DNA Methylation as Clinically Useful Biomarkers—Light at the End of the Tunnel

{kind=link}
Abstract
:1. Introduction
2. cfcDNA as the Source of Genomic DNA for Analysis
3. DNA Methylation as a Biomarker
3.1. Location
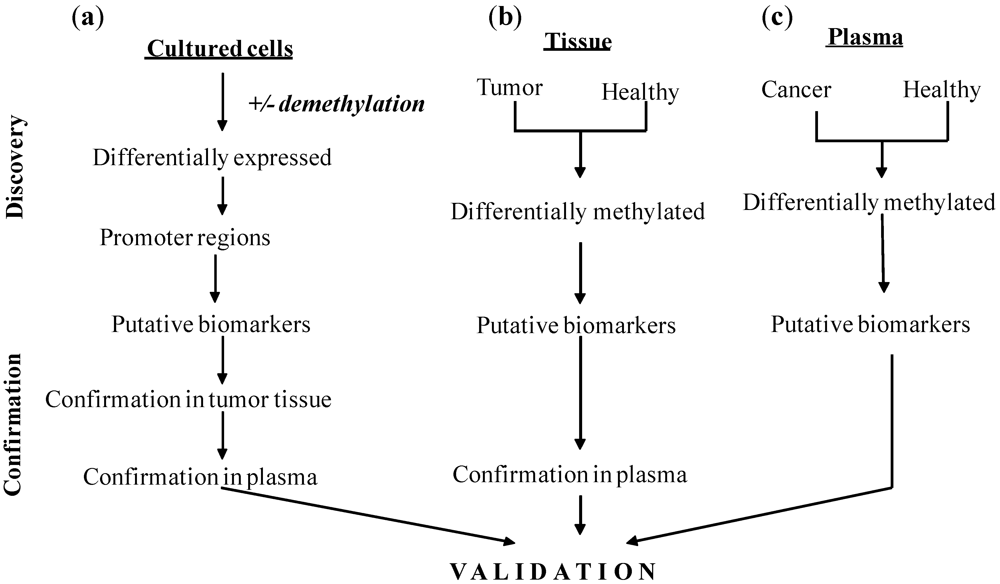
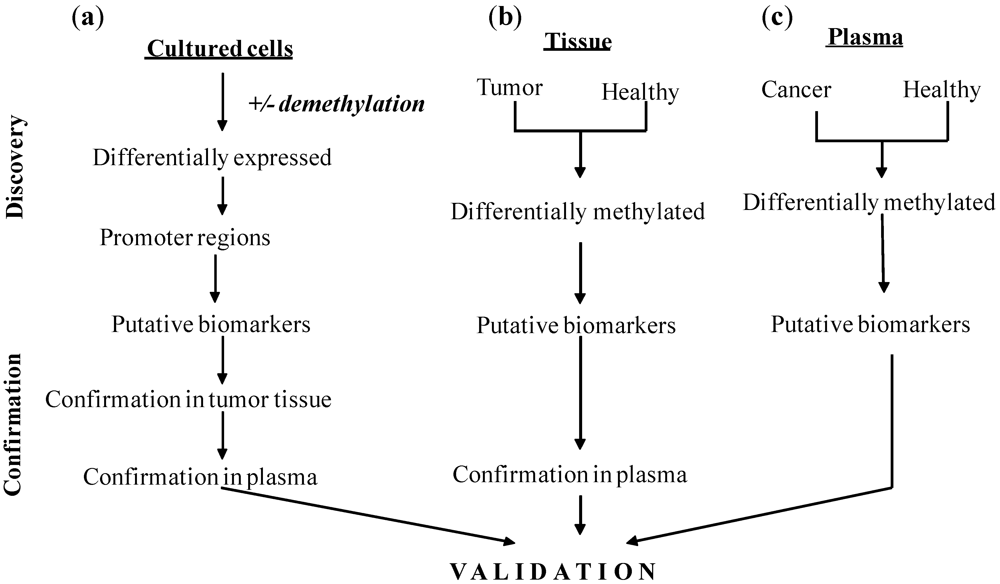
3.2. Development of Biomarkers—Approaches and Techniques
3.3. Development of Biomarkers—Different Requirements for Different Biomarkers
3.4. Blood-Based DNA Methylation Biomarkers for Detection and Diagnosis
3.5. Blood-Based DNA Methylation Biomarkers for Other Diseases
3.6. Blood-Based DNA Methylation Biomarkers for Monitoring of Drug Treatment
4. Conclusions
Acknowledgements
References
- Gibson, D.G.; Glass, J.I.; Lartigue, C.; Noskov, V.N.; Chuang, R.Y.; Algire, M.A.; Benders, G.A.; Montague, M.G.; Ma, L.; Moodie, M.M.; et al. Creation of a bacterial cell controlled by a chemically synthesized genome. Science 2010, 329, 52–56. [Google Scholar]
- Warner, E.; Causer, P.A.; Wong, J.W.; Wright, F.C.; Jong, R.A.; Hill, K.A.; Messner, S.J.; Yaffe, M.J.; Narod, S.A.; Plewes, D.B. Improvement in DCIS detection rates by MRI over time in a high-risk breast screening study. Breast J. 2011, 17, 9–17. [Google Scholar] [CrossRef]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar]
- Ordonez, D.; Sanchez, A.J.; Martinez-Rodriguez, J.E.; Cisneros, E.; Ramil, E.; Romo, N.; Moraru, M.; Munteis, E.; Lopez-Botet, M.; Roquer, J.; et al. Multiple sclerosis associates with LILRA3 deletion in Spanish patients. Genes Immun. 2009, 10, 579–585. [Google Scholar] [CrossRef]
- Kauffman, M.A.; Gonzalez-Moron, D.; Garcea, O.; Villa, A.M. TNFSFR1A R92Q mutation, autoinflammatory symptoms and multiple sclerosis in a cohort from Argentina. Mol. Biol. Rep. 2011, 39, 117–121. [Google Scholar]
- Campbell, G.R.; Ziabreva, I.; Reeve, A.K.; Krishnan, K.J.; Reynolds, R.; Howell, O.; Lassmann, H.; Turnbull, D.M.; Mahad, D.J. Mitochondrial DNA deletions and neurodegeneration in multiple sclerosis. Ann. Neurol. 2011, 69, 481–492. [Google Scholar] [CrossRef]
- Baronica, K.B.; Mlinac, K.; Ozretic, D.; Vladic, A.; Bognar, S.K. Arylsulfatase a gene polymorphisms in relapsing remitting multiple sclerosis: Genotype-phenotype correlation and estimation of disease progression. Coll. Antropol. 2011, 35, S11–S16. [Google Scholar]
- Kubarenko, A.V.; Ranjan, S.; Rautanen, A.; Mills, T.C.; Wong, S.; Vannberg, F.; Neumaier, M.; Bekeredjian-Ding, I.; Hill, A.V.; Ahmad-Nejad, P.; et al. A naturally occurring variant in human TLR9, P99L, is associated with loss of CpG oligonucleotide responsiveness. J. Biol. Chem. 2010, 285, 36486–36494. [Google Scholar]
- Segat, L.; Catamo, E.; Fabris, A.; Padovan, L.; Morgutti, M.; Crovella, S. HLA-G 3' UTR haplotypes and HIV vertical transmission. AIDS 2009, 23, 1916–1918. [Google Scholar] [CrossRef]
- Issa, J.P.; Kantarjian, H.M. Targeting DNA methylation. Clin. Cancer Res. 2009, 15, 3938–3946. [Google Scholar] [CrossRef]
- Matei, D.E.; Nephew, K.P. Epigenetic therapies for chemoresensitization of epithelial ovarian cancer. Gynecol. Oncol. 2010, 116, 195–201. [Google Scholar] [CrossRef]
- Liggett, T.E.; Melnikov, A.A.; Marks, J.R.; Levenson, V.V. Methylation patterns in cell-free plasma DNA reflect removal of the primary tumor and drug treatment of breast cancer patients. Int. J. Cancer 2011, 128, 492–499. [Google Scholar] [CrossRef]
- Shivapurkar, N.; Gazdar, A.F. DNA methylation based biomarkers in non-invasive cancer screening. Curr. Mol. Med. 2010, 10, 123–132. [Google Scholar] [CrossRef]
- Gahan, P.B.; Swaminathan, R. Circulating nucleic acids in plasma and serum. Recent developments. Ann. NY Acad. Sci. 2008, 1137, 1–6. [Google Scholar]
- Atamaniuk, J.; Vidotto, C.; Kinzlbauer, M.; Bachl, N.; Tiran, B.; Tschan, H. Cell-free plasma DNA and purine nucleotide degradation markers following weightlifting exercise. Eur. J. Appl. Physiol. 2010, 110, 695–701. [Google Scholar] [CrossRef]
- Gahan, P.B.; Anker, P.; Stroun, M. Metabolic DNA as the origin of spontaneously released DNA? Ann. NY Acad. Sci. 2008, 1137, 7–17. [Google Scholar] [CrossRef]
- van der Vaart, M.; Pretorius, P.J. Circulating DNA. Its origin and fluctuation. Ann. NY Acad. Sci. 2008, 1137, 18–26. [Google Scholar] [CrossRef]
- Anker, P.; Stroun, M.; Maurice, P.A. Spontaneous release of DNA by human blood lymphocytes as shown in an in vitro system. Cancer Res. 1975, 35, 2375–2382. [Google Scholar]
