siRNA Genome Screening Approaches to Therapeutic Drug Repositioning




Abstract
:1. Introduction
1.1. RNAi Dependent Gene Silencing Pathways
1.2. Using Human Genome Data to Generate siGenome Libraries
1.3. Considerations for RNAi Screens
1.4. miRNA Screens
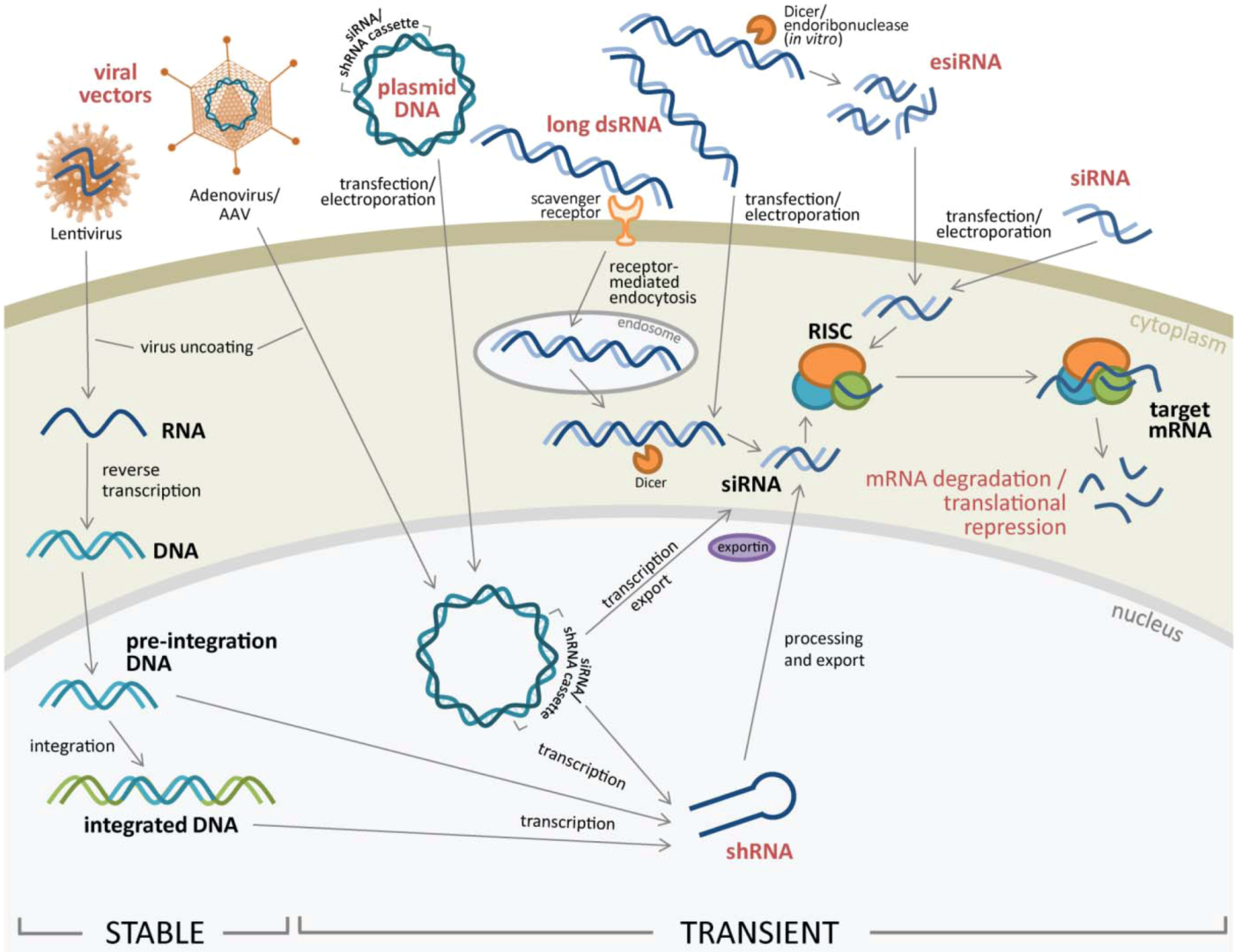
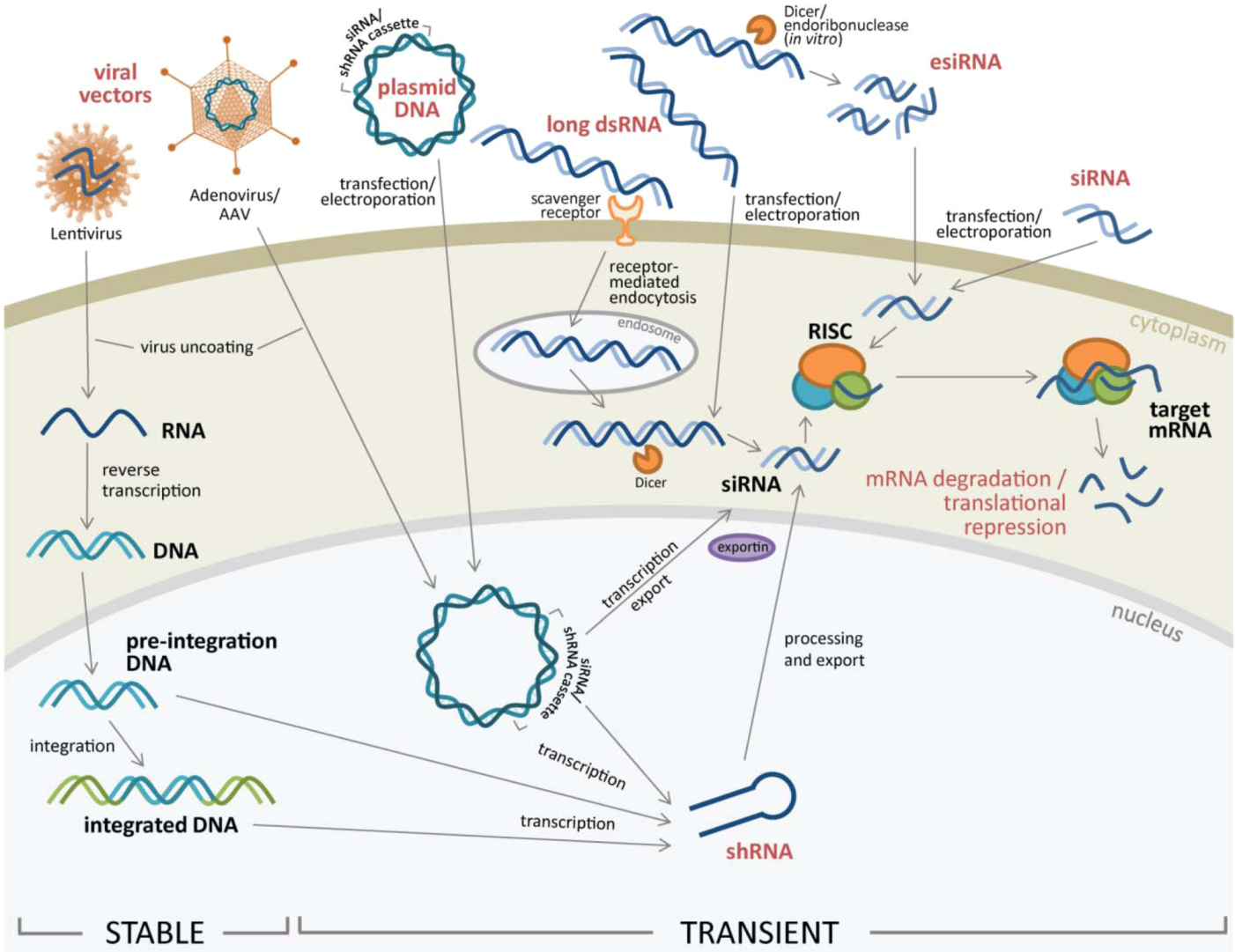
2. RNAi Delivery to Cells
2.1. RNAi Delivery Methods into Target Cells

{kind=link}
{kind=link}
| Transient RNA interference | Stable RNA interference | ||||
| Cell type | Lipid-based transfection | Electroporation | Adenovirus/AAV vectors | Lentivirus vectors | Retrovirus vectors |
| Most secondary and transformed cell lines (adherent or suspension) | X | X | X | X | X |
| Difficult to transfect cells | X | X | X | X | |
| Primary non-transformed cells (dividing) | X | X | X | X | |
| Primary non-transformed cells (non-dividing) | (nucleofection) | X | X | ||
| Growth-arrested and contact-inhibited cells | X | ||||
2.1.1. Lipid-Based Transfection and Electroporation of Nucleic Acids
2.1.2. Viral Vectors to Transfer RNAi into Target Cells
2.2. Different Types of RNAi Introduced for RNA Silencing
2.2.1. siRNA and esiRNA
2.2.2. Long dsRNA
2.2.3. Expression Vectors Containing RNAi Cassettes
2.2.4. Stable Gene Expression Silencing using Lentivirus or Retrovirus Vectors
3. Unraveling the Biological Implications of Pathogens using HTS Screening
3.1. RNAi Screens for Human Bacterial Pathogens
| Pathogen | Screen size | Gene family target | Validated candidates | Major pathways identified | Ref. |
|---|---|---|---|---|---|
| Bacterial | |||||
| L. monocytogenes | ~21,300 | Whole genome | 305 | Protein biosynthesis, proteasomal degradation, cytoskeletal networks | [68] |
| 779 | Kinome | 7 | Kinase networks | [69] | |
| M. fortuitum | ~21,300 | Whole genome | 2 | Vesicular transport and cytoskeletal networks | [68] |
| M. fortuitum and other species | ~21,000 | Whole genome | 86 | Lipid metabolism, chromatin organization, proton transport, vesicular transport, actin cytoskeleton, and signal transduction | [70] |
| M. marinum, M. tuberculosis | 1,000 | n.a | 1 | β-hexosaminidase | [71] |
| M. tuberculosis H37Rv | 744 + 288 | Kinases + phosphatases | 41 | Signaling networks | [72] |
| M. tuberculosis | 18,174 | Whole genome | 275 | Multiple pathways | [73] |
| C. caviae | 16,128 | Whole genome | 54 | Multiple pathways | [74] |
| 7,216 | Partial genome | 226 | Kinases Abl and PDGFR | [75] | |
| P. aeruginosa | 80 | Actin cytoskeleton associated genes | 4 | Abl kinase, Crk adaptor protein, Rac1 small GTPase, Cdc42, and p21 kinase components | [78] |
| S. typhimurium | 6,978 | SopE-associated host proteins | 72 | COPI complex, lipid biosynthesis | [79] |
| ~22,000 | Whole genome | 252 | Cellular development, cellular growth, carbohydrate metabolism | [80] | |
| Brucella spp. | 240 | ER associated proteins | 52 | Inositol metabolism, eukaryotic unfolded protein response (UPR) | [81] |
| F. tularensis | ~47,400 | Whole genome | ~200 | Multiple pathways | [82] |
| Fungal | |||||
| C. albicans | 7,216 | Genes shared among metazoans | 184 | Multiple pathways | [83] |
| C. neoformans | 410 | Targeted subset of multiple pathways | 57 | Multiple pathways | [84] |
| Protozoal | |||||
| P. falciparum | 727 | Kinome | 5 | Signaling networks | [85] |
| n.a. | Gene specific | 1 | Scavenger receptor B1 | [86] | |
| Plasmodium spp. | Gene specific | 3 | oxr1, argK & prs1 | [87] | |
| T. cruzi | 21,127 | Whole genome and gene specific studies | 162 | Multiple pathways | [88,89,90,91] |
| Viral | |||||
| Drosophila C virus | 21,000 | Whole genome | 66 | Ribosomal proteins, translation | [116] |
| Human Immunodeficiency virus (HIV) | n.a. | Gene specific | 1 | Human Spt5 transcription elongation factor | [94] |
| n.a. | Gene specific | 1 | DBR1 splicing factor | [95] | |
| 5,000 | Druggable gene targets | 4 | Multiple pathways; kinases | [96] | |
| 21,121 | Genome wide | 273 | Multiple pathways | [97] | |
| 19,709 | Genome wide | 311 | Multiple pathways | [98] | |
| Human Immunodeficiency virus (HIV) | 622 + 180 | Human kinase + phosphatase shRNAs | 14 | Multiple pathways | [99] |
| 30 | Targeted genes | 15 | Kinases, vesicular transport, and others | [100] | |
| 232 | DNA repair pathway | 35 | Base excision pathway repair | [101] | |
| 19,121 | Whole genome | 114 | PAF1 complex | [102] | |
| 12 | Autophagy pathway targeted shRNAs | 5 | Autophagy pathway components | [103] | |
| Influenza virus | 13,071 | Drosophila whole genome | ~100 | Multiple pathways | [104] |
| 17,877 | Human whole genome siRNA library | 120 | Multiple pathways | [105] | |
| 1,745 | Targeted influenza protein interactors | 616 | Multiple pathways | [106] | |
| 22,843 | Human whole genome | 287 | Multiple pathways | [107] | |
| 19,628 | Human whole genome | 295 | Multiple pathways | [108] | |
| 481 | Human protease siRNA library | 5 | c-AMP , NF-κb, and apoptosis | [109] | |
| Hepatitis C virus (HCV) | ~4,000 | Druggable targets | 9 | Multiple pathways | [117] |
| 62 | HCV–host interactions | 26 | Multiple pathways including Dicer | [118] | |
| 510 | Human kinase library | 3 | Csk, Jak1, and Vrk1 | [119] | |
| 140 | Membrane trafficking family | 16 | Clathrin coated pit proteins, actin polymerization, AP2 adaptor, ubiquitin ligase, ER/Golgi trafficking | [110] | |
| 140 | Membrane trafficking family | 7 | Endosomal trafficking, lipid organization, and actin polymerization | [120] | |
| 21,094 | Human whole genome | 96 | Multiple pathways | [121] | |
| Vesicular stomatitis virus (VSV), lymphocytic choriomeningitis (LCMV), and parainfluenza virus (PIV) 5 | 22,909 | Human whole genome | 72 | Coatomer complex 1 and other pathways | [111] |
| Ebola virus | 720 | Kinases and phosphorylases | ~190 | Multiple pathways | [112] |
| Vaccinia virus | 7,000 | Drosophila druggable genes library | 188 | Multiple pathways | [114] |
