Cell Penetrating Peptoids (CPPos): Synthesis of a Small Combinatorial Library by Using IRORI MiniKans

 and
and 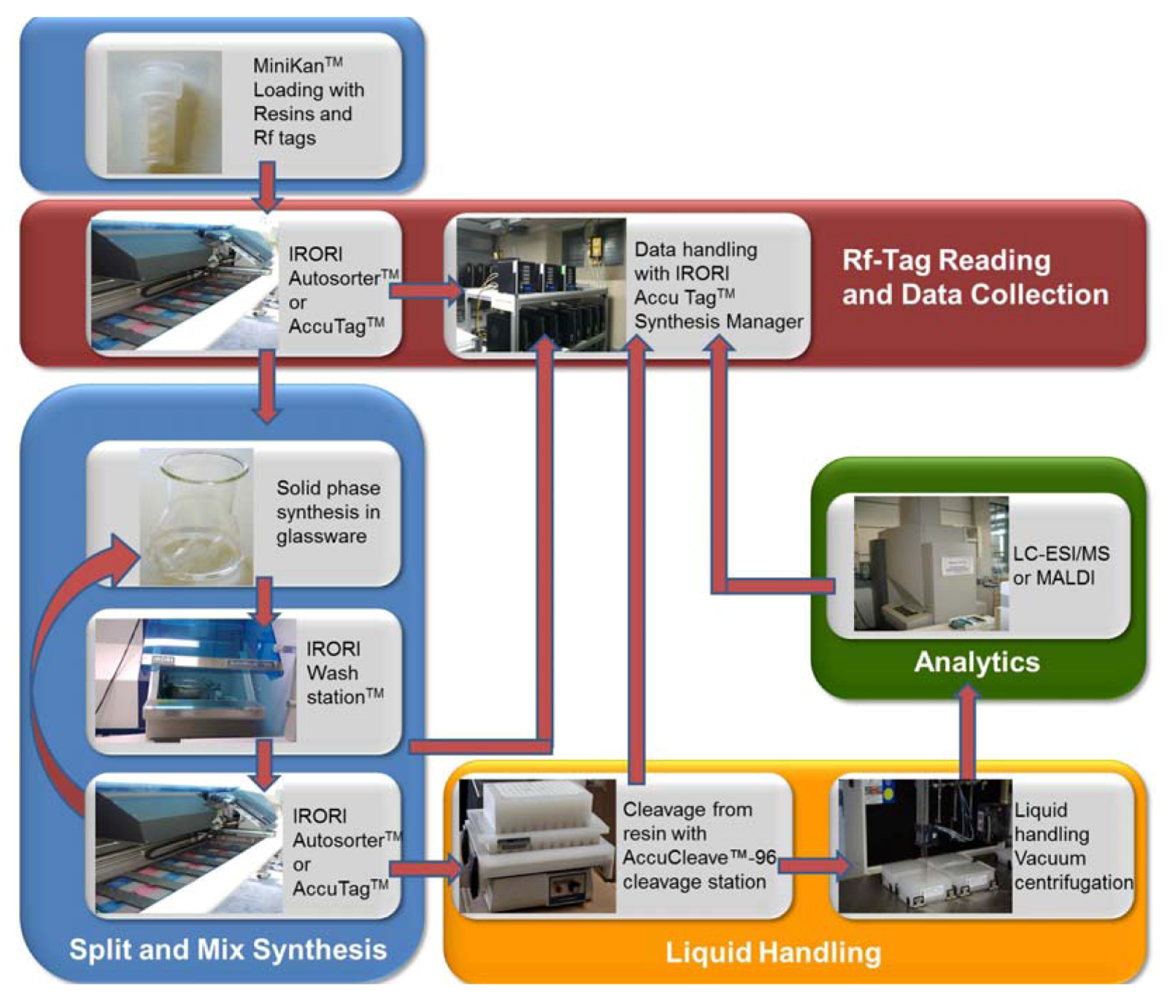{kind=link}
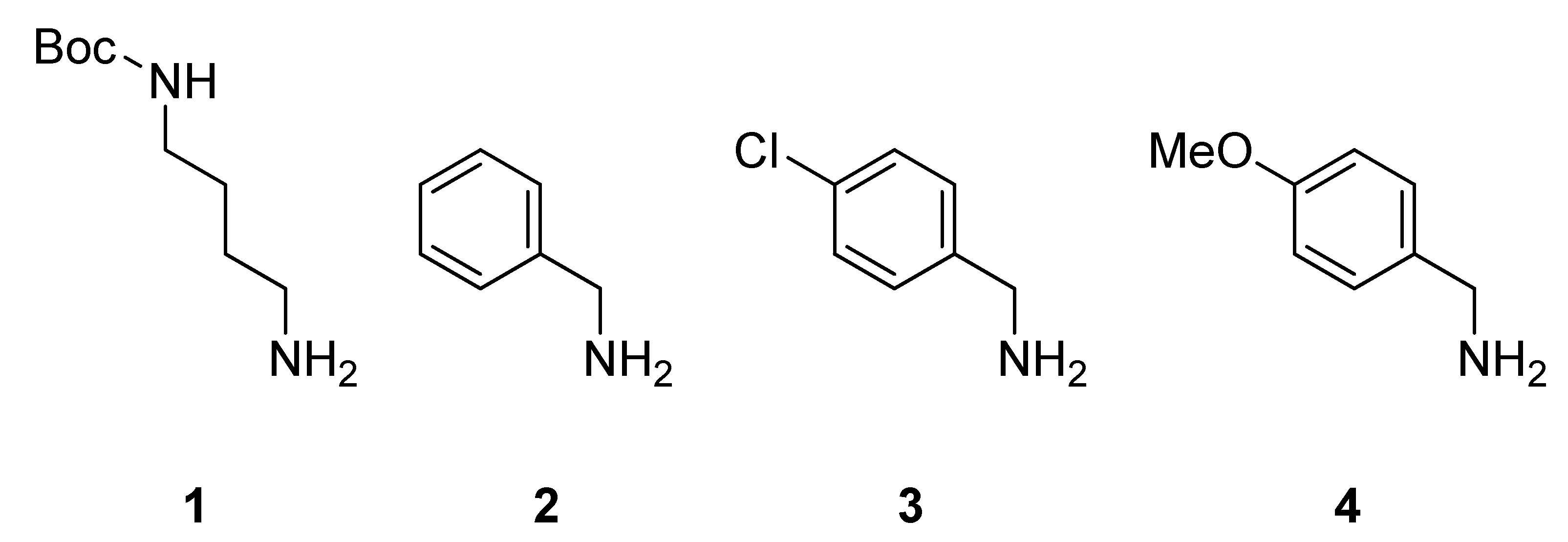{kind=link}
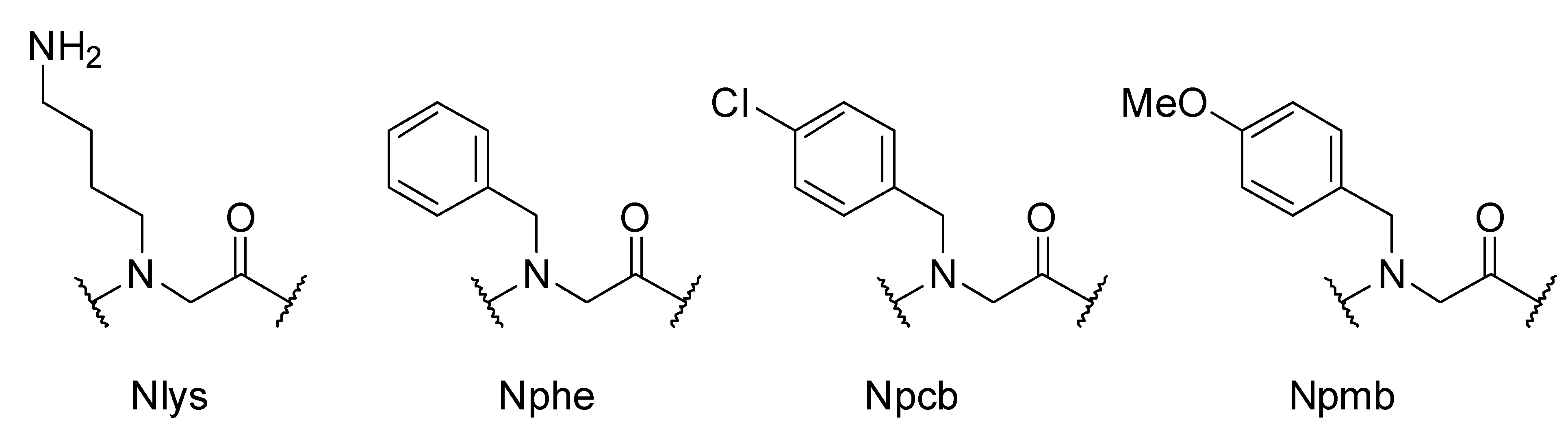{kind=link}
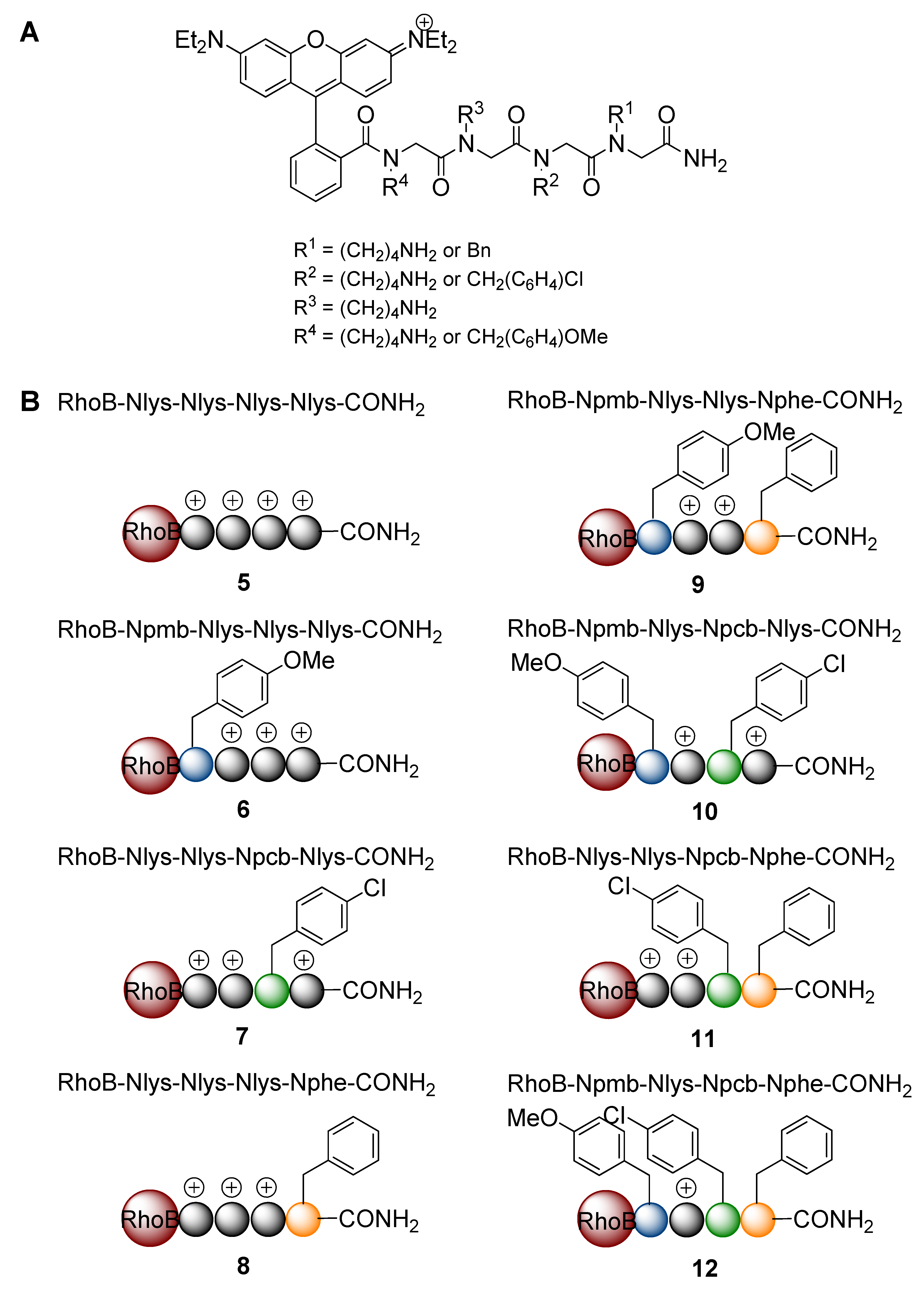{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
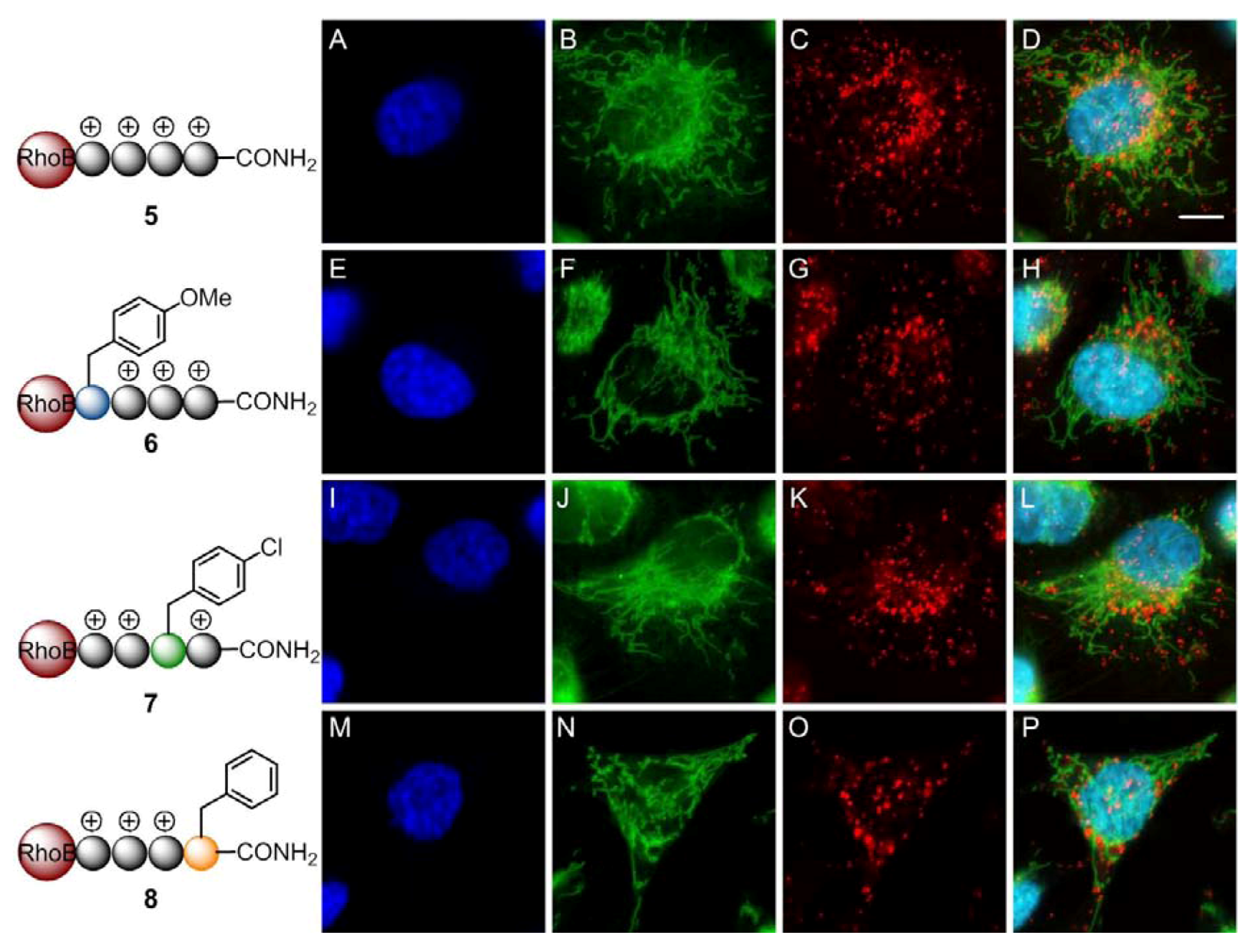
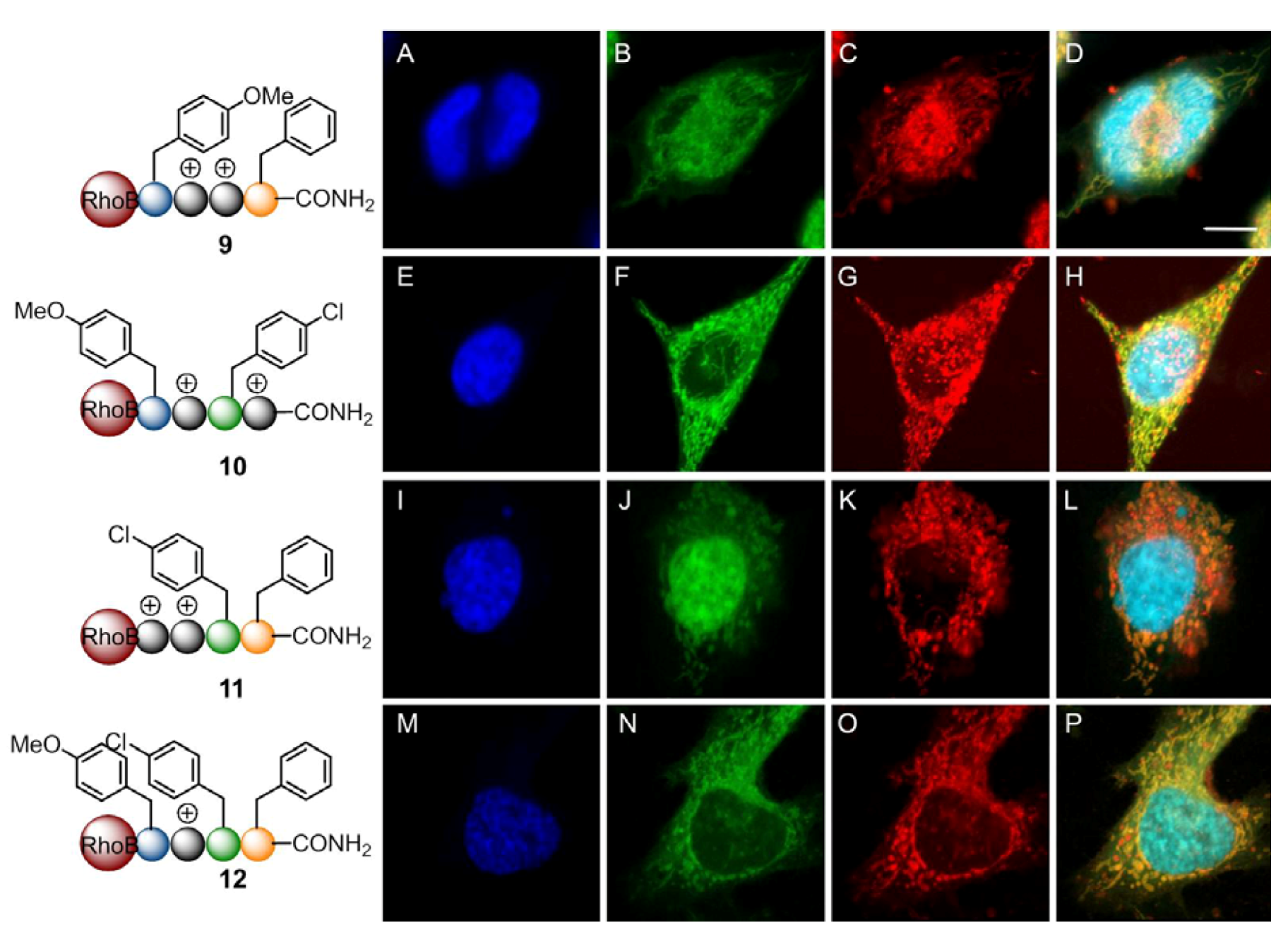
2. Results and Discussion
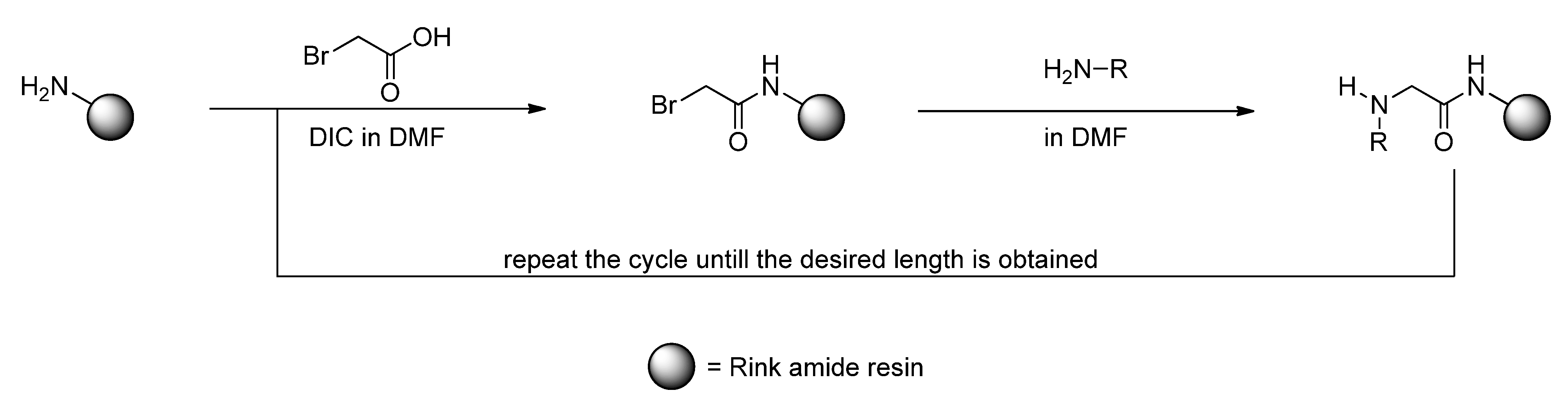
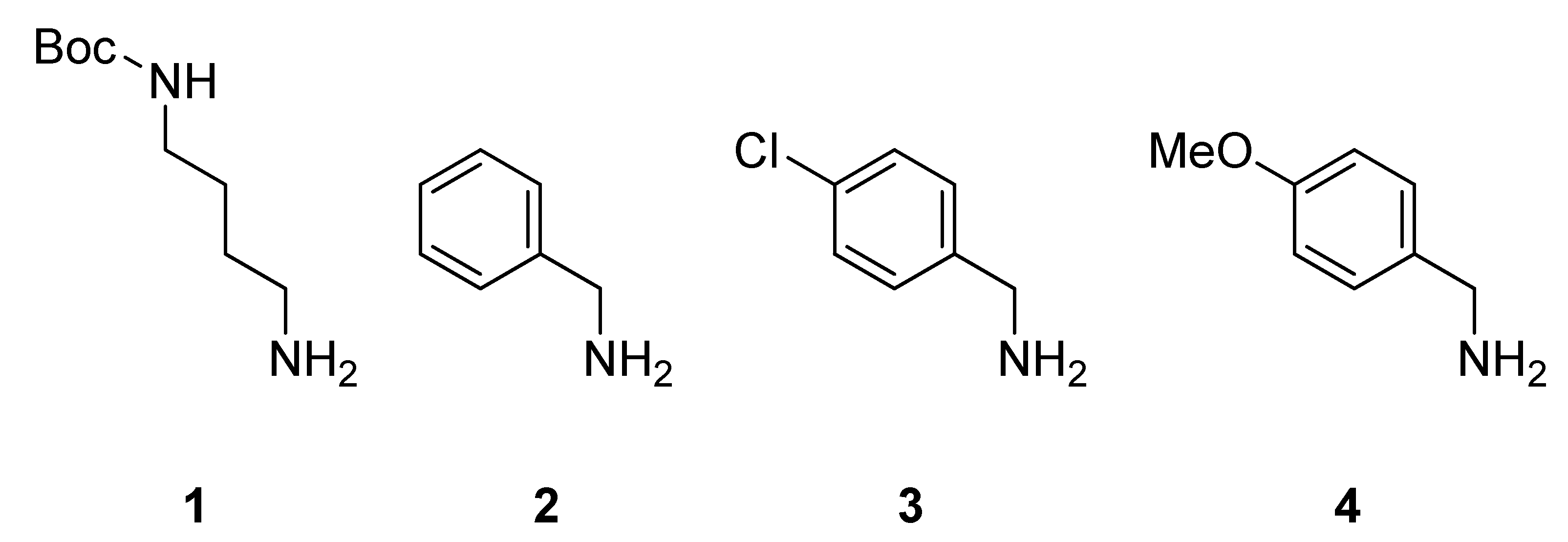
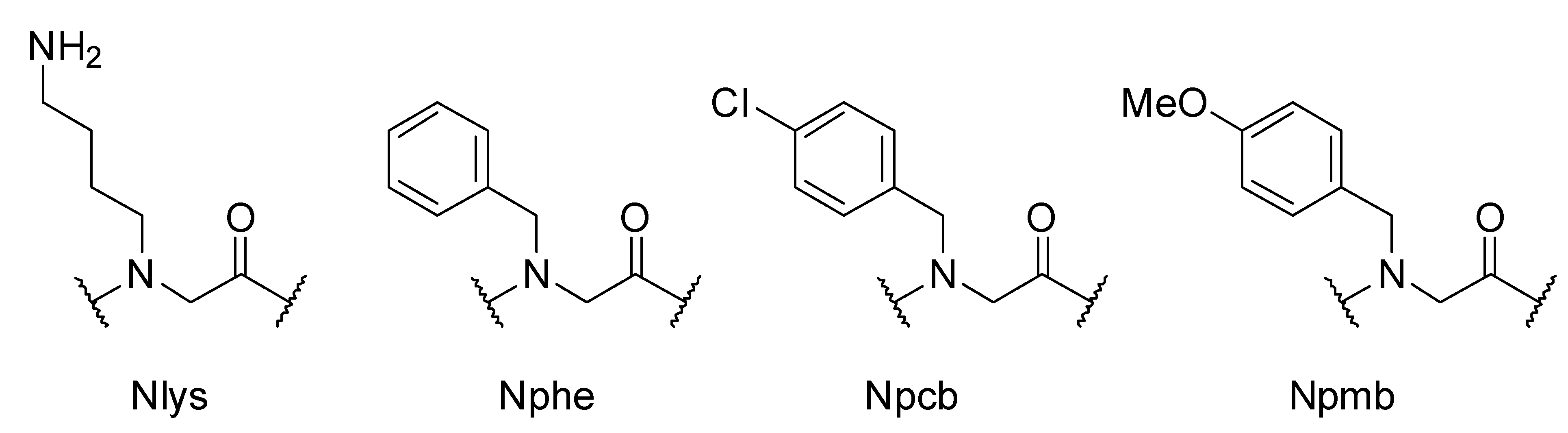
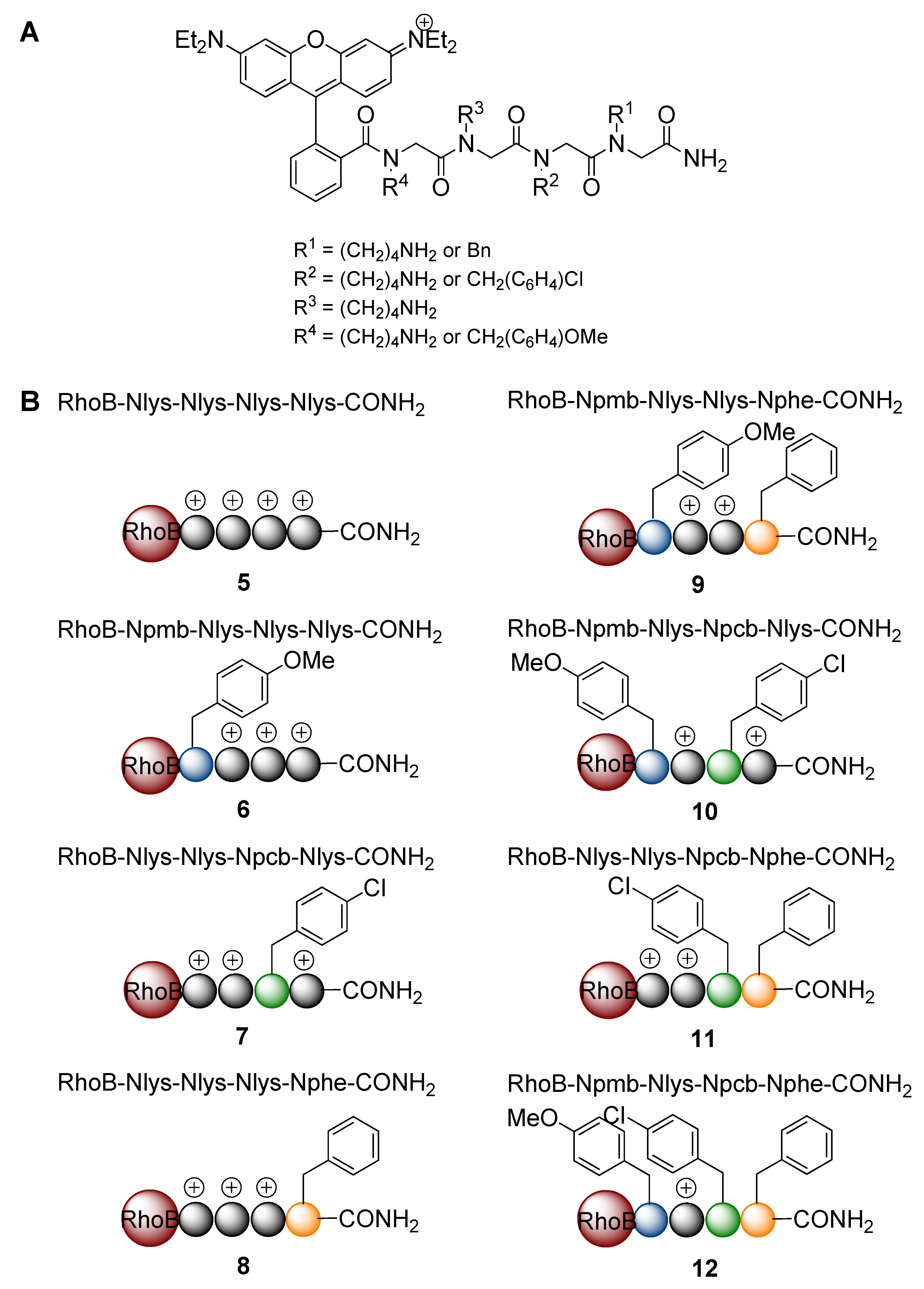
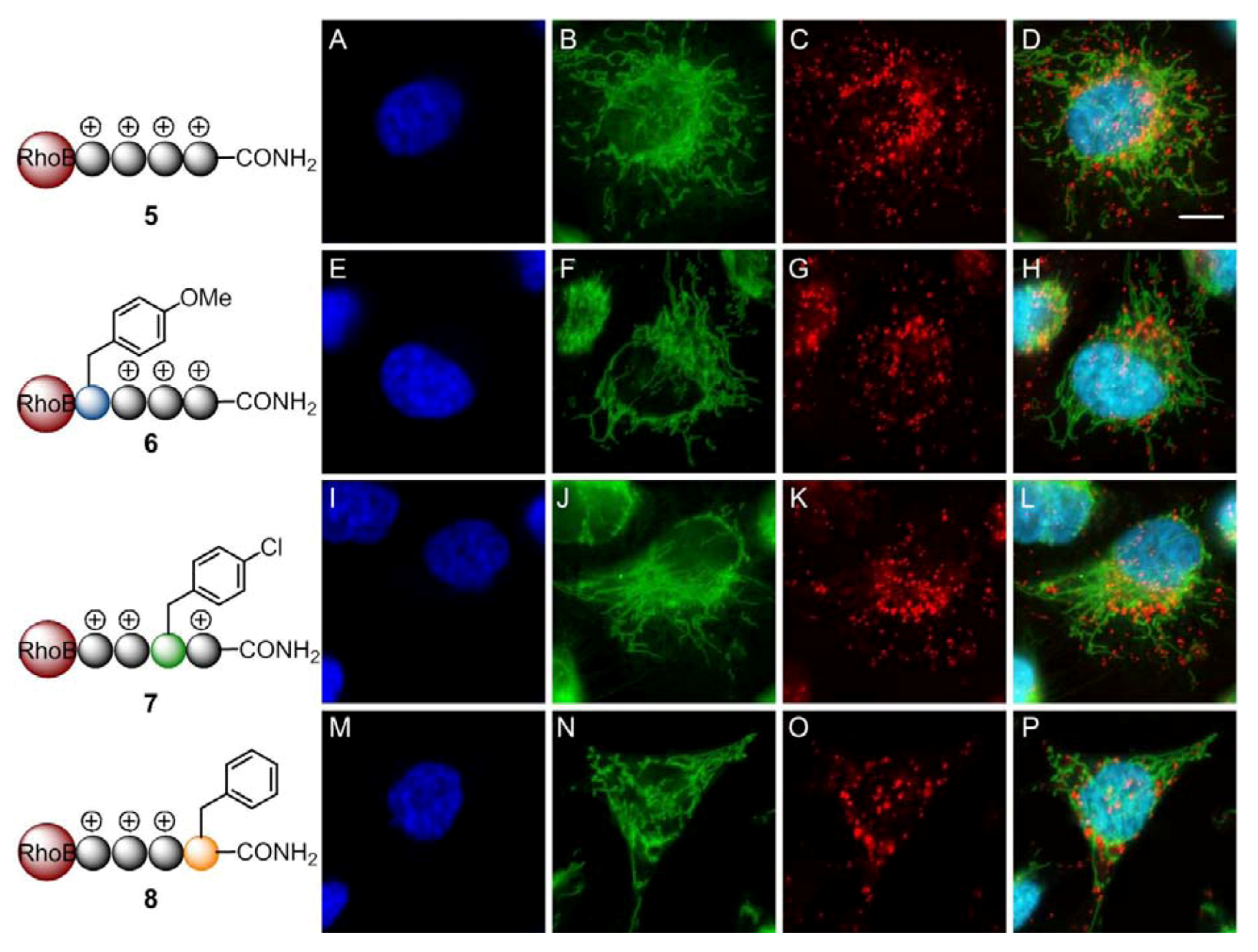
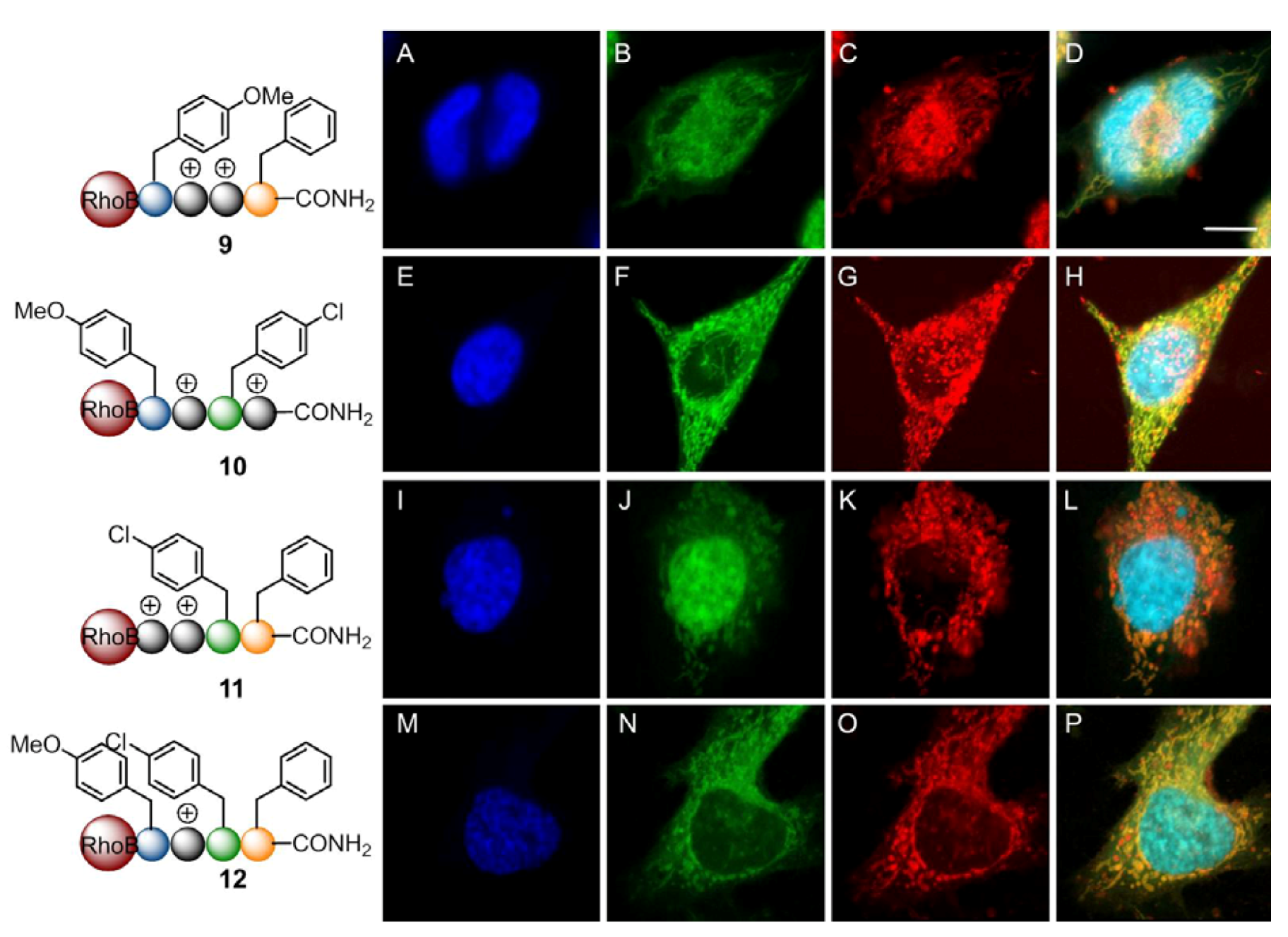
3. Experimental
3.1. General Remarks
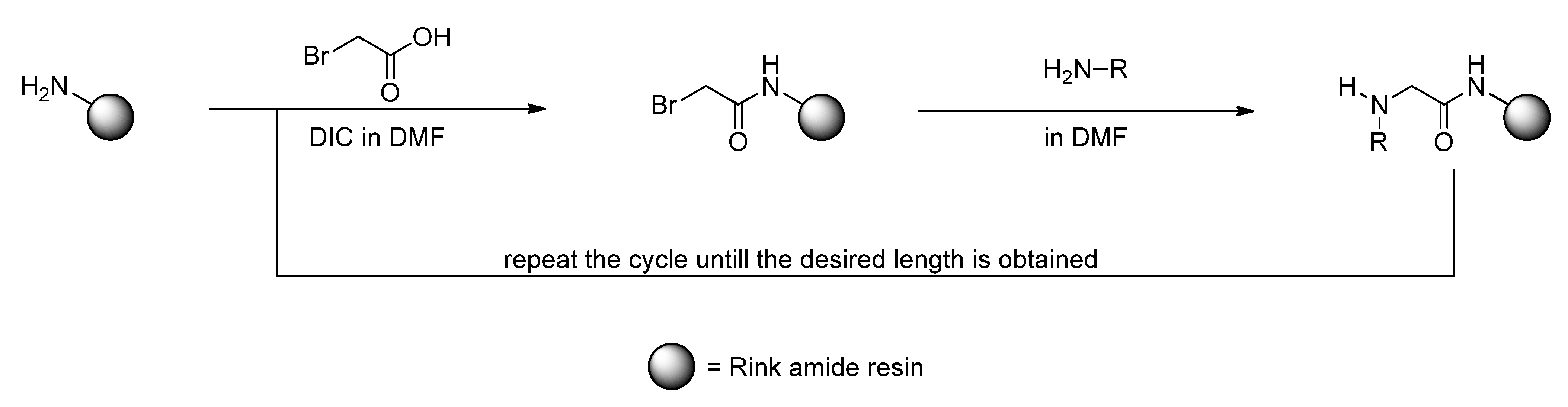
3.2. Sub-Monomer Synthesis
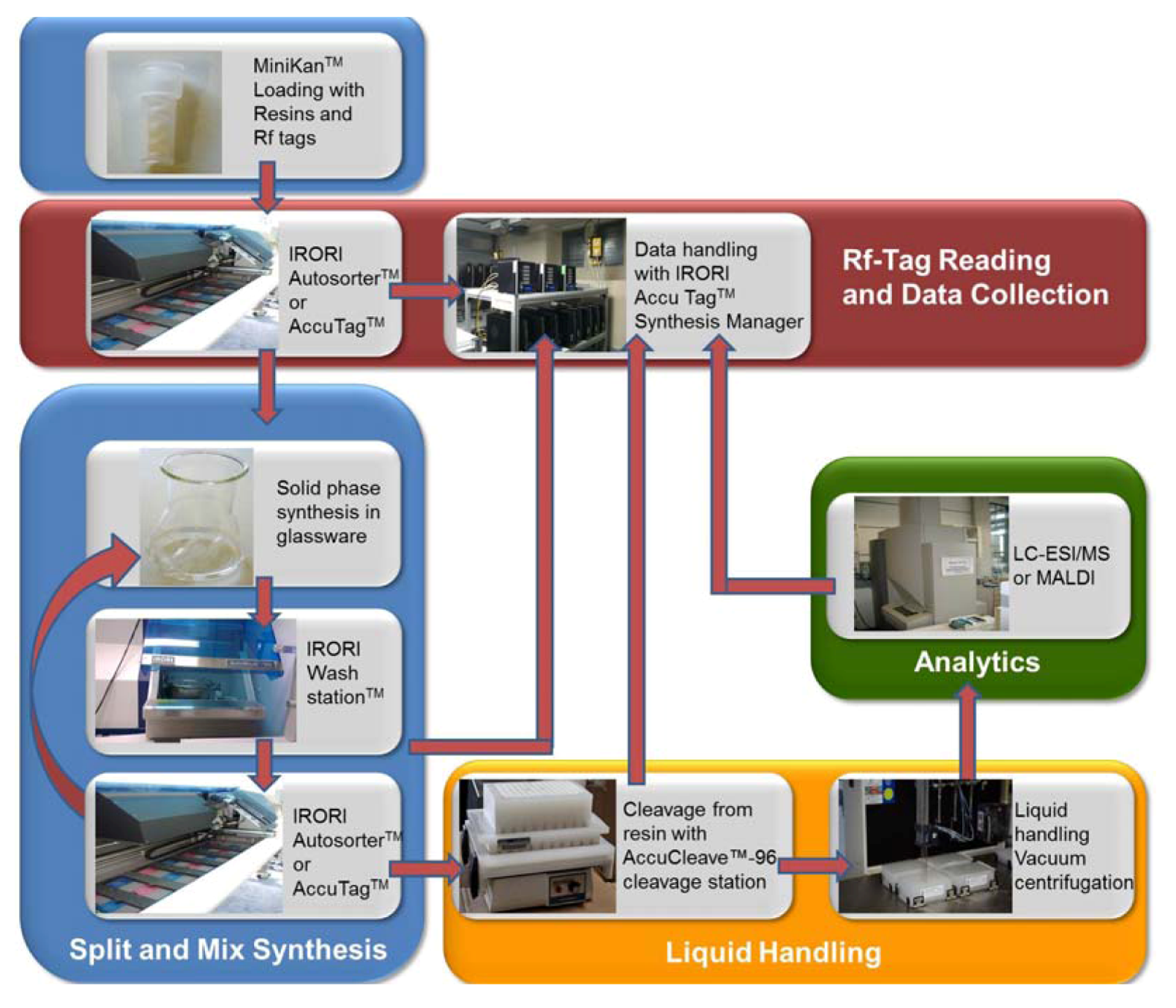
3.3. Synthesis of the Peptoid Library by using IRORI MiniKans (Data are given for 32 MiniKans in a 300 mL Erlenmeyer Flask)
3.4. Cell Culture Techniques for Mammalian Cells
3.5. Treatment of HeLa Cells with Rhodamine B Coupled Peptoids
3.6. Live Imaging by Confocal Microscopy
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Wadia, J.S.; Dowdy, S.F. Protein transduction technology. Curr. Opin. Biotechnol. 2002, 13, 52–56. [Google Scholar] [CrossRef]
- Vives, E.; Richard, J.P.; Rispal, C.; Lebleu, B. TAT peptide internalization: Seeking the mechanism of entry. Curr. Protein. Pept. Sci. 2003, 4, 125–132. [Google Scholar] [CrossRef]
- Simon, R.J.; Kania, R.S.; Zuckermann, R.N.; Huebner, V.D.; Jewell, D.A.; Banville, S.; Ng, S.; Wang, L.; Rosenberg, S.; Marlowe, C.K.; et al. Peptoids: A modular approach to drug discovery. Proc. Natl. Acad. Sci. USA 1992, 89, 9367–9371. [Google Scholar]
