Mitochondrial and Cell Death Mechanisms in Neurodegenerative Diseases

Department of Pathology, Division of Neuropathology and Department of Neuroscience, Johns Hopkins University School of Medicine, 558 Ross Building, 720 Rutland Avenue, Baltimore, Maryland 21205-2196, USA
Pharmaceuticals 2010, 3(4), 839-915; https://doi.org/10.3390/ph3040839
Submission received: 31 December 2009
/
Revised: 22 March 2010
/
Accepted: 23 March 2010
/
Published: 25 March 2010
(This article belongs to the Special Issue Mitochondrial Drugs for Neurodegenerative Diseases)
Abstract
:Alzheimer’s disease (AD), Parkinson’s disease (PD) and amyotrophic lateral sclerosis (ALS) are the most common human adult-onset neurodegenerative diseases. They are characterized by prominent age-related neurodegeneration in selectively vulnerable neural systems. Some forms of AD, PD, and ALS are inherited, and genes causing these diseases have been identified. Nevertheless, the mechanisms of the neuronal cell death are unresolved. Morphological, biochemical, genetic, as well as cell and animal model studies reveal that mitochondria could have roles in this neurodegeneration. The functions and properties of mitochondria might render subsets of selectively vulnerable neurons intrinsically susceptible to cellular aging and stress and overlying genetic variations, triggering neurodegeneration according to a cell death matrix theory. In AD, alterations in enzymes involved in oxidative phosphorylation, oxidative damage, and mitochondrial binding of Aβ and amyloid precursor protein have been reported. In PD, mutations in putative mitochondrial proteins have been identified and mitochondrial DNA mutations have been found in neurons in the substantia nigra. In ALS, changes occur in mitochondrial respiratory chain enzymes and mitochondrial cell death proteins. Transgenic mouse models of human neurodegenerative disease are beginning to reveal possible principles governing the biology of selective neuronal vulnerability that implicate mitochondria and the mitochondrial permeability transition pore. This review summarizes how mitochondrial pathobiology might contribute to neuronal death in AD, PD, and ALS and could serve as a target for drug therapy.
Introduction
Pathologists conceived the concept of cell death as a mechanism of disease to aid in diagnosis and therapy [1]. Pathological stimuli can be extrinsic or intrinsic and can cause abrupt or delayed cell death or inactivate normal cell survival or cell death networks. Many neurological disorders are characterized by undesirable cell death [2], while the absence of precise control of cell number in tissues causes cancer (impaired apoptosis is a central step toward neoplasia) [3]. It is compelling that a goal of human disease management and treatment is, on one hand, to prevent cell death in neurological disease [2] and, on the other hand, to stimulate cell death in malignancy [4]. Thus, the study of cell death is fundamental to human pathobiology and disease mechanisms and the identification of therapeutic targets for disease treatment.
An exciting new understanding of mitochondrial biology has emerged over the past two decades from multiple disciplines that is likely to be very relevant to adult-onset neurodegenerative disorders of the central nervous system (CNS). [5]. Mitochondria are multi-functional organelles (Figure 1) [6].
In addition to their critical role in the production of ATP through the electron transport chain (Figure 1), these organelles function in intracellular Ca2+ homeostasis, synthesis of steroids, heme and iron-sulfur clusters, and programmed cell death (PCD) [6,7,8]. Mitochondria are also sites of formation of reactive oxygen species (ROS), including superoxide anion (O2•-) [9] and the highly reactive hydroxyl radical (•OH) or its intermediates [10], and reactive nitrogen species such as nitric oxide (•NO) [6]. Mitochondria generate endogenous ROS as by-products of oxidative phosphorylation (Figure 1) [8]. Oxygen- and proton pump-driven ATP production by the electron transport chain (Figure 1, lower left) is one function. The respiratory chain proteins (complex I-IV) establish an electrochemical gradient across the inner mitochondrial membrane (IMM) by extruding protons out of the matrix into the intermembrane space, thereby creating an energy gradient that drives the production of ATP by complex V (Figure 1, lower left). Superoxide (O2•-) is produced as a by-product in the process of electron transport. Electrons in the electron carriers, such as the unpaired electron of ubisemiquinone bound to coenzyme Q binding sites of complexes I, II, and III, can be donated directly to O2 to generate O2•- [8]. O2•- does not easily pass through biological membranes and thus must be inactivated in compartments where it is generated [9]. The mitochondrial matrix enzyme manganese superoxide dismutase (MnSOD or SOD2) or copper/zinc SOD (Cu/ZnSOD or SOD1) in the mitochondrial intermembrane space and cytosol convert O2•- to hydrogen peroxide (H2O2) in the reaction O2•- + O2•- + 2H+→ H2O2 + O2 (Figure 1) [9]. H2O2 is more stable than O2•- and can diffuse from mitochondria into the cytosol and nucleus. H2O2 is detoxified by glutathione peroxidase in mitochondria and the cytosol and by catalase in peroxisomes.
Figure 1.
Mitochondria (upper right) are multi-functional organelles and regulate neuronal cell life and death (adapted from an earlier form [5]). See text for descriptions.
