NMDA Receptor Modulators in the Treatment of Drug Addiction

{kind=link}
{kind=link}
{kind=link}
Abstract
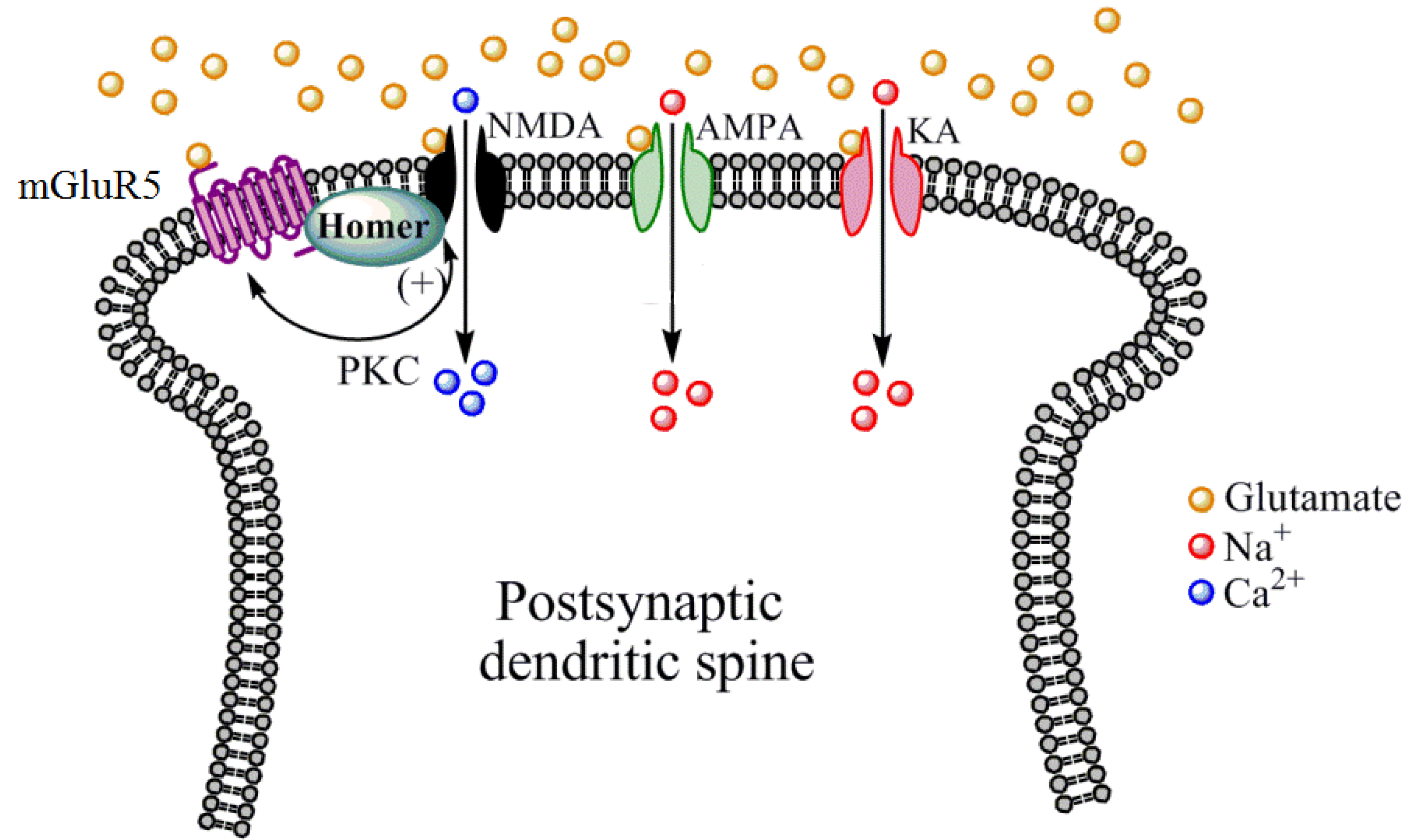
:1. Introduction
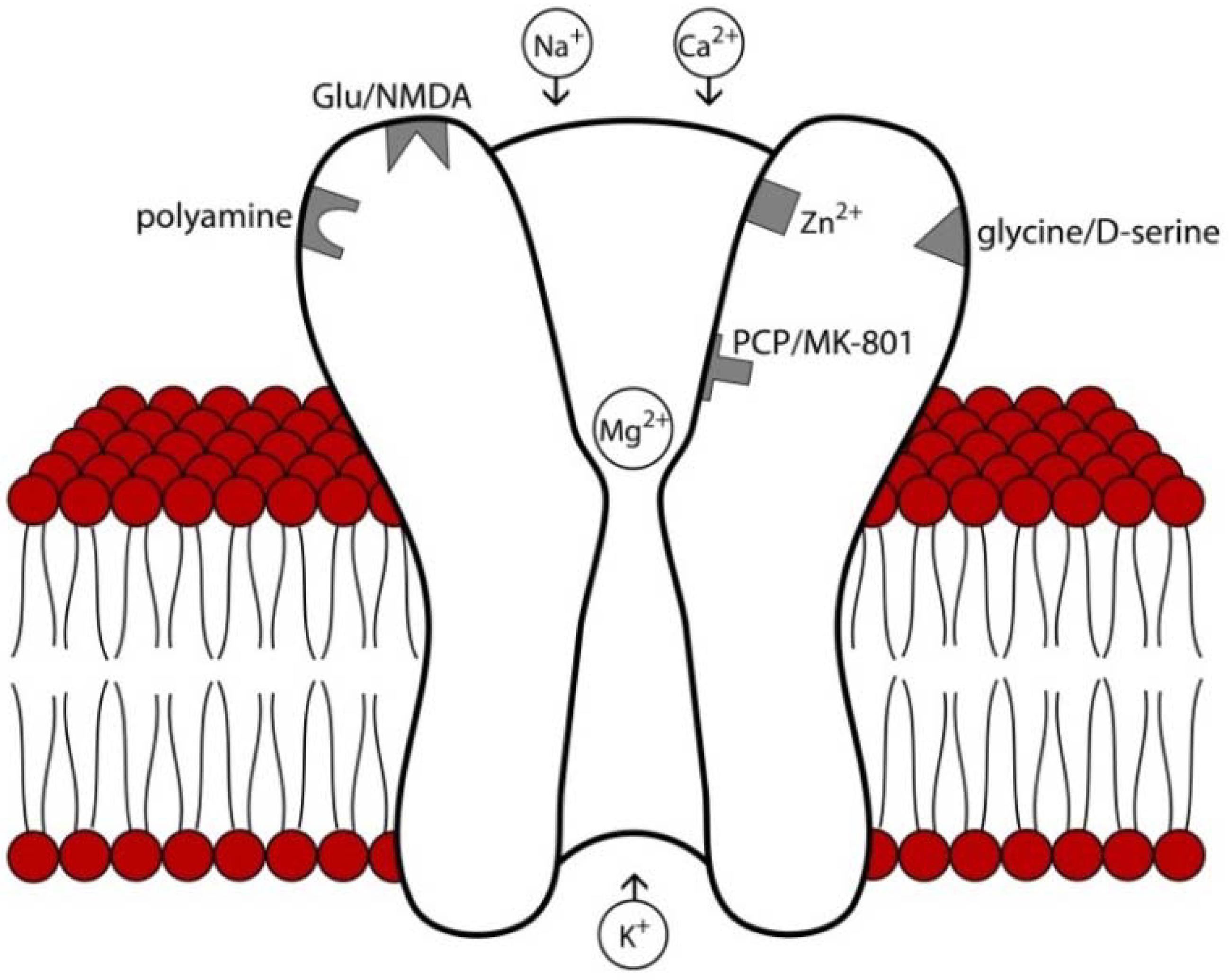
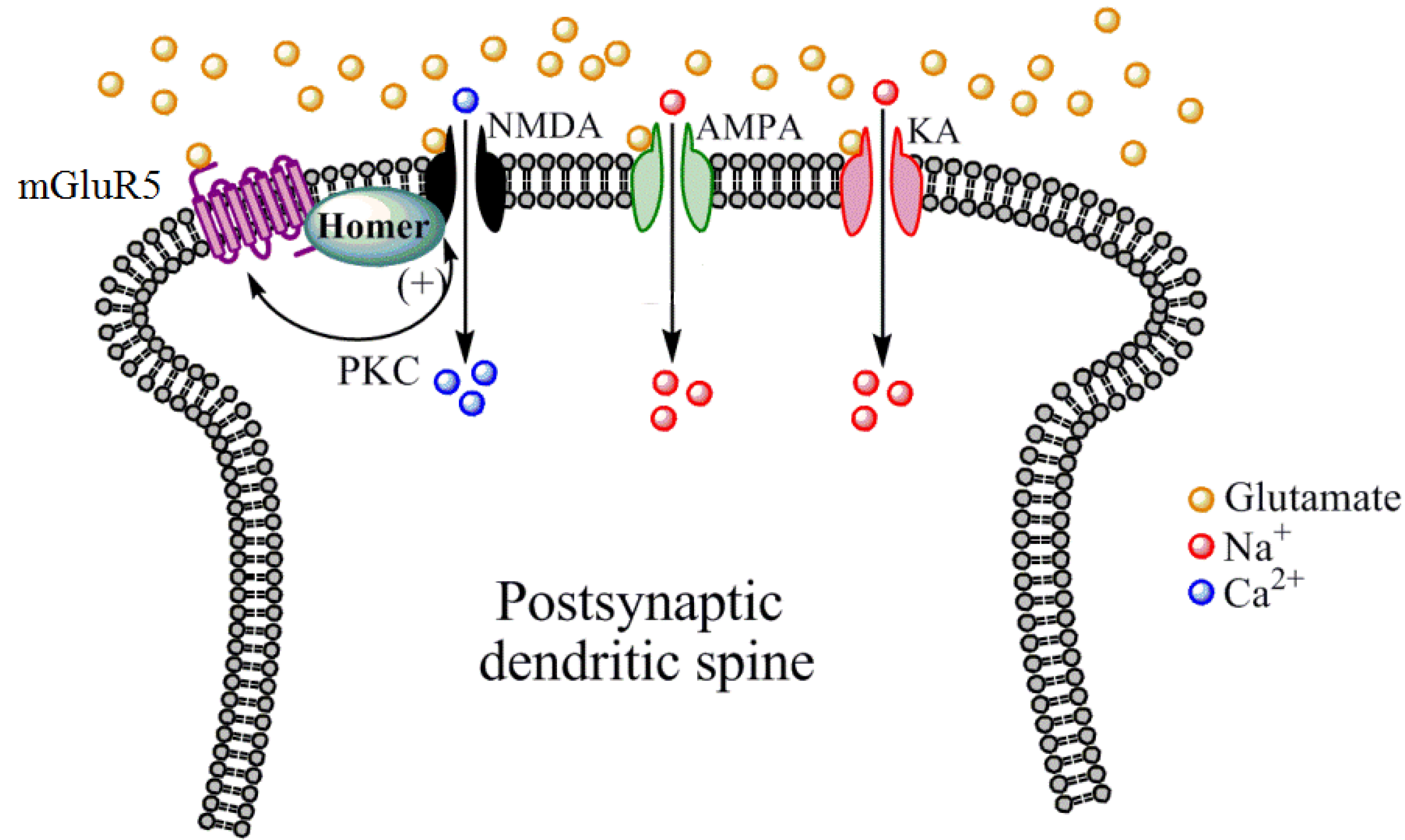
2. NMDA Receptor Structure, Expression Patterns, and Pharmacology
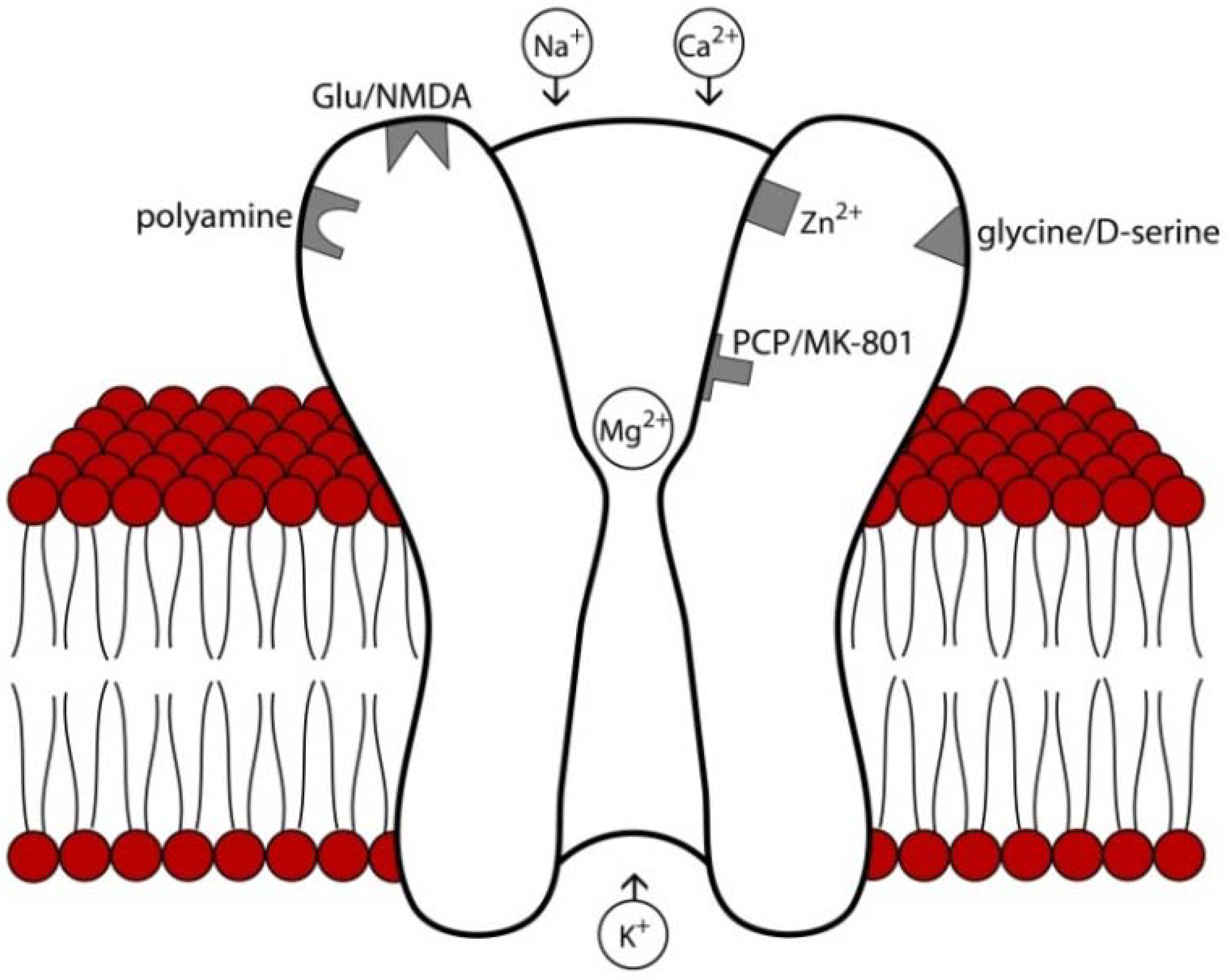
3. NMDA Receptor Modulators
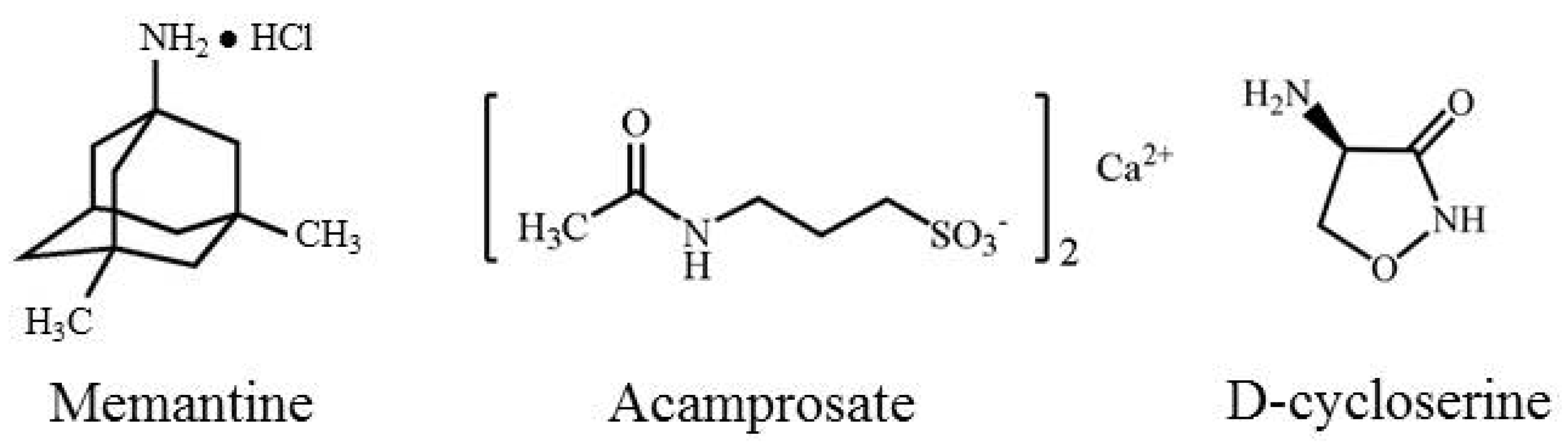
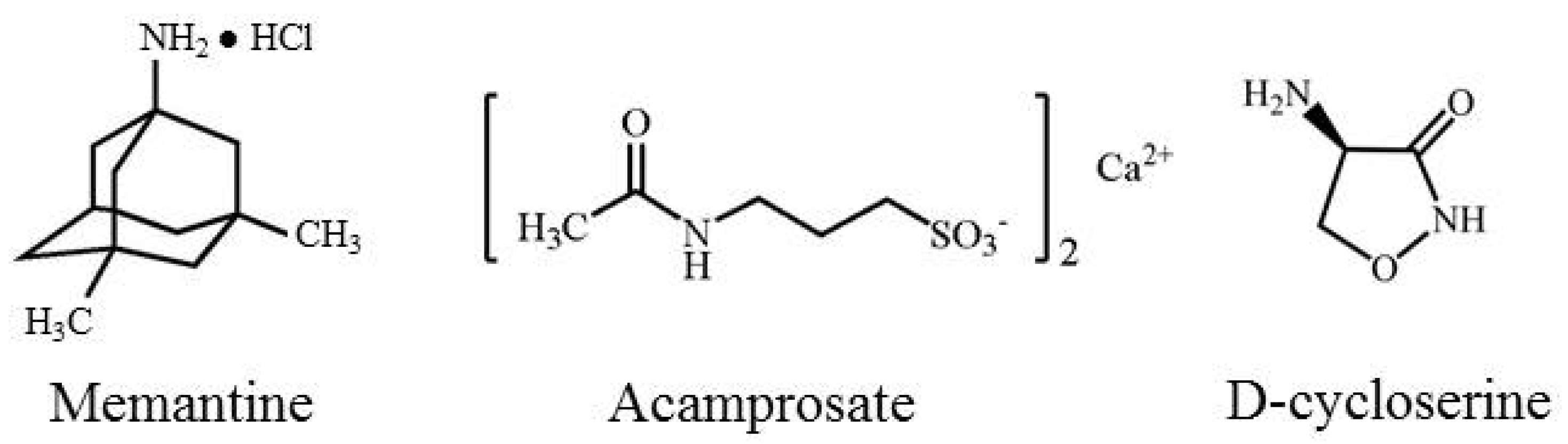
3.1. Memantine
3.1.1. Mechanism of Action
3.1.2. Preclinical Findings
3.1.3. Clinical Efficacy
3.1.4. Adverse Side Effects
3.2. Acamprosate
3.2.1. Mechanism of Action
3.2.2. Preclinical Findings
3.2.3. Clinical Efficacy
3.2.4. Adverse Side Effects
3.3. d-Cycloserine (DCS)
3.3.1. Mechanism of Action
3.3.2. Preclinical Findings
3.3.3. Clinical Efficacy
3.3.4. Adverse Side Effects
4. Conclusions
Acknowledgments
Conflict of Interest
References
- United Nations Office on Drugs and Crime, World Drug Report; United Nations Pulication: Vienna, Austria, 2009.
- Results from the 2009 National Survey on Drug Use and Health, Volume 1. Summary of National Findings; U.S. Department of Health and Human Services, Substance Abuse and Mental Health Services Administration, Center for Mental Health Services: Rockville, MD, USA, 2010.
- Kalivas, P.W.; Volkow, N.D. New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol. Psychiatry 2011, 16, 974–986. [Google Scholar] [CrossRef]
- American Psychiatric Association, Diagnostic and Statistical Manual of Mental Disorders, 4th ed; American Psychiatric Press: Washington DC, USA, 2002.
- Koob, G.F.; Volkow, N.D. Neurocircuitry of addiction. Neuropsychopharmacology 2010, 35, 217–238. [Google Scholar] [CrossRef]
- Grant, B.F.; Dawson, D.A. Age of onset of drug use and its association with DSM-IV drug abuse and dependence: results from the National Longitudinal Alcohol Epidemiologic Survey. J. Subst. Abuse 1998, 10, 163–173. [Google Scholar] [CrossRef]
- Grant, B.F.; Dawson, D.A.; Stinson, F.S.; Chou, S.P.; Dufour, M.C.; Pickering, R.P. The 12-month prevalence and trends in DSM-IV alcohol abuse and dependence: United States, 1991–1992 and 2001–2002. Drug Alcohol Depend. 2004, 74, 223–234. [Google Scholar] [CrossRef]
- Nestler, E.J. Molecular neurobiology of addiction. Am. J. Addict. 2001, 10, 201–217. [Google Scholar] [CrossRef]
- Olive, M.F.; Cleva, R.M.; Kalivas, P.W.; Malcolm, R.J. Glutamatergic medications for the treatment of drug and behavioral addictions. Pharmacol. Biochem. Behav. 2012, 100, 801–810. [Google Scholar] [CrossRef]
- Grant, J.E.; Potenza, M.N.; Weinstein, A.; Gorelick, D.A. Introduction to behavioral addictions. Am. J. Drug Alcohol Abuse 2010, 36, 233–241. [Google Scholar] [CrossRef]
- Sussman, S.; Lisha, N.; Griffiths, M. Prevalence of the addictions: a problem of the majority or the minority? Eval. Health Prof. 2011, 34, 3–56. [Google Scholar] [CrossRef]
- Lee, H.W.; Choi, J.S.; Shin, Y.C.; Lee, J.Y.; Jung, H.Y.; Kwon, J.S. Impulsivity in internet addiction: a comparison with pathological gambling. Cyberpsychol. Behav. Soc. Netw 2012, 15, 373–377. [Google Scholar] [CrossRef]