- Chan, K.C.; Leung, S.F.; Yeung, S.W.; Chan, A.T.; Lo, Y.M. Quantitative analysis of the transrenal excretion of circulating EBV DNA in nasopharyngeal carcinoma patients. Clin. Cancer Res. 2008, 14, 4809–4813. [Google Scholar]
- Lo, Y.M.; Rainer, T.H.; Chan, L.Y.; Hjelm, N.M.; Cocks, R.A. Plasma DNA as a prognostic marker in trauma patients. Clin. Chem. 2000, 46, 319–323. [Google Scholar]
- Giacona, M.B.; Ruben, G.C.; Iczkowski, K.A.; Roos, T.B.; Porter, D.M.; Sorenson, G.D. Cell-free DNA in human blood plasma: Length measurements in patients with pancreatic cancer and healthy controls. Pancreas 1998, 17, 89–97. [Google Scholar] [CrossRef]
- Liggett, T.; Melnikov, A.; Yi, Q.L.; Replogle, C.; Brand, R.; Kaul, K.; Talamonti, M.; Abrams, R.A.; Levenson, V. Differential methylation of cell-free circulating DNA among patients with pancreatic cancer versus chronic pancreatitis. Cancer 2010, 116, 1674–1680. [Google Scholar]
- Rainer, T.H.; Wong, L.K.; Lam, W.; Yuen, E.; Lam, N.Y.; Metreweli, C.; Lo, Y.M. Prognostic use of circulating plasma nucleic acid concentrations in patients with acute stroke. Clin. Chem. 2003, 49, 562–569. [Google Scholar] [CrossRef]
- Goebel, G.; Zitt, M.; Zitt, M.; Muller, H.M. Circulating nucleic acids in plasma or serum (CNAPS) as prognostic and predictive markers in patients with solid neoplasias. Dis. Markers 2005, 21, 105–120. [Google Scholar]
- Liggett, T.E.; Melnikov, A.; Yi, Q.; Replogle, C.; Hu, W.; Rotmensch, J.; Kamat, A.; Sood, A.K.; Levenson, V. Distinctive DNA methylation patterns of cell-free plasma DNA in women with malignant ovarian tumors. Gynecol. Oncol. 2011, 120, 113–120. [Google Scholar] [CrossRef]
- Lee, T.H.; Montalvo, L.; Chrebtow, V.; Busch, M.P. Quantitation of genomic DNA in plasma and serum samples: Higher concentrations of genomic DNA found in serum than in plasma. Transfusion 2001, 41, 276–282. [Google Scholar] [CrossRef]
- Thijssen, M.A.; Swinkels, D.W.; Ruers, T.J.; de Kok, J.B. Difference between free circulating plasma and serum DNA in patients with colorectal liver metastases. Anticancer Res. 2002, 22, 421–425. [Google Scholar]
- Holdenrieder, S.; Stieber, P.; Chan, L.Y.; Geiger, S.; Kremer, A.; Nagel, D.; Lo, Y.M. Cell-free DNA in serum and plasma: Comparison of ELISA and quantitative PCR. Clin. Chem. 2005, 51, 1544–1546. [Google Scholar] [CrossRef]
- Zhong, X.Y.; Hahn, S.; Kiefer, V.; Holzgreve, W. Is the quantity of circulatory cell-free DNA in human plasma and serum samples associated with gender, age and frequency of blood donations? Ann. Hematol. 2007, 86, 139–143. [Google Scholar]
- Holdenrieder, S.; Burges, A.; Reich, O.; Spelsberg, F.W.; Stieber, P. DNA integrity in plasma and serum of patients with malignant and benign diseases. Ann. NY Acad. Sci. 2008, 1137, 162–170. [Google Scholar] [CrossRef]
- Mancuso, R.; Hernis, A.; Cavarretta, R.; Caputo, D.; Calabrese, E.; Nemni, R.; Ferrante, P.; Delbue, S.; Clerici, M. Detection of viral DNA sequences in the cerebrospinal fluid of patients with multiple sclerosis. J. Med. Virol. 2010, 82, 1051–1057. [Google Scholar] [CrossRef]
- Jeong, B.H.; Lee, Y.J.; Carp, R.I.; Kim, Y.S. The prevalence of human endogenous retroviruses in cerebrospinal fluids from patients with sporadic Creutzfeldt-Jakob disease. J. Clin. Virol. 2010, 47, 136–142. [Google Scholar] [CrossRef]
- Al-Yatama, M.K.; Mustafa, A.S.; Ali, S.; Abraham, S.; Khan, Z.; Khaja, N. Detection of Y chromosome-specific DNA in the plasma and urine of pregnant women using nested polymerase chain reaction. Prenat. Diagn. 2001, 21, 399–402. [Google Scholar] [CrossRef]
- Majer, S.; Bauer, M.; Magnet, E.; Strele, A.; Giegerl, E.; Eder, M.; Lang, U.; Pertl, B. Maternal urine for prenatal diagnosis—An analysis of cell-free fetal DNA in maternal urine and plasma in the third trimester. Prenat. Diagn. 2007, 27, 1219–1223. [Google Scholar] [CrossRef]
- Shekhtman, E.M.; Anne, K.; Melkonyan, H.S.; Robbins, D.J.; Warsof, S.L.; Umansky, S.R. Optimization of transrenal DNA analysis: Detection of fetal DNA in maternal urine. Clin. Chem. 2009, 55, 723–729. [Google Scholar] [CrossRef]
- Baylin, S.B.; Herman, J.G.; Graff, J.R.; Vertino, P.M.; Issa, J.P. Alterations in DNA methylation: A fundamental aspect of neoplasia. Adv. Cancer Res. 1998, 72, 141–196. [Google Scholar]
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetic gene silencing in cancer: The DNA hypermethylome. Hum. Mol. Genet. 2007, 16, R50–R59. [Google Scholar] [CrossRef]