- Peretto, I.; Sanchez-Martin, R.M.; Wang, X.H.; Ellard, J.; Mittoo, S.; Bradley, M. Cell penetrable peptoid carrier vehicles: Synthesis and evaluation. Chem. Commun. 2003, 2312–2313. [Google Scholar]
- Sawant, R.; Torchilin, V. Intracellular transduction using cell-penetrating peptides. Mol. Biosyst. 2010, 6, 628–640. [Google Scholar] [CrossRef]
- Ezzat, K.; Zaghloul, E.M.; El Andaloussi, S.; Lehto, T.; El-Sayed, R.; Magdy, T.; Smith, C.I.; Langel, U. Solid formulation of cell-penetrating peptide nanocomplexes with siRNA and their stability in simulated gastric conditions. J. Control. Release 2012, 162, 1–8. [Google Scholar]
- Holm, T.; Raagel, H.; Andaloussi, S.E.; Hein, M.; Mae, M.; Pooga, M.; Langel, U. Retro-inversion of certain cell-penetrating peptides causes severe cellular toxicity. Biochim. Biophys. Acta 2011, 1808, 1544–1551. [Google Scholar] [CrossRef]
- Yin, H.; Moulton, H.M.; Betts, C.; Seow, Y.; Boutilier, J.; Iverson, P.L.; Wood, M.J. A fusion peptide directs enhanced systemic dystrophin exon skipping and functional restoration in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2009, 18, 4405–4414. [Google Scholar] [CrossRef]
- Umezawa, N.; Gelman, M.A.; Haigis, M.C.; Raines, R.T.; Gellman, S.H. Translocation of a beta-peptide across cell membranes. J. Am. Chem. Soc. 2002, 124, 368–369. [Google Scholar]
- Nakase, I.; Takeuchi, T.; Tanaka, G.; Futaki, S. Methodological and cellular aspects that govern the internalization mechanisms of arginine-rich cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 598–607. [Google Scholar] [CrossRef]
- Gademann, K.; Hintermann, T.; Schreiber, J.V. Beta-peptides: Twisting and turning. Curr. Med. Chem. 1999, 6, 905–925. [Google Scholar]
- Murphy, J.E.; Uno, T.; Hamer, J.D.; Cohen, F.E.; Dwarki, V.; Zuckermann, R.N. A combinatorial approach to the discovery of efficient cationic peptoid reagents for gene delivery. Proc. Natl. Acad. Sci. USA 1998, 95, 1517–1522. [Google Scholar] [CrossRef]