- Gass, J.T.; Olive, M.F. Glutamatergic substrates of drug addiction and alcoholism. Biochem. Pharmacol. 2008, 75, 218–265. [Google Scholar] [CrossRef]
- Nemirovsky, N.E.; Olive, M.F. Medications for the treatment of cocaine addiction: focus on glutamatergic compounds. In Cocaine Abuse: Pharmacology, Treatment and Relapse Prevention; Fang, X.C., Yue, L., Eds.; Nova Biomedical: Hauppauge, NY, USA, 2012; pp. 115–129. [Google Scholar]
- Kalivas, P.W. The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci. 2009, 10, 561–572. [Google Scholar] [CrossRef]
- Myers, K.M.; Carlezon, W.A., Jr.; Davis, M. Glutamate receptors in extinction and extinction-based therapies for psychiatric illness. Neuropsychopharmacology 2011, 36, 274–293. [Google Scholar] [CrossRef]
- Seeburg, P.H. The molecular biology of mammalian glutamate receptor channels. Trends Neurosci. 1993, 16, 359–365. [Google Scholar] [CrossRef]
- Nicoll, R.A.; Malenka, R.C. Expression mechanisms underlying NMDA receptor-dependent long-term potentiation. Ann. N. Y. Acad. Sci. 1999, 868, 515–525. [Google Scholar] [CrossRef]
- Tang, Y.P.; Shimizu, E.; Dube, G.R.; Rampon, C.; Kerchner, G.A.; Zhuo, M.; Liu, G.; Tsien, J.Z. Genetic enhancement of learning and memory in mice. Nature 1999, 401, 63–69. [Google Scholar]
- Lau, C.G.; Zukin, R.S. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat. Rev. Neurosci. 2007, 8, 413–426. [Google Scholar]
- Albensi, B.C. The NMDA receptor/ion channel complex: A drug target for modulating synaptic plasticity and excitotoxicity. Curr. Pharm. Des. 2007, 13, 3185–3194. [Google Scholar] [CrossRef]
- Hardingham, G.E.; Bading, H. The Yin and Yang of NMDA receptor signalling. Trends Neurosci. 2003, 26, 81–89. [Google Scholar] [CrossRef]
- Haberny, K.A.; Paule, M.G.; Scallet, A.C.; Sistare, F.D.; Lester, D.S.; Hanig, J.P.; Slikker, W.J. Ontogeny of the N-methyl-D-aspartate (NMDA) receptor system and susceptibility to neurotoxicity. Toxicol. Sci. 2002, 68, 9–17. [Google Scholar] [CrossRef]
- Smith, P.F. Therapeutic N-methyl-D-aspartate receptor antagonists: will reality meet expectation? Curr. Opin. Investig. Drugs 2003, 4, 826–832. [Google Scholar]
- Kew, J.N.C. Positive and negative allosteric modulation of metabotropic glutamate receptors: emerging therapeutic potential. Pharmacol. Ther. 2004, 104, 233–244. [Google Scholar] [CrossRef]
- Chen, H.S.; Lipton, S.A. The chemical biology of clincially tolerated NMDA receptor antagonists. J. Neurochem. 2006, 97, 1611–1626. [Google Scholar] [CrossRef]
- Waxman, E.A.; Lynch, D.R. N-methyl-D-aspartate receptor subtypes: multiple roles in excitotoxicity and neurological disease. Neuroscientist 2005, 11, 37–49. [Google Scholar] [CrossRef]
- Dingledine, R.; Borges, K.; Bowie, D.; Traynelis, S.F. The glutamate receptor ion channels. Pharmacol. Rev. 1999, 51, 7–62. [Google Scholar]
- Law, A.J.; Weickert, C.S.; Webster, M.J.; Herman, M.M.; Kleinman, J.E.; Harrison, P.J. Expression of NMDA receptor NR1, NR2A and NR2B subunit mRNAs during development of the human hippocampal formation. Eur. J. Neurosci. 2003, 18, 1197–1205. [Google Scholar]