Figure 1.
Mitochondria (upper right) are multi-functional organelles and regulate neuronal cell life and death (adapted from an earlier form [5]). See text for descriptions.
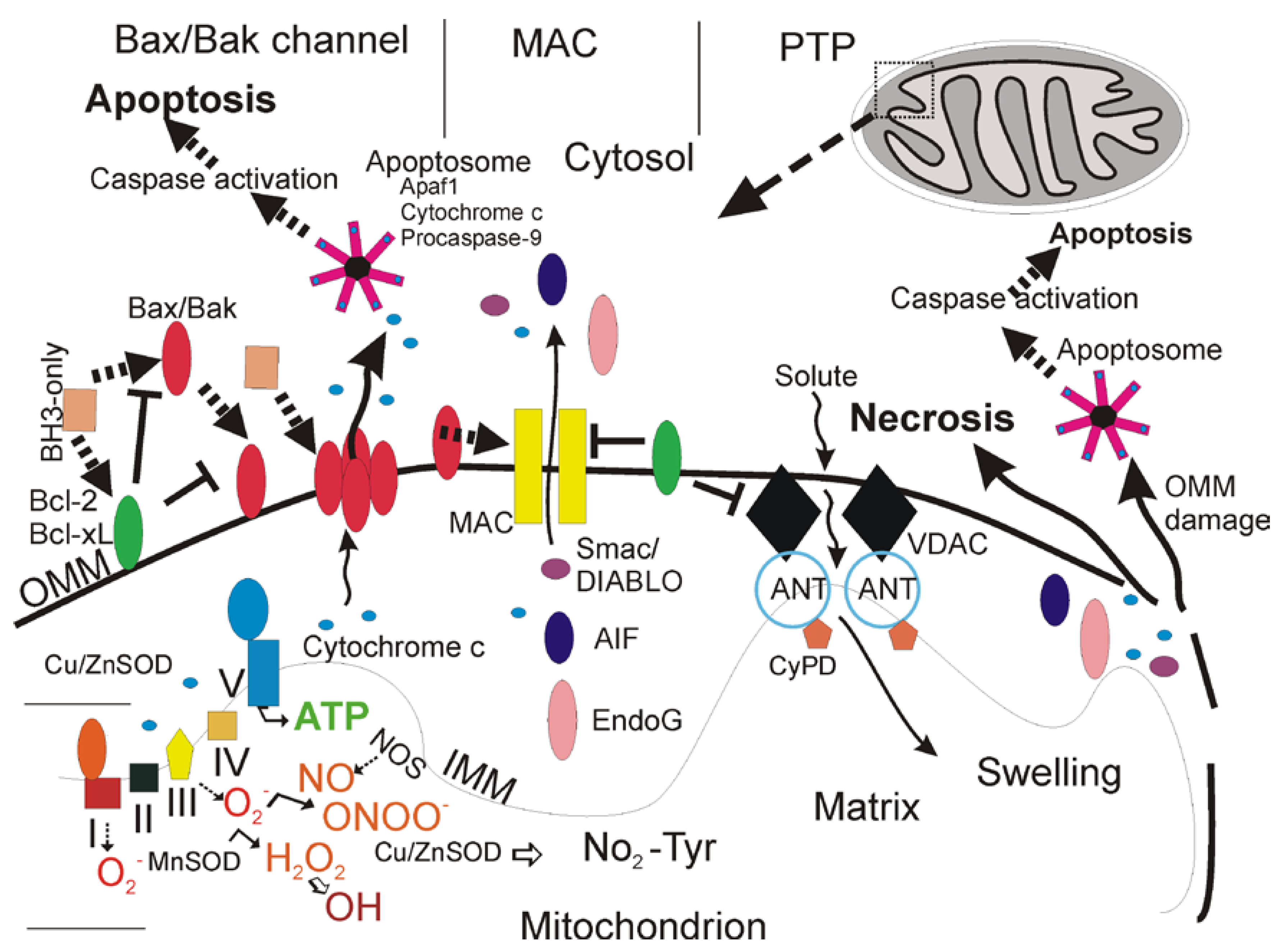
Because many mitochondrial proteins possess iron-sulfur clusters for oxidation-reduction reactions and because mitochondrial DNA (mtDNA) lacks protective histones, these macromolecules are particularly vulnerable to ROS attack [8]. In pathological settings that can trigger cell senescence and death, H2O2 in the presence of reduced transitional metals (Fe2+) can be converted to hydroxyl radical (•OH) or hydroxyl-like intermediates [10]. O2•- can react with the diffusible gas nitric oxide (•NO), synthesized by three isoforms of nitric oxide synthase (NOS) enzymes [11], to form the potent nucleophile oxidant and nitrating agent peroxynitrite (ONOO-) (Figure 1) [12]. ONOO- or products of ONOO- can damage proteins by nitration [12]. ONOO- is genotoxic directly to neurons by causing single- and double-strand breaks in DNA [13]. Cu/ZnSOD can use ONOO- to catalyze the nitration (NO2-Tyr) of mitochondrial protein tyrosine residues (Figure 1, bottom center) such as cyclophilin D (CyPD) and the adenine nucleotide translocator (ANT) which are core components of the mitochondrial permeability transition pore (PTP, another critical function of mitochondria). •NO can be produced in mitochondria [14] and has direct effects in mitochondria. •NO at nanomolar concentrations can inhibit rapidly and reversibly mitochondrial respiration by nitration or nitrosylation [15].
