J.; Shah, R.; Jones, D.R.; Charoonpatrapong, K.; Hock, J.M.; Watt, A.J. Involvement of the nuclear matrix in the control of skeletal genes: The NMP1 (YY1), NMP2 (Cbfa1), and NMP4 (Nmp4/CIZ) transcription factors. Crit. Rev. Eukaryot. Gene Expr. 2001, 11, 279–297. [Google Scholar]
- Girod, P.A.; Zahn-Zabal, M.; Mermod, N. Use of the chicken lysozyme 5' matrix attachment region to generate high producer CHO cell lines. Biotechnol. Bioeng. 2005, 91, 1–11. [Google Scholar] [CrossRef]
- Kim, J.M.; Kim, J.S.; Park, D.H.; Kang, H.S.; Yoon, J.; Baek, K.; Yoon, Y. Improved recombinant gene expression in CHO cells using matrix attachment regions. J. Biotechnol. 2004, 107, 95–105. [Google Scholar]
- Kim, J.D.; Yoon, Y.; Hwang, H.Y.; Park, J.S.; Yu, S.; Lee, J.; Baek, K.; Yoon, J. Efficient selection of stable chinese hamster ovary (CHO) cell lines for expression of recombinant proteins by using human interferon beta SAR element. Biotechnol. Prog. 2005, 21, 933–937. [Google Scholar]
- Zahn-Zabal, M.; Kobr, M.; Girod, P.A.; Imhof, M.; Chatellard, P.; de Jesus, M.; Wurm, F.; Mermod, N. Development of stable cell lines for production or regulated expression using matrix attachment regions. J. Biotechnol. 2001, 87, 29–42. [Google Scholar]
- Benton, T.; Chen, T.; McEntee, M.; Fox, B.; King, D.; Crombie, R.; Thomas, T.C.; Bebbington, C. The use of UCOE vectors in combination with a preadapted serum free, suspension cell line allows for rapid production of large quantities of protein. Cytotechnology 2002, 38, 43–46. [Google Scholar]
- De Poorter, J.J.; Lipinski, K.S.; Nelissen, R.G.; Huizinga, T.W.; Hoeben, R.C. Optimization of short-term transgene expression by sodium butyrate and ubiquitous chromatin opening elements (UCOEs). J. Gene Med. 2007, 9, 639–648. [Google Scholar] [CrossRef]
- Ye, J.; Alvin, K.; Latif, H.; Hsu, A.; Parikh, V.; Whitmer, T.; Tellers, M.; de la Cruz Edmonds, M.C.; Ly, J.; Salmon, P.; Markusen, J.F. Rapid protein production using CHO stable transfection pools. Biotechnol. Prog. 2010, 26, 1431–1437. [Google Scholar] [CrossRef]
- Jia, Q.; Wu, H.; Zhou, X.; Gao, J.; Zhao, W.; Aziz, J.; Wei, J.; Hou, L.; Wu, S.; Zhang, Y.; et al. A “GC-rich” method for mammalian gene expression: A dominant role of non-coding DNA GC content in regulation of mammalian gene expression. Sci. China Life Sci. 2010, 53, 94–100. [Google Scholar] [CrossRef]
- Cao, H.; Widlund, H.R.; Simonsson, T.; Kubista, M. TGGA repeats impair nucleosome formation. J. Mol. Biol. 1998, 281, 253–260. [Google Scholar] [CrossRef]
- Lowary, P.T.; Widom, J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol. 1998, 276, 19–42. [Google Scholar] [CrossRef]
- Levitsky, V.G. RECON: A program for prediction of nucleosome formation potential. Nucleic Acids Res. 2004, 32, W346–W349. [Google Scholar] [CrossRef]
- Huang, Y.; Li, Y.; Wang, Y.G.; Gu, X.; Wang, Y.; Shen, B.F. An efficient and targeted gene integration system for high-level antibody expression. J. Immunol. Methods 2007, 322, 28–39. [Google Scholar] [CrossRef]
- Little, P. Genetics. Small and perfectly formed. Nature 1993, 366, 204–205. [Google Scholar] [CrossRef]
- Branda, C.S.; Dymecki, S.M. Talking about a revolution: The impact of site-specific recombinases on genetic analyses in mice. Dev. Cell 2004, 6, 7–28. [Google Scholar] [CrossRef]
- Groth, A.C.; Fish, M.; Nusse, R.; Calos, M.P. Construction of transgenic Drosophila by using the site-specific integrase from phage phiC31. Genetics 2004, 166, 1775–1782. [Google Scholar] [CrossRef]