- Uno, T.; Beausoleil, E.; Goldsmith, R.A.; Levine, B.H.; Zuckermann, R.N. New submonomers for poly N-substituted glycines (peptoids). Tetrahedron Lett. 1999, 40, 1475–1478. [Google Scholar]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar]
- Wright, L.R.; Rothbard, J.B.; Wender, P.A. Guanidinium rich peptide transporters and drug delivery. Curr. Prot. Pept. Sci. 2003, 4, 105–124. [Google Scholar] [CrossRef]
- Schröder, T.; Schmitz, K.; Niemeier, N.; Balaban, T.S.; Krug, H.F.; Schepers, U.; Bräse, S. Solid-phase synthesis, bioconjugation, and toxicology of novel cationic oligopeptoids for cellular drug delivery. Bioconjug. Chem. 2007, 18, 342–354. [Google Scholar] [CrossRef]
- Schröder, T.; Niemeier, N.; Afonin, S.; Ulrich, A.S.; Krug, H.F.; Bräse, S. Peptoidic amino- and guanidinium-carrier systems: targeted drug delivery into the cell cytosol or the nucleus. J. Med. Chem. 2008, 51, 376–379. [Google Scholar]
- Eggenberger, K.; Birtalan, E.; Schröder, T.; Bräse, S.; Nick, P. Passage of Trojan peptoids into plant cells. ChemBioChem 2009, 10, 2504–2512. [Google Scholar]
- Rudat, B.; Birtalan, E.; Thomé, I.; Kölmel, D.K.; Horhoiu, V.L.; Wissert, M.D.; Lemmer, U.; Eisler, H.J.; Balaban, T.S.; Bräse, S. Novel pyridinium dyes that enable investigations of peptoids at the single-molecule level. J. Phys. Chem. B 2010, 114, 13473–13480. [Google Scholar]
- Birtalan, E.; Rudat, B.; Kölmel, D.K.; Fritz, D.; Vollrath, S.B.L.; Schepers, U.; Bräse, S. Investigating rhodamine B-Labeled peptoids: Scopes and limitations of its applications. Biopolymers 2011, 96, 694–701. [Google Scholar] [CrossRef]
- Lee, M.M.; French, J.M.; Disney, M.D. Influencing uptake and localization of aminoglycoside-functionalized peptoids. Mol. Biosyst. 2011, 7, 2441–2451. [Google Scholar] [CrossRef]