Mitochondrial perturbations are known to participate in the mechanisms of human neuropathology, particularly disorders involving acute interruptions in O2 and substrate delivery to the brain and bioenergetic failure as seen in tissue ischemia and toxic exposures [6]. Optic atrophy type 1 (OPA1), a hereditary optic neuropathy, is one example of a chronic neurodegenerative disease caused by mutations in the OPA1 gene that encodes a mitochondrial dynamin-related GTPase that functions in maintenance of mitochondrial morphology, including fusion, and metabolism [16]. The properties and functions of mitochondria (Figure 1) might confer an intrinsic susceptibility of subsets of long-lived post-mitotic cells such as neurons to aging and stresses, including mutations and environmental toxins. This review summarizes the contributions of the different forms of cell death to three human neurodegenerative diseases (Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis), the evidence for mitochondrial involvement, and their animal and cell models. In this regard varying degrees of mitochondrial dysfunction and intrinsic mitochondrial-mediated cell death mechanisms could be critical determinants in the regulation of disease and neuronal cell death ranging from necrosis and apoptosis to autophagy [17,18,19]; thus, targeting mitochondrial properties or entities, such as the mitochondrial PTP (Figure 1) [20,21,22,23], could be important for developing new mechanism-based pharmaco-therapies for neurodegenerative diseases.
Types of Cell Death
Cells can die by different processes [24]. These processes have been classified canonically into two distinct categories, called necrosis and apoptosis. These forms of cellular degeneration were classified originally as different because they appeared different morphologically under a microscope (Figure 2).
Necrosis is a lytic destruction of individual or groups of cells, while apoptosis (derived from a Greek word for ‘dropping of leaves from trees’) is an orderly and compartmental dismantling of single cells or groups of cells into consumable components for nearby cells. Apoptosis is an example of programmed cell death (PCD) that is an ATP-driven (sometimes gene transcription-requiring) form of cell suicide often committed by demolition enzymes called caspases, but other apoptotic and non-apoptotic, caspase-independent forms of PCD exist [25]. Apoptotic PCD is instrumental in developmental organogenesis and histogenesis and adult tissue homeostasis, functioning to eliminate excess cells [26]. In healthy people, estimates reveal that between 50 to 70 billion cells in adults and 20 to 30 billion cells in a child between the ages of 8 and 14 die each day due to apoptosis [26]. Another form of cell degeneration is called autophagy [27]. Autophagy is an intracellular catabolic process that occurs by lysosomal degradation of damaged or expendable organelles. Necrosis and apoptosis both differ morphologically (Figure 2) and mechanistically from autophagy [25,27].
Figure 2.
Cell death matrix (modified from its original form [194]). This diagram summarizes in linear (top) and 3-dimensional matrix (bottom) formats the concept of the apoptosis-necrosis continuum of cell death. See text for descriptions.
Figure 2.
Cell death matrix (modified from its original form [194]). This diagram summarizes in linear (top) and 3-dimensional matrix (bottom) formats the concept of the apoptosis-necrosis continuum of cell death. See text for descriptions.
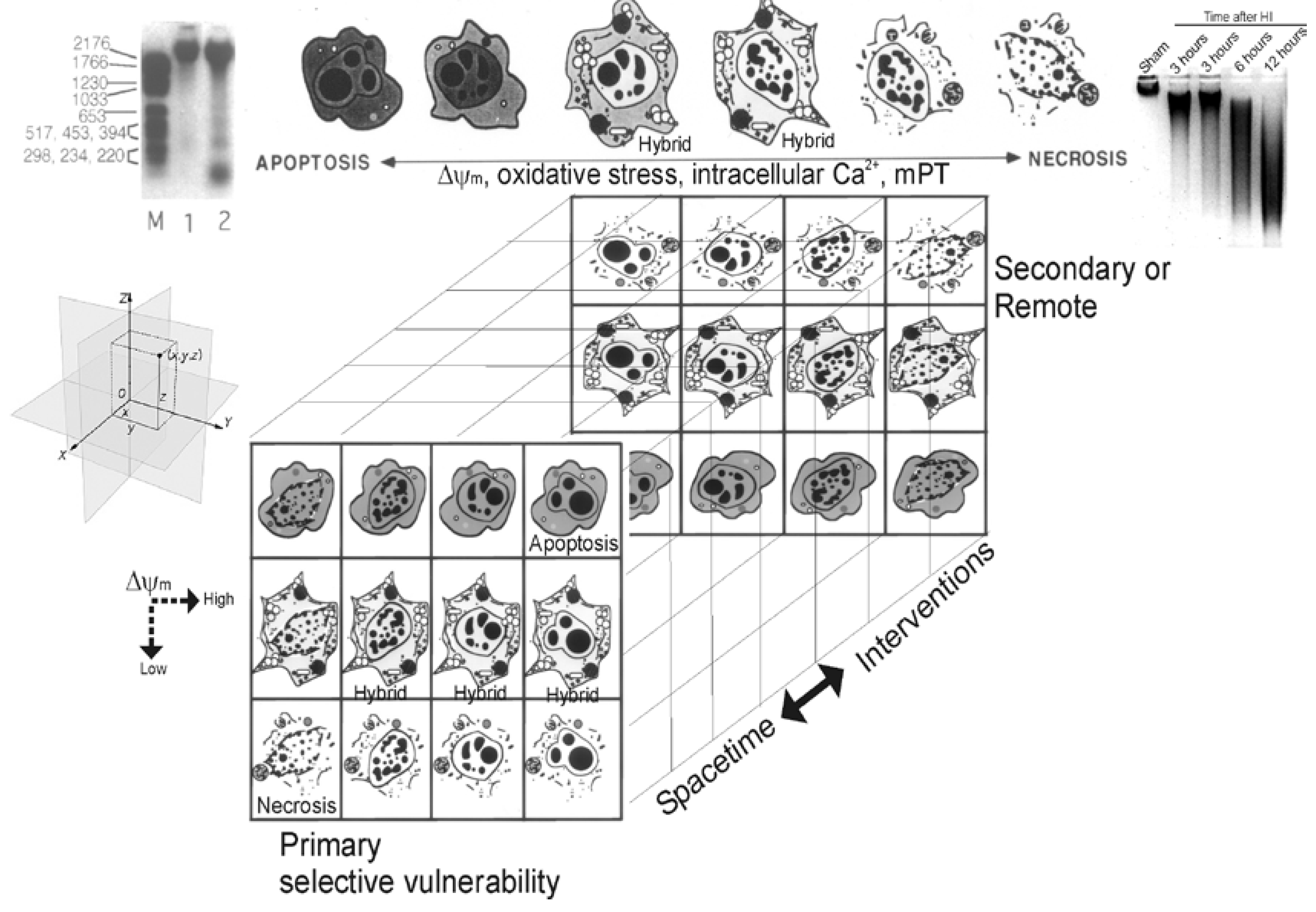
