Lp-PLA2 Inhibition—The Atherosclerosis Panacea?

Department of Internal Medicine II-Cardiology, University of Ulm Medical Center, Ulm, Germany
*
Author to whom correspondence should be addressed.
Pharmaceuticals 2010, 3(5), 1360-1373; https://doi.org/10.3390/ph3051360
Submission received: 3 February 2010
/
Accepted: 21 April 2010
/
Published: 29 April 2010
(This article belongs to the Special Issue Biomarkers)
Abstract
:Based on the complex pathophysiology of atherosclerosis, a large number of biomarkers that relate to lipids, inflammation, immunity, thrombosis and hemostasis, have been investigated experimentally, in epidemiologic studies and in clinical trials. Interest focuses on their potential role to aid in risk stratification, as possible surrogate markers of atherosclerosis, and potential targets for therapy. More recently, one lipid associated biomarker, lipoprotein-associated phospholipase A2 (Lp-PLA2), has gained considerable interest. In addition to a plausible pathophysiological role by generating pro-inflammatory and pro-atherogenic compounds from oxidized LDL in the vessel wall, there is a large, fairly consistent epidemiological database indicating that increased levels of Lp-PLA2 mass or activity are associated with increased risk for cardiovascular outcomes; such data further suggest that it might improve risk stratification. In addition, clinical studies indicate that increased Lp-PLA2 levels are associated with endothelial dysfunction. Moreover, it may also serve as an interesting therapeutic target, since a specific inhibitor of the enzyme is available with promising animal data and initial positive data in humans. Recent experimental data from a hyperlipidemic diabetic pig model strongly suggest that increased Lp-PLA2 in the vessel wall is associated with a more vulnerable plaque phenotype which can be modulated by inhibiting Lp-PLA2 activity. A biomarker study in more than 1,000 patients with CHD over three months has demonstrated a positive effect on various inflammatory molecules. In addition, an imaging study using IVUS based modalities (greyscale, virtual histology, and palpography) together with a panel of biomarkers (IBIS-2) has been done in more than 300 patients with CHD treated over 12 months and results indicate that the progression of the necrotic core of the plaque can be retarded. Inhibition of the pro-atherogenic and pro-inflammatory effects of Lp-PLA2 may therefore contribute to decrease the residual risk in high risk patients already on polypharmacotherapy. This hypothesis is now being tested in two large phase 3 clinical trials. Thus, Lp-PLA2 indeed may represent a biomarker and a promising target for intervention.
1. Biochemistry and Biology
Based on the complex pathophysiology of atherosclerosis, a large number of biomarkers that relate to lipids, inflammation, immunity, thrombosis and hemostasis, have been investigated experimentally, in epidemiologic studies, and in clinical trials. Interest focuses on their potential role to aid in risk stratification, as possible surrogate markers of atherosclerosis, and potential targets for therapy. More recently, one lipid associated biomarker, lipoprotein-associated phospholipase A2 (Lp-PLA2), has gained considerable interest.
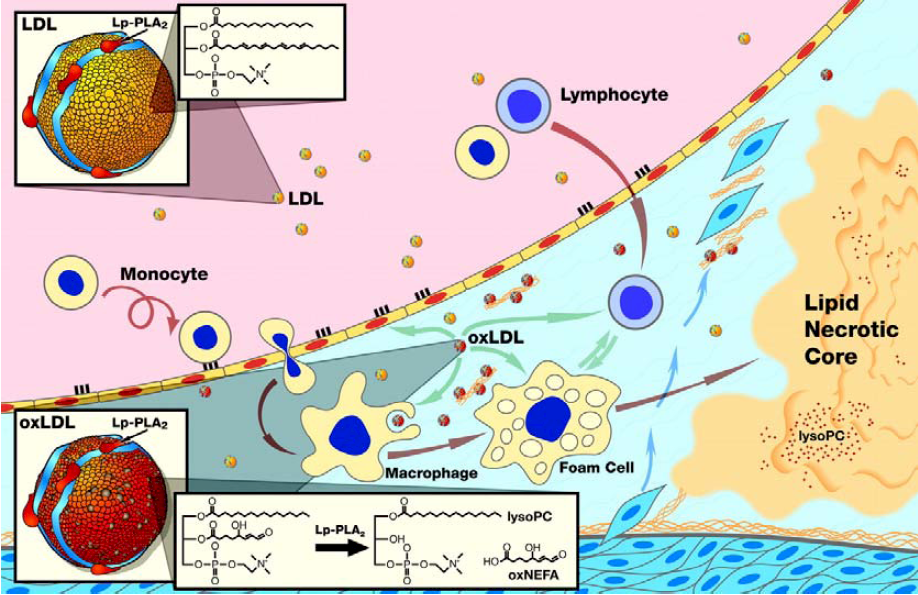
Lp-PLA2, also known as secretory phospholipase A2 group VII (sPLA2-VII) and as platelet activating factor acetylhydrolase (PAF-AH), is widely expressed in cells involved in atherosclerosis, such as macrophages, T-cells, lymphocytes, and mast cells [1,2]. It is a calcium-independent serin lipase that hydrolyzes phospholipids at the sn-2 position and acts preferentially on water-soluble polar phospholipids, particularly those with oxidatively truncated fatty acids [3,4]. Via a specific protein-protein interaction between the N-terminus of Lp-PLA2 and the C-terminus of apolipoprotein B (apoB) two-third of the Lp-PLA2 circulates primarily bound to LDL cholesterol while the remaining third is distributed between high-density lipoprotein (HDL) cholesterol and very-low-density lipoproteins (VLDL) [5]. The oxidation of LDL cholesterol within the arterial wall provides the substrate for the hydrolytic action of Lp-PLA2 -a short acyl group at the sn-2 position of phospholipids. By cleaving an oxidized phosphatidylcholine component of the lipoprotein particle, Lp-PLA2 generates potent proinflammatory and proatherogenic mediators like oxidized nonesterified fatty acids (ox-FA), arachidonic acid and lysophosphatidylcholine (Lyso-PC) [6] (Figure 1). Since inflammation plays a major role at all stages of atherogenesis, from endothelial dysfunction to plaque development and ultimately to plaque rupture, Lp-PLA2 may contribute to atherosclerosis by generation of various proinflammatory lipid mediators, including Lyso-PC, ox-FA, and arachidonic acid [7]. Ox-FA´s promote atherosclerosis by directly and indirectly increasing oxidative stress and the presence of oxidized LDL and other lipoproteins in the plasma and arterial walls, thereby initiating fatty streak formation [8]. Cyclooxygenase converts arachidonic acid to inflammatory mediators like thromboxanes and leukotrienes [9,10]. Lyso-PCs act pro-atherogenic in various early steps of atherosclerosis, are expressed by macrophages in human atherosclerotic lesions, and are increased 5-fold in oxidized LDL compared to normal LDL [11,12,13]. In the arterial wall they upregulate adhesive molecules like vascular cell adhesion molecule (VCAM)-1 and intercellular adhesion molecule (ICAM)-1 [14,15]. Furthermore they promote monocyte migration by inducing monocyte chemotactic protein (MCP)-1 and stimulate Interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α production and scavenger receptor expression in macrophages in a concentration-dependent manner [16,17,18]. Furthermore, Lyso-PCs also upregulate Lp-PLA2 activity, resulting in a viscous cycle, thereby pro-inflammatory mediators are becoming increasingly upregulated, contributing to plaque progression and destabilization [19].