- Seo, J.; Ren, G.; Liu, H.; Miao, Z.; Park, M.; Wang, Y.; Miller, T.M.; Barron, A.E.; Cheng, Z. In Vivo biodistribution and small animal PET of (64)Cu-Labeled antimicrobial peptoids. Bioconjug. Chem. 2012, 23, 1069–1079. [Google Scholar]
- Huang, W.; Seo, J.; Lin, J.S.; Barron, A.E. Peptoid transporters: Effects of cationic, amphipathic structure on their cellular uptake. Mol. Biosyst. 2012, 8, 2626–2628. [Google Scholar]
- Tan, N.C.; Yu, P.; Kwon, Y.U.; Kodadek, T. High-throughput evaluation of relative cell permeability between peptoids and peptides. Bioorg. Med. Chem. 2008, 16, 5853–5861. [Google Scholar] [CrossRef]
- Aditya, A.; Kodadek, T. Incorporation of heterocycles into the backbone of peptoids to generate diverse peptoid-inspired one bead one compound libraries. ACS Comb. Sci. 2012, 14, 164–169. [Google Scholar] [CrossRef]
- Diaz-Mochon, J.J.; Fara, M.A.; Sanchez-Martin, R.M.; Bradley, M. Peptoid dendrimers-microwave-assisted solid-phase synthesis and transfection agent evaluation. Tetrahedron Lett. 2008, 49, 923–926. [Google Scholar]
- Fara, M.A.; Diaz-Mochon, J.J.; Bradley, M. Microwave-assisted coupling with DIC/HOBt for the synthesis of difficult peptoids and fluorescently labelled peptides—a gentle heat goes a long way. Tetrahedron Lett. 2006, 47, 1011–1014. [Google Scholar] [CrossRef]
- Li, S.; Bowerman, D.; Marthandan, N.; Klyza, S.; Luebke, K.J.; Garner, H.R.; Kodadek, T. Photolithographic synthesis of peptoids. J. Am. Chem. Soc. 2004, 126, 4088–4089. [Google Scholar]
- Olivos, H.J.; Alluri, P.G.; Reddy, M.M.; Salony, D.; Kodadek, T. Microwave-assisted solid-phase synthesis of peptoids. Org. Lett. 2002, 4, 4057–4059. [Google Scholar] [CrossRef]
- Goodman, B. Managing the workflow of a high-throughput organic synthesis laboratory: A marriage of automation and information management technologies. J. Lab. Automation 1999, 4, 48–52. [Google Scholar]