- Curran, H.V.; Monaghan, L. In and out of the K-hole: a comparison of the acute and residual effects of ketamine in frequent and infrequent ketamine users. Addiction 2001, 96, 749–760. [Google Scholar] [CrossRef]
- Kotermanski, S.E.; Wood, J.T.; Johnson, J.W. Memantine binding to a superficial site on NMDA receptors contributes to partial trapping. J. Physiol. 2009, 587, 4589–4604. [Google Scholar] [CrossRef]
- Naassila, M.; Hammoumi, S.; Legrand, E.; Durbin, P.; Daoust, M. Mechanism of action of acamprosate. Part I. Characterization of spermidine-sensitive acamprosate binding site in rat brain. Alcohol. Clin. Exp. Res. 1998, 22, 802–809. [Google Scholar] [CrossRef]
- Hood, W.F.; Compton, R.P.; Monahan, J.B. D-Cycloserine: a ligand for the N-methyl-D-aspartate coupled glycine receptor has partial agonist characteristics. Neurosci. Lett. 1989, 98, 91–95. [Google Scholar] [CrossRef]
- Nestler, E.J.; Aghajanian, G.K. Molecular and cellular basis of addiction. Science 1997, 278, 58–63. [Google Scholar] [CrossRef]
- Tzschentke, T.M.; Schmidt, W.J. N-methyl-D-aspartic acid-receptor antagonists block morphine-induced conditioned place preference in rats. Neurosci. Lett. 1995, 193, 37–40. [Google Scholar] [CrossRef]
- Schenk, S.; Valadez, A.; Worley, C.M.; McNamara, C. Blockade of the acquisition of cocaine self-administration by the NMDA antagonist MK-801 (dizocilpine). Behav. Pharmacol. 1993, 4, 652–659. [Google Scholar]
- Zdanys, K.; Tampi, R.R. A systematic review of off-label uses of memantine for psychiatric disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1362–1374. [Google Scholar] [CrossRef]
- Robinson, D.M.; Keating, G.M. Memantine: A review of its use in alzheimerʼs disease. Drugs 2006, 66, 1515–1534. [Google Scholar] [CrossRef]
- Semenova, S.; Danysz, W.; Bespalov, A. Low-affinity NMDA receptor channel blockers inhibit acquisition of intravenous morphine self-administration in naive mice. Eur. J. Pharmacol. 1999, 378, 1–8. [Google Scholar] [CrossRef]
- Hyytia, P.; Backstrom, P.; Liljequist, S. Site-specific NMDA receptor antagonists produce differential effects on cocaine self-administration in rats. Eur. J. Pharmacol. 1999, 378, 9–16. [Google Scholar] [CrossRef]
- Blokhina, E.A.; Kashkin, V.A.; Zvartau, E.E.; Danysz, W.; Bespalov, A.Y. Effects of nicotinic and NMDA receptor channel blockers on intravenous cocaine and nicotine self-administration in mice. Eur. Neuropsychopharmacol. 2005, 15, 219–225. [Google Scholar] [CrossRef]
- Popik, P.; Danysz, W. Inhibition of reinforcing effects of morphine and motivational aspects of naloxone-precipitated opioid withdrawal by N-methyl-D-aspartate receptor antagonist, memantine. J. Pharmacol. Exp. Ther. 1997, 280, 854–865. [Google Scholar]
- Kotlinska, J.; Biala, G. Memantine and ACPC affect conditioned place preference induced by cocaine in rats. Pol. J. Pharmacol. 2000, 52, 179–185. [Google Scholar]