- Golic, M.M.; Rong, Y.S.; Petersen, R.B.; Lindquist, S.L.; Golic, K.G. FLP-mediated DNA mobilization to specific target sites in Drosophila chromosomes. Nucleic Acids Res. 1997, 25, 3665–3671. [Google Scholar] [CrossRef]
- Voziyanov, Y.; Pathania, S.; Jayaram, M. A general model for site-specific recombination by the integrase family recombinases. Nucleic Acids Res. 1999, 27, 930–941. [Google Scholar] [CrossRef]
- Wirth, D.; Gama-Norton, L.; Riemer, P.; Sandhu, U.; Schucht, R.; Hauser, H. Road to precision: Recombinase-based targeting technologies for genome engineering. Curr. Opin. Biotechnol. 2007, 18, 411–419. [Google Scholar] [CrossRef]
- O’Gorman, S.; Fox, D.T.; Wahl, G.M. Recombinase-mediated gene activation and site-specific integration in mammalian cells. Science 1991, 251, 1351–1355. [Google Scholar]
- Voziyanov, Y.; Konieczka, J.H.; Stewart, A.F.; Jayaram, M. Stepwise manipulation of DNA specificity in Flp recombinase: Progressively adapting Flp to individual and combinatorial mutations in its target site. J. Mol. Biol. 2003, 326, 65–76. [Google Scholar] [CrossRef]
- Kito, M.; Itami, S.; Fukano, Y.; Yamana, K.; Shibui, T. Construction of engineered CHO strains for high-level production of recombinant proteins. Appl. Microbiol. Biotechnol. 2002, 60, 442–448. [Google Scholar] [CrossRef]
- Kameyama, Y.; Kawabe, Y.; Ito, A.; Kamihira, M. An accumulative site-specific gene integration system using Cre recombinase-mediated cassette exchange. Biotechnol. Bioeng. 2010, 105, 1106–1114. [Google Scholar]
- Smith, M.C.; Thorpe, H.M. Diversity in the serine recombinases. Mol. Microbiol. 2002, 44, 299–307. [Google Scholar] [CrossRef]
- Russell, J.P.; Chang, D.W.; Tretiakova, A.; Padidam, M. Phage Bxb1 integrase mediates highly efficient site-specific recombination in mammalian cells. Biotechniques 2006, 40, 460, 462, 464. [Google Scholar]
- Campbell, M.; Corisdeo, S.; McGee, C.; Kraichely, D. Utilization of site-specific recombination for generating therapeutic protein producing cell lines. Mol. Biotechnol. 2010, 45, 199–202. [Google Scholar] [CrossRef]
- Kennard, M.L. Engineered mammalian chromosomes in cellular protein production: Future prospects. Methods Mol. Biol. 2011, 738, 217–238. [Google Scholar] [CrossRef]
- Kennard, M.L.; Goosney, D.L.; Monteith, D.; Zhang, L.; Moffat, M.; Fischer, D.; Mott, J. The generation of stable, high MAb expressing CHO cell lines based on the artificial chromosome expression (ACE) technology. Biotechnol. Bioeng. 2009, 104, 540–553. [Google Scholar] [CrossRef]
- Dejong, G.; Telenius, A.H.; Telenius, H.; Perez, C.F.; Drayer, J.I.; Hadlaczky, G. Mammalian artificial chromosome pilot production facility: Large-scale isolation of functional satellite DNA-based artificial chromosomes. Cytometry 1999, 35, 129–133. [Google Scholar] [CrossRef]
- Cost, G.J.; Freyvert, Y.; Vafiadis, A.; Santiago, Y.; Miller, J.C.; Rebar, E.; Collingwood, T.N.; Snowden, A.; Gregory, P.D. BAK and BAX deletion using zinc-finger nucleases yields apoptosis-resistant CHO cells. Biotechnol. Bioeng. 2010, 105, 330–340. [Google Scholar] [CrossRef]
- Hwang, S.O.; Lee, G.M. Effect of Akt overexpression on programmed cell death in antibody-producing Chinese hamster ovary cells. J. Biotechnol. 2009, 139, 89–94. [Google Scholar] [CrossRef]
- Dreesen, I.A.; Fussenegger, M. Ectopic expression of human mTOR increases viability, robustness, cell size, proliferation, and antibody production of chinese hamster ovary cells. Biotechnol. Bioeng. 2011, 108, 853–866. [Google Scholar] [CrossRef]