| 440 | Kinases + phosphatases + regulator factors | 7 | AMPK kinase, endocytosis | [113] | |
| Human papillomavirus (HPV) | 21,121 | Human whole genome library | 96 | DNA demethylation, histone acetyl transferases | [122] |
| Coxsackie and polio virus | 5,492 | Human druggable genome library | 117 | Rab GTPases, Src tyrosine kinases, and phosphatase networks | [123] |
| West Nile virus (WNV) | 21,121 | Human whole genome library | 305 | Multiple pathways | [124] |
| Dengue Virus | 22,632 | Drosophila whole genome | 116 | Multiple pathways | [125] |
| 119 | membrane trafficking genes | 6 | Clathrin-mediated endocytosis | [126] | |
| Drug | Original indication | New indication | Clinical trial stage | Ref. |
|---|---|---|---|---|
| Infectious Diseases | ||||
| Anti-bacterial | ||||
| PNU-100480 | MRSA | Tuberculosis | Phase I clinical trail | [145] |
| Sulphamethaxazole + Trimethoprim | Generic antibacterial | Tuberculosis | Clinical use | [146] |
| Raloxifen | Osteoporosis + breast cancer | P. aeruginosa | Preclinical | [147] |
| Anti-protozoal | ||||
| Astemizole | Antihistamine | Malaria | preclinical | [148] |
| Dapsone | Leprosy | Malaria | phase 3 completed | [117] |
| Amphotericin | Antifungal | Leishmaniasis | phase 3 completed | [117] |
| DB289 | Pneumocystis | Malaria and African trypanosomiasis | phase 2 completed | [117] |
| Eflornithine | Cancer | African trypanosomiasis | phase 3 completed | [117] |
| Fosmidomycin | Urinary-tract infections | Malaria | phase 2 completed | [117] |
| Harmine | Cancer | Malaria | preclinical | [148] |
| Miltefosine | Cancer | Visceral and cutaneous leishmaniasis | phase 2 completed | [117,149] |
| Paromomycin | Antiamebic | Visceral leishmaniasis | phase 4 completed | [117] |
| Pentamidine | Pneumonia (Pneumocystis carinii) | Trypanosomiasis and antimony-resistant leishmaniasis | phase 2 completed | [117] |
| Auranofin | Rheumatoid Arthritis | Amebiasis | Clinical use | [150] |
| Anti-parasitic | ||||
| Closantel | Antihelminthic | Onchocerciasis | preclinical | [148] |
| Anti-prion disease | ||||
| Quinacrine | Malaria | Creutzfeldt-Jakob Disease | phase 2 completed | [117] |
| Others | ||||
| Arsenic | Tuberculosis and syphilis | Acute promyelocytic leukemia | phase 2 completed, phase 3 active | [117] |
| Digoxin | Congestive heart failure and arrhythmia | Cancer | phase 1 completed, recruiting subjects for phase 2 | [148] |
| Fumagillin | Antiamebic | Cancer (angiogenesis inhibitor) | preclinical | [117] |
| Gemcitabine | Antiviral | Cancer | phase 2 active | [149] |
| Itraconazole | Antifungal | Angiogenesis inhibitor | phase 2 active | [148] |
| Glefenine | Analgesic | Chemotherapeutics for tumor resistance | preclinical | [148] |
| Mycophenolic acid | Immunosuppresive drug | Angiogenesis inhibitor | phase 2 active | [148] |
| Nitroxoline | Urinary-tract infections | Angiogenesis inhibitor | preclinical | [148] |
| Retinoic acid | Acne | Acute promyelocytic leukemia | phase 2 completed | [117] |
| Riluzole | Amyotrophic lateral sclerosis | Melanoma and other cancers | phase 2 active | [148] |
| Thalidomide | Sedative / antiemetic | Cancer (angiogenesis inhibitor), erythema nodosum leprosum | phase 2 active | [117,149] |
| Bupropion | Antidepressant | Smoking cessation | phase 3 completed | [149] |
| Ceftriaxone | β-lactamase antibiotic | Amyotrophic lateral sclerosis | phase 3 completed | [117] |
| Dapoxetine | Antidepressant, analgesic | Premature ejaculation | phase 3 completed | [149] |
| Doxepin | Antidepressant | Insomnia, antipruritic | phase 2 completed | [149] |
| Duloxetine | Antidepressant | Urinary incontinence (stress-related) | phase 3 completed | [149] |
| Finasteride | Benign prostatic hyperplasia | Male baldness | phase 3 completed | [149] |
| Fluoxetine | Antidepressant | Premenstrual dysphoria | phase 4 completed | [149] |
| Hydroxychloroquine | Antiparasitic | Arthritis, systemic lupus erythematosus | recruiting subjects for phase 3 | [149] |
| Milnacipran | Antidepressant | Fibromyalgia | phase 3 completed | [149] |
| Minocycline | Antibiotic | Amyotrophic lateral sclerosis | phase 3 completed | [117] |
| Mycophenolate mofetil | Immunosuppresive drug (transplant rejection) | Renal symptoms of systemic lupus erythematosus | phase 3 completed | [149] |
| Naltrexone | Opioid addiction | Alcohol withdrawal | phase 4 completed | [149] |
| Pioglitazone | Type-II diabetes | Nonalcoholic steatohepatitis | phase 2 completed | [149] |
| Raloxifene | Breast cancer | Osteoporosis | phase 3 completed | [149] |
| Ropinirole | Antihypertensive | Parkinson's disease, restless legs syndrome | phase 3 completed | [149] |
3.2. RNAi Screens for Human Fungal Pathogens
3.3. RNAi Screens for Human Protozoan Pathogens
3.4. RNAi Screens for Human Viral Infections
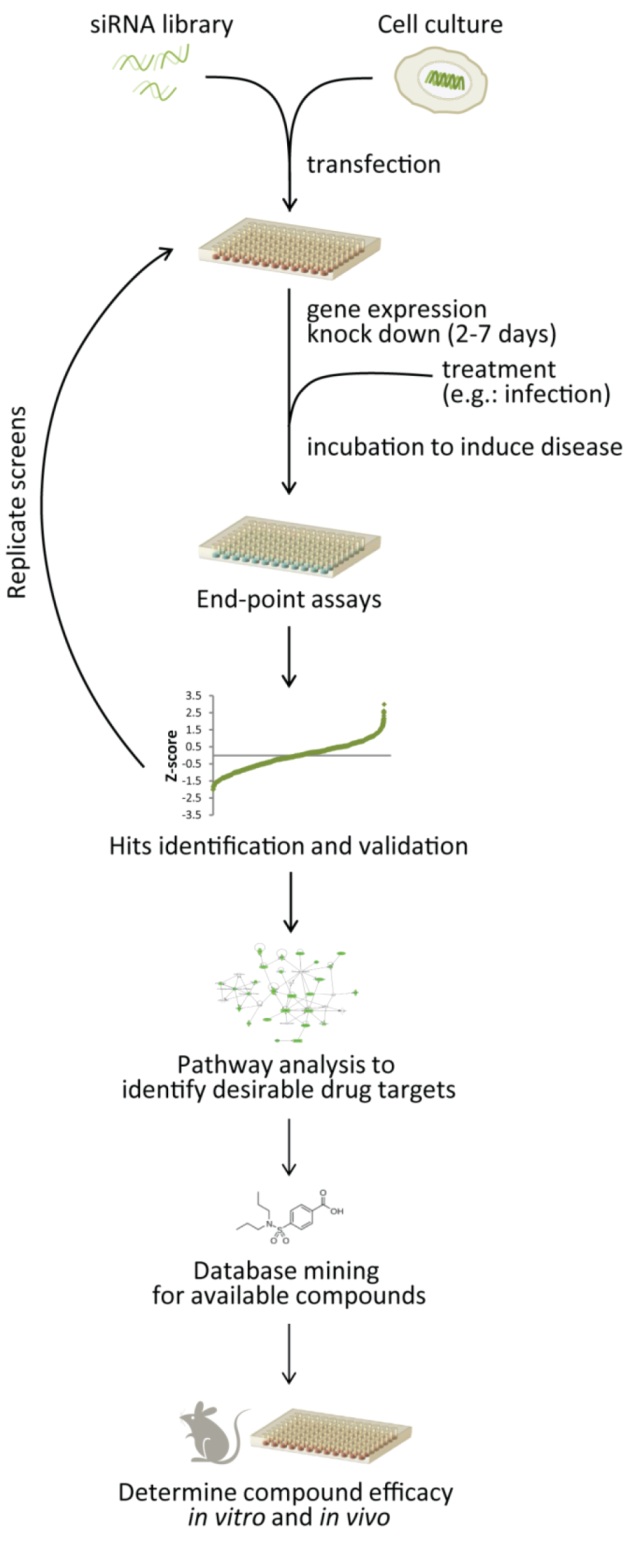
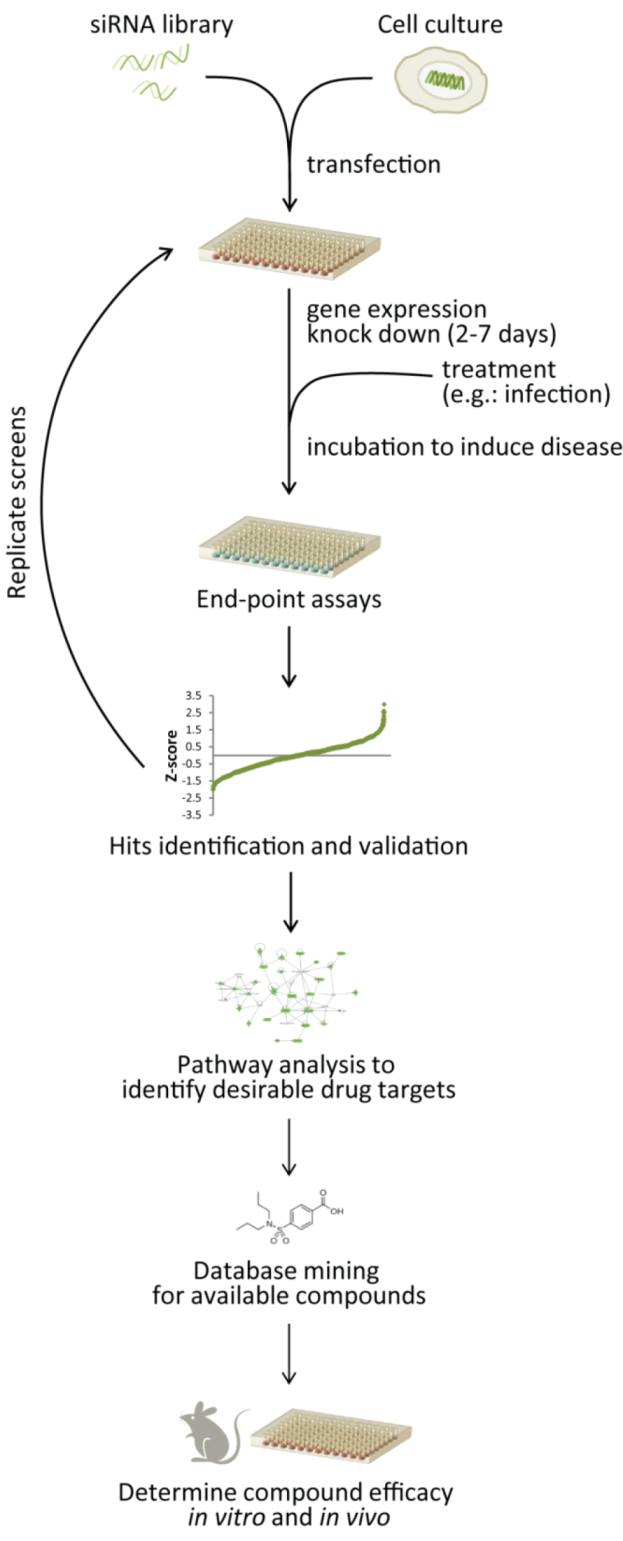
4. Combining Strategies to Target Host Genes
5. Rescuing and Repurposing Drugs
5.1. RNAi Screening to Identify Drug Targets and Drug Repurposing
| Name | Website address | Contents | Ref. |
|---|---|---|---|
| PROMISCUOUS | http://bioinformatics.charite.de/promiscuous | A database containing 25,000 annotated withdrawn or experimental drugs searchable by name, target, or pathway. | [141] |
| ChemSpider | http://www.chemspider.com | Free drug database containing 28 million structures searchable by calculated properties, structures, or drug ligands. | [152] |
| DrugBank | http://www.drugbank.ca | A comprehensive database hosted by the University of Alberta that contains 4,800 drugs including FDA-approved small drugs, natural agents, and experimental drugs and their sequence, structure, and target pathway. | [140] |
6. Case Studies for RNAi Screening towards Drug Repurposing
6.1. Identification of OAT3 as Pro-influenza A Host Factor and Repurposing of OAT3 Inhibitor Probenecid