More recently the morphological and molecular regulatory distinctions between the different forms of cell death have become blurred and uncertain due to observations made on degenerating neurons in animal models and to a new concept that attempts to accommodate these observations [24,28,29]. This concept, in its original form, posited that cell death exists as a continuum with necrosis and apoptosis at opposite ends of a spectrum with hybrid forms of degeneration manifesting in between (Figure 2) [17,24,28,29]. For example, a hypothetical dying neuron in the CNS is illustrated at coordinates (x,y,z) in the Euclidian coordinate system (Figure 2, at left). The degeneration of this neuron in diseased or damaged animal nervous systems is not always strictly necrosis or apoptosis, according to the traditional binary classification of cell death, but also occurs as intermediate or hybrid forms with coexisting morphological and biochemical characteristics that lie in a structural continuum (Figure 2) [17,24,28,29]. Apoptosis with internucleosomal fragmentation of DNA (Figure 2, top left) and necrosis with random digestion of DNA (Figure 2, top right) are at the extremes and different syncretic hybrid forms are in between (Figure 2, top). The front matrix of the cube shows some of the numerous possible structures of neuronal cell death near or at the terminal stages of degeneration. Combining different nuclear morphologies and cytoplasmic morphologies generates a nonlinear matrix of possible cell death structures. In the cell at the extreme upper right corner (Figure 2), nuclear and cytoplasmic morphologies combine to form an apoptotic neuron that is typical of naturally occurring PCD during nervous system development. This death is classical apoptosis. In contrast, in other cells (Figure 2, extreme lower left corner), the merging of necrotic nuclear and necrotic cytoplasmic morphologies forms a typical necrotic neuron resulting from N-methyl-D-aspartate (NMDA) receptor excitotoxicity and cerebral ischemia (stroke and cardiac arrest). Between these two extremes, hybrids of cell death can be produced with varying contributions of apoptosis and necrosis to the nuclear and cytoplasmic morphologies. Thus, neuronal cell death can be syncretic in a manner similar to that described in fibroblastic cells [30]. The typical apoptosis-necrosis hybrid cell death structure is best exemplified by neurons in the neonatal CNS dying from ischemia or non-NMDA glutamate receptor-mediated excitotoxicity. The death forms shown in the front matrix of the cube represent only a small number of the possible forms of cell death that can be envisioned to fill the empty cells of the matrix. Neuronal maturity and the subtypes of glutamate receptors that are over-activated are known to influence where an injured/degenerating neuron falls within the matrix. The types and levels of DNA damage that are sustained by a cell and concurrent graded activation of mitochondrial permeability transition (mPT) and the levels of oxidative stress and Ca2+ might also influence the position of a degenerating cell within the death matrix and in the brain Euclidian coordinate system. The back panel represents the possible cell death forms occurring in space-time over a delayed period after injury or after administration of therapeutic interventions. The matrix predicts that the cell death patterns might change over time from apoptosis to apoptosis-necrosis variants or necrosis and from necrosis to apoptosis-necrosis variants or apoptosis. This prediction could have critical relevance to the idea of neuroprotection. This concept may also be relevant to cell death in general, and thus may be widely applicable to cell biology outside the nervous system.