- Flanagan, J.M.; Munoz-Alegre, M.; Henderson, S.; Tang, T.; Sun, P.; Johnson, N.; Fletcher, O.; Dos Santos Silva, I.; Peto, J.; Boshoff, C.; et al. Gene-body hypermethylation of ATM in peripheral blood DNA of bilateral breast cancer patients. Hum. Mol. Genet. 2009, 18, 1332–1342. [Google Scholar]
- Kurukuti, S.; Tiwari, V.K.; Tavoosidana, G.; Pugacheva, E.; Murrell, A.; Zhao, Z.; Lobanenkov, V.; Reik, W.; Ohlsson, R. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc. Natl. Acad. Sci. USA 2006, 103, 10684–10689. [Google Scholar]
- Choufani, S.; Shapiro, J.S.; Susiarjo, M.; Butcher, D.T.; Grafodatskaya, D.; Lou, Y.; Ferreira, J.C.; Pinto, D.; Scherer, S.W.; Shaffer, L.G.; et al. A novel approach identifies new differentially methylated regions (DMRs) associated with imprinted genes. Genome Res. 2011, 21, 465–476. [Google Scholar]
- Eggermann, T.; Leisten, I.; Binder, G.; Begemann, M.; Spengler, S. Disturbed methylation at multiple imprinted loci: An increasing observation in imprinting disorders. Epigenomics 2011, 3, 625–637. [Google Scholar] [CrossRef]
- Szpakowski, S.; Sun, X.; Lage, J.M.; Dyer, A.; Rubinstein, J.; Kowalski, D.; Sasaki, C.; Costa, J.; Lizardi, P.M. Loss of epigenetic silencing in tumors preferentially affects primate-specific retroelements. Gene 2009, 448, 151–167. [Google Scholar] [CrossRef]
- Estecio, M.R.; Gallegos, J.; Vallot, C.; Castoro, R.J.; Chung, W.; Maegawa, S.; Oki, Y.; Kondo, Y.; Jelinek, J.; Shen, L.; et al. Genome architecture marked by retrotransposons modulates predisposition to DNA methylation in cancer. Genome Res. 2010, 20, 1369–1382. [Google Scholar] [CrossRef]
- Akimoto, K.; Katakami, H.; Kim, H.J.; Ogawa, E.; Sano, C.M.; Wada, Y.; Sano, H. Epigenetic inheritance in rice plants. Ann. Bot. 2007, 100, 205–217. [Google Scholar] [CrossRef]
- Maksakova, I.A.; Mager, D.L.; Reiss, D. Keeping active endogenous retroviral-like elements in heck: The epigenetic perspective. Cell. Mol. Life Sci. 2008, 65, 3329–3347. [Google Scholar] [CrossRef]
- Levenson, V.V. DNA methylation as a universal biomarker. Expert Rev. Mol. Diagn. 2010, 10, 481–488. [Google Scholar] [CrossRef]
- Tsai, H.C.; Baylin, S.B. Cancer epigenetics: Linking basic biology to clinical medicine. Cell Res. 2011, 21, 502–517. [Google Scholar] [CrossRef]
- Rodriguez-Paredes, M.; Esteller, M. Cancer epigenetics reaches mainstream oncology. Nat. Med. 2011, 17, 330–339. [Google Scholar] [CrossRef]
- Virmani, A.K.; Rathi, A.; Sathyanarayana, U.G.; Padar, A.; Huang, C.X.; Cunnigham, H.T.; Farinas, A.J.; Milchgrub, S.; Euhus, D.M.; Gilcrease, M.; et al. Aberrant methylation of the adenomatous polyposis coli (APC) gene promoter 1A in breast and lung carcinomas. Clin. Cancer Res. 2001, 7, 1998–2004. [Google Scholar]
- Xing, M.; Usadel, H.; Cohen, Y.; Tokumaru, Y.; Guo, Z.; Westra, W.B.; Tong, B.C.; Tallini, G.; Udelsman, R.; Califano, J.A.; et al. Methylation of the thyroid-stimulating hormone receptor gene in epithelial thyroid tumors: A marker of malignancy and a cause of gene silencing. Cancer Res. 2003, 63, 2316–2321. [Google Scholar]
- Machida, E.O.; Brock, M.V.; Hooker, C.M.; Nakayama, J.; Ishida, A.; Amano, J.; Picchi, M.A.; Belinsky, S.A.; Herman, J.G.; Taniguchi, S.; et al. Hypermethylation of ASC/TMS1 is a sputum marker for late-stage lung cancer. Cancer Res. 2006, 66, 6210–6218. [Google Scholar]
- Anglim, P.P.; Galler, J.S.; Koss, M.N.; Hagen, J.A.; Turla, S.; Campan, M.; Weisenberger, D.J.; Laird, P.W.; Siegmund, K.D.; Laird-Offringa, I.A. Identification of a panel of sensitive and specific DNA methylation markers for squamous cell lung cancer. Mol. Cancer 2008, 7, 62. [Google Scholar] [CrossRef]
- Weaver, K.D.; Grossman, S.A.; Herman, J.G. Methylated tumor-specific DNA as a plasma biomarker in patients with glioma. Cancer Invest. 2006, 24, 35–40. [Google Scholar] [CrossRef]
- Huang, T.H.; Perry, M.R.; Laux, D.E. Methylation profiling of CpG islands in human breast cancer cells. Hum. Mol. Genet. 1999, 8, 459–470. [Google Scholar] [CrossRef]
- Shames, D.S.; Girard, L.; Gao, B.; Sato, M.; Lewis, C.M.; Shivapurkar, N.; Jiang, A.; Perou, C.M.; Kim, Y.H.; Pollack, J.R.; et al. A genome-wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies. PLoS Med. 2006, 3, e486. [Google Scholar]
- Shen, L.; Kondo, Y.; Guo, Y.; Zhang, J.; Zhang, L.; Ahmed, S.; Shu, J.; Chen, X.; Waterland, R.A.; Issa, J.P. Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters. PLoS Genet. 2007, 3, 2023–2036. [Google Scholar]