6.2. Identification of Host Proteins in Phosphatidylinositol-3-Kinase and Calcium/Calmodulin Kinase-Related Pathways Important for Zaire Ebola Virus Entry and Its Inhibition by Known Inhibitors
6.3. siRNA Screen Reveals Chemical Inhibitor to Prevent MYC-Driven Oncogenesis
7. Going Forward with RNAi Screens and Drug Repurposing
8. Conclusions
Acknowledgments
Conflict of Interest
References
- Mayr, L.M.; Bojanic, D. Novel trends in high-throughput screening. Curr. Opin. Pharmacol. 2009, 9, 580–588. [Google Scholar] [CrossRef]
- Waszkowycz, B. Towards improving compound selection in structure-based virtual screening. Drug Discov. Today 2008, 13, 219–226. [Google Scholar] [CrossRef]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Drews, J. What's in a number? Nat. Rev. Drug Discov. 2006, 5, 975–975. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- Liu, C.; Bai, B.; Skogerbo, G.; Cai, L.; Deng, W.; Zhang, Y.; Bu, D.; Zhao, Y.; Chen, R. NONCODE: an integrated knowledge database of non-coding RNAs. Nucleic Acids Res. 2005, 33, D112–115. [Google Scholar] [CrossRef]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar]
- Eddy, S.R. Non-coding RNA genes and the modern RNA world. Nat. Rev. Genet. 2001, 2, 919–929. [Google Scholar] [CrossRef]
- Reinhart, B.J.; Slack, F.J.; Basson, M.; Pasquinelli, A.E.; Bettinger, J.C.; Rougvie, A.E.; Horvitz, H.R.; Ruvkun, G. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature 2000, 403, 901–906. [Google Scholar] [CrossRef]
- Mo, Y.-Y. MicroRNA regulatory networks and human disease. Cell. Mol. Life Sci. 2012, 69, 3529–3531. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A., et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar]
- Jackson, A.L.; Bartz, S.R.; Schelter, J.; Kobayashi, S.V.; Burchard, J.; Mao, M.; Li, B.; Cavet, G.; Linsley, P.S. Expression profiling reveals off-target gene regulation by RNAi. Nat. Biotechnol. 2003, 21, 635–637. [Google Scholar] [CrossRef]
- Reynolds, A.; Leake, D.; Boese, Q.; Scaringe, S.; Marshall, W.S.; Khvorova, A. Rational siRNA design for RNA interference. Nat. Biotechnol. 2004, 22, 326–330. [Google Scholar] [CrossRef]
- Jackson, A.L.; Burchard, J.; Schelter, J.; Chau, B.N.; Cleary, M.; Lim, L.; Linsley, P.S. Widespread siRNA "off-target" transcript silencing mediated by seed region sequence complementarity. RNA 2006, 12, 1179–1187. [Google Scholar] [CrossRef]
- Lai, E.C. Micro RNAs are complementary to 3' UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 2002, 30, 363–364. [Google Scholar] [CrossRef]
- Li, L.; Xu, J.; Yang, D.; Tan, X.; Wang, H. Computational approaches for microRNA studies: a review. Mamm. Genome 2010, 21, 1–12. [Google Scholar] [CrossRef]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X., et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.C.; Munschauer, M., et al. PAR-CliP--a method to identify transcriptome-wide the binding sites of RNA binding proteins. J. Vis. Exp. 2010, 41. pii: 2034. [Google Scholar]
- Aza-Blanc, P.; Cooper, C.L.; Wagner, K.; Batalov, S.; Deveraux, Q.L.; Cooke, M.P. Identification of Modulators of TRAIL-Induced Apoptosis via RNAi-Based Phenotypic Screening. Mol. Cell 2003, 12, 627–637. [Google Scholar] [CrossRef]
- Berns, K.; Hijmans, E.M.; Mullenders, J.; Brummelkamp, T.R.; Velds, A.; Heimerikx, M.; Kerkhoven, R.M.; Madiredjo, M.; Nijkamp, W.; Weigelt, B., et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 2004, 428, 431–437. [Google Scholar] [CrossRef]
- Paddison, P.J.; Silva, J.M.; Conklin, D.S.; Schlabach, M.; Li, M.; Aruleba, S.; Balija, V.; O'Shaughnessy, A.; Gnoj, L.; Scobie, K., et al. A resource for large-scale RNA-interference-based screens in mammals. Nature 2004, 428, 427–431. [Google Scholar]
- Kittler, R.; Putz, G.; Pelletier, L.; Poser, I.; Heninger, A.-K.; Drechsel, D.; Fischer, S.; Konstantinova, I.; Habermann, B.; Grabner, H.; et al. An endoribonuclease-prepared siRNA screen in human cells identifies genes essential for cell division. Nature 2004, 432, 1036–1040. [Google Scholar] [CrossRef]
- Sachse, C.; Krausz, E.; Krönke, A.; Hannus, M.; Walsh, A.; Grabner, A.; Ovcharenko, D.; Dorris, D.; Trudel, C.; Sönnichsen, B., et al. High-Throughput RNA Interference Strategies for Target Discovery and Validation by Using Synthetic Short Interfering RNAs: Functional Genomics Investigations of Biological Pathways. Methods Enzymol. 2005, 392, 242–277. [Google Scholar] [CrossRef]
- Whitehurst, A.W.; Bodemann, B.O.; Cardenas, J.; Ferguson, D.; Girard, L.; Peyton, M.; Minna, J.D.; Michnoff, C.; Hao, W.; Roth, M.G., et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 2007, 446, 815–819. [Google Scholar] [CrossRef]
- Simpson, K.J.; Davis, G.M.; Boag, P.R. Comparative high-throughput RNAi screening methodologies in C. elegans and mammalian cells. N. Biotechnol. 2012, 29, 459–470. [Google Scholar] [CrossRef]
- Mohr, S.; Bakal, C.; Perrimon, N. Genomic screening with RNAi: results and challenges. Annu. Rev. Biochem. 2010, 79, 37–64. [Google Scholar] [CrossRef]
- Mohr, S.E.; Perrimon, N. RNAi screening: new approaches, understandings, and organisms. Wiley Interdiscip. Rev. RNA 2012, 3, 145–158. [Google Scholar] [CrossRef]
- Russ, A.P.; Lampel, S. The druggable genome: an update. Drug Discov. Today 2005, 10, 1607–1610. [Google Scholar] [CrossRef]
- Conrad, C.; Gerlich, D.W. Automated microscopy for high-content RNAi screening. J. Cell Biol. 2010, 188, 453–461. [Google Scholar] [CrossRef]
- Ghildiyal, M.; Zamore, P.D. Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 2009, 10, 94–108. [Google Scholar] [CrossRef]
- Griffiths-Jones, S.; Saini, H.K.; van Dongen, S.; Enright, A.J. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008, 36, D154–D158. [Google Scholar] [CrossRef]
- Friedman, R.C.; Farh, K.K.-H.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Research 2009, 19, 92–105. [Google Scholar]