The in vivo reality of a neuronal cell death continuum was revealed first in rat models of glutamate receptor excitotoxicity [28,29]. The hybrid cells can be distinguished cytopathologically by the progressive compaction of the nuclear chromatin into few, discrete, large, irregularly shaped clumps (Figure 2). This morphology contrasts with the formation of few, uniformly shaped, dense, round masses in classic apoptosis and the formation of numerous, smaller, irregularly shaped chromatin clumps in classic necrosis. The cytoplasmic organelle pathology in hybrid cells has a basic pattern that appears more similar to necrosis than to apoptosis but is lower in amplitude than in necrosis (e.g., mitochondrial swelling). Toxicological studies of cultured cells have shown that stimulus intensity influences the mode of cell death [31,32,33], such that apoptosis can be induced by injurious stimuli of lesser amplitude than insults causing necrosis [34], but the cell death modes were still considered distinct [33].
The molecular mechanisms of cell death are becoming known [35,36,37,38], and, with this knowledge, the distinctiveness of the different cell death processes and the potential superposition among different cell death mechanisms are being realized. Experimental studies on cell death mechanisms, and particularly the cell death continuum, are important because they could lead to the rational development of molecular mechanism-based therapies for treating neurodegenerative disorders. The different categories of cell death are discussed below.
Cell Necrosis and the Mitochondrial Permeability Transition Pore
Cell death caused by cytoplasmic swelling, nuclear dissolution (karyolysis), and lysis has been classified traditionally as necrosis (Figure 2) [39,40]. Cell necrosis (sometimes termed oncosis) [40] can result from rapid and severe failure to sustain cellular homeostasis, notably cell volume control [41]. The process of necrosis involves damage to the structural and functional integrity of the cell plasma membrane and associated enzymes, for example Na+,K+ ATPase, abrupt influx and overload of ions (e.g., Na+ and Ca2+) and H2O, and rapid mitochondrial damage and bioenergetic collapse [33,42,43,44]. Metabolic inhibition, anoxia, and oxidative stress from ROS can trigger necrosis. Inhibitory crosstalk between ion pumps causes pro-necrotic effects when Na+,K+ ATPase ‘steals’ ATP from the plasma membrane Ca2+ ATPase, contributing to Ca2+ overload and mitochondrial damage [45].
The morphology and some biochemical features of classic necrosis in neurons are distinctive (Figure 2) [24]. The main features are swelling and vacuolation/vesiculation of organelles, destruction of membrane integrity, digestion of chromatin, and dissolution of the cell. The overall profile of the moribund cell is maintained generally as it degrades into the surrounding tissue parenchyma. The debris instigates an inflammatory reaction in tissue. In necrosis, dying cells do not bud to form discrete, membrane-bound fragments. The nuclear pyknosis and karyolysis appear as condensation of chromatin into many irregularly shaped, small clumps, sharply contrasting with the formation of few, uniformly dense and regularly shaped chromatin aggregates that occurs in apoptosis. In cells undergoing necrosis, genomic DNA is digested globally, because proteases that digest histones, which protect DNA, and DNases are co-activated to generate many randomly sized fragments seen as a DNA ‘smear’ by gel electrophoresis (Figure 2, top right). These cytoplasmic and nuclear changes in pure necrosis are thought to be very diagnostic (Figure 2).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Function |
|---|---|
| Bcl-2* | Anti-apoptotic, blocks Bax/Bak channel formation |
| Bcl-XL | Anti-apoptotic, blocks Bax/Bak channel formation |
| Bax* | Pro-apoptotic, forms pores for cytochrome c release |
| Bak* | Pro-apoptotic, forms pores for cytochrome c release |
| Bad | Pro-apoptotic, decoy for Bcl-2/Bcl-XL promoting Bax/Bak pore formation |
| Bid | Pro-apoptotic, decoy for Bcl-2/Bcl-XL promoting Bax/Bak pore formation |
| Noxa | Pro-apoptotic, decoy for Bcl-2/Bcl-XL promoting Bax/Bak pore formation |
| Puma | Pro-apoptotic, decoy for Bcl-2/Bcl-XL promoting Bax/Bak pore formation |
| p53* | Antagonizes activity of Bcl-2/Bcl-XL, promotes Bax/Bak oligomerization |
| Cytochrome c | Activator of apoptosome |
| Smac/DIABLO | IAP inhibitor |
| AIF | Antioxidant flavoprotein/released from mitochondria to promote nuclear DNA fragmentation |
| Endonuclease G | Released from mitochondria to promote nuclear DNA fragmentation |
| HtrA2/Omi | IAP inhibitor |
| VDAC | mPTP component in outer mitochondrial membrane |
| ANT+ | mPTP component in inner mitochondrial membrane |
| Cyclophilin D+ | mPTP component in mitochondrial matrix |
| TSPO (peripheral benzodiazepine receptor) | Modulator of mPTP |
| Hexokinase | Modulator of VDAC |
* Changes have been reported in human ALS (see below)+ A reported target of oxidative modification in mouse ALS (see below)
Recent work has shown that cell necrosis might not be as chaotic, random, and incomprehensible as envisioned originally but can involve the activation of specific signaling pathways to eventuate in cell death [46,47,48]. This idea is very important for developing new mechanism-based therapeutics to block cell necrosis. For example, DNA damage can lead to poly(ADP-ribose) polymerase activation and ATP depletion, energetic collapse, and necrosis [49]. Other pathways for ‘programmed’ necrosis involve death receptor signaling through NADPH oxidase, receptor-interacting protein 1 (RIP1), and mPT (Figure 1 and Figure 2) [47,48,50,51].