- Astley, K.; Al-Rubeai, M. The role of Bcl-2 and its combined effect with p21CIP1 in adaptation of CHO cells to suspension and protein-free culture. Appl. Microbiol. Biotechnol. 2008, 78, 391–399. [Google Scholar] [CrossRef]
- Zhou, M.; Crawford, Y.; Ng, D.; Tung, J.; Pynn, A.F.; Meier, A.; Yuk, I.H.; Vijayasankaran, N.; Leach, K.; Joly, J.; et al. Decreasing lactate level and increasing antibody production in Chinese Hamster Ovary cells (CHO) by reducing the expression of lactate dehydrogenase and pyruvate dehydrogenase kinases. J. Biotechnol. 2011, 153, 27–34. [Google Scholar]
- Peng, R.W.; Abellan, E.; Fussenegger, M. Differential effect of exocytic SNAREs on the production of recombinant proteins in mammalian cells. Biotechnol. Bioeng. 2011, 108, 611–620. [Google Scholar]
- Datta, P.; Linhardt, R.J.; Sharfstein, S.T. An 'omics approach towards CHO cell engineering. Biotechnol. Bioeng. 2013, 110, 1255–1271. [Google Scholar] [CrossRef]
- Mohan, C.; Kim, Y.G.; Koo, J.; Lee, G.M. Assessment of cell engineering strategies for improved therapeutic protein production in CHO cells. Biotechnol. J. 2008, 3, 624–630. [Google Scholar] [CrossRef]
- Becker, E.; Florin, L.; Pfizenmaier, K.; Kaufmann, H. Evaluation of a combinatorial cell engineering approach to overcome apoptotic effects in XBP-1(s) expressing cells. J. Biotechnol. 2010, 146, 198–206. [Google Scholar] [CrossRef]
- Shaffer, A.L.; Shapiro-Shelef, M.; Iwakoshi, N.N.; Lee, A.H.; Qian, S.B.; Zhao, H.; Yu, X.; Yang, L.; Tan, B.K.; Rosenwald, A.; et al. XBP1, downstream of Blimp-1, expands the secretory apparatus and other organelles, and increases protein synthesis in plasma cell differentiation. Immunity 2004, 21, 81–93. [Google Scholar] [CrossRef]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar]
- Mussolino, C.; Cathomen, T. TALE nucleases: Tailored genome engineering made easy. Curr. Opin. Biotechnol. 2012, 23, 644–650. [Google Scholar] [CrossRef]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Miller, J.C.; Holmes, M.C.; Wang, J.; Guschin, D.Y.; Lee, Y.L.; Rupniewski, I.; Beausejour, C.M.; Waite, A.J.; Wang, N.S.; Kim, K.A.; et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007, 25, 778–785. [Google Scholar] [CrossRef]
- Fan, L.; Kadura, I.; Krebs, L.E.; Hatfield, C.C.; Shaw, M.M.; Frye, C.C. Improving the efficiency of CHO cell line generation using glutamine synthetase gene knockout cells. Biotechnol. Bioeng. 2012, 109, 1007–1015. [Google Scholar] [CrossRef]
- Chevalier, B.S.; Stoddard, B.L. Homing endonucleases: Structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res. 2001, 29, 3757–3774. [Google Scholar] [CrossRef]
- Cabaniols, J.P.; Ouvry, C.; Lamamy, V.; Fery, I.; Craplet, M.L.; Moulharat, N.; Guenin, S.P.; Bedut, S.; Nosjean, O.; Ferry, G.; et al. Meganuclease-driven targeted integration in CHO-K1 cells for the fast generation of HTS-compatible cell-based assays. J. Biomol. Screen. 2010, 15, 956–967. [Google Scholar] [CrossRef]
- De Oliveira Dal’Molin, C.G.; Quek, L.E.; Palfreyman, R.W.; Brumbley, S.M.; Nielsen, L.K. AraGEM, a genome-scale reconstruction of the primary metabolic network in Arabidopsis. Plant Physiol. 2010, 152, 579–589. [Google Scholar] [CrossRef]
- Quek, L.E.; Nielsen, L.K. On the reconstruction of the Mus musculus genome-scale metabolic network model. Genome Inform. 2008, 21, 89–100. [Google Scholar] [CrossRef]