- Leung, R.K.; Whittaker, P.A. RNA interference: from gene silencing to gene-specific therapeutics. Pharmacol. Ther. 2005, 107, 222–239. [Google Scholar] [CrossRef]
- Meliopoulos, V.A.; Andersen, L.E.; Birrer, K.F.; Simpson, K.J.; Lowenthal, J.W.; Bean, A.G.; Stambas, J.; Stewart, C.R.; Tompkins, S.M.; van Beusechem, V.W., et al. Host gene targets for novel influenza therapies elucidated by high-throughput RNA interference screens. FASEB J 2012, 26, 1372–1386. [Google Scholar]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Sakurai, K.; Chomchan, P.; Rossi, J.J. Silencing of gene expression in cultured cells using small interfering RNAs. Curr. Protoc. Cell Biol. 2010. Chapter 27, Unit 27 21 21-28. [Google Scholar]
- Gopalakrishnan, B.; Wolff, J. siRNA and DNA transfer to cultured cells. Methods Mol. Biol. 2009, 480, 31–52. [Google Scholar] [CrossRef]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef]
- Recillas-Targa, F. Multiple strategies for gene transfer, expression, knockdown, and chromatin influence in mammalian cell lines and transgenic animals. Mol. Biotechnol. 2006, 34, 337–354. [Google Scholar] [CrossRef]
- Bukrinsky, M.I.; Sharova, N.; Dempsey, M.P.; Stanwick, T.L.; Bukrinskaya, A.G.; Haggerty, S.; Stevenson, M. Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc. Natl. Acad. Sci. USA 1992, 89, 6580–6584. [Google Scholar]
- Manjunath, N.; Wu, H.; Subramanya, S.; Shankar, P. Lentiviral delivery of short hairpin RNAs. Adv. Drug. Deliv. Rev. 2009, 61, 732–745. [Google Scholar] [CrossRef]
- Sliva, K.; Schnierle, B.S. Selective gene silencing by viral delivery of short hairpin RNA. Virol. J. 2010, 7, 248. [Google Scholar] [CrossRef]
- Lewis, P.; Hensel, M.; Emerman, M. Human immunodeficiency virus infection of cells arrested in the cell cycle. EMBO J. 1992, 11, 3053–3058. [Google Scholar]
- Lewis, P.F.; Emerman, M. Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 1994, 68, 510–516. [Google Scholar]
- Miller, D.G.; Adam, M.A.; Miller, A.D. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol. Cell Biol. 1990, 10, 4239–4242. [Google Scholar]
- Mitani, K.; Kubo, S. Adenovirus as an integrating vector. Curr. Gene Ther. 2002, 2, 135–144. [Google Scholar] [CrossRef]
- Grimm, D.; Pandey, K.; Kay, M.A. Adeno-associated virus vectors for short hairpin RNA expression. Methods Enzymol. 2005, 392, 381–405. [Google Scholar] [CrossRef]
- Ewert, K.K.; Zidovska, A.; Ahmad, A.; Bouxsein, N.F.; Evans, H.M.; McAllister, C.S.; Samuel, C.E.; Safinya, C.R. Cationic liposome-nucleic acid complexes for gene delivery and silencing: pathways and mechanisms for plasmid DNA and siRNA. Top. Curr. Chem. 2010, 296, 191–226. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Kim, V.N.; Han, J.; Siomi, M.C. Biogenesis of small RNAs in animals. Nat. Rev. Mol. Cell Biol. 2009, 10, 126–139. [Google Scholar] [CrossRef]
- Saxena, S.; Jonsson, Z.O.; Dutta, A. Small RNAs with imperfect match to endogenous mRNA repress translation. Implications for off-target activity of small inhibitory RNA in mammalian cells. J. Biol. Chem. 2003, 278, 44312–44319. [Google Scholar]
- Theis, M.; Buchholz, F. MISSION esiRNA for RNAi Screening in Mammalian Cells. J. Vis. Exp. 2010, 39. pii: 2008. [Google Scholar]
- Reynolds, A.; Anderson, E.M.; Vermeulen, A.; Fedorov, Y.; Robinson, K.; Leake, D.; Karpilow, J.; Marshall, W.S.; Khvorova, A. Induction of the interferon response by siRNA is cell type- and duplex length-dependent. RNA 2006, 12, 988–993. [Google Scholar] [CrossRef]
- Kim, D.H.; Longo, M.; Han, Y.; Lundberg, P.; Cantin, E.; Rossi, J.J. Interferon induction by siRNAs and ssRNAs synthesized by phage polymerase. Nat. Biotechnol. 2004, 22, 321–325. [Google Scholar]
- de Veer, M.J.; Sledz, C.A.; Williams, B.R.G. Detection of foreign RNA: Implications for RNAi. Immunol Cell Biol 2005, 83, 224–228. [Google Scholar] [CrossRef]
- Bartlett, D.W.; Davis, M.E. Effect of siRNA nuclease stability on the in vitro and in vivo kinetics of siRNA-mediated gene silencing. Biotechnol. Bioeng. 2007, 97, 909–921. [Google Scholar] [CrossRef]
- Eberle, F.; Giessler, K.; Deck, C.; Heeg, K.; Peter, M.; Richert, C.; Dalpke, A.H. Modifications in small interfering RNA that separate immunostimulation from RNA interference. J. Immunol. 2008, 180, 3229–3237. [Google Scholar]
- Robbins, M.; Judge, A.; Liang, L.; McClintock, K.; Yaworski, E.; MacLachlan, I. 2'-O-methyl-modified RNAs act as TLR7 antagonists. Mol. Ther. 2007, 15, 1663–1669. [Google Scholar] [CrossRef]
- Saleh, M.-C.; van Rij, R.P.; Hekele, A.; Gillis, A.; Foley, E.; O'Farrell, P.H.; Andino, R. The endocytic pathway mediates cell entry of dsRNA to induce RNAi silencing. Nat. Cell Biol. 2006, 8, 793–802. [Google Scholar] [CrossRef]
- Dieudonne, A.; Torres, D.; Blanchard, S.; Taront, S.; Jeannin, P.; Delneste, Y.; Pichavant, M.; Trottein, F.; Gosset, P. Scavenger receptors in human airway epithelial cells: role in response to double-stranded RNA. PLoS One 2012, 7, e41952. [Google Scholar]
- Takahashi, Y.; Nishikawa, M.; Takakura, Y. Nonviral vector-mediated RNA interference: Its gene silencing characteristics and important factors to achieve RNAi-based gene therapy. Adv. Drug Deliv. Rev. 2009, 61, 760–766. [Google Scholar] [CrossRef]
- Chiu, Y.L.; Rana, T.M. RNAi in human cells: basic structural and functional features of small interfering RNA. Mol. Cell 2002, 10, 549–561. [Google Scholar] [CrossRef]
- Singh, S.; Narang, A.S.; Mahato, R.I. Subcellular fate and off-target effects of siRNA, shRNA, and miRNA. Pharm. Res. 2011, 28, 2996–3015. [Google Scholar] [CrossRef]
- Goff, S.P. Retroviridae: The Retroviruses and Their Replication. In Fields Virology; Knipe, D. M., Howley, P. M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, 2001; Vol. 2, pp. 1871–1940. [Google Scholar]
- Gupta, S.; Schoer, R.A.; Egan, J.E.; Hannon, G.J.; Mittal, V. Inducible, reversible, and stable RNA interference in mammalian cells. Proc. Natl. Acad. Sci. USA 2004, 101, 1927–1932. [Google Scholar]