mPT is a mitochondrial state in which the proton-motive force is disrupted reversibly or irreversibly [23,50,51,52,53]. Conditions of intra-mitochondrial Ca2+ overload, excessive oxidative stress, and decreased electrochemical gradient (Δψm), ADP, and ATP can favor mPT. This alter condition of mitochondria involves the mitochondrial permeability transition pore (mPTP) that functions as a voltage, thiol, and Ca2+ sensor [23,50,51,52,53]. The mPTP is believed to be a poly-protein transmembrane channel (Figure 1) formed at the contact sites between the outer mitochondrial membrane (OMM) and inner mitochondrial membrane (IMM). The collective components of the mPTP are still controversial, but the voltage-gated anion channel (VDAC, or porin) in the OMM, the adenine nucleotide translocator (ANT, or solute carrier family 25) in the IMM, and cyclophilin D (CyPD) in the matrix are believed to be the core components (Figure 1 and Table 1) [23,51,53]. Other components or modulators of the mPTP appear to be hexokinase, creatine kinase, translocator protein 18 kDa (TSPO, or peripheral benzodiazepine receptor), and Bcl-2 family members (Table 1) [53].
The VDAC family in human and mouse cells consists of three proteins of ~31 kDa (VDAC1-3) encoded by three different genes [54]. VDACs are the major transport proteins in the OMM, functioning in ATP rationing, Ca2+ homeostasis, oxidative stress response, and cell death [54]. Monomeric VDAC serves as the functional channel, although oligomerization of VDAC into dimers and tetramers can occur and might function in cell death [54]. The VDAC adopts an open conformation at low or zero membrane potentials and a closed conformation at potentials above 30–40 mV making the OMM permeable to most small hydrophilic molecules up to 1.3 kDa for free exchange of respiratory chain substrates [55]. Most data implicating VDAC opening or closing as an important regulator of cell death are based on in vitro conditions, while limited in vivo evidence is available [56]. VDAC1 binds Bcl-2-antagonist/killer 1 (Bak1, see below for description of Bcl-2 family members), hexokinase, gelsolin, and ANT1/ANT2; VDAC2 binds Bak1, hexokinase, cytochrome c, glycerol kinase and ANT1/ANT2; VDAC3 binds glycerol kinase, CyPD, and ANT1-3 [54]. In human tissues, VDAC1 and VDAC2 isoforms are expressed more abundantly than VDAC3; highest levels are found in kidney, heart, skeletal muscle, and brain [57]. The effects of selective knockout of VDAC isoforms are not equivalent, implying different functions. Mice deficient in either VDAC1 or VDAC3 are viable [58,59,60], but VDAC2 deficiency causes embryonic lethality [61]. Lack of both VDAC1 and VDAC3 causes growth retardation [60]. VDAC null mouse tissues exhibit deficits in mitochondrial respiration and abnormalities in mitochondrial ultrastructure [58]. However, mitochondria without VDAC1 have an intact mPT response [62,63]. VDAC2 deletion, but not lack of the more abundant VDAC1, results in enhanced activation of the mitochondrial apoptosis pathway and enforced activation of Bak1 at mitochondria [61], consistent with the idea that VDAC2 is a key inhibitor of Bak1-mediated apoptosis [60]. However, other data show that cells lacking individual VDACs or combinations of VDACs have normal death responses to Bax and Bid [63]. Recent work in yeast has revealed that SOD1 is necessary for proper functioning of VDAC; specifically, SOD1 regulates VDAC channel activity and protein levels in mitochondria [64].