Although results from the aforementioned experimental studies seem to be fairly consistent, there has been some controversy regarding the biological role of Lp-PLA2 in atherosclerosis, since initially, it was thought to be atheroprotective [20,21]. Adenoviral gene transfer of human Lp-PLA2 in ApoE-/- mice reduced VLDL-induced ex vivo macrophage adhesion and in vivo macrophage homing, thereby resulting in reduced atherosclerosis [22]. Furthermore, the pretreatment of an electronegative LDL cholesterol subfraction from hypercholesterolemic human plasma with recombinant Lp-PLA2 completely prevented the pro-apoptotic effects of the LDL subfraction on vascular endothelial cells [23].
Figure 1.
Schematic representation of the proposed pro-atherogenic mechanism of Lp-PLA2 in the vessel wall.
Figure 1.
Schematic representation of the proposed pro-atherogenic mechanism of Lp-PLA2 in the vessel wall.
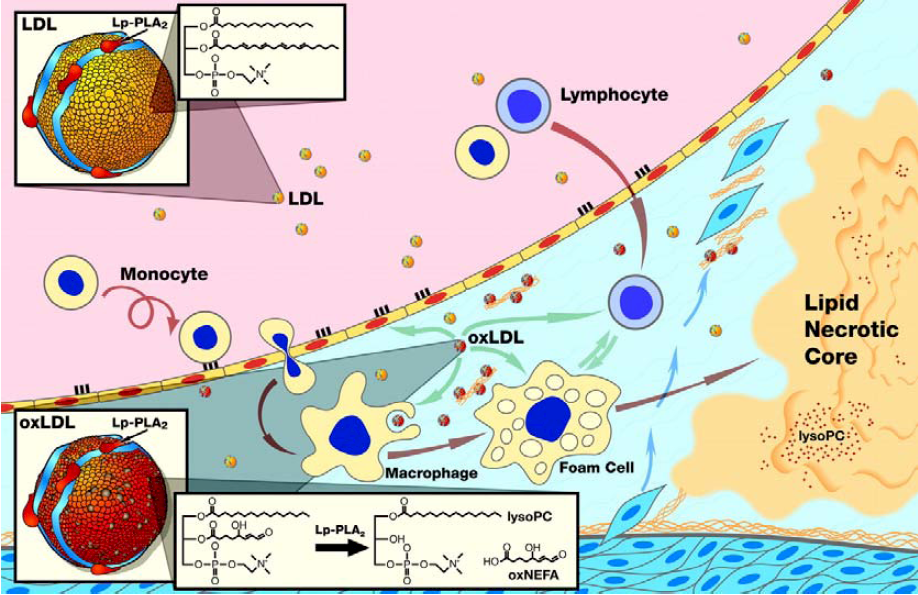
1.1. Pathoanatomical Evidence
In an atherosclerotic diabetes/hypercholesterolemia swine model which develops advanced coronary lesions within six months, expression of 59 genes in the vasculature, related to cholesterol metabolism, inflammation, and insulin signaling pathways were characterized [25]. Inflammatory genes were more markedly upregulated in coronary arteries than in thoracic aortae and carotids. At six months Lp-PLA2 gene was significantly upregulated, indicating the potential role that this molecule plays in the development and progression of atherosclerosis. Mannheim et al. [26] determined the expression of Lp-PLA2 in 167 carotid artery plaques by immunoblotting and immunostaining. Symptomatic carotid artery plaques were characterized by increased levels of Lp-PLA2 and its product Lyso-PC in correlation with markers of tissue oxidative stress, inflammation, and instability, strongly supporting a role for Lp-PLA2 in the pathophysiology and clinical presentation of cerebrovascular disease. In a recently published study [27] carotid artery plaque expression of Lp-PLA2 was quantified in 162 consecutive patients undergoing elective carotid endarterectomy. Follow-up for cardiac death and non-fatal acute myocardial infarction was accomplished over a period of 48 ± 14 months. Carotid plaque Lp-PLA2 expression above the median constituted a more than three times higher risk for cardiac events [HR 3.39 (1.13–10.17), P = 0.03]. The relative expression of Lp-PLA2 in coronary plaque phenotypes, including unstable lesions, has first been established by Kolodgie et al. [28]. They prospectively collected coronary segments (n = 30) from 25 sudden coronary death patients for immunolocalization of Lp-PLA2, and showed that Lp-PLA2 was strongly expressed within the necrotic core and surrounding macrophages of vulnerable and ruptured plaques, with relatively weak staining in less advanced lesions, suggesting a potential role in promoting plaque instability.
A study of nuclear families attributed 62% variance of Lp-PLA2 activity to heritability, although until now few genetic determinants of Lp-PLA2 have been identified, and the data for these genetic factors are inconsistent [29]. Most recently, a study in monozygotic and dizygotic twins of Caucasian origin was conducted, to investigate the heritability of plasma levels (mass) and activity of Lp-PLA2 [30]. The authors reported that when phenotypic covariance was partitioned into additive genetic effects, environmental effects common to co-twins, and error variance components, a non-negligible component of both Lp-PLA2 mass and activity was accounted for by genetic effects, suggesting that both Lp-PLA2 activity and mass variance may be genetically determined, although the heritability estimates were only significant for Lp-PLA2 activity. Interestingly, these heritability estimates were remarkably similar to the above mentioned estimate variance in US nuclear families.