- Popik, P.; Wrobel, M.; Rygula, R.; Bisaga, A.; Bespalov, A.Y. Effects of memantine, an NMDA receptor antagonist, on place preference conditioned with drug and nondrug reinforcers in mice. Behav. Pharmacol. 2003, 14, 237–244. [Google Scholar] [CrossRef]
- Ribeiro Do Couto, B.; Aguilar, M.A.; Manzanedo, C.; Rodriguez-Arias, M.; Minarro, J. Effects of NMDA receptor antagonists (MK-801 and memantine) on the acquisition of morphine-induced conditioned place preference in mice. Prog. Neuropsychopharmacol. Biol. Psychiatry 2004, 28, 1035–1043. [Google Scholar] [CrossRef]
- Ribeiro Do Couto, B.; Aguilar, M.A.; Manzanedo, C.; Rodriguez-Arias, M.; Minarro, J. NMDA glutamate but not dopamine antagonists blocks drug-induced reinstatement of morphine place preference. Brain Res. Bull. 2005, 64, 493–503. [Google Scholar] [CrossRef]
- Popik, P.; Wrobel, M.; Bisaga, A. Reinstatement of morphine-conditioned reward is blocked by memantine. Neuropsychopharmacology 2006, 31, 160–170. [Google Scholar]
- Maldonado, C.; Rodriguez-Arias, M.; Castillo, A.; Aguilar, M.A.; Minarro, J. Effect of memantine and CNQX in the acquisition, expression and reinstatement of cocaine-induced conditioned place preference. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 932–939. [Google Scholar]
- Aguilar, M.A.; Manzanedo, C.; Ribiero Do Couto, B.; Rodriguez-Arias, M.; Minarro, J. Memantine blocks sensitization to the rewarding effects of morphine. Brain Res. 2009, 1288, 95–104. [Google Scholar] [CrossRef]
- Bespalov, A.Y.; Zvartau, E.E.; Balster, R.L.; Beardsley, P.M. Effects of N-methyl-D-aspartate receptor antagonists on reinstatement of cocaine-seeking behavior by priming injections of cocaine or exposures to cocaine-associated cues in rats. Behav. Pharmacol. 2000, 11, 37–44. [Google Scholar] [CrossRef]
- Bisaga, A.; Evans, S.M. Acute effects of memantine in combination with alcohol in moderate drinkers. Psychopharmacology 2004, 16–24. [Google Scholar] [CrossRef]
- Krupitsky, E.M.; Neznanova, O.; Masalov, D.; Burakov, A.M.; Didenko, T.; Romanova, T.; Tsoy, M.; Bespalov, A.; Slavina, T.; Grinenko, A.A.; et al. Effect of memantine on cue-induced alcohol craving in recovering alcohol-dependent patients. Psychiatry 2007, 519–523. [Google Scholar]
- Arias, A.J.; Feinn, R.; Covault, J.; Kranzler, H.R. Memantine for alcohol dependence: an open-label pilot study. Addict. Disord. Treat. 2007, 77–83. [Google Scholar]
- Evans, S.M.; Levin, F.R.; Brooks, D.J.; Garawi, F. A pilot double-blind treatment trial of memantine for alcohol dependence. Alcohol. Clin. Exp. Res. 2007, 31, 775–782. [Google Scholar] [CrossRef]
- Hart, C.L.; Haney, M.; Foltin, R.W.; Fischman, M.W. Effects of the NMDA antagonist memantine on human methamphetamine discrimination. Psychopharmacology 2002, 164, 376–384. [Google Scholar] [CrossRef]
- Kiefer, F.; Mann, K. Acamprosate: how, where, and for whom does it work? Mechanism of action, treatment targets, and individualized therapy. Curr. Pharm. Des. 2010, 16, 2098–2102. [Google Scholar] [CrossRef]
- Boismare, F.; Daoust, M.; Moore, N.; Saligaut, C.; Lhuintre, J.P.; Chretien, P.; Durlach, J. A homotaurine derivative reduces the voluntary intake of ethanol by rats: are cerebral GABA receptors involved? Pharmacol. Biochem. Behav. 1984, 21, 787–789. [Google Scholar] [CrossRef]