- Hammill, L.; Welles, J.; Carson, G.R. The gel microdrop secretion assay: Identification of a low productivity subpopulation arising during the production of human antibody in CHO cells. Cytotechnology 2000, 34, 27–37. [Google Scholar] [CrossRef]
- Underwood, P.A.; Bean, P.A. Hazards of the limiting-dilution method of cloning hybridomas. J. Immunol. Methods 1988, 107, 119–128. [Google Scholar] [CrossRef]
- Yang, G.; Withers, S.G. Ultrahigh-throughput FACS-based screening for directed enzyme evolution. Chembiochem 2009, 10, 2704–2715. [Google Scholar] [CrossRef]
- Black, C.B.; Duensing, T.D.; Trinkle, L.S.; Dunlay, R.T. Cell-based screening using high-throughput flow cytometry. Assay Drug Dev. Technol. 2011, 9, 13–20. [Google Scholar] [CrossRef]
- Meng, Y.G.; Liang, J.; Wong, W.L.; Chisholm, V. Green fluorescent protein as a second selectable marker for selection of high producing clones from transfected CHO cells. Gene 2000, 242, 201–207. [Google Scholar] [CrossRef]
- Yoshikawa, T.; Nakanishi, F.; Ogura, Y.; Oi, D.; Omasa, T.; Katakura, Y.; Kishimoto, M.; Suga, K.I. Flow cytometry: An improved method for the selection of highly productive gene-amplified CHO cells using flow cytometry. Biotechnol. Bioeng. 2001, 74, 435–442. [Google Scholar] [CrossRef]
- Atochina, O.; Mylvaganam, R.; Akselband, Y.; McGrath, P. Comparison of results using the gel microdrop cytokine secretion assay with ELISPOT and intracellular cytokine staining assay. Cytokine 2004, 27, 120–128. [Google Scholar] [CrossRef]
- Gray, F.; Kenney, J.S.; Dunne, J.F. Secretion capture and report web: Use of affinity derivatized agarose microdroplets for the selection of hybridoma cells. J. Immunol. Methods 1995, 182, 155–163. [Google Scholar] [CrossRef]
- Powell, K.T.; Weaver, J.C. Gel microdroplets and flow cytometry: Rapid determination of antibody secretion by individual cells within a cell population. Biotechnology (NY) 1990, 8, 333–337. [Google Scholar]
- Manz, R.; Assenmacher, M.; Pfluger, E.; Miltenyi, S.; Radbruch, A. Analysis and sorting of live cells according to secreted molecules, relocated to a cell-surface affinity matrix. Proc. Natl. Acad. Sci. USA 1995, 92, 1921–1925. [Google Scholar]
- Holmes, P.; Al-Rubeai, M. Improved cell line development by a high throughput affinity capture surface display technique to select for high secretors. J. Immunol. Methods 1999, 230, 141–147. [Google Scholar] [CrossRef]
- Brezinsky, S.C.; Chiang, G.G.; Szilvasi, A.; Mohan, S.; Shapiro, R.I.; MacLean, A.; Sisk, W.; Thill, G. A simple method for enriching populations of transfected CHO cells for cells of higher specific productivity. J. Immunol. Methods 2003, 277, 141–155. [Google Scholar] [CrossRef]
- Lee, C.; Ly, C.; Sauerwald, T.; Kelly, T.; Moore, G. High-throughput screening of cell lines expressing monoclonal antibodies. BioProcess Int. 2006, 4, 32–35. [Google Scholar]
- Dharshanan, S.; Chong, H.; Hung, C.S.; Zamrod, Z.; Kamal, N. Rapid automated selection of mammalian cell line secreting high level of humanized monoclonal antibody using Clone Pix FL system and the correlation between exterior median intensity and antibody productivity. Electron. J. Biotechnol. 2011, 14. [Google Scholar] [CrossRef]
- Serpieri, F.; Inocencio, A.; de Oliveira, J.M.; Pimenta, A.A., Jr.; Garbuio, A.; Kalil, J.; Brigido, M.M.; Moro, A.M. Comparison of humanized IgG and FvFc anti-CD3 monoclonal antibodies expressed in CHO cells. Mol. Biotechnol. 2010, 45, 218–225. [Google Scholar] [CrossRef]