- Estecio, M.R.; Yan, P.S.; Ibrahim, A.E.; Tellez, C.S.; Shen, L.; Huang, T.H.; Issa, J.P. High-throughput methylation profiling by MCA coupled to CpG island microarray. Genome Res. 2007, 17, 1529–1536. [Google Scholar] [CrossRef]
- Ordway, J.M.; Budiman, M.A.; Korshunova, Y.; Maloney, R.K.; Bedell, J.A.; Citek, R.W.; Bacher, B.; Peterson, S.; Rohlfing, T.; Hall, J.; et al. Identification of novel high-frequency DNA methylation changes in breast cancer. PLoS One 2007, 2, e1314. [Google Scholar]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar]
- Mori, Y.; Olaru, A.V.; Cheng, Y.; Agarwal, R.; Yang, J.; Luvsanjav, D.; Yu, W.; Selaru, F.M.; Hutfless, S.; Lazarev, M.; et al. Novel candidate colorectal cancer biomarkers identified by methylation microarray-based scanning. Endocr. Relat. Cancer 2011, 18, 465–478. [Google Scholar] [CrossRef]
- Guerrero-Preston, R.; Soudry, E.; Acero, J.; Orera, M.; Moreno-Lopez, L.; Macia-Colon, G.; Jaffe, A.; Berdasco, M.; Ili-Gangas, C.; Brebi-Mieville, P.; et al. NID2 and HOXA9 promoter hypermethylation as biomarkers for prevention and early detection in oral cavity squamous cell carcinoma tissues and saliva. Cancer Prev. Res. (Phila.) 2011, 4, 1061–1072. [Google Scholar] [CrossRef]
- Sova, P.; Feng, Q.; Geiss, G.; Wood, T.; Strauss, R.; Rudolf, V.; Lieber, A.; Kiviat, N. Discovery of novel methylation biomarkers in cervical carcinoma by global demethylation and microarray analysis. Cancer Epidemiol. Biomarkers Prev. 2006, 15, 114–123. [Google Scholar] [CrossRef]
- Ibanez de Caceres, I.; Dulaimi, E.; Hoffman, A.M.; Al-Saleem, T.; Uzzo, R.G.; Cairns, P. Identification of novel target genes by an epigenetic reactivation screen of renal cancer. Cancer Res. 2006, 66, 5021–5028. [Google Scholar]
- Khulan, B.; Thompson, R.F.; Ye, K.; Fazzari, M.J.; Suzuki, M.; Stasiek, E.; Figueroa, M.E.; Glass, J.L.; Chen, Q.; Montagna, C.; et al. Comparative isoschizomer profiling of cytosine methylation: The HELP assay. Genome Res. 2006, 16, 1046–1055. [Google Scholar] [CrossRef]
- Yan, P.S.; Chen, C.M.; Shi, H.; Rahmatpanah, F.; Wei, S.H.; Huang, T.H. Applications of CpG island microarrays for high-throughput analysis of DNA methylation. J. Nutr. 2002, 132, 2430–2434. [Google Scholar]
- Gebhard, C.; Schwarzfischer, L.; Pham, T.H.; Schilling, E.; Klug, M.; Andreesen, R.; Rehli, M. Genome-wide profiling of CpG methylation identifies novel targets of aberrant hypermethylation in myeloid leukemia. Cancer Res. 2006, 66, 6118–6128. [Google Scholar]
- Weng, Y.I.; Huang, T.H.; Yan, P.S. Methylated DNA immunoprecipitation and microarray-based analysis: Detection of DNA methylation in breast cancer cell lines. Methods Mol. Biol. 2009, 590, 165–176. [Google Scholar] [CrossRef]
- Melnikov, A.A.; Gartenhaus, R.B.; Levenson, A.S.; Motchoulskaia, N.A.; Levenson Chernokhvostov, V.V. MSRE-PCR for analysis of gene-specific DNA methylation. Nucleic Acids Res. 2005, 33, e93. [Google Scholar] [CrossRef]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.H.; LeProust, E.M.; Park, I.H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar]
- Guo, J.U.; Ma, D.K.; Mo, H.; Ball, M.P.; Jang, M.H.; Bonaguidi, M.A.; Balazer, J.A.; Eaves, H.L.; Xie, B.; Ford, E.; et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011, 14, 1345–1351. [Google Scholar]
- Kim, J.H.; Dhanasekaran, S.M.; Prensner, J.R.; Cao, X.; Robinson, D.; Kalyana-Sundaram, S.; Huang, C.; Shankar, S.; Jing, X.; Iyer, M.; et al. Deep sequencing reveals distinct patterns of DNA methylation in prostate cancer. Genome Res. 2011, 21, 1028–1041. [Google Scholar] [CrossRef]
- Laird, P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Umbricht, C.B.; Evron, E.; Gabrielson, E.; Ferguson, A.; Marks, J.; Sukumar, S. Hypermethylation of 14-3-3 sigma (stratifin) is an early event in breast cancer. Oncogene 2001, 20, 3348–3353. [Google Scholar] [CrossRef]
- Issa, J.P.; Ahuja, N.; Toyota, M.; Bronner, M.P.; Brentnall, T.A. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001, 61, 3573–3577. [Google Scholar]
- Guo, M.; House, M.G.; Hooker, C.; Han, Y.; Heath, E.; Gabrielson, E.; Yang, S.C.; Baylin, S.B.; Herman, J.G.; Brock, M.V. Promoter hypermethylation of resected bronchial margins: A field defect of changes? Clin. Cancer Res. 2004, 10, 5131–5136. [Google Scholar]