- Madamba, S.G.; Schweitzer, P.; Zieglgänsberger, W.; Siggins, G.R. Acamprosate (calcium acetylhomotaurinate) enhances the N-methyl-D-aspartate component of excitatory neurotransmission in rat hippocampal CA1 neurons in vitro. Alcohol. Clin. Exp. Res. 1996, 20, 651–658. [Google Scholar] [CrossRef]
- Berton, F.; Francesconi, W.G.; Madamba, S.G.; Zieglgänsberger, W.; Siggins, G.R. Acamprosate enhances N-methyl-D-apartate receptor-mediated neurotransmission but inhibits presynaptic GABAB receptors in nucleus accumbens neurons. Alcohol. Clin. Exp. Res. 1998, 22, 183–191. [Google Scholar] [CrossRef]
- Zeise, M.L.; Kasparov, S.; Capogna, M.; Zieglgänsberger, W. Acamprosate (calciumacetylhomotaurinate) decreases postsynaptic potentials in the rat neocortex: possible involvement of excitatory amino acid receptors. Eur. J. Pharmacol. 1993, 231, 47–52. [Google Scholar] [CrossRef]
- Zeise, M.L.; Kasparow, S.; Capogna, M.; Zieglgänsberger, W. Calcium diacetylhomotaurinate (CA-AOTA) decreases the action of excitatory amino acids in the rat neocortex in vitro. Prog. Clin. Biol. Res. 1990, 351, 237–242. [Google Scholar]
- Rammes, G.; Mahal, B.; Putzke, J.; Parsons, C.; Spielmanns, P.; Pestel, E.; Spanagel, R.; Zieglgansberger, W.; Schadrack, J. The anti-craving compound acamprosate acts as a weak NMDA-receptor antagonist, but modulates NMDA-receptor subunit expression similar to memantine and MK-801. Neuropharmacology 2001, 40, 749–760. [Google Scholar] [CrossRef]
- Allgaier, C.; Franke, H.; Sobottka, H.; Scheibler, P. Acamprosate inhibits Ca2+ influx mediated by NMDA receptors and voltage-sensitive Ca2+ channels in cultured rat mesencephalic neurones. Naunyn-Schmied. Arch. Pharmacol. 2000, 362, 440–443. [Google Scholar] [CrossRef]
- Popp, R.L.; Lovinger, D.M. Interaction of acamprosate with ethanol and spermine on NMDA receptors in primary cultured neurons. Eur. J. Pharmacol. 2000, 394, 221–231. [Google Scholar] [CrossRef]
- De Witte, P.; Littleton, J.; Parot, P.; Koob, G. Neuroprotective and abstinence-promoting effects of acamprosate : elucidating the mechanism of action. CNS Drugs 2005, 19, 517–537. [Google Scholar] [CrossRef]
- Al Qatari, M.; Bouchenafa, O.; Littleton, J. Mechanism of action of acamprosate. Part II. Ethanol dependence modifies effects of acamprosate on NMDA receptor binding in membranes from rat cerebral cortex. Alcohol. Clin. Exp. Res. 1998, 22, 810–814. [Google Scholar] [CrossRef]
- Lhuintre, J.P.; Daoust, M.; Moore, N.D.; Chretien, P.; Saligaut, C.; Tran, G.; Bosimare, F.; Hillemand, B. Ability of calcium bis acetyl homotaurine, a GABA agonist, to prevent relapse in weaned alcoholics. Lancet 1985, 1, 1014–1016. [Google Scholar]
- Mann, K.; Kiefer, F.; Spanagel, R.; Littleton, J. Acamprosate: recent findings and future research directions. Alcohol. Clin. Exp. Res. 2008, 32, 1105–1110. [Google Scholar] [CrossRef]