- Agaisse, H.; Burrack, L.S.; Philips, J.A.; Rubin, E.J.; Perrimon, N.; Higgins, D.E. Genome-wide RNAi screen for host factors required for intracellular bacterial infection. Science 2005, 309, 1248–1251. [Google Scholar] [CrossRef]
- Chong, R.; Squires, R.; Swiss, R.; Agaisse, H. RNAi screen reveals host cell kinases specifically involved in Listeria monocytogenes spread from cell to cell. PLoS One 2011, 6, e23399. [Google Scholar]
- Philips, J.A.; Rubin, E.J.; Perrimon, N. Drosophila RNAi screen reveals CD36 family member required for mycobacterial infection. Science 2005, 309, 1251–1253. [Google Scholar] [CrossRef]
- Koo, I.C.; Ohol, Y.M.; Wu, P.; Morisaki, J.H.; Cox, J.S.; Brown, E.J. Role for lysosomal enzyme beta-hexosaminidase in the control of mycobacteria infection. Proc. Natl. Acad. Sci. USA 2008, 105, 710–715. [Google Scholar]
- Jayaswal, S.; Kamal, M.A.; Dua, R.; Gupta, S.; Majumdar, T.; Das, G.; Kumar, D.; Rao, K.V. Identification of host-dependent survival factors for intracellular Mycobacterium tuberculosis through an siRNA screen. PLoS Pathog. 2010, 6, e1000839. [Google Scholar] [CrossRef]
- Kumar, D.; Nath, L.; Kamal, M.A.; Varshney, A.; Jain, A.; Singh, S.; Rao, K.V. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell 2010, 140, 731–743. [Google Scholar] [CrossRef]
- Derre, I.; Pypaert, M.; Dautry-Varsat, A.; Agaisse, H. RNAi screen in Drosophila cells reveals the involvement of the Tom complex in Chlamydia infection. PLoS Pathog. 2007, 3, 1446–1458. [Google Scholar]
- Elwell, C.A.; Ceesay, A.; Kim, J.H.; Kalman, D.; Engel, J.N. RNA interference screen identifies Abl kinase and PDGFR signaling in Chlamydia trachomatis entry. PLoS Pathog. 2008, 4, e1000021. [Google Scholar] [CrossRef]
- Gurumurthy, R.K.; Maurer, A.P.; Machuy, N.; Hess, S.; Pleissner, K.P.; Schuchhardt, J.; Rudel, T.; Meyer, T.F. A loss-of-function screen reveals ras- and raf-independent mek-erk signaling during chlamydia trachomatis infection. Sci. Signal. 2010, 3, ra21. [Google Scholar] [CrossRef]
- Garvis, S.; Munder, A.; Ball, G.; de Bentzmann, S.; Wiehlmann, L.; Ewbank, J.J.; Tummler, B.; Filloux, A. Caenorhabditis elegans semi-automated liquid screen reveals a specialized role for the chemotaxis gene cheb2 in pseudomonas aeruginosa virulence. PLoS Pathog. 2009, 5, e1000540. [Google Scholar] [CrossRef]
- Pielage, J.F.; Powell, K.R.; Kalman, D.; Engel, J.N. RNAi screen reveals an Abl kinase-dependent host cell pathway involved in Pseudomonas aeruginosa internalization. PLoS Pathog. 2008, 4, e1000031. [Google Scholar] [CrossRef]
- Misselwitz, B.; Dilling, S.; Vonaesch, P.; Sacher, R.; Snijder, B.; Schlumberger, M.; Rout, S.; Stark, M.; von Mering, C.; Pelkmans, L.; et al. RNAi screen of Salmonella invasion shows role of COPI in membrane targeting of cholesterol and Cdc42. Mol. Syst. Biol. 2011, 7, 474. [Google Scholar]
- Thornbrough, J.M.; Hundley, T.; Valdivia, R.; Worley, M.J. Human genome-wide RNAi screen for host factors that modulate intracellular Salmonella growth. PLoS One 2012, 7, e38097. [Google Scholar]
- Qin, Q.M.; Pei, J.; Ancona, V.; Shaw, B.D.; Ficht, T.A.; de Figueiredo, P. RNAi screen of endoplasmic reticulum-associated host factors reveals a role for IRE1alpha in supporting Brucella replication. PLoS Pathog. 2008, 4, e1000110. [Google Scholar] [CrossRef]
- Zhou, H.; DeLoid, G.; Browning, E.; Gregory, D.J.; Tan, F.; Bedugnis, A.S.; Imrich, A.; Koziel, H.; Kramnik, I.; Lu, Q., et al. Genome-wide RNAi screen in IFN-gamma-treated human macrophages identifies genes mediating resistance to the intracellular pathogen Francisella tularensis. PLoS One 2012, 7, e31752. [Google Scholar]
- Stroschein-Stevenson, S.L.; Foley, E.; O'Farrell, P.H.; Johnson, A.D. Identification of Drosophila gene products required for phagocytosis of Candida albicans. PLoS Biol. 2006, 4, e4. [Google Scholar] [CrossRef]
- Qin, Q.M.; Luo, J.; Lin, X.; Pei, J.; Li, L.; Ficht, T.A.; de Figueiredo, P. Functional analysis of host factors that mediate the intracellular lifestyle of Cryptococcus neoformans. PLoS Pathog. 2011, 7, e1002078. [Google Scholar] [CrossRef]
- Prudencio, M.; Rodrigues, C.D.; Hannus, M.; Martin, C.; Real, E.; Goncalves, L.A.; Carret, C.; Dorkin, R.; Rohl, I.; Jahn-Hoffmann, K.; et al. Kinome-wide RNAi screen implicates at least 5 host hepatocyte kinases in Plasmodium sporozoite infection. PLoS Pathog. 2008, 4, e1000201. [Google Scholar] [CrossRef]
- Rodrigues, C.D.; Hannus, M.; Prudencio, M.; Martin, C.; Goncalves, L.A.; Portugal, S.; Epiphanio, S.; Akinc, A.; Hadwiger, P.; Jahn-Hofmann, K.; et al. Host scavenger receptor SR-BI plays a dual role in the establishment of malaria parasite liver infection. Cell Host Microbe 2008, 4, 271–282. [Google Scholar] [CrossRef]
- Chertemps, T.; Mitri, C.; Perrot, S.; Sautereau, J.; Jacques, J.C.; Thiery, I.; Bourgouin, C.; Rosinski-Chupin, I. Anopheles gambiae PRS1 modulates Plasmodium development at both midgut and salivary gland steps. PLoS One 2010, 5, e11538. [Google Scholar]
- Genovesio, A.; Giardini, M.A.; Kwon, Y.J.; de Macedo Dossin, F.; Choi, S.Y.; Kim, N.Y.; Kim, H.C.; Jung, S.Y.; Schenkman, S.; Almeida, I.C.; et al. Visual genome-wide RNAi screening to identify human host factors required for Trypanosoma cruzi infection. PLoS One 2011, 6, e19733. [Google Scholar]
- Nde, P.N.; Simmons, K.J.; Kleshchenko, Y.Y.; Pratap, S.; Lima, M.F.; Villalta, F. Silencing of the laminin gamma-1 gene blocks Trypanosoma cruzi infection. Infect. Immun. 2006, 74, 1643–1648. [Google Scholar] [CrossRef]
- Simmons, K.J.; Nde, P.N.; Kleshchenko, Y.Y.; Lima, M.F.; Villalta, F. Stable RNA interference of host thrombospondin-1 blocks Trypanosoma cruzi infection. FEBS Lett. 2006, 580, 2365–2370. [Google Scholar] [CrossRef]