- Cheung, A.W.; Qi, L.; Gore, V.; Chu, X.J.; Bartkovitz, D.; Kurylko, G.; Swistok, J.; Danho, W.; Chen, L.; Yagaloff, K. Preparation of human Melanocortin-4 receptor agonist libraries: Linear peptides X-Y-DPhe7-Arg8-Trp(or 2-Nal)9-Z-NH2. Bioorg. Med. Chem. Lett. 2005, 15, 5504–5508. [Google Scholar] [CrossRef]
- Xiao, X.Y.; Li, R.S.; Zhuang, H.; Ewing, B.; Karunaratne, K.; Lillig, J.; Brown, R.; Nicolaou, K.C. Solid-phase combinatorial synthesis using MicroKan reactors, Rf tagging, and directed sorting. Biotechnol. Bioeng. 2000, 71, 44–50. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Xiao, X.Y.; Parandoosh, Z.; Senyei, A.; Nova, M.P. Radiofrequency encoded combinatorial chemistry. Angew. Chem. Int. Ed. 1995, 34, 2289–2291. [Google Scholar]
- Nagai, K.; Doi, T.; Sekiguchi, T.; Namatame, I.; Sunazuka, T.; Tomoda, H.; Omura, S.; Takahashi, T. Synthesis and biological evaluation of a beauveriolide analogue library. J. Comb. Chem. 2006, 8, 103–109. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Vourloumis, D.; Li, T.H.; Pastor, J.; Winssinger, N.; He, Y.; Ninkovic, S.; Sarabia, F.; Vallberg, H.; Roschangar, F.; et al. Designed epothilones: Combinatorial synthesis, tubulin assembly properties, and cytotoxic action against taxol-resistant tumor cells. Angew. Chem. Int. Ed. 1997, 36, 2097–2103. [Google Scholar] [CrossRef]
- Marsault, E.; Hoveyda, H.R.; Peterson, M.L.; Gagnon, R.; Vezina, M.; Pinault, J.F.; Landry, A.; Saint-Louis, C.; Ouellet, L.G.; Beauchemin, S.; et al. High throughput solid phase parallel synthesis of macrocyclic peptidomimetics. Adv. Exp. Med. Biol. 2009, 611, 15–16. [Google Scholar] [CrossRef]
- Zuckermann, R.N.; Kerr, J.M.; Kent, S.B.H.; Moos, W.H. Efficient method for the preparation of peptoids [Oligo(N-Substituted Glycines)] by submonomer solid-phase synthesis. J. Am. Chem. Soc. 1992, 114, 10646–10647. [Google Scholar]