2. The Epidemiologic Evidence
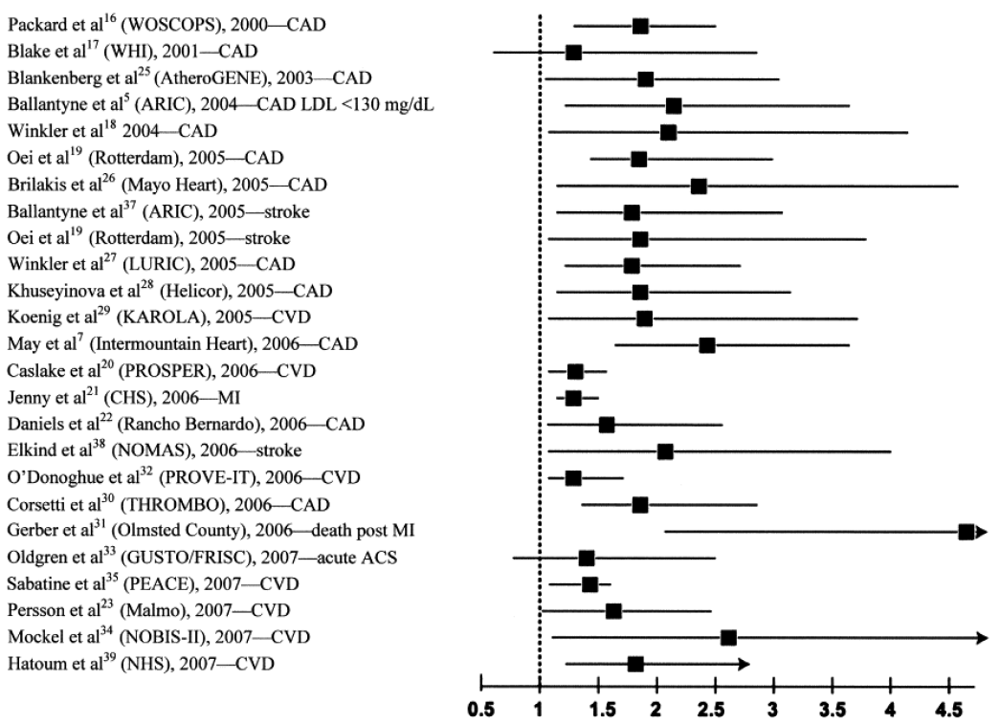
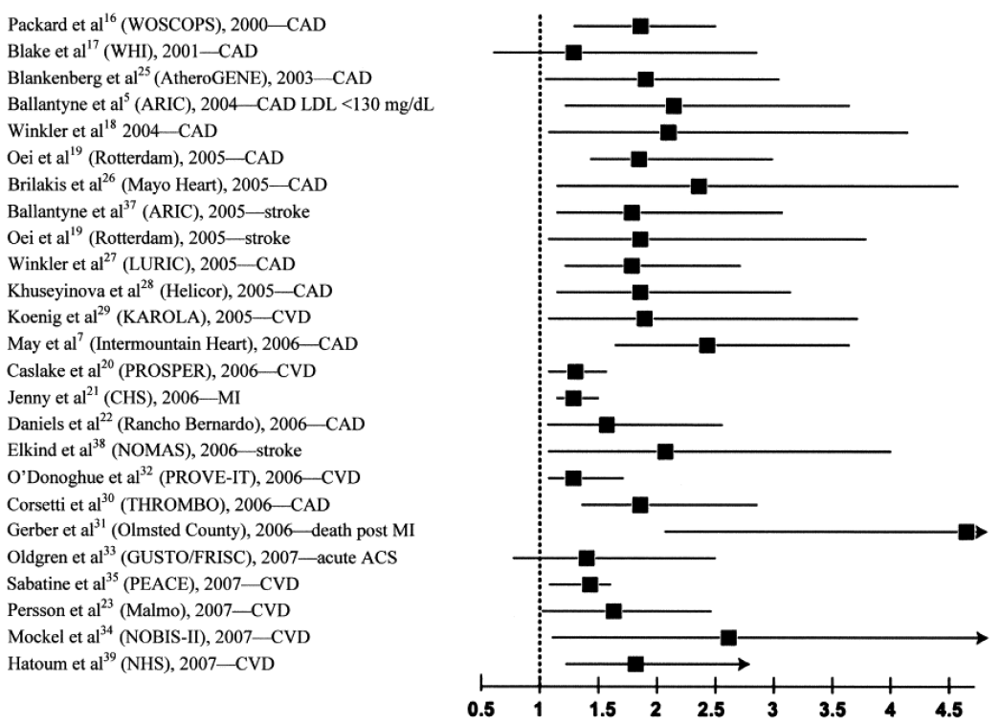
To date, the vast majority of prospective studies analyzing the association between Lp-PLA2 and subsequent cardiovascular (CV) events demonstrated a strong, positive, statistically significant and independent association between increased Lp-PLA2 mass or elevated activity and future CV risk, including a wide variety of clinical settings, i.e., apparently healthy men and women, elderly subjects, patients with acute coronary syndrome (ACS), and with stable CHD (Figure 2). Garza et al. conducted a meta-analysis summarizing the results of 14 prospective long-term studies with a total of 20,549 participants [31]. They found a significant and independent association between elevated Lp-PLA2 concentrations and activity and risk of CVD, resulting in a summary odds ratio (OR) of 1.60 (95%CI, 1.36–1.89) after adjustment for conventional CV risk factors.
2.1. Lp-PLA2 in Apparently Healthy, Middle-Aged Subjects
The potential association between Lp-PLA2 and cardiovascular outcome was first demonstrated in WOSCOPS (West of Scotland Coronary Prevention Study), where 580 hypercholesterolemic middle-aged, initial healthy men, who developed a CHD event over a 4.9-year follow-up (FU), served as cases and were compared to 1,160 matched event-free controls [32]. A one standard deviation (SD) increase in Lp-PLA2 concentrations was independently associated with a relative risk (RR) of 1.18 (95%CI 1.05–1.33) for a future CHD event after multivariate adjustment. By contrast, the subsequent Women’s Health Study (WHS), that had included only 123 cases and 123 controls in a nested case-control design, failed to confirm a significant association [33].
Figure 2.
Elevated lipoprotein-associated phospholipase A2 (Lp-PLA2) is consistently associated with a doubling of risk for cardiovascular disease (CVD).
Figure 2.
Elevated lipoprotein-associated phospholipase A2 (Lp-PLA2) is consistently associated with a doubling of risk for cardiovascular disease (CVD).