- Olejniczak, E.T.; Ruan, Q.; Ziemann, R.N.; Birkenmeyer, L.G.; Saldana, S.C.; Tetin, S.Y. Rapid determination of antigenic epitopes in human NGAL using NMR. Biopolymers 2010, 93, 657–667. [Google Scholar] [CrossRef]
- Lobito, A.A.; Ramani, S.R.; Tom, I.; Bazan, J.F.; Luis, E.; Fairbrother, W.J.; Ouyang, W.; Gonzalez, L.C. Murine insulin growth factor-like (IGFL) and human IGFL1 proteins are induced in inflammatory skin conditions and bind to a novel tumor necrosis factor receptor family member, IGFLR1. J. Biol. Chem. 2011, 286, 18969–18981. [Google Scholar] [CrossRef]
- Hanania, E.G.; Fieck, A.; Stevens, J.; Bodzin, L.J.; Palsson, B.O.; Koller, M.R. Automated in situ measurement of cell-specific antibody secretion and laser-mediated purification for rapid cloning of highly-secreting producers. Biotechnol. Bioeng. 2005, 91, 872–876. [Google Scholar] [CrossRef]
- Koller, M.R.; Hanania, E.G.; Stevens, J.; Eisfeld, T.M.; Sasaki, G.C.; Fieck, A.; Palsson, B.O. High-throughput laser-mediated in situ cell purification with high purity and yield. Cytometry A 2004, 61, 153–161. [Google Scholar]
- Richardson, G.; Lin, N.; Lacy, K.; Davis, L.; Gray, M.; Cresswell, J.; Gerber, M.; Caple, M.; Kayser, K. Cell Xpress™ Technology Facilitates High-Producing Chinese Hamster Ovary Cell Line Generation Using Glutamine Synthetase Gene Expression System. In Cells and Culture; Noll, T., Ed.; Noll, T.: Berlin, Heidelberg, Germany, 2010; Volume 4, pp. 45–4. [Google Scholar]
- Yang, W.; Xia, W.; Mao, J.; Xu, D.; Chen, J.; Feng, S.; Wang, J.; Li, H.; Theisen, C.F.; Petersen, J.M.; et al. High level expression, purification and activation of human dipeptidyl peptidase I from mammalian cells. Protein Expr. Purif. 2011, 76, 59–64. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, X.; Yang, H.; Yu, X.; Li, Y.; Xing, J.; Chen, Z. New strategy for large-scale preparation of the extracellular domain of tumor-associated antigen HAb18G/CD147 (HAb18GED). J. Biosci. Bioeng. 2011, 111, 1–6. [Google Scholar] [CrossRef]
- Xu, X.; Nagarajan, H.; Lewis, N.E.; Pan, S.; Cai, Z.; Liu, X.; Chen, W.; Xie, M.; Wang, W.; Hammond, S.; et al. The genomic sequence of the Chinese hamster ovary (CHO)-K1 cell line. Nat. Biotechnol. 2011, 29, 735–741. [Google Scholar] [CrossRef]
- Melville, M.; Doolan, P.; Mounts, W.; Barron, N.; Hann, L.; Leonard, M.; Clynes, M.; Charlebois, T. Development and characterization of a Chinese hamster ovary cell-specific oligonucleotide microarray. Biotechnol. Lett. 2011, 33, 1773–1779. [Google Scholar] [CrossRef]
- Wlaschin, K.F.; Nissom, P.M.; Gatti Mde, L.; Ong, P.F.; Arleen, S.; Tan, K.S.; Rink, A.; Cham, B.; Wong, K.; Yap, M.; et al. EST sequencing for gene discovery in Chinese hamster ovary cells. Biotechnol. Bioeng. 2005, 91, 592–606. [Google Scholar] [CrossRef]
- Baik, J.Y.; Lee, M.S.; An, S.R.; Yoon, S.K.; Joo, E.J.; Kim, Y.H.; Park, H.W.; Lee, G.M. Initial transcriptome and proteome analyses of low culture temperature-induced expression in CHO cells producing erythropoietin. Biotechnol. Bioeng. 2006, 93, 361–371. [Google Scholar]
- De Leon Gatti, M.; Wlaschin, K.F.; Nissom, P.M.; Yap, M.; Hu, W.S. Comparative transcriptional analysis of mouse hybridoma and recombinant Chinese hamster ovary cells undergoing butyrate treatment. J. Biosci. Bioeng. 2007, 103, 82–91. [Google Scholar] [CrossRef]
- Baycin-Hizal, D.; Tabb, D.L.; Chaerkady, R.; Chen, L.; Lewis, N.E.; Nagarajan, H.; Sarkaria, V.; Kumar, A.; Wolozny, D.; Colao, J.; et al. Proteomic analysis of Chinese hamster ovary cells. J. Proteome Res. 2012, 11, 5265–5276. [Google Scholar] [CrossRef]