- Culf, A.S.; Ouellette, R.J. Solid-phase synthesis of N-substituted glycine oligomers (alpha-peptoids) and derivatives. Molecules 2010, 15, 5282–5335. [Google Scholar] [CrossRef]
- Goun, E.A.; Shinde, R.; Dehnert, K.W.; Adams-Bond, A.; Wender, P.A.; Contag, C.H.; Franc, B.L. Intracellular cargo delivery by an octaarginine transporter adapted to target prostate cancer cells through cell surface protease activation. Bioconjug. Chem. 2006, 17, 787–796. [Google Scholar] [CrossRef]
- Wender, P.A.; Jessop, T.C.; Pattabiraman, K.; Pelkey, E.T.; VanDeusen, C.L. An efficient, scalable synthesis of the molecular transporter octaarginine via a segment doubling strategy. Org. Lett. 2001, 3, 3229–3232. [Google Scholar]
- Schröder, T.; Quintilla, A.; Setzler, J.; Birtalan, E.; Wenzel, W.; Bräse, S. Joint experimental and theoretical investigation of the propensity of peptoids as drug carriers. WSEAS Trans. Biol. Biomed. 2007, 4, 145–148. [Google Scholar]
- Chen, C.L.; Qi, J.; Zuckermann, R.N.; DeYoreo, J.J. Engineered biomimetic polymers as tunable agents for controlling CaCO3 mineralization. J. Am. Chem. Soc. 2011, 133, 5214–5217. [Google Scholar]
- Thakkar, A.; Cohen, A.S.; Connolly, M.D.; Zuckermann, R.N.; Pei, D. High-throughput sequencing of peptoids and peptide-peptoid hybrids by partial edman degradation and mass spectrometry. J. Comb. Chem. 2009, 11, 294–302. [Google Scholar] [CrossRef]
- Simpson, L. S.; Kodadek, T. A cleavable scaffold strategy for the synthesis of one-bead one-compound cyclic peptoid libraries that can be sequenced by tandem mass spectrometry. Tetrahedron Lett. 2012, 53, 2341–2344. [Google Scholar] [CrossRef]
- Aquino, C.; Sarkar, M.; Chalmers, M.J.; Mendes, K.; Kodadek, T.; Micalizio, G.C. A biomimetic polyketide-inspired approach to small-molecule ligand discovery. Nat. Chem. 2012, 4, 99–104. [Google Scholar]