- Mcgeehan, A.J.; Olive, M.F. The anti-relapse compound acamprosate inhibits the development of a conditioned place preference to ethanol and cocaine but not morphine. Br. J. Pharmacol. 2003, 138, 9–12. [Google Scholar] [CrossRef]
- Mcgeehan, A.J.; Olive, M.F. Attenuation of cocaine-induced reinstatement of cocaine conditioned place preference by acamprosate. Behav. Pharmacol. 2006, 17, 363–367. [Google Scholar] [CrossRef]
- Bowers, M.S.; Chen, B.T.; Chou, J.K.; Osborne, M.P.H.; Gass, J.T.; See, R.E.; Bonci, A.; Janak, P.H.; Olive, M.F. Acamprosate attenuates cocaine and cue-induced reinstatement of cocaine-seeking behavior in rats. Psychopharmacology 2007, 195, 397–406. [Google Scholar] [CrossRef]
- Spanagel, R.; Sillaber, I.; Zieglgansberger, W.; Corrigall, W.A.; Stewart, J.; Shaham, Y. Acamprosate suppresses the expression of morphine-induced sensitization in rats but does not affect heroin self-administration or relapse induced by heroin or stress. Psychopharmacology 1998, 139, 391–401. [Google Scholar] [CrossRef]
- Mason, B.J.; Heyser, C.J. The neurobiology, clinical efficacy and safety of acamprosate in the treatment of alcohol dependence. Expert Opin. Drug Saf. 2010, 177–188. [Google Scholar]
- Kiefer, F.; Mann, K. Acamprosate: How, Where, and for Whom does it work? Mechanism of Action, Treatment Targets and Individualized Therapy. Current Pharmaceut. Des. 2010, 2098–2102. [Google Scholar] [CrossRef]
- Anton, R.F.; OʼMalley, S.S.; Ciraulo, D.A.; Cisler, R.A.; Couper, D.; Donovan, D.M.; Gastfriend, D.R.; Hosking, J.D.; Johnson, B.A.; LoCastro, J.S.; et al. Combined pharmacotherapies and behavioral interventions for alcohol dependence—The COMBINE study: a randomized controlled trial. JAMA 2006, 295, 2003–2017. [Google Scholar] [CrossRef]
- Mason, B.J.; Heyser, C.J. The neurobiology, clinical efficacy and safety of acamprosate in the treatment of alcohol dependence. Expert Opin. Drug Saf. 2010, 9, 177–188. [Google Scholar] [CrossRef]
- Kampman, K.M.; Dackis, C.; Pettinati, H.M.; Lynch, K.G.; Sparkman, T.; O'Brien, C.P. A double-blind, placebo-controlled pilot trial of acamprosate for the treatment of cocaine dependence. Addict. Behav. 2011, 36, 217–221. [Google Scholar] [CrossRef]
- Myers, K.M.; Carlezon, W.A.J. D-Cycloserine effects on extinction of conditioned responses to drug-related cues. Biol. Psychiatry 2012, 71, 947–955. [Google Scholar] [CrossRef]
- Sheinin, A.; Shavit, S.; Benveniste, M. Subunit specificity and mechanism of action of NMDA partial agonist D-Cycloserine. Neuropharmacology 2001, 41, 151–158. [Google Scholar] [CrossRef]
- Davis, M.; Ressler, K.; Rothbaum, B.O.; Richardson, R. Effects of D-Cycloserine on extinction: translation from preclinical to clinical work. Biol. Psychiatry 2006, 60, 369–375. [Google Scholar] [CrossRef]
- Botreau, F.; Paolone, G.; Stewart, J. d-Cycloserine facilitates extinction of a cocaine-induced conditioned place preference. Behav. Brain Res. 2006, 172, 173–178. [Google Scholar] [CrossRef]
- Thanos, P.K.; Bermeo, C.; Wang, G.J.; Volkow, N.D. D-Cycloserine accelerates the extinction of cocaine-induced conditioned place preference in C57BL/c mice. Behav. Brain Res. 2009, 199, 345–349. [Google Scholar] [CrossRef]