- Selvarasu, S.; Ho, Y.S.; Chong, W.P.; Wong, N.S.; Yusufi, F.N.; Lee, Y.Y.; Yap, M.G.; Lee, D.Y. Combined in silico modeling and metabolomics analysis to characterize fed-batch CHO cell culture. Biotechnol. Bioeng. 2012, 109, 1415–1429. [Google Scholar] [CrossRef]
- Chong, W.P.; Goh, L.T.; Reddy, S.G.; Yusufi, F.N.; Lee, D.Y.; Wong, N.S.; Heng, C.K.; Yap, M.G.; Ho, Y.S. Metabolomics profiling of extracellular metabolites in recombinant Chinese Hamster Ovary fed-batch culture. Rapid Commun. Mass Spectrom. 2009, 23, 3763–3771. [Google Scholar] [CrossRef]
- Dietmair, S.; Hodson, M.P.; Quek, L.E.; Timmins, N.E.; Chrysanthopoulos, P.; Jacob, S.S.; Gray, P.; Nielsen, L.K. Metabolite profiling of CHO cells with different growth characteristics. Biotechnol. Bioeng. 2012, 109, 1404–1414. [Google Scholar] [CrossRef]
- Hackl, M.; Jadhav, V.; Jakobi, T.; Rupp, O.; Brinkrolf, K.; Goesmann, A.; Puhler, A.; Noll, T.; Borth, N.; Grillari, J. Computational identification of microRNA gene loci and precursor microRNA sequences in CHO cell lines. J. Biotechnol. 2012, 158, 151–155. [Google Scholar]
- Hammond, S.; Swanberg, J.C.; Polson, S.W.; Lee, K.H. Profiling conserved microRNA expression in recombinant CHO cell lines using Illumina sequencing. Biotechnol. Bioeng. 2012, 109, 1371–1375. [Google Scholar] [CrossRef]
- Barron, N.; Kumar, N.; Sanchez, N.; Doolan, P.; Clarke, C.; Meleady, P.; O’Sullivan, F.; Clynes, M. Engineering CHO cell growth and recombinant protein productivity by overexpression of miR-7. J. Biotechnol. 2011, 151, 204–211. [Google Scholar]
- Jadhav, V.; Hackl, M.; Bort, J.A.; Wieser, M.; Harreither, E.; Kunert, R.; Borth, N.; Grillari, J. A screening method to assess biological effects of microRNA overexpression in Chinese hamster ovary cells. Biotechnol. Bioeng. 2012, 109, 1376–1385. [Google Scholar] [CrossRef]
- Hackl, M.; Jakobi, T.; Blom, J.; Doppmeier, D.; Brinkrolf, K.; Szczepanowski, R.; Bernhart, S.H.; Honer Zu Siederdissen, C.; Bort, J.A.; Wieser, M.; et al. Next-generation sequencing of the Chinese hamster ovary microRNA transcriptome: Identification, annotation and profiling of microRNAs as targets for cellular engineering. J. Biotechnol. 2011, 153, 62–75. [Google Scholar]
- Muller, D.; Katinger, H.; Grillari, J. MicroRNAs as targets for engineering of CHO cell factories. Trends Biotechnol. 2008, 26, 359–365. [Google Scholar] [CrossRef]
- Umana, P.; Jean-Mairet, J.; Moudry, R.; Amstutz, H.; Bailey, J.E. Engineered glycoforms of an antineuroblastoma IgG1 with optimized antibody-dependent cellular cytotoxic activity. Nat. Biotechnol. 1999, 17, 176–180. [Google Scholar]
- Baik, J.Y.; Gasimli, L.; Yang, B.; Datta, P.; Zhang, F.; Glass, C.A.; Esko, J.D.; Linhardt, R.J.; Sharfstein, S.T. Metabolic engineering of Chinese hamster ovary cells: Towards a bioengineered heparin. Metab. Eng. 2012, 14, 81–90. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lai, T.; Yang, Y.; Ng, S.K. Advances in Mammalian Cell Line Development Technologies for Recombinant Protein Production. Pharmaceuticals 2013, 6, 579-603. https://doi.org/10.3390/ph6050579
Lai T, Yang Y, Ng SK. Advances in Mammalian Cell Line Development Technologies for Recombinant Protein Production. Pharmaceuticals. 2013; 6(5):579-603. https://doi.org/10.3390/ph6050579
Chicago/Turabian StyleLai, Tingfeng, Yuansheng Yang, and Say Kong Ng. 2013. "Advances in Mammalian Cell Line Development Technologies for Recombinant Protein Production" Pharmaceuticals 6, no. 5: 579-603. https://doi.org/10.3390/ph6050579