- Claser, C.; Curcio, M.; de Mello, S.M.; Silveira, E.V.; Monteiro, H.P.; Rodrigues, M.M. Silencing cytokeratin 18 gene inhibits intracellular replication of Trypanosoma cruzi in HeLa cells but not binding and invasion of trypanosomes. BMC Cell Biol. 2008, 9, 68. [Google Scholar] [CrossRef]
- Berkhout, B. A new Houdini act: Multiple routes for HIV-1 escape from rnai-mediated inhibition. Future Microbiol. 2009, 4, 151–154. [Google Scholar] [CrossRef]
- Leonard, J.N.; Shah, P.S.; Burnett, J.C.; Schaffer, D.V. HIV evades RNA interference directed at TAR by an indirect compensatory mechanism. Cell Host Microbe 2008, 4, 484–494. [Google Scholar] [CrossRef]
- Ping, Y.H.; Chu, C.Y.; Cao, H.; Jacque, J.M.; Stevenson, M.; Rana, T.M. Modulating HIV-1 replication by RNA interference directed against human transcription elongation factor SPT5. Retrovirology 2004, 1, 46. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; De Leon, J.; Yokoyama, N.; Naidu, Y.; Camerini, D. DBR1 siRNA inhibition of HIV-1 replication. Retrovirology 2005, 2, 63. [Google Scholar] [CrossRef]
- Nguyen, D.G.; Wolff, K.C.; Yin, H.; Caldwell, J.S.; Kuhen, K.L. "UnPAKing" human immunodeficiency virus (HIV) replication: using small interfering RNA screening to identify novel cofactors and elucidate the role of group I PAKs in HIV infection. J. Virol. 2006, 80, 130–137. [Google Scholar] [CrossRef]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef]
- Rato, S.; Maia, S.; Brito, P.M.; Resende, L.; Pereira, C.F.; Moita, C.; Freitas, R.P.; Moniz-Pereira, J.; Hacohen, N.; Moita, L.F.; et al. Novel HIV-1 knockdown targets identified by an enriched kinases/phosphatases shRNA library using a long-term iterative screen in Jurkat T-cells. PLoS One 2010, 5, e9276. [Google Scholar]
- Eekels, J.J.; Geerts, D.; Jeeninga, R.E.; Berkhout, B. Long-term inhibition of HIV-1 replication with RNA interference against cellular co-factors. Antiviral Res. 2011, 89, 43–53. [Google Scholar] [CrossRef]
- Espeseth, A.S.; Fishel, R.; Hazuda, D.; Huang, Q.; Xu, M.; Yoder, K.; Zhou, H. siRNA screening of a targeted library of DNA repair factors in HIV infection reveals a role for base excision repair in HIV integration. PLoS One 2011, 6, e17612. [Google Scholar]
- Liu, L.; Oliveira, N.M.; Cheney, K.M.; Pade, C.; Dreja, H.; Bergin, A.M.; Borgdorff, V.; Beach, D.H.; Bishop, C.L.; Dittmar, M.T.; et al. A whole genome screen for HIV restriction factors. Retrovirology 2011, 8, 94. [Google Scholar] [CrossRef] [Green Version]
- Eekels, J.J.; Sagnier, S.; Geerts, D.; Jeeninga, R.E.; Biard-Piechaczyk, M.; Berkhout, B. Inhibition of HIV-1 replication with stable RNAi-mediated knockdown of autophagy factors. Virol. J. 2012, 9, 69. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.; Sakurai, A.; Watanabe, T.; Sorensen, E.; Nidom, C.A.; Newton, M.A.; Ahlquist, P.; Kawaoka, Y. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 2008, 454, 890–893. [Google Scholar] [CrossRef]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef]
- Shapira, S.D.; Gat-Viks, I.; Shum, B.O.V.; Dricot, A.; de Grace, M.M.; Wu, L.; Gupta, P.B.; Hao, T.; Silver, S.J.; Root, D.E.; et al. A Physical and Regulatory Map of Host-Influenza Interactions Reveals Pathways in H1N1 Infection. Cell 2009, 139, 1255–1267. [Google Scholar] [CrossRef] [Green Version]
- Karlas, A.; Machuy, N.; Shin, Y.; Pleissner, K.-P.; Artarini, A.; Heuer, D.; Becker, D.; Khalil, H.; Ogilvie, L.A.; Hess, S.; et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–822. [Google Scholar] [CrossRef]
- Konig, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef]
- Meliopoulos, V.A.; Andersen, L.E.; Brooks, P.; Yan, X.; Bakre, A.; Coleman, J.K.; Tompkins, S.M.; Tripp, R.A. MicroRNA regulation of human protease genes essential for influenza virus replication. PLoS One 2012, 7, e37169. [Google Scholar]
- Coller, K.E.; Berger, K.L.; Heaton, N.S.; Cooper, J.D.; Yoon, R.; Randall, G. RNA interference and single particle tracking analysis of hepatitis C virus endocytosis. PLoS Pathog. 2009, 5, e1000702. [Google Scholar] [CrossRef]
- Panda, D.; Das, A.; Dinh, P.X.; Subramaniam, S.; Nayak, D.; Barrows, N.J.; Pearson, J.L.; Thompson, J.; Kelly, D.L.; Ladunga, I.; et al. RNAi screening reveals requirement for host cell secretory pathway in infection by diverse families of negative-strand RNA viruses. Proc. Natl. Acad. Sci. USA 2011, 108, 19036–19041. [Google Scholar]
- Kolokoltsov, A.A.; Saeed, M.F.; Freiberg, A.N.; Holbrook, M.R.; Davey, R.A. Identification of novel cellular targets for therapeutic intervention against Ebola virus infection by siRNA screening. Drug Dev. Res. 2009, 70, 255–265. [Google Scholar] [CrossRef]
- Moser, T.S.; Jones, R.G.; Thompson, C.B.; Coyne, C.B.; Cherry, S. A kinome RNAi screen identified AMPK as promoting poxvirus entry through the control of actin dynamics. PLoS Pathog. 2010, 6, e1000954. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.; Snijder, B.; Sacher, R.; Burkard, C.; Bleck, C.K.; Stahlberg, H.; Pelkmans, L.; Helenius, A. RNAi Screening Reveals Proteasome- and Cullin3-Dependent Stages in Vaccinia Virus Infection. Cell Rep. 2012, 2, 1036–1047. [Google Scholar] [CrossRef]
- Mukherjee, S.; Hanley, K.A. RNA interference modulates replication of dengue virus in Drosophila melanogaster cells. BMC Microbiol. 2010, 10, 127. [Google Scholar]
- Cherry, S.; Doukas, T.; Armknecht, S.; Whelan, S.; Wang, H.; Sarnow, P.; Perrimon, N. Genome-wide RNAi screen reveals a specific sensitivity of IRES-containing RNA viruses to host translation inhibition. Genes Dev. 2005, 19, 445–452. [Google Scholar] [CrossRef]
- Chong, C.R.; Sullivan, D.J., Jr. New uses for old drugs. Nature 2007, 448, 645–646. [Google Scholar] [CrossRef]
- Randall, G.; Panis, M.; Cooper, J.D.; Tellinghuisen, T.L.; Sukhodolets, K.E.; Pfeffer, S.; Landthaler, M.; Landgraf, P.; Kan, S.; Lindenbach, B.D., et al. Cellular cofactors affecting hepatitis C virus infection and replication. Proc. Natl. Acad. Sci. USA 2007, 104, 12884–12889. [Google Scholar]