- Thanos, P.K.; Bermeo, C.; Wang, G.J.; Volkow, N.D. D-Cycloserine facilitates extinction of cocaine self-administration in rats. Synapse 2011, 65, 938–944. [Google Scholar] [CrossRef]
- Nic Dhonnchadha, B.A.; Szalay, J.J.; Achat-Mendes, C.; Platt, D.M.; Otto, M.W.; Spealman, R.D.; Kantak, K.M. D-Cycloserine deters reacquisition of cocaine self-administration by augmenting extinction learning. Neuropsychopharmacology 2010, 35, 357–367. [Google Scholar] [CrossRef]
- Torregrossa, M.M.; Sanchez, H.; Taylor, J.R. D-Cycloserine reduces the context specificity of Pavlovian extinction of cocaine cues through actions in the nucleus accumbens. J. Neurosci. 2010, 30, 10526–10533. [Google Scholar] [CrossRef]
- Lee, J.L.; Gardner, R.J.; Butler, V.J.; Everitt, B.J. D-Cycloserine potentiates the reconsolidation of cocaine-associated memories. Learn. Mem. 2009, 16, 82–85. [Google Scholar] [CrossRef]
- Myers, K.M.; Davis, M. Mechanisms of fear extinction. Mol. Psychiatry 2007, 12, 120–150. [Google Scholar] [CrossRef]
- Childress, A.R.; McLellan, A.T.; OʼBrien, C.P. Role of conditioning factors in the development of drug dependence. Psychiatr. Clin. North. Am. 1986, 9, 413–425. [Google Scholar]
- Siegel, S.; Ramos, B.M. Applying laboratory research: drug anticipation and the treatment of drug addiction. Exp. Clin. Psychopharmacol. 2002, 10, 162–183. [Google Scholar] [CrossRef]
- Santa Ana, E.J.; Rounsaville, B.J.; Frankforter, T.L.; Nich, C.; Babuscio, T.; Poling, J.; Gonsai, K.; Hill, K.P.; Carroll, K.M. D-Cycloserine attenuates reactivity to smoking cues in nicotine dependent smokers: a pilot investigation. Drug Alcohol Depend. 2009, 104, 220–227. [Google Scholar] [CrossRef]
- Kamboj, S.K.; Joye, A.; Das, R.K.; Gibson, A.J.; Morgan, C.J.; Curran, H.V. Cue exposure and response prevention with heavy smokers: a laboratory-based randomised placebo-controlled trial examining the effects of D-Cycloserine on cue reactivity and attentional bias. Psychopharmacology 2012, 221, 273–284. [Google Scholar] [CrossRef]
- Price, K.L.; McRae-Clark, A.L.; Saladin, M.E.; Maria, M.M.; DeSantis, S.M.; Back, S.E.; Brady, K.T. D-Cycloserine and cocaine cue reactivity: preliminary findings. Am. J. Drug Alcohol Abuse 2009, 35, 434–438. [Google Scholar] [CrossRef]
- Price, K.L.; Baker, N.L.; McRae-Clark, A.L.; Saladin, M.E.; Desantis, S.M.; Santa Ana, E.J.; Brady, K.T. A randomized, placebo-controlled laboratory study of the effects of D-cycloserine on craving in cocaine-dependent individuals. Psychopharmacology 2012, in press. [Google Scholar]
- Watson, B.J.; Wilson, S.; Griffin, L.; Kalk, N.J.; Taylor, L.G.; Munafo, M.R.; Lingford-Hughes, A.R.; Nutt, D.J. A pilot study of the effectiveness of D-Cycloserine during cue-exposure therapy in abstinent alcohol-dependent subjects. Psychopharmacology 2011, 216, 121–129. [Google Scholar] [CrossRef]
- Hofmann, S.G.; Huweler, R.; MacKillop, J.; Kantak, K.M. Effects of D-Cycloserine on craving to alcohol cues in problem drinkers: preliminary findings. Am. J. Drug Alcohol Abuse 2012, 38, 101–107. [Google Scholar] [CrossRef]