The mitochondrial ANT family in human consists of 3 members (ANT1-3, or solute carrier family 25, members 4, 5, and 6) encoded by three different genes, but in mouse only two isoforms of the ANT are present [65]. The proteins are ~33 kDa and function as homodimers [65]. They are multi-pass membrane proteins, with odd-numbered transmembrane helices that mediate exchange of cytosolic ADP for mitochondrial ATP across the inner membrane utilizing the electrochemical gradient [66]. These helices have kinks because of proline residues [66]. ANT1 binds VDAC1, CyPD, Bax, twinkle (ataxin-8), and cyclophilin-40; ANT2 binds VDAC1-3 and cyclophilin-40; ANT3 binds VDAC1, steroid sulfatase, and translocase of inner mitochondrial membrane-13 (TIMM13) and TIMM23 [65]. The ANT isoforms are expressed differentially in tissue- and species-specific patterns [67]. ANT1 is expressed highly in human and mouse heart and skeletal muscle; human brain has low ANT1 mRNA but high ANT3 mRNA, while mouse brain has high ANT1 mRNA [67]. ANT2 mRNA is very low or not expressed in most adult human and mouse tissues, except kidney [67]. In tissue mitochondria where more than one ANT isoform is expressed, it is ANT1 that binds preferentially to CyPD to form the mPTP at contact sites between the IMM and OMM (Figure 1) [68]. It has been proposed that, in the presence of high mitochondrial Ca2+, the binding of CyPD to proline residue 61 (Pro61) in loop 1 of ANT1 results in a conformation that converts the ANT into a non-specific pore [65]. Non-conditional ANT1 null mice are viable and grow normally but develop mitochondrial skeletal myopathy and cardiomyopathy [66]. Ablation both ANT isoforms in mouse liver surprisingly did not change fundamentally mPT and cell death in hepatocytes [69], and some ANT ligands induce mitochondrial dysfunction and cytochrome c release independent of mPT [70]. Thus, the mechanisms of ANT-mediated cell deaths need further study.
CyPD (also named cyclophilin F, peptidyl prolyl isomerase F) is encoded by a single gene [23,51,71]. Despite confusing nomenclature, there is only one isoform of CyPD (EC 5.2.1.8, ppif gene product) in mouse and human. The ~20 kDa protein encoded by this gene is a member of the peptidyl-prolyl cis-trans isomerase (PPIase) family. PPIases catalyze the cis-trans isomerization of proline imidic peptide bonds in oligopeptides and accelerate the folding of proteins. CyPD binds ANT1 [66].
During normal mitochondrial function the OMM and the IMM are separated by the intermembrane space, and the VDAC and the ANT do not interact [21,22,23,51]. Permeability transition is activated by the formation of the mPTP (Figure 1); the IMM loses its integrity and the ANT changes its conformation from its native state into a non-selective pore [21,22,23,51]. This process is catalyzed by CyPD functioning as a protein cis-trans isomerase and chaperone [20]. The ANT and CyPD interact directly in this process [72]. The amount of CyPD (in heart mitochondria) is much lower than the ANT concentration (< 5%); thus, under normal conditions only a minor fraction of the ANT can be in a complex with CyPD [53,72,73]. When this occurs, small ions and metabolites permeate freely across the IMM and oxidation of metabolites by O2 proceeds with electron flux not coupled to proton pumping, resulting in collapse of ΔP, dissipation of ATP production, elevated production of ROS, equilibration of ions between the matrix and cytosol, increased matrix volume, and mitochondrial swelling [52,55].
Very few studies have been published on the localizations of mPTP components in the mammalian CNS; thus, details about the cellular expressions in different nervous system cell types are lacking. VDAC expression patterns are complicated by alternative splicing that generates two different VDAC1 mRNAs, three different VDAC2 mRNAs, and two different VDAC3 mRNAs [54]. Studies of nervous tissue have found VDAC in neurons and glial cells [74] and associated with mitochondria, the endoplasmic reticulum (ER), and the plasma membrane [54,76]. Non-mitochondrial localizations of VDAC have been disputed [77]. Information on the cellular localizations of ANT in nervous tissue is scarce. ANT appears to be expressed in reactive astrocytes [78]. The few published studies on CyPD localization in mammalian CNS have found it enriched in subsets of neurons in adult rat brain, with some interneurons being positive [79], and relative low levels in astrocytes [80]. In mouse spinal cord, the core components of the mPTP (VDAC, ANT, and CyPD) are enriched in motor neurons as determined by immunohistochemistry [81]. The specific isoforms of ANT and VDAC in motor neurons have not been determined. CyPD, ANT, and VDAC have mitochondrial and non-mitochondrial localizations in motor neurons [81]. They are all nuclear-encoded mitochondrial-targeted proteins, thus a possible explanation for their non-mitochondrial localizations is that they are pre-mitochondrial forms. Some cyclophilins are located in the cytoplasm [82], such as cyclophilin A, but CyPD immunoreactivity is annulled in ppif-/- mice, demonstrating that the antibody is detecting only CyPD [81]. Spinal cord, brainstem, and forebrain had similar levels of CyPD, as well as similar levels of ANT and VDAC [81]. Thus, differences in the levels of individual mPTP components cannot explain the intrinsic differences in the sensitivity to Ca2+-induced mPT seen in isolated mitochondria from spinal cord and brain [83,84]. Not all mitochondria within individual motor neurons contain CyPD, ANT, and VDAC [81]; this observation supports the idea that mitochondria in individual cells are not only heterogeneous in shape [85,86] but also in biochemical composition, metabolism [87] and genetics [8].