In the large Atherosclerosis Risk in Communities (ARIC) study, conducted in 608 men and women with incident CHD and 740 controls, and followed for at least six years, after multivariable adjustments, Lp-PLA2 was not associated with an increased risk for CHD, except in subjects with LDL cholesterol below the median of 130 mg/dL [35]. In this subgroup, Lp-PLA2 significantly and independently predicted CHD (hazard ratio (HR) 2.08; 95%CI 1.20– 3.62), suggesting that it might be a useful marker for identifying patients at risk in those with low and intermediate cardiovascular risk. We had determined Lp-PLA2 concentrations in 934 initially healthy, middle-aged men in the MONICA-Augsburg cohort study [36]. Baseline levels of Lp-PLA2 were higher in subjects who experienced a coronary event (295 ± 113 vs. 263 ±79 ng/mL; p < 0.01), and after multivariate adjustment, including the TC/HDL-C ratio as the strongest lipoprotein variable, a one SD increase in Lp-PLA2 was strongly and independently related to a first-ever event (HR 1.23; 95%CI 1.02–1.47). Importantly, like in the ARIC study we evaluated the potential additive value of Lp-PLA2 to high-sensitive (hs) CRP in predicting risk. For this purpose, elevated hs CRP was defined according to a recent AHA/CDC consensus document as >3.0 mg/L, and for Lp-PLA2 the upper tertile cut-point was used (422 ng/mL in ARIC and 290.8 ng/mL in MONICA). In ARIC, individuals with high Lp-PLA2 and high CRP exhibited a threefold increased risk for CHD (HR 2.95; 95%CI 1.47–5.94), whereas in our study the combination of elevated Lp-PLA2 and elevated CRP resulted in a HR of 1.93 (95%CI 1.09–3.40) compared with both markers not being increased in the fully adjusted model. Furthermore, in the Rotterdam study, Oei et al. [37] measured Lp-PLA2 activity in 308 CHD cases and a random sample of 1,820 subjects and followed them for a median of 7.2 years. After controlling for a variety of potential confounders, a one SD increase in Lp-PLA2 activity was strongly and independently related to a first-ever CV event (HR 1.20; 95%CI 1.04–1.39), almost identical to findings in the MONICA Augsburg cohort. Kiechl et al. [38], in, a population-based survey of 765 men and women aged 40–79 years, the Bruneck study, who were followed over a 10-year period demonstrated in multivariable analysis a HR of Lp-PLA2 for subsequent coronary events of 1.4 per one SD change in enzyme activity.
2.2. Lp-PLA2 in the Elderly Population
Although the predictive value of Lp-PLA2 seems to be slightly smaller in the elderly population, it still has been found to be a potent predictor of CVD [34]. The Rancho Bernardo Study [39] enrolled 1,077 initially healthy men and women with a mean age of 72 years, and showed a 60% to 90% increased risk for incident CHD across extreme quartiles of the Lp-PLA2 distribution after multivariable adjustment. Data from the Cardiovascular Health Study (CHS), an elderly population without a history of vascular disease at baseline, partially confirmed these results [40]. Only increased Lp-PLA2 mass, but not an elevated Lp-PLA2 activity was found to be associated with an increased 10-year risk of myocardial infarction (MI) independently of traditional cardiovascular risk factors. Similarly, in the PROSPER (The Prospective Study of Pravastatin in the Elderly at Risk) trial only Lp-PLA2 mass was found to be significantly related to future CHD risk, while no association was found for enzyme activity after controlling for various confounders [41].
2.3. Lp-PLA2 in Patients with ACS
Data on the predictive value of Lp-PLA2 in the setting of an ACS still remains controversial. While several studies investigating the association between baseline Lp-PLA2 activity and -mass and CV events yielded fairly strong associations, like the German NOBIS-II study and data from Olmsted County, Minnesota, the PROVE IT-TIMI 22 trial, the FRISC II trial, and the GUSTO IV ACS study failed to establish baseline levels of Lp-PLA2 activity and -mass as an independent risk marker of recurrent CV events [42,43,44,45]. Different time intervals between the index event and blood sampling most likely account for these differences.
2.4. Lp-PLA2 in Patients with Stable CHD
By contrast, data regarding the role of Lp-PLA2 in patients with stable CHD seems fairly consistent. Brilakis et al. [46] were the first to report on Lp-PLA2 in patients with pre-existing CHD. They enrolled 466 consecutive patients, who were followed for a median of four years. In multivariable analyses, the RR for a future event for a one SD increase in Lp-PLA2 mass was found to be 1.28 (95% CI 1.06–1.54). In the KAROLA (Langzeiterfolge der KARdiOLogischen Anschlussheilbehandlung) study, Lp-PLA2 mass and activity were measured on the average 43 days after an acute event in a cohort of 1,051 patients aged 30–70 years with CHD [47]. In multivariable analyses after four years of FU, Lp-PLA2 mass was shown to possess prognostic value, whereas Lp-PLA2 activity became only borderline significant. Even after adjusting for markers of renal function (i.e. cystatin C), and hemodynamic stress (NT-proBNP) there was still an 2-fold increased risk for future CVD events in patients in the upper two tertiles of Lp-PLA2 mass compared with the bottom tertile (HR 2.09; 95% CI 1.10 to 3.96). The THROMBO (Thrombogenic Factors and Recurrent Coronary Events) study further confirmed these findings in 766 post-MI patients, who were followed for 26 months [48]. In the large PEACE (Prevention of Events with Angiotensin-Converting Enzyme Inhibition) trial, Lp-PLA2 mass was measured in 3,766 patients with documented CHD. After five years of FU elevated Lp-PLA2 concentrations predicted adverse CV outcomes [49]. Interestingly, these effects were more pronounced for the prediction of non-fatal events such as revascularization and unstable angina pectoris (UAP). Within the LURIC (Ludwigshafen Risk and Cardiovascular Health) study Lp-PLA2 activity predicted risk for cardiac and total mortality over 5.5 years in 2513 patients with angiographically confirmed CHD and 719 without, and added prognostic information in patients with low and medium CRP concentration with regard to 5-year cardiac mortality independently of established risk factors [50].
3. Clinical Studies
3.1. Lp-PLA2 and Endothelial Dysfunction
Recent studies have demonstrated that Lp-PLA2 is associated with endothelial dysfunction and early atherosclerosis. Yang et al. [51] recruited 172 patients without significant CAD in whom coronary endothelial function was assessed in response to intracoronary acetylcholine. The OR for presence of coronary endothelial dysfunction in patients with Lp-PLA2 in the highest tertile was 3.3 (95% CI, 1.6 to 6.6). In another study, coronary angiography, blood flow, flow reserve, endothelial function, and intravascular ultrasound with volumetric analysis were performed in 15 patients with mild coronary atherosclerosis and in 15 control subjects [52]. Plasma samples were collected simultaneously from the left main coronary artery and coronary sinus for measurement of Lp-PLA2, Lyso-PC, and CRP. While CRP was not significantly different between the groups, net production of Lp-PLA2 and Lyso-PC in the coronary circulation was higher in patients compared with control subjects, and correlated with coronary endothelial dysfunction.
3.2. Lp-PLA2 as A Target for Pharmacologic Intervention
Since there is increasing evidence for a pivotal role of inflammation in atherothrombosis, and the main downstream product of Lp-PLA2, Lyso-PC represents a potent pro-inflammatory molecule, Lp-PLA2 may not only be a predictor of CVD risk as discussed above but may also become a therapeutic target.