- Wolff, E.M.; Chihara, Y.; Pan, F.; Weisenberger, D.J.; Siegmund, K.D.; Sugano, K.; Kawashima, K.; Laird, P.W.; Jones, P.A.; Liang, G. Unique DNA methylation patterns distinguish noninvasive and invasive urothelial cancers and establish an epigenetic field defect in premalignant tissue. Cancer Res. 2010, 70, 8169–8178. [Google Scholar]
- Califano, J.; van der Riet, P.; Westra, W.; Nawroz, H.; Clayman, G.; Piantadosi, S.; Corio, R.; Lee, D.; Greenberg, B.; Koch, W.; et al. Genetic progression model for head and neck cancer: Implications for field cancerization. Cancer Res. 1996, 56, 2488–2492. [Google Scholar]
- Nguyen, H.; Loustaunau, C.; Facista, A.; Ramsey, L.; Hassounah, N.; Taylor, H.; Krouse, R.; Payne, C.M.; Tsikitis, V.L.; Goldschmid, S.; et al. Deficient Pms2, ERCC1, Ku86, CcOI in field defects during progression to colon cancer. J. Vis. Exp. 2010, 41, 1931. [Google Scholar]
- Bhusari, S.; Yang, B.; Kueck, J.; Huang, W.; Jarrard, D.F. Insulin-like growth factor-2 (IGF2) loss of imprinting marks a field defect within human prostates containing cancer. Prostate 2011, 71, 1621–1630. [Google Scholar] [CrossRef]
- Hegi, M.E.; Liu, L.; Herman, J.G.; Stupp, R.; Wick, W.; Weller, M.; Mehta, M.P.; Gilbert, M.R. Correlation of O6-methylguanine methyltransferase (MGMT) promoter methylation with clinical outcomes in glioblastoma and clinical strategies to modulate MGMT activity. J. Clin. Oncol. 2008, 26, 4189–4199. [Google Scholar]
- Ahn, J.B.; Chung, W.B.; Maeda, O.; Shin, S.J.; Kim, H.S.; Chung, H.C.; Kim, N.K.; Issa, J.P. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer 2011, 117, 1847–1854. [Google Scholar]
- Dai, W.; Teodoridis, J.M.; Zeller, C.; Graham, J.; Hersey, J.; Flanagan, J.M.; Stronach, E.; Millan, D.W.; Siddiqui, N.; Paul, J.; et al. Systematic CpG islands methylation profiling of genes in the Wnt pathway in epithelial ovarian cancer identifies biomarkers of progression-free survival. Clin. Cancer Res. 2011, 17, 4052–4062. [Google Scholar]
- Ibragimova, I.; Cairns, P. Assays for hypermethylation of the BRCA1 gene promoter in tumor cells to predict sensitivity to PARP-inhibitor therapy. Methods Mol. Biol. 2011, 780, 277–291. [Google Scholar] [CrossRef]
- Oldenhuis, C.N.; Oosting, S.F.; Gietema, J.A.; de Vries, E.G. Prognostic versus predictive value of biomarkers in oncology. Eur. J. Cancer 2008, 44, 946–953. [Google Scholar] [CrossRef]
- Misek, D.E.; Patwa, T.H.; Lubman, D.M.; Simeone, D.M. Early detection and biomarkers in pancreatic cancer. J. Natl. Compr. Cancer Netw. 2007, 5, 1034–1041. [Google Scholar]
- Urban, N.; Thorpe, J.D.; Bergan, L.A.; Forrest, R.M.; Kampani, A.V.; Scholler, N.; O'Briant, K.C.; Anderson, G.L.; Cramer, D.W.; Berg, C.D.; et al. Potential role of HE4 in multimodal screening for epithelial ovarian cancer. J. Natl. Cancer Inst. 2011, 103, 1630–1634. [Google Scholar] [CrossRef]
- Cho, Y.H.; Yazici, H.; Wu, H.C.; Terry, M.B.; Gonzalez, K.; Qu, M.; Dalay, N.; Santella, R.M. Aberrant promoter hypermethylation and genomic hypomethylation in tumor, adjacent normal tissues and blood from breast cancer patients. Anticancer Res. 2010, 30, 2489–2496. [Google Scholar]
- Pedersen, K.S.; Bamlet, W.R.; Oberg, A.L.; de Andrade, M.; Matsumoto, M.E.; Tang, H.; Thibodeau, S.N.; Petersen, G.M.; Wang, L. Leukocyte DNA methylation signature differentiates pancreatic cancer patients from healthy controls. PLoS One 2011, 6, e18223. [Google Scholar]
- Sapienza, C.; Lee, J.; Powell, J.; Erinle, O.; Yafai, F.; Reichert, J.; Siraj, E.S.; Madaio, M. DNA methylation profiling identifies epigenetic differences between diabetes patients with ESRD and diabetes patients without nephropathy. Epigenetics 2011, 6, 20–28. [Google Scholar] [CrossRef]
- Hoque, M.O.; Feng, Q.; Toure, P.; Dem, A.; Critchlow, C.W.; Hawes, S.E.; Wood, T.; Jeronimo, C.; Rosenbaum, E.; Stern, J.; et al. Detection of aberrant methylation of four genes in plasma DNA for the detection of breast cancer. J. Clin. Oncol. 2006, 24, 4262–4269. [Google Scholar]
- Vinayanuwattikun, C.; Sriuranpong, V.; Tanasanvimon, S.; Chantranuwat, P.; Mutirangura, A. Epithelial-specific methylation marker: A potential plasma biomarker in advanced non-small cell lung cancer. J. Thorac. Oncol. 2011, 6, 1818–1825. [Google Scholar] [CrossRef]
- Kneip, C.; Schmidt, B.; Seegebarth, A.; Weickmann, S.; Fleischhacker, M.; Liebenberg, V.; Field, J.K.; Dietrich, D. SHOX2 DNA methylation is a biomarker for the diagnosis of lung cancer in plasma. J. Thorac. Oncol. 2011, 6, 1632–1638. [Google Scholar]