- Supekova, L.; Supek, F.; Lee, J.; Chen, S.; Gray, N.; Pezacki, J.P.; Schlapbach, A.; Schultz, P.G. Identification of human kinases involved in hepatitis C virus replication by small interference RNA library screening. J. Biol. Chem. 2008, 283, 29–36. [Google Scholar]
- Berger, K.L.; Cooper, J.D.; Heaton, N.S.; Yoon, R.; Oakland, T.E.; Jordan, T.X.; Mateu, G.; Grakoui, A.; Randall, G. Roles for endocytic trafficking and phosphatidylinositol 4-kinase III alpha in hepatitis C virus replication. Proc. Natl. Acad. Sci. USA 2009, 106, 7577–7582. [Google Scholar]
- Tai, A.W.; Benita, Y.; Peng, L.F.; Kim, S.S.; Sakamoto, N.; Xavier, R.J.; Chung, R.T. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 2009, 5, 298–307. [Google Scholar] [CrossRef]
- Smith, J.A.; White, E.A.; Sowa, M.E.; Powell, M.L.; Ottinger, M.; Harper, J.W.; Howley, P.M. Genome-wide siRNA screen identifies SMCX, EP400, and Brd4 as E2-dependent regulators of human papillomavirus oncogene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 3752–3757. [Google Scholar]
- Coyne, C.B.; Bozym, R.; Morosky, S.A.; Hanna, S.L.; Mukherjee, A.; Tudor, M.; Kim, K.S.; Cherry, S. Comparative RNAi screening reveals host factors involved in enterovirus infection of polarized endothelial monolayers. Cell Host Microbe 2011, 9, 70–82. [Google Scholar] [CrossRef]
- Krishnan, M.N.; Ng, A.; Sukumaran, B.; Gilfoy, F.D.; Uchil, P.D.; Sultana, H.; Brass, A.L.; Adametz, R.; Tsui, M.; Qian, F., et al. RNA interference screen for human genes associated with West Nile virus infection. Nature 2008, 455, 242–245. [Google Scholar] [CrossRef]
- Sessions, O.M.; Barrows, N.J.; Souza-Neto, J.A.; Robinson, T.J.; Hershey, C.L.; Rodgers, M.A.; Ramirez, J.L.; Dimopoulos, G.; Yang, P.L.; Pearson, J.L.; et al. Discovery of insect and human dengue virus host factors. Nature 2009, 458, 1047–1050. [Google Scholar] [CrossRef]
- Ang, F.; Wong, A.P.; Ng, M.M.; Chu, J.J. Small interference RNA profiling reveals the essential role of human membrane trafficking genes in mediating the infectious entry of dengue virus. Virol. J. 2010, 7, 24. [Google Scholar] [CrossRef] [Green Version]
- Boutros, M.; Bras, L.P.; Huber, W. Analysis of cell-based RNAi screens. Genome Biol. 2006, 7, R66. [Google Scholar] [CrossRef]
- Tolopko, A.N.; Sullivan, J.P.; Erickson, S.D.; Wrobel, D.; Chiang, S.L.; Rudnicki, K.; Rudnicki, S.; Nale, J.; Selfors, L.M.; Greenhouse, D.; et al. Screensaver: an open source lab information management system (LIMS) for high throughput screening facilities. BMC Bioinformatics 2010, 11, 260. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols 2008, 4, 44–57. [Google Scholar] [CrossRef]
- Khatri, P.; Sirota, M.; Butte, A.J. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput. Biol. 2012, 8, e1002375. [Google Scholar] [CrossRef]
- Viswanathan, G.A.; Seto, J.; Patil, S.; Nudelman, G.; Sealfon, S.C. Getting started in biological pathway construction and analysis. PLoS Comput. Biol. 2008, 4, e16. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: new estimates of drug development costs. J. Health. Econ. 2003, 22, 151–185. [Google Scholar] [CrossRef]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G.; Lasagna, L. Research and development costs for new drugs by therapeutic category. A study of the US pharmaceutical industry. Pharmacoeconomics 1995, 7, 152–169. [Google Scholar] [CrossRef]
- Collins, F.S. Mining for therapeutic gold. Nat. Rev. Drug Discov. 2011, 10, 397. [Google Scholar] [CrossRef]
- Fischl, M.A.; Richman, D.D.; Grieco, M.H.; Gottlieb, M.S.; Volberding, P.A.; Laskin, O.L.; Leedom, J.M.; Groopman, J.E.; Mildvan, D.; Schooley, R.T.; et al. The efficacy of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. A double-blind, placebo-controlled trial. N. Engl. J. Med. 1987, 317, 185–191. [Google Scholar]
- Yarchoan, R.; Broder, S. Development of antiretroviral therapy for the acquired immunodeficiency syndrome and related disorders. A progress report. N. Engl. J. Med. 1987, 316, 557–564. [Google Scholar] [CrossRef]
- Allison, M. NCATS launches drug repurposing program. Nat. Biotechnol. 2012, 30, 571–572. [Google Scholar] [CrossRef]
- Huang, R.; Southall, N.; Wang, Y.; Yasgar, A.; Shinn, P.; Jadhav, A.; Nguyen, D.T.; Austin, C.P. The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci. Transl. Med. 2011, 3, 80ps16. [Google Scholar] [CrossRef]
- Grinnon, S.T.; Miller, K.; Marler, J.R.; Lu, Y.; Stout, A.; Odenkirchen, J.; Kunitz, S. National Institute of Neurological Disorders and Stroke Common Data Element Project - approach and methods. Clin. Trials 2012, 9, 322–329. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: a knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar]
- von Eichborn, J.; Murgueitio, M.S.; Dunkel, M.; Koerner, S.; Bourne, P.E.; Preissner, R. PROMISCUOUS: a database for network-based drug-repositioning. Nucleic Acids Res. 2011, 39, D1060–D1066. [Google Scholar] [CrossRef]
- Terrett, N.K.; Bell, A.S.; Brown, D.; Ellis, P. Sildenafil (VIAGRATM), a potent and selective inhibitor of type 5 cGMP phosphodiesterase with utility for the treatment of male erectile dysfunction. Bioorg. Med. Chem. Lett. 1996, 6, 1819–1824. [Google Scholar] [CrossRef]
- Goldenberg, M.M. Safety and efficacy of sildenafil citrate in the treatment of male erectile dysfunction. Clin. Ther. 1998, 20, 1033–1048. [Google Scholar] [CrossRef]
- Raja, S.G.; Nayak, S.H. Sildenafil: Emerging Cardiovascular Indications. Ann. Thorac. Surg. 2004, 78, 1496–1506. [Google Scholar] [CrossRef]
- Wallis, R.S.; Jakubiec, W.M.; Kumar, V.; Silvia, A.M.; Paige, D.; Dimitrova, D.; Li, X.; Ladutko, L.; Campbell, S.; Friedland, G., et al. Pharmacokinetics and whole-blood bactericidal activity against Mycobacterium tuberculosis of single doses of PNU-100480 in healthy volunteers. J. Infect. Dis. 2010, 202, 745–751. [Google Scholar]