{kind=link}
{kind=link}
| Author [Ref.] | Phase | Mass or activity | Cohort | N | Treatment | Primary Endpoint | Significant effects |
|---|---|---|---|---|---|---|---|
| Johnson et al. [56] | II | Lp-PLA2 activity | Patients before elective endarterectomy | 59 | 14 days | Lp-PLA2 activity | 80% reduction in enzyme activity |
| Mohler et al. [57] | II | Lp-PLA2 activity | CHD and CHD-risk equivalent patients receiving atorvastatin | 959 | 12 weeks | Lp-PLA2 activity and biomarkers | 43-66% reduction in enzyme activity, 13% reduction of IL-6, and CRP (p = 0.028 and p = 0.15) |
| Serruys et al. [58] | II | Lp-PLA2 activity | Patients with angiographically documented CHD | 330 | 12 months | Coronary atheroma plaque cap deformability; CRP | 59% reduction in enzyme activity, halt of necrotic core expansion (-5.2 mm3; p = 0.012) |
This Table summarizes results from three phase II clinical trials. Modified with friendly permission after [53].
Darapladib, a selective inhibitor of Lp-PLA2 is a small molecule which was developed in 2003 by GlaxoSmithKline (GSK) [54]. Darapladib convincingly demonstrated beneficial effects in a diabetic/hypercholesterolemic pig model. These animals were randomly assigned either to a control group or a treatment group receiving 10 mg/kg darapladib per day [55]. After 24 weeks, Lp-PLA2 activity in plasma was reduced by 89% in the treatment group (p < 0.00001 vs. placebo), and coronary gene expression analyses revealed a substantial reduction of the expression of 24 genes associated with macrophage and T-cell function. Furthermore, the study indicated that selective Lp-PLA2 inhibition may promote lesion stabilization. The median plaque area in the left anterior descending coronary artery was significantly reduced from 0.222 mm2 to 0.086 mm2 (p < 0.05), and seven out of 17 control pigs showed a fibrous or thin fibrous cap atheroma compared to only two out of twenty in the darapladib group (41% vs. 10%; p = 0.05). In addition, the necrotic area from the arterial section with the greatest plaque area was significantly reduced in the treatment group (0.87 ± 0.33 mm2 vs. 0.03 ± 0.003 mm2; p = 0.015 vs. placebo). To date darapladib has demonstrated efficacy in three multicenter, randomized, double-blind, placebo-controlled trials [56,57,58] (Table 1).
In an early phase II clinical trial [56], the administration of two different doses of darapladib for 14 days before elective carotid endarterectomy in 59 patients resulted in a significant systemic inhibition of Lp-PLA2 plasma activity by 80%, and a significantly reduced local Lp-PLA2 activity in atherosclerotic plaque. Furthermore, IL-18 levels and activity of the pro-apoptotic caspase-3 and caspase-8 were attenuated compared to placebo.
Mohler et al. [57] tested the effects of darapladib (40, 80 and 160 mg, respectively) on Lp-PLA2 activity and on biomarkers of CV risk in 959 CHD and CHD-risk equivalent patients receiving aggressive lipid-lowering therapy (atorvastatin 20 or 80 mg per day) (NCT00269048). After 12 weeks of therapy, darapladib inhibited Lp-PLA2 activity in a dose-dependent manner by approximately 43%, 55%, and 66% compared with placebo (p < 0.001 vs. placebo). Furthermore, IL-6 and CRP displayed a strong decrease in the high-dose treatment group (12.6% and 13.0% decrease, respectively; p = 0.028 and p = 0.15 vs. placebo, respectively). In contrast, levels of total cholesterol, LDL- and HDL cholesterol were not modified as compared with placebo. Of special note, no major safety concerns were noted after 12 weeks of treatment.
The international, multicenter, randomized, double-blind, placebo-controlled IBIS-2 (Integrated Biomarker and Imaging Study-2, [58]) tested the effect of 12 months of treatment with darapladib 160 mg daily in 330 patients with angiographically documented CHD. After 12 months, treatment with darapladib resulted in significantly reduced Lp-PLA2 activity levels (59% inhibition, p < 0.001 vs. placebo), while the primary endpoints of the study -atheroma deformability measured by palpography (p = 0.22 vs. placebo) and plasma CRP lowering (p = 0.35 vs. placebo) were not met. Also, HDL- and LDL cholesterol were unaffected by treatment. However, despite adherence to a high level of standard-of-care treatment, in the placebo-treated group the necrotic core volume increased significantly (4.5 ± 17.9 mm3; p = 0.009), whereas darapladib halted this increase (-0.5 ± 13.9 mm3; 0.71) in the intervention group, resulting in a significant treatment difference of –5.2 mm3 (p = 0.012). These intra-plaque compositional changes occurred without a significant treatment difference in total plaque volume or calcification (p = 0.95). Regarding clinical safety, higher systolic casual blood pressure in the darapladib group (3.0 mmHg, 95%CI 0.3-5.7; p = 0.031) was not consistent with results of the prior clinical study, and additional comparison of the intra-arterial blood pressure also revealed no differences between groups. Treatment-emergent adverse effects did not significantly differ from placebo.
In December 2008 GSK initiated the STABILITY trial (NCT00799903), a phase III, randomized, double-blind, placebo-controlled, parallel-assigned, multicenter clinical outcome trial in 15,500 patients with chronic CHD [59]. In this event driven trial, darapladib is assessed on top of standard CHD pharmacotherapy, including high dose statins. The primary outcome measure is determined as time to first occurrence of any component of the composite of major adverse cardiovascular events, consisting of cardiovascular death, non-fatal myocardial infarction and non-fatal stroke. Final results of the study will be available by 2012, or until around 1,500 major adverse CV events have occurred. This trial will test the inflammation hypothesis and will provide more insight into whether or not targeting inflammation carries clinical benefit for the high risk patient with CVD. In spring 2010 another randomized controlled clinical trial of similar size, SOLID, will be launched in post-ACS patients.
4. Disclosures
M. Karakas declares that he has no financial or personal relations to other parties whose interests could have affected the content of this article in any way, either positively or negatively. W. Koenig declares that he is a member of the Steering Committee of the STABILITY Trial, and has received honoraria for lectures from GSK.