- Begum, S.; Brait, M.; Dasgupta, S.; Ostrow, K.L.; Zahurak, M.; Carvalho, A.L.; Califano, J.A.; Goodman, S.N.; Westra, W.H.; Hoque, M.O.; et al. An epigenetic marker panel for detection of lung cancer using cell-free serum DNA. Clin. Cancer Res. 2011, 17, 4494–4503. [Google Scholar]
- Hoque, M.O.; Kim, M.S.; Ostrow, K.L.; Liu, J.; Wisman, G.B.; Park, H.L.; Poeta, M.L.; Jeronimo, C.; Henrique, R.; Lendvai, A.; et al. Genome-wide promoter analysis uncovers portions of the cancer methylome. Cancer Res. 2008, 68, 2661–2670. [Google Scholar]
- Shao, C.; Sun, W.; Tan, M.; Glazer, C.A.; Bhan, S.; Zhong, X.; Fakhry, C.; Sharma, R.; Westra, W.H.; Hoque, M.O.; et al. Integrated, genome-wide screening for hypomethylated oncogenes in salivary gland adenoid cystic carcinoma. Clin. Cancer Res. 2011, 17, 4320–4330. [Google Scholar] [CrossRef]
- Liang, G.; Salem, C.E.; Yu, M.C.; Nguyen, H.D.; Gonzales, F.A.; Nguyen, T.T.; Nichols, P.W.; Jones, P.A. DNA methylation differences associated with tumor tissues identified by genome scanning analysis. Genomics 1998, 53, 260–268. [Google Scholar] [CrossRef]
- Toyota, M.; Ho, C.; Ahuja, N.; Jair, K.W.; Li, Q.; Ohe-Toyota, M.; Baylin, S.B.; Issa, J.P. Identification of differentially methylated sequences in colorectal cancer by methylated CpG island amplification. Cancer Res. 1999, 59, 2307–2312. [Google Scholar]
- Lofton-Day, C.; Model, F.; Devos, T.; Tetzner, R.; Distler, J.; Schuster, M.; Song, X.; Lesche, R.; Liebenberg, V.; Ebert, M.; et al. DNA methylation biomarkers for blood-based colorectal cancer screening. Clin. Chem. 2008, 54, 414–423. [Google Scholar]
- Church, T.R.; Wandell, M.; Lofton-Day, C.; Mongin, S.; Blumenstein, B.A.; Allen, J.I.; Roesch, T.; Snover, D.; Day, R.; Ransohoff, D.F. Prospective clinical validation of an assay for methylated SEPT9 DNA in human plasma as a colorectal cancer screening tool in average risk men and women ≥50 years. Gastroenterology 2010, 139, e18. [Google Scholar]
- Montagna, C.; Lyu, M.S.; Hunter, K.; Lukes, L.; Lowther, W.; Reppert, T.; Hissong, B.; Weaver, Z.; Ried, T. The Septin 9 (MSF) gene is amplified and overexpressed in mouse mammary gland adenocarcinomas and human breast cancer cell lines. Cancer Res. 2003, 63, 2179–2187. [Google Scholar]
- Connolly, D.; Yang, Z.; Castaldi, M.; Simmons, N.; Oktay, M.H.; Coniglio, S.; Fazzari, M.J.; Verdier-Pinard, P.; Montagna, C. Septin 9 isoform expression, localization and epigenetic changes during human and mouse breast cancer progression. Breast Cancer Res. 2011, 13, R76. [Google Scholar]
- Burrows, J.F.; Chanduloy, S.; McIlhatton, M.A.; Nagar, H.; Yeates, K.; Donaghy, P.; Price, J.; Godwin, A.K.; Johnston, P.G.; Russell, S.E. Altered expression of the septin gene, SEPT9, in ovarian neoplasia. J. Pathol. 2003, 201, 581–588. [Google Scholar] [CrossRef]
- Scott, M.; McCluggage, W.G.; Hillan, K.J.; Hall, P.A.; Russell, S.E. Altered patterns of transcription of the septin gene, SEPT9, in ovarian tumorigenesis. Int. J. Cancer 2006, 118, 1325–1329. [Google Scholar]
- Bennett, K.L.; Karpenko, M.; Lin, M.T.; Claus, R.; Arab, K.; Dyckhoff, G.; Plinkert, P.; Herpel, E.; Smiraglia, D.; Plass, C. Frequently methylated tumor suppressor genes in head and neck squamous cell carcinoma. Cancer Res. 2008, 68, 4494–4499. [Google Scholar]
- Bennett, K.L.; Lee, W.; Lamarre, E.; Zhang, X.; Seth, R.; Scharpf, J.; Hunt, J.; Eng, C. HPV status-independent association of alcohol and tobacco exposure or prior radiation therapy with promoter methylation of FUSSEL18, EBF3, IRX1, and SEPT9, but not SLC5A8, in head and neck squamous cell carcinomas. Genes Chromosomes Cancer 2010, 49, 319–326. [Google Scholar]
- Scott, M.; Hyland, P.L.; McGregor, G.; Hillan, K.J.; Russell, S.E.; Hall, P.A. Multimodality expression profiling shows SEPT9 to be overexpressed in a wide range of human tumours. Oncogene 2005, 24, 4688–4700. [Google Scholar]
- Melnikov, A.; Scholtens, D.; Godwin, A.; Levenson, V. Differential methylation profile of ovarian cancer in tissues and plasma. J. Mol. Diagn. 2009, 11, 60–65. [Google Scholar] [CrossRef]
- Chang, H.; Yi, B.; Li, L.; Zhang, H.Y.; Sun, F.; Dong, S.Q.; Cao, Y. Methylation of tumor associated genes in tissue and plasma samples from liver disease patients. Exp. Mol. Pathol. 2008, 85, 96–100. [Google Scholar] [CrossRef]