- Hooks, J.C.; Matharage, J.P.; Udugamasooriya, D.G. Development of homomultimers and heteromultimers of lung cancer-specific peptoids. Biopolymers 2011, 96, 567–577. [Google Scholar] [CrossRef]
- Klimkait, T.; Felder, E.R.; Albrecht, G.; Hamy, F. Rational optimization of a HIV-1 Tat inhibitor: rapid progress on combinatorial lead structures. Biotechnol. Bioeng. 1998, 61, 155–168. [Google Scholar]
- D'Souza, G.G.; Cheng, S.M.; Boddapati, S.V.; Horobin, R.W.; Weissig, V. Nanocarrier-assisted sub-cellular targeting to the site of mitochondria improves the pro-apoptotic activity of paclitaxel. J. Drug Target. 2008, 16, 578–585. [Google Scholar] [CrossRef]
- Boddapati, S.V.; D'Souza, G.G.; Erdogan, S.; Torchilin, V.P.; Weissig, V. Organelle-targeted nanocarriers: Specific delivery of liposomal ceramide to mitochondria enhances its cytotoxicity in vitro and in vivo. Nano Lett. 2008, 8, 2559–2563. [Google Scholar] [CrossRef]
- Weissig, V. Targeted drug delivery to mammalian mitochondria in living cells. Expert Opin. Drug. Deliv. 2005, 2, 89–102. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar]
- Chinnery, P.F.; Turnbull, D.M. Mitochondrial DNA mutations in the pathogenesis of human disease. Mol. Med. Today 2000, 6, 425–432. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondria and cancer: Warburg addressed. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 363–374. [Google Scholar] [CrossRef]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef]
- Horton, K.L.; Kelley, S.O. Engineered apoptosis-inducing peptides with enhanced mitochondrial localization and potency. J. Med. Chem. 2009, 52, 3293–3299. [Google Scholar] [CrossRef]
- Horton, K.L.; Stewart, K.M.; Fonseca, S.B.; Guo, Q.; Kelley, S.O. Mitochondria-penetrating peptides. Chem. Biol. 2008, 15, 375–382. [Google Scholar] [CrossRef]