References and Notes
- Asano, K.; Okamoto, S.; Fukunaga, K.; Shiomi, T.; Mori, T.; Iwata, M.; Ikeda, Y.; Yamaguchi, K. Cellular sources of platelet-activating-factor acetylhydrolase activity in plasma. Biochem. Biophys. Res. Commun. 1999, 261, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Stafforini, D.M.; Elstad, M.R.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Human macrophages secrete platelet-activating factor acetylhydrolase. J. Biol. Chem. 1990, 265, 9682–9687. [Google Scholar]
- Venable, M.E.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Platelet-activating factor: a phospholipid autacoid with diverse actions. J. Lipid Res. 1993, 34, 691–702. [Google Scholar]
- Min, J.H.; Jain, M.K.; Wilder, C.; Paul, L.; Apitz-Castro, R.; Aspleaf, D.C.; Gelb, M.H. Membrane-bound plasma platelet activating factor acetylhydrolase acts on substrate in the aqueous phase. Biochemistry 1999, 38, 12935–12942. [Google Scholar]
- Stafforini, D.M.; Tjoelker, L.W.; McCormick, S.P.; Vaitkus, D.; McIntyre, T.M.; Gray, P.W.; Young, S.G.; Prescott, S.M. Molecular basis of the interaction between plasma platelet-activating factor acetylhydrolase and low-density lipoprotein. J. Biol. Chem. 1999, 274, 7018–7024. [Google Scholar]
- Macphee, C.H.; Moores, K.E.; Boyd, H.F.; Dhanak, D.; Ife, R.J.; Leach, C.A.; Leake, D.S.; Milliner, K.J.; Patterson, R.A.; Suckling, K.E.; Tew, D.G.; Hickey, D.M. Lipoprotein-associated phospholipase A2, platelet-activating factor acetylhydrolase, generates two bioactive products during the oxidation of low-density lipoprotein: use of a novel inhibitor. Biochem. J. 1999, 338, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Hannson, G.K. Mechanisms of disease—Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med. 2005, 352, 1685–1695. [Google Scholar] [CrossRef] [PubMed]
- Khan-Merchant, N.; Penumetcha, M.; Meilhac, O.; Parthasarathy, S. Oxidized fatty acids promote atherosclerosis only in the presence of dietary cholesterol in low-density lipoprotein receptor knockout mice. J. Nutr. 2002, 132, 3256–3262. [Google Scholar] [PubMed]
- Bäck, M. Leukotriene signaling in atherosclerosis and ischemia. Cardiovasc. Drugs Ther. 2009, 23, 41–48. [Google Scholar]
- Balsinde, J.; Winstead, M.V.; Dennis, E.A. Phospholipase A (2) regulation of arachidonic acid mobilization. FEBS Lett. 2002, 531, 2–6. [Google Scholar]
- Matsumo, T.; Kobayashi, T.; Kamata, K. Role of lysophoshatidylcholine (LPC) in atheroclerosis. Curr. Med. Chem. 2007, 14, 3209–3220. [Google Scholar]
- Schmitz, G.; Ruebsaamen, K. Metabolism and atherogenic disease association of lysophosphatidylcholine. Atherosclerosis 2009, 10–18. [Google Scholar]
- Hakkinen, T.; Luoma, J.S.; Hiltunen, M.O.; Macphee, C.H.; Milliner, K.J.; Patel, L.; Rice, S.Q.; Tew, D.G.; Karkola, K.; Ylä-Herttuala, S. Lipoprotein-associated phospholipase 2, a platelet-activating factor acetylhydrolase, is expressed by macrophages in human and rabbit atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2909–2917. [Google Scholar] [PubMed]
- Kume, N.; Cybulski, M.I.; Gimbrone, M.A., Jr. Lysophosphatidylcholine, a component of atherogenic lipoproteins, induces mononuclear leukocyte adhesion molecules in cultured human and rabbit arterial endothelial cells. J. Clin. Invest. 1992, 90, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Berman, J.W.; Taubman, M.B.; Fisher, E.A. Lysophosphatidylcholine stimulates monocyte chemoattractant protein-1 gene expression in rat aortic smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1617–1623. [Google Scholar]
- Takahara, N.; Kashiwaga, A.; Maegawa, H.; Shigeta, Y. Lysophosphatidylcholine stimulates the expression and production of MCP-1 by human vascular endothelial cells. Metabolism 1996, 45, 559–564. [Google Scholar]
- Liu-Wu, Y.; Hurt-Camejo, E.; Wiklund, O. Lysophosphatidylcholine induces the production of IL-1beta by human monocytes. Atherosclerosis 1998, 137, 351–357. [Google Scholar]
- Ousman, S.S.; David, S. Lysophosphatidylcholine induces rapid recruitment and activation of macrophages in the adult mouse spinal cord. Glia 2000, 30, 92–104. [Google Scholar]
- Macphee, C.H.; Nelsonj, J.J.; Zalewski, A. Lipoprotein-associated phospholipase A2 as a target of therapy. Curr. Opin. Lipidol. 2005, 16, 4442–4446. [Google Scholar]
- Tjoelker, L.W.; Wilder, C.; Eberhardt, C.; Stafforini, D.M.; Dietsch, G.; Schimpf, B.; Hooper, S.; Le Trong, H.; Cousens, L.S.; Zimmerman, G.A. Anti-inflammatory properties of a platelet-activating factor acetylhydrolase. Nature 1995, 374, 549–553. [Google Scholar]
- Lerman, A.; McConnell, J.P. Lipoprotein-associated phospholipase A2: a risk marker or a risk factor? Am. J. Cardiol. 2008, 101, 11F–22F. [Google Scholar] [PubMed]
- Theilmeier, G.; De Geest, B.; Van Veldhofen, P.P.; Stengel, D.; Michiels, C.; Lox, M.; Landeloos, M.; Chapman, M.J.; Ninio, E.; Collen, D.; Himpens, B.; Holvoet, P. HDL-associated PAF-AH reduces endothelial adhesiveness in apoE-/- mice. FASEB J. 2000, 14, 2032–2039. [Google Scholar]
- Chen, C.H.; Jiang, T.; Yang, J.H.; Jiang, W.; Lu, J.; Marathe, G.K.; Pownall, H.J.; Ballantyne, C.M.; McIntyre, T.M.; Henry, P.D.; Yang, C.Y. Low-density lipoprotein in hypercholesterolemic human plasma induces vascular endothelial cell apoptosis by inhibiting fibroblast growth factor 2 transcription. Circulation 2003, 107, 2102–2108. [Google Scholar]
- Zalewski, A.; Macphee, C. Role of lipoprotein-associated phospholipase A2 in atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 923–931. [Google Scholar]