- Iyer, P.; Zekri, A.R.; Hung, C.W.; Schiefelbein, E.; Ismail, K.; Hablas, A.; Seifeldin, I.A.; Soliman, A.S. Concordance of DNA methylation pattern in plasma and tumor DNA of Egyptian hepatocellular carcinoma patients. Exp. Mol. Pathol. 2010, 88, 107–111. [Google Scholar] [CrossRef]
- Vaissiere, T.; Cuenin, C.; Paliwal, A.; Vineis, P.; Hoek, G.; Krzyzanowski, M.; Airoldi, L.; Dunning, A.; Garte, S.; Hainaut, P.; et al. Quantitative analysis of DNA methylation after whole bisulfitome amplification of a minute amount of DNA from body fluids. Epigenetics 2009, 4, 221–230. [Google Scholar]
- Levenson, V.V.; Melnikov, A.A. The MethDet: A technology for biomarker development. Expert Rev. Mol. Diagn. 2011, 11, 807–812. [Google Scholar] [CrossRef]
- Melnikov, A.A.; Scholtens, D.; Talamonti, M.S.; Bentrem, D.J.; Levenson, V.V. Methylation profile of circulating plasma DNA in patients with pancreatic cancer. J. Surg. Oncol. 2009, 99, 119–122. [Google Scholar] [CrossRef]
- Liggett, T.; Melnikov, A.; Tilwalli, S.; Yi, Q.; Chen, H.; Replogle, C.; Feng, X.; Reder, A.; Stefoski, D.; Balabanov, R.; et al. Methylation patterns of cell-free plasma DNA in relapsing-remitting multiple sclerosis. J. Neurol. Sci. 2010, 290, 16–21. [Google Scholar] [CrossRef]
- Cassinotti, E.; Melson, J.; Liggett, T.; Melnikov, A.; Yi, Q.; Replogle, C.; Mobarhan, S.; Boni, L.; Segato, S.; Levenson, V. DNA methylation patterns in blood of patients with colorectal cancer and adenomatous colorectal polyps. Int. J. Cancer 2011. [Google Scholar] [CrossRef]
- Papageorgiou, E.A.; Karagrigoriou, A.; Tsaliki, E.; Velissariou, V.; Carter, N.P.; Patsalis, P.C. Fetal-specific DNA methylation ratio permits noninvasive prenatal diagnosis of trisomy 21. Nat. Med. 2011, 17, 510–513. [Google Scholar]
- Mill, J.; Tang, T.; Kaminsky, Z.; Khare, T.; Yazdanpanah, S.; Bouchard, L.; Jia, P.; Assadzadeh, A.; Flanagan, J.; Schumacher, A.; et al. Epigenomic profiling reveals DNA-methylation changes associated with major psychosis. Am. J. Hum. Genet. 2008, 82, 696–711. [Google Scholar] [CrossRef]
- Kaminsky, Z.; Tochigi, M.; Jia, P.; Pal, M.; Mill, J.; Kwan, A.; Ioshikhes, I.; Vincent, J.B.; Kennedy, J.L.; Strauss, J.; et al. A multi-tissue analysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol. Psychiatry 2011. [Google Scholar] [CrossRef]
- Carrard, A.; Salzmann, A.; Malafosse, A.; Karege, F. Increased DNA methylation status of the serotonin receptor 5HTR1A gene promoter in schizophrenia and bipolar disorder. J. Affect. Disord. 2011, 132, 450–453. [Google Scholar] [CrossRef]
- Sharma, P.; Kumar, J.; Garg, G.; Kumar, A.; Patowary, A.; Karthikeyan, G.; Ramakrishnan, L.; Brahmachari, V.; Sengupta, S. Detection of altered global DNA methylation in coronary artery disease patients. DNA Cell Biol. 2008, 27, 357–365. [Google Scholar] [CrossRef]
- Baccarelli, A.; Wright, R.; Bollati, V.; Litonjua, A.; Zanobetti, A.; Tarantini, L.; Sparrow, D.; Vokonas, P.; Schwartz, J. Ischemic heart disease and stroke in relation to blood DNA methylation. Epidemiology 2010, 21, 819–828. [Google Scholar] [CrossRef]
- Kim, M.; Long, T.I.; Arakawa, K.; Wang, R.; Yu, M.C.; Laird, P.W. DNA methylation as a biomarker for cardiovascular disease risk. PLoS One 2010, 5, e9692. [Google Scholar]
- Cash, H.L.; McGarvey, S.T.; Houseman, E.A.; Marsit, C.J.; Hawley, N.L.; Lambert-Messerlian, G.M.; Viali, S.; Tuitele, J.; Kelsey, K.T. Cardiovascular disease risk factors and DNA methylation at the LINE-1 repeat region in peripheral blood from Samoan Islanders. Epigenetics 2011, 6, 1257–1264. [Google Scholar] [CrossRef]
- Kaur, P.; Paliwal, A.; Durantel, D.; Hainaut, P.; Scoazec, J.Y.; Zoulim, F.; Chemin, I.; Herceg, Z. DNA methylation of hepatitis B virus (HBV) genome associated with the development of hepatocellular carcinoma and occult HBV infection. J. Infect. Dis. 2010, 202, 700–704. [Google Scholar]
- Li, W.; Sun, W.; Liu, L.; Yang, F.; Li, Y.; Chen, Y.; Fang, J.; Zhang, W.; Wu, J.; Zhu, Y. IL-32: A host proinflammatory factor against influenza viral replication is upregulated by aberrant epigenetic modifications during influenza A virus infection. J. Immunol. 2010, 185, 5056–5065. [Google Scholar]
- Leonard, S.; Wei, W.; Anderton, J.; Vockerodt, M.; Rowe, M.; Murray, P.G.; Woodman, C.B. Epigenetic and transcriptional changes which follow Epstein-Barr virus infection of germinal center B cells and their relevance to the pathogenesis of Hodgkin’s lymphoma. J. Virol. 2011, 85, 9568–9577. [Google Scholar]