- Groblewski, P.A.; Lattal, K.M.; Cunningham, C.L. Effects of D-Cycloserine on extinction and reconditioning of ethanol-seeking behavior in mice. Alcohol. Clin. Exp. Res. 2009, 33, 772–782. [Google Scholar] [CrossRef]
- Das, R.K.; Kamboj, S.K. Maintaining clinical relevance: considerations for the future of research into d-cycloserine and cue exposure therapy for addiction. Biol. Psychiatry 2012, 72, e29–30. [Google Scholar] [CrossRef]
- Gass, J.T.; Olive, M.F. Positive allosteric modulation of mGluR5 receptors facilitates extinction of a cocaine contextual memory. Biol. Psychiatry 2009, 65, 717–720. [Google Scholar] [CrossRef]
- Cleva, R.M.; Hicks, M.P.; Gass, J.T.; Wischerath, K.C.; Plasters, E.T.; Widholm, J.J.; Olive, M.F. mGluR5 positive allosteric modulation enhances extinction learning following cocaine self-administration. Behav. Neurosci. 2011, 125, 10–19. [Google Scholar] [CrossRef]
- Kufahl, P.R.; Hood, L.E.; Nemirovsky, N.E.; Barabas, P.; Halstengard, C.; Villa, A.; Moore, E.; Watterson, L.R.; Olive, M.F. Positive allosteric modulation of mGluR5 accelerates extinction learning but not relearning following methamphetamine self-administration. Front. Pharmacol. 2012, 3, 194. [Google Scholar]
- Reichel, C.M.; Schwendt, M.; McGinty, J.F.; Olive, M.F.; See, R.E. Loss of object recognition memory produced by exteded access to methamphetamine self-administration is reversed by positive allosteric modulation of metabotropic glutamate receptor 5. Neuropsychopharmacology 2011, 36, 782–792. [Google Scholar] [CrossRef]
- Niswender, C.M.; Conn, P.J. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef]
- Bird, M.K.; Lawrence, A.J. The promiscuous mGlu5 receptor - a range of partners for therapeutic possibilities? Trends Pharmacol. Sci. 2009, 30, 617–623. [Google Scholar] [CrossRef]
- Olive, M.F. Cognitive effects of Group I metabotropic glutamate receptor ligands in the context of drug addiction. Eur. J. Pharmacol. 2010, 639, 47–58. [Google Scholar] [CrossRef]
- Olive, M.F. Metabotropic glutamate receptor ligands as potential therapeutics for drug addiction. Curr. Drug Abuse Rev. 2009, 2, 83–98. [Google Scholar] [CrossRef]
- Carroll, F.I. Antagonists at metabotropic glutamate receptor subtype 5: structure activity relationships and therapeutic potential for addiction. Ann. NY Acad. Sci. 2008, 1141, 221–232. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tomek, S.E.; Lacrosse, A.L.; Nemirovsky, N.E.; Olive, M.F. NMDA Receptor Modulators in the Treatment of Drug Addiction. Pharmaceuticals 2013, 6, 251-268. https://doi.org/10.3390/ph6020251
Tomek SE, Lacrosse AL, Nemirovsky NE, Olive MF. NMDA Receptor Modulators in the Treatment of Drug Addiction. Pharmaceuticals. 2013; 6(2):251-268. https://doi.org/10.3390/ph6020251
Chicago/Turabian StyleTomek, Seven E., Amber L. Lacrosse, Natali E. Nemirovsky, and M. Foster Olive. 2013. "NMDA Receptor Modulators in the Treatment of Drug Addiction" Pharmaceuticals 6, no. 2: 251-268. https://doi.org/10.3390/ph6020251