- Mohler, E.R., III; Sarov-Blat, L.; Shi, Y.; Hamamdzic, D.; Zalewski, A.; Macphee, C.; Llano, R.; Pelchovitz, D.; Mainigi, S.K.; Osman, H.; et al. Site-specific atherogenic gene expression correlates with subsequent variable lesion development in coronary and peripheral vasculature. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 850–855. [Google Scholar] [PubMed]
- Mannheim, D.; Herrmann, J.; Versari, D.; Gössl, M.; Meyer, F.B.; McConnell, J.P.; Lerman, L.O.; Lerman, A. Enhanced expression of Lp-PLA2 and lysophosphatidylcholine in symptomatic carotid atherosclerotic plaques. Stroke 2008, 39, 1448–1455. [Google Scholar]
- Herrmann, J.; Mannheim, D.; Wohlert, C.; Versari, D.; Meyer, F.B.; McConnell, J.P.; Gössl, M.; Lerman, L.O.; Lerman, A. Expression of lipoprotein-associated phospholipase A2 in carotid artery plaques predicts long-term cardiac outcome. Eur. Heart J. 2009, 30, 2930–2938. [Google Scholar]
- Kolodgie, F.D.; Burke, A.P.; Skorija, K.S.; Ladich, E.; Kutys, R.; Makuria, A.T.; Virmani, R. Lipoprotein-associated phospholipase A2 protein expression in the natural progression of human coronary atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2523–2529. [Google Scholar]
- Guerra, R.; Zhao, B.; Mooser, V.; Stafforini, D.; Johnston, J.M.; Cohen, J.C. Determinants of plasma platelet-activating factor acetylhydrolase: heritability and relationship to plasma lipoproteins. J. Lipid Res. 1997, 38, 2281–2288. [Google Scholar]
- Lenzini, L.; Antezza, K.; Caroccia, B.; Wolfert, R.L.; Szczech, R.; Cesari, M.; Narkiewicz, K.; Williams, C.J.; Rossi, G.P. A twin study of heritability of plasma lipoprotein-associated phospholipase A2 (Lp-PLA2) mass and activity. Atherosclerosis 2009, 205, 181–185. [Google Scholar]
- Garza, C.A.; Montori, V.M.; McConnell, J.P.; Somers, V.K.; Kullo, I.J.; Lopez-Jimenez, F. Association between lipoprotein-associated phospholipase A2 and cardiovascular disease: A systematic review. Mayo Clin. Proc. 2007, 82, 159–165. [Google Scholar]
- Packard, C.J.; O’Reilly, D.S.; Caslake, M.J.; McMahon, A.D.; Cooney, J.; Macphee, C.H.; Suckling, K.E.; Krishna, M.; Wilkinson, F.E.; Rumley, A.; Lowe, G.D. Lipoprotein-associated phospholipase A2 as an independent predictor of coronary heart disease. N. Engl. J. Med. 2000, 343, 1148–1155. [Google Scholar]
- Blake, G.J.; Dada, N.; Fox, J.C.; Manson, J.E.; Ridker, P.M. A prospective evaluation of lipoprotein-associated phospholipase A2 levels and the risk of future cardiovascular events in women. J. Am. Coll. Cardiol. 2001, 38, 1302–1306. [Google Scholar]
- Koenig, W.; Khuseyinova, N. Lipoprotein-associated and secretory phospholipase A2 in cardiovascular disease: the epidemiologic evidence. Cardiovasc. Drugs Ther. 2009, 23, 85–92. [Google Scholar]
- Ballantyne, C.M.; Hoogeveen, R.C.; Bang, H.; Coresh, J.; Folsom, A.R.; Heiss, G.; Sharrett, A.R. Lipoprotein-associated phospholipase A2, high-sensitivity C-reactive protein, and risk for incident coronary heart disease in middle-aged men and women in the Atherosclerosis Risk in Communities (ARIC) study. Circulation 2004, 109, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Koenig, W.; Khuseyinova, N.; Lowel, H.; Trischler, G.; Meisinger, C. Lipoprotein-associated phospholipase A2 adds to risk prediction of incident coronary events by C-reactive protein in apparently healthy middle-aged men from the general population: results from the 14-year follow-up of a large cohort from southern Germany. Circulation 2004, 110, 1903–1908. [Google Scholar]
- Oei, H.H.; van der Meer, I.M.; Hofman, A.; Koudstaal, P.J.; Stijnen, T.; Breteler, M.M.; Witteman, J.C. Lipoprotein-associated phospholipase A2 activity is associated with risk of coronary heart disease and ischemic stroke: the Rotterdam Study. Circulation 2005, 111, 570–575. [Google Scholar]
- Kiechl, S.; Willeit, J.; Mayr, M.; Viehweider, B.; Oberhollenzer, M.; Kronenberg, F.; Wiedemann, C.J.; Oberthaler, S.; Xu, Q.; Witztum, J.L.; Tsimikas, S. Oxidized phospholipids, lipoprotein (a), lipoprotein-associated phospholipase A2 activity, and 10-year cardiovascular outcomes: prospective results from the Bruneck study. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1788–1795. [Google Scholar] [PubMed]
- Daniels, L.B.; Laughlin, G.A.; Sarno, M.J.; Bettencourt, R.; Wolfert, R.L.; Barrett-Connor, E. Lipoprotein-associated phospholipase A2 is an independent predictor of incident coronary heart disease in an apparently healthy older population: the Rancho Bernardo Study. J. Am. Coll. Cardiol. 2008, 51, 913–919. [Google Scholar]
- Jenny, N.S.; Solomon, C.; Cushman, M.; Nelson, J.J.; Tracy, R.P.; Psaty, B.M.; Furberg, C.D. Lipoprotein-associated phospholipase A2 and cardiovascular disease: results from the Cardiovascular Health Study. Circulation 2006, 113, E332, Abstract. [Google Scholar]
- Caslake, M.J.; Cooney, J.; Murray, E.; Bedford, D.; Robertson, M.; Nelson, J.J; Packard, C.J. Lipoprotein-associated phospholipase A2 as a risk factor for coronary vascular disease in the elderly. Atherosclerosis. 2006, 7 (Suppl.), 484, Abstract. [Google Scholar]
- Möckel, M.; Müller, R.; Vollert, J.O.; Müller, C.; Danne, O.; Gareis, R.; Störk, T.; Dietz, R.; Koenig, W. Lipoprotein-associated phospholipase A2 for early risk stratification in patients with suspected acute coronary syndrome: a multi-marker approach: the North Wuerttemberg and Berlin Infarction Study-II (NOBIS-II). Clin. Res. Cardiol. 2007, 96, 604–612. [Google Scholar]
- Gerber, Y.; McConnell, J.P.; Jaffe, A.S.; Weston, S.A.; Killian, J.M.; Roger, V.L. Lipoprotein-associated phospholipase A2 and prognosis after myocardial infarction in the community. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2517–2522. [Google Scholar]