- Horton, K.L.; Pereira, M.P.; Stewart, K.M.; Fonseca, S.B.; Kelley, S.O. Tuning the activity of mitochondria-penetrating peptides for delivery or disruption. ChemBioChem 2012, 13, 476–485. [Google Scholar] [CrossRef]
- Kelley, S.O.; Stewart, K.M.; Mourtada, R. Development of novel peptides for mitochondrial drug delivery: Amino acids featuring delocalized lipophilic cations. Pharm. Res. 2011, 28, 2808–2819. [Google Scholar] [CrossRef]
- Yousif, L.F.; Stewart, K.M.; Horton, K.L.; Kelley, S.O. Mitochondria-penetrating peptides: sequence effects and model cargo transport. ChemBioChem 2009, 10, 2081–2088. [Google Scholar] [CrossRef]
- Yousif, L.F.; Stewart, K.M.; Kelley, S.O. Targeting mitochondria with organelle-specific compounds: Strategies and applications. ChemBioChem 2009, 10, 1939–1950. [Google Scholar] [CrossRef]
- Furka, A.; Sebestyen, F.; Asgedom, M.; Dibo, G. General method for rapid synthesis of multicomponent peptide mixtures. Int. J. Pept. Protein Res. 1991, 37, 487–493. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kölmel, D.K.; Fürniss, D.; Susanto, S.; Lauer, A.; Grabher, C.; Bräse, S.; Schepers, U. Cell Penetrating Peptoids (CPPos): Synthesis of a Small Combinatorial Library by Using IRORI MiniKans. Pharmaceuticals 2012, 5, 1265-1281. https://doi.org/10.3390/ph5121265
Kölmel DK, Fürniss D, Susanto S, Lauer A, Grabher C, Bräse S, Schepers U. Cell Penetrating Peptoids (CPPos): Synthesis of a Small Combinatorial Library by Using IRORI MiniKans. Pharmaceuticals. 2012; 5(12):1265-1281. https://doi.org/10.3390/ph5121265
Chicago/Turabian StyleKölmel, Dominik K., Daniel Fürniss, Steven Susanto, Andrea Lauer, Clemens Grabher, Stefan Bräse, and Ute Schepers. 2012. "Cell Penetrating Peptoids (CPPos): Synthesis of a Small Combinatorial Library by Using IRORI MiniKans" Pharmaceuticals 5, no. 12: 1265-1281. https://doi.org/10.3390/ph5121265