- Shin, S.H.; Park, S.Y.; Ko, J.S.; Kim, N.; Kang, G.H. Aberrant CpG island hypermethylation in pediatric gastric mucosa in association with Helicobacter pylori infection. Arch. Pathol. Lab. Med. 2011, 135, 759–765. [Google Scholar]
- Uddin, M.; Aiello, A.E.; Wildman, D.E.; Koenen, K.C.; Pawelec, G.; de Los Santos, R.; Goldmann, E.; Galea, S. Epigenetic and immune function profiles associated with posttraumatic stress disorder. Proc. Natl. Acad. Sci. USA 2010, 107, 9470–9475. [Google Scholar]
- de Rooij, S.R.; Costello, P.M.; Veenendaal, M.V.; Lillycrop, K.A.; Gluckman, P.D.; Hanson, M.A.; Painter, R.C.; Roseboom, T.J. Associations between DNA methylation of a glucocorticoid receptor promoter and acute stress responses in a large healthy adult population are largely explained by lifestyle and educational differences. Psychoneuroendocrinology 2011, in press. [Google Scholar]
- Li, C.; Xu, M.; Wang, S.; Yang, X.; Zhou, S.; Zhang, J.; Liu, Q.; Sun, Y. Lead exposure suppressed ALAD transcription by increasing methylation level of the promoter CpG islands. Toxicol. Lett. 2011, 203, 48–53. [Google Scholar] [CrossRef]
- Madrigano, J.; Baccarelli, A.; Mittleman, M.A.; Wright, R.O.; Sparrow, D.; Vokonas, P.S.; Tarantini, L.; Schwartz, J. Prolonged exposure to particulate pollution, genes associated with glutathione pathways, and DNA methylation in a cohort of older men. Environ. Health Perspect. 2011, 119, 977–982. [Google Scholar] [CrossRef]
- Mathers, J.C.; Strathdee, G.; Relton, C.L. Induction of epigenetic alterations by dietary and other environmental factors. Adv. Genet. 2010, 71, 3–39. [Google Scholar] [CrossRef]
- Cheng, J.C.; Weisenberger, D.J.; Gonzales, F.A.; Liang, G.; Xu, G.L.; Hu, Y.G.; Marquez, V.E.; Jones, P.A. Continuous zebularine treatment effectively sustains demethylation in human bladder cancer cells. Mol. Cell. Biol. 2004, 24, 1270–1278. [Google Scholar]
- Appleton, K.; Mackay, H.J.; Judson, I.; Plumb, J.A.; McCormick, C.; Strathdee, G.; Lee, C.; Barrett, S.; Reade, S.; Jadayel, D.; et al. Phase I and pharmacodynamic trial of the DNA methyltransferase inhibitor decitabine and carboplatin in solid tumors. J. Clin. Oncol. 2007, 25, 4603–4609. [Google Scholar]
- Fang, F.; Balch, C.; Schilder, J.; Breen, T.; Zhang, S.; Shen, C.; Li, L.; Kulesavage, C.; Snyder, A.J.; Nephew, K.P.; et al. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer 2010, 116, 4043–4053. [Google Scholar] [CrossRef]
- Zambrano, P.; Segura-Pacheco, B.; Perez-Cardenas, E.; Cetina, L.; Revilla-Vazquez, A.; Taja-Chayeb, L.; Chavez-Blanco, A.; Angeles, E.; Cabrera, G.; Sandoval, K.; et al. A phase I study of hydralazine to demethylate and reactivate the expression of tumor suppressor genes. BMC Cancer 2005, 5, 44. [Google Scholar]
- Chuang, J.C.; Yoo, C.B.; Kwan, J.M.; Li, T.W.; Liang, G.; Yang, A.S.; Jones, P.A. Comparison of biological effects of non-nucleoside DNA methylation inhibitors versus 5-aza-2'-deoxycytidine. Mol. Cancer Ther. 2005, 4, 1515–1520. [Google Scholar] [CrossRef]
- Whitaker, J.W.; McConkey, G.A.; Westhead, D.R. The transferome of metabolic genes explored: Analysis of the horizontal transfer of enzyme encoding genes in unicellular eukaryotes. Genome Biol. 2009, 10, R36. [Google Scholar] [CrossRef]
- Schaack, S.; Gilbert, C.; Feschotte, C. Promiscuous DNA: Horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol. Evol. 2010, 25, 537–546. [Google Scholar] [CrossRef]
- Dunning Hotopp, J.C. Horizontal gene transfer between bacteria and animals. Trends Genet. 2011, 27, 157–163. [Google Scholar] [CrossRef]
- Garcia-Olmo, D.C.; Dominguez, C.; Garcia-Arranz, M.; Anker, P.; Stroun, M.; Garcia-Verdugo, J.M.; Garcia-Olmo, D. Cell-free nucleic acids circulating in the plasma of colorectal cancer patients induce the oncogenic transformation of susceptible cultured cells. Cancer Res. 2010, 70, 560–567. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Levenson, V.V.; Melnikov, A.A. DNA Methylation as Clinically Useful Biomarkers—Light at the End of the Tunnel. Pharmaceuticals 2012, 5, 94-113. https://doi.org/10.3390/ph5010094
Levenson VV, Melnikov AA. DNA Methylation as Clinically Useful Biomarkers—Light at the End of the Tunnel. Pharmaceuticals. 2012; 5(1):94-113. https://doi.org/10.3390/ph5010094
Chicago/Turabian StyleLevenson, Victor V., and Anatoliy A. Melnikov. 2012. "DNA Methylation as Clinically Useful Biomarkers—Light at the End of the Tunnel" Pharmaceuticals 5, no. 1: 94-113. https://doi.org/10.3390/ph5010094