- Huang, T.S.; Kunin, C.M.; Yan, B.S.; Chen, Y.S.; Lee, S.S.; Syu, W., Jr. Susceptibility of Mycobacterium tuberculosis to sulfamethoxazole, trimethoprim and their combination over a 12 year period in Taiwan. J. Antimicrob. Chemother. 2012, 67, 633–637. [Google Scholar] [CrossRef]
- Ho Sui, S.J.; Lo, R.; Fernandes, A.R.; Caulfield, M.D.; Lerman, J.A.; Xie, L.; Bourne, P.E.; Baillie, D.L.; Brinkman, F.S. Raloxifene attenuates Pseudomonas aeruginosa pyocyanin production and virulence. Int. J. Antimicrob. Agents 2012, 40, 246–251. [Google Scholar] [CrossRef]
- Ekins, S.; Williams, A.J.; Krasowski, M.D.; Freundlich, J.S. In silico repositioning of approved drugs for rare and neglected diseases. Drug Discov. Today 2011, 16, 298–310. [Google Scholar] [CrossRef]
- Henriksen, K.; Christiansen, C.; Karsdal, M.A. Serological biochemical markers of surrogate efficacy and safety as a novel approach to drug repositioning. Drug Discov. Today 2011, 16, 967–975. [Google Scholar] [CrossRef]
- Debnath, A.; Parsonage, D.; Andrade, R.M.; He, C.; Cobo, E.R.; Hirata, K.; Chen, S.; Garcia-Rivera, G.; Orozco, E.; Martinez, M.B.; et al. A high-throughput drug screen for Entamoeba histolytica identifies a new lead and target. Nat. Med. 2012, 18, 956–960. [Google Scholar] [CrossRef]
- Kaimal, V.; Bardes, E.E.; Tabar, S.C.; Jegga, A.G.; Aronow, B.J. ToppCluster: a multiple gene list feature analyzer for comparative enrichment clustering and network-based dissection of biological systems. Nucleic Acids Res. 2010, 38, W96–W102. [Google Scholar] [CrossRef]
- Williams, A.J. Public chemical compound databases. Curr. Opin. Drug Discov. Devel. 2008, 11, 393–404. [Google Scholar]
- Thompson, W.W.; Shay, D.K.; Weintraub, E.; Brammer, L.; Cox, N.; Anderson, L.J.; Fukuda, K. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 2003, 289, 179–186. [Google Scholar] [CrossRef]
- Beigel, J.; Bray, M. Current and future antiviral therapy of severe seasonal and avian influenza. Antiviral Res. 2008, 78, 91–102. [Google Scholar] [CrossRef]
- Dreitlein, W.B.; Maratos, J.; Brocavich, J. Zanamivir and oseltamivir: two new options for the treatment and prevention of influenza. Clin. Ther. 2001, 23, 327–355. [Google Scholar] [CrossRef]
- Kandel, R.; Hartshorn, K.L. Novel strategies for prevention and treatment of influenza. Expert Opin. Ther. Targets 2005, 9, 1–22. [Google Scholar] [CrossRef]
- Beigel, J.H. Antiviral Compounds In the Pipeline to Tackle H1N1 Influenza Infection. Drugs Future 2010, 35, 385–392. [Google Scholar] [CrossRef]
- Sui, B.; Bamba, D.; Weng, K.; Ung, H.; Chang, S.; Van Dyke, J.; Goldblatt, M.; Duan, R.; Kinch, M.S.; Li, W.-B. The use of Random Homozygous Gene Perturbation to identify novel host-oriented targets for influenza. Virology 2009, 387, 473–481. [Google Scholar] [CrossRef]
- Perwitasari, O.; Yan, X.; Johnson, S.; White, C.; Brooks, P.; Tompkins, S.M.; Tripp, R.A. Targeting the Organic Anion Transporter-3 (OAT3) with Probenecid as a Novel Anti-Influenza A Virus Strategy. Antimicrob. Agents Chemother. 2012. [Google Scholar] [CrossRef]
- Dantzler, W.H.; Evans, K.K.; Wright, S.H. Kinetics of interactions of para-aminohippurate, probenecid, cysteine conjugates and N-acetyl cysteine conjugates with basolateral organic anion transporter in isolated rabbit proximal renal tubules. J. Pharmacol. Exp. Ther. 1995, 272, 663–672. [Google Scholar]
- Stamp, L.K.; O'Donnell, J.L.; Chapman, P.T. Emerging therapies in the long-term management of hyperuricaemia and gout. Intern. Med. J. 2007, 37, 258–266. [Google Scholar] [CrossRef]
- Hill, G.; Cihlar, T.; Oo, C.; Ho, E.S.; Prior, K.; Wiltshire, H.; Barrett, J.; Liu, B.; Ward, P. The Anti-Influenza Drug Oseltamivir Exhibits Low Potential to Induce Pharmacokinetic Drug Interactions via Renal Secretion-Correlation of in Vivo and in Vitro Studies. Drug Metab. Dispos. 2002, 30, 13–19. [Google Scholar] [CrossRef]
- Holodniy, M.; Penzak, S.R.; Straight, T.M.; Davey, R.T.; Lee, K.K.; Goetz, M.B.; Raisch, D.W.; Cunningham, F.; Lin, E.T.; Olivo, N.; et al. Pharmacokinetics and tolerability of oseltamivir combined with probenecid. Antimicrob. Agents Chemother. 2008, 52, 3013–3021. [Google Scholar] [CrossRef]
- Rayner, C.R.; Chanu, P.; Gieschke, R.; Boak, L.M.; Jonsson, E.N. Population pharmacokinetics of oseltamivir when coadministered with probenecid. J. Clin. Pharmacol. 2008, 48, 935–947. [Google Scholar] [CrossRef]
- MacNeil, A.; Rollin, P.E. Ebola and Marburg hemorrhagic fevers: neglected tropical diseases? PLoS Negl. Trop. Dis. 2012, 6, e1546. [Google Scholar] [CrossRef]
- Huggins, J.W. Prospects for treatment of viral hemorrhagic fevers with ribavirin, a broad-spectrum antiviral drug. Rev. Infect. Dis. 1989, 11 Suppl 4, S750–S761. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef]
- Hermeking, H. The MYC oncogene as a cancer drug target. Curr. Cancer Drug Targets 2003, 3, 163–175. [Google Scholar] [CrossRef]
- Toyoshima, M.; Howie, H.L.; Imakura, M.; Walsh, R.M.; Annis, J.E.; Chang, A.N.; Frazier, J.; Chau, B.N.; Loboda, A.; Linsley, P.S.; et al. Functional genomics identifies therapeutic targets for MYC-driven cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 9545–9550. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Perwitasari, O.; Bakre, A.; Tompkins, S.M.; Tripp, R.A. siRNA Genome Screening Approaches to Therapeutic Drug Repositioning. Pharmaceuticals 2013, 6, 124-160. https://doi.org/10.3390/ph6020124
Perwitasari O, Bakre A, Tompkins SM, Tripp RA. siRNA Genome Screening Approaches to Therapeutic Drug Repositioning. Pharmaceuticals. 2013; 6(2):124-160. https://doi.org/10.3390/ph6020124
Chicago/Turabian StylePerwitasari, Olivia, Abhijeet Bakre, S. Mark Tompkins, and Ralph A. Tripp. 2013. "siRNA Genome Screening Approaches to Therapeutic Drug Repositioning" Pharmaceuticals 6, no. 2: 124-160. https://doi.org/10.3390/ph6020124