- O’Donoghue, M.; Morrow, D.A.; Sabatine, M.S.; Murphy, S.A.; McCabe, C.H.; Cannon, C.P.; Braunwald, E. Lipoprotein-associated phospholipase A2 and its association with cardiovascular outcomes in patients with acute coronary syndromes in the PROVE IT-TIMI 22 (PRavastatin Or atorVastatin Evaluation and Infection Therapy-Thrombolysis In Myocardial Infarction) trial. Circulation 2006, 113, 1745–1752. [Google Scholar]
- Oldgren, J.; James, S.K.; Siegbahn, A.; Wallentin, L. Lipoprotein-associated phospholipase A2 does not predict mortality or new ischaemic events in acute coronary syndrome patients. Eur. Heart. J. 2007, 28, 699–704. [Google Scholar]
- Brilakis, E.S.; McConnell, J.P.; Lennon, R.J.; Elesber, A.A.; Meyer, J.G.; Berger, P.B. Association of lipoprotein-associated phospholipase A2 levels with coronary artery disease risk factors, angiographic coronary artery disease, and major adverse events at follow-up. Eur. Heart J. 2005, 26, 137–144. [Google Scholar] [PubMed]
- Koenig, W.; Twardella, D.; Brenner, H.; Rothenbacher, D. Lipoprotein-associated phospholipase A2 predicts future cardiovascular events in patients with coronary heart disease independently of traditional risk factors, markers of inflammation, renal function and hemodynamic stress. Arterioscler Thromb. Vasc. Biol. 2006, 26, 1586–1593. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, J.P.; Rainwater, D.L.; Moss, A.J.; Zareba, W.; Sparks, C.E. High lipoprotein-associated phospholipase A2 is a risk factor for recurrent coronary events in postinfarction patients. Clin. Chem. 2006, 52, 1331–1338. [Google Scholar]
- Sabatine, M.S.; Morrow, D.A.; O’Donoghue, M.; Jablonski, K.; Rice, M.M.; Solomon, S.; Rosenberg, Y.; Domanski, M.J.; Hsia, J. PEACE Investigators. Prognostic utility of lipoprotein-associated phospholipase A2 for cardiovascular outcomes in patients with stable coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2463–2469. [Google Scholar] [CrossRef] [PubMed]
- Winkler, K.; Hoffmann, M.M.; Winkelmann, B.R.; Friedrich, I.; Schäfer, G.; Seelhorst, U.; Wellnitz, B.; Wieland, H.; Boehm, B.O.; März, W. Lipoprotein-associated phospholipase A2 predicts 5-year cardiac mortality independently of established risk factors and adds prognostic information in patients with low and medium high-sensitivity C-reactive protein (the Ludwigshafen risk and cardiovascular health study). Clin. Chem. 2007, 53, 1440–1447. [Google Scholar]
- Yang, E.H.; McConnell, J.P.; Lennon, R.J.; Barsness, G.W.; Pumper, G.; Hartman, S.J.; Rihal, C.S.; Lerman, L.O.; Lerman, A. Lipoprotein-associated phospholipase A2 is an independent marker for coronary endothelial dysfunction in humans. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 106–111. [Google Scholar]
- Lavi, S.; McConnell, J.; P Rihal, C.S.; Prasad, A.; Mathew, V.; Lerman, L.O.; Lerman, A. Local production of lipoprotein-associated phospholipase A2 and lysophosphatidlcholine in the coronary circulation: association with early coronary atherosclerosis and endothelial dysfunction in humans. Circulation 2007, 115, 2715–2721. [Google Scholar]
- Karakas, M.; Koenig, W. Phospholipase A2 as a therapeutic target for atherosclerosis. Clin. Lip. 2010, 5, 43–56. [Google Scholar]
- Rosenson, R.S. Future role for selective phospolipase A2 inhibitors in the prevention of atherosclerotic cardiovascular disease. Cardiovasc. Drugs Ther. 2009, 23, 93–101. [Google Scholar]
- Wilensky, R.L.; Shi, Y.; Mohler, E.R.; Hamamdzic, D.; Burgert, M.E.; Li, J.; Postle, A.; Fenning, R.S.; Bollinger, J.G.; Hoffmann, B.E.; et al. Inhibition of lipoprotein-associated phospholipase A2 reduces complex coronary atherosclerotic plaque development. Nat. Med. 2008, 14, 1015–1016. [Google Scholar] [PubMed]
- Johnson, A.; Zalewski, A.; Janmohamed, S.; Sawyer, J.; Rolfe, T.; Staszkiewicz, W.; Alvarez, S. Lipoprotein-associated phospholipase A2 (Lp-PLA2) activity, an emerging CV risk marker, can be inhibited in atherosclerotic lesions and plasma by novel pharmacologic intervention: the results of a multicenter clinical study. Circulation 2004, 110, III-590. [Google Scholar]
- Mohler, E.R.; Ballantyne, C.M.; Davidson, M.H.; Hanefeld, M.; Ruilope, L.M.; Johnson, J.L.; Zalewski, A. The effect of darapladib on plasma lipoprotein-associated phospholipase A2 activity and cardiovascular biomarkers in patients with stable coronary heart disease risk equivalent: the results of a multicenter, randomized, double-blind, placebo-controlled study. J. Am. Coll. Cardiol. 2008, 51, 1632–1641. [Google Scholar] [CrossRef] [PubMed]
- Serruys, P.W.; Garcia-Garcia, H.M.; Buszman, P.; Erne, P.; Verheye, S.; Aschermann, M.; Duckers, H.; Bleie, O.; Dudek, D.; Botker, H.E.; von Birgelen, C.; DÁmico, D.; Hutchinson, T.; Zambanini, A.; Mastik, F.; van Ees, G.A.; van der Steen, A.F.; Vince, D.G.; Ganz, P.; Hamm, C.W.; Wijns, W.; Zalewski, A. Effects of the direct lipoprotein-associated phospholipase A2 inhibitor darapladib on human coronary atherosclerotic plaque. Circulation 2008, 118, 1172–1182. [Google Scholar]
- McCullough, P.A. Darapladib and atherosclerotic plaque: should lipoprotein-associated phospholipase A2 be a therapeutic target? Curr. Atheroscler. Rep. 2009, 11, 334–337. [Google Scholar] [CrossRef] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Karakas, M.; Koenig, W. Lp-PLA2 Inhibition—The Atherosclerosis Panacea? Pharmaceuticals 2010, 3, 1360-1373. https://doi.org/10.3390/ph3051360
AMA Style
Karakas M, Koenig W. Lp-PLA2 Inhibition—The Atherosclerosis Panacea? Pharmaceuticals. 2010; 3(5):1360-1373. https://doi.org/10.3390/ph3051360
Chicago/Turabian StyleKarakas, Mahir, and Wolfgang Koenig. 2010. "Lp-PLA2 Inhibition—The Atherosclerosis Panacea?" Pharmaceuticals 3, no. 5: 1360-1373. https://doi.org/10.3390/ph3051360