Apoptosis
Apoptosis is a form of PCD because it is carried out by active, intrinsic transcription-dependent [88] or transcription-independent mechanisms [89] that involve specific molecules (Table 1 and Table 2; Figure 1); the predominance of the different mechanisms in neurons appears to be dictated in part by their maturation state [90]. Kerr and colleagues were the first to describe apoptosis in liver pathology settings [91], but many descriptions cell death different from necrosis were made prior to this time in studies of developing animal systems. Apoptosis should not be used as a synonym for PCD because non-apoptotic forms of PCD exist [92,93]. Apoptosis is only one example of PCD.
Apoptosis is critical for the normal growth and differentiation of organ systems in vertebrates and invertebrates (see [94] regarding Ernst’s 1926 discovery of developmental PCD) [95,96,97]. In physiological settings in adult tissues, apoptosis is a normal process, occurring continuously in populations of cells that undergo slow proliferation (e.g., liver and adrenal gland) or rapid proliferation (e.g., epithelium of intestinal crypts) [98,99]. Apoptosis is a normal event in the immune system when lymphocyte clones are deleted after an immune response [100]. The structure of apoptosis is similar to the Type I form of PCD described by Clarke [101].
Classical apoptosis has a distinctive structural appearance (Figure 2). The cell condenses and is dismantled in an organized way into small packages that can be consumed by nearby cells. Nuclear breakdown is orderly. The DNA is digested in a specific pattern of internucleosomal fragments (Figure 2), and the chromatin is packaged into sharply delineated, uniformly dense masses that appear as crescents abutting the nuclear envelope or as smooth, round masses within the nucleus (Figure 2). The execution of apoptosis is linked to Ca2+-activated DNases [102], one being DNA fragmentation factor 45 (DFF45) [103], that digests genomic DNA at internucleosomal sites only (because proteases that digest histones remain inactivated and the DNA at these sites is protected from DNases) to generate a DNA ‘ladder’ (Figure 2, top left) [102]. However, the emergence of the apoptotic nuclear morphology can be independent of the degradation of chromosomal DNA [104]. Cytoplasmic breakdown during apoptosis is also orderly. The cytoplasm condenses, (as reflected by a darkening of the cell in electron micrographs), and subsequently the cell shrinks in size, while the plasma membrane remains intact. During the course of these events, it is believed that the mitochondria are required for ATP-dependent processes. Subsequently, the nuclear and plasma membranes become convoluted, and, then the cell undergoes a process called budding. In this process, the nucleus, containing smooth, uniform masses of condensed chromatin, undergoes fragmentation in association with the condensed cytoplasm, forming cellular debris (called apoptotic bodies) composed of pieces of nucleus surrounded by cytoplasm with closely packed and apparently intact organelles. Apoptotic cells display surface markers (e.g., phosphatidylserine or sugars) for recognition by phagocyte cells. Phagocytosis of cellular debris by adjacent cells is the final phase of apoptosis in vivo.
Variants of classical apoptosis or non-classical apoptosis can occur during nervous system development [101,105] and also frequently in pathophysiological settings of nervous system injury and disease [24,28,29]. Axonal damage (axotomy) and target deprivation in the mature nervous system can induce apoptosis in neurons that is similar structurally, but not identical, to developmental PCD [17]. Excitotoxins and cerebral hypoxia-ischemia can induce readily and robustly non-classical forms of apoptosis in neurons in rodent brain [28,29,106,107,108].
Cells can die by PCD through mechanisms that are distinct from apoptosis [92,93]. The structure of non-apoptotic PCD is similar to the Type II or Type III forms of cell death described by Clarke [101]. Interestingly, there is no internucleosomal fragmentation of genomic DNA in some forms of non-apoptotic PCD [92,93].
Autophagy
Autophagy is a mechanism whereby eukaryotic cells degrade their own cytoplasm and organelles [27]. Autophagy functions as a homeostatic non-lethal stress response mechanism for recycling proteins to protect cells from low supplies of nutrients and as a cell death mechanism. Autophagy is also called Type II PCD [101]. This degradation of organelles and long-lived proteins is carried out by the lysosomal system; thus, a hallmark of autophagy is accumulation of autophagic vacuoles of lysosomal origin. Autophagy has been seen in developmental and pathological conditions. For example, insect metamorphosis involves autophagy [25], and developing neurons can use autophagy as a PCD mechanism [109,110]. Degeneration of Purkinje neurons in the mouse mutant Lucher appears to be a form of autophagy, thus linking excitotoxic constitutive activation of the GluRδ2 glutamate receptor to autophagic cell death [110]. However, loss of basal autophagic function in the CNS causes neurodegeneration in mice [112,113]. This finding could be a testimonial to the importance of Parkin, a ubiquitin kinase encoded by PD-related PARK2, which functions to promote autophagic turnover of mitochondria [114].
The molecular controls of autophagy appear common in eukaryotic cells from yeast to human, and autophagy may have evolved before apoptosis [35]. However, most of the work has been done on yeast, but detailed work on autophagy in mammalian cells emerging [115]. Double-membrane autophagosomes for sequestration of cytoplasmic components are derived from the ER or the plasma membrane. Tor kinase, phosphatidylinositol 3 (PI3)-kinase, a family of cysteine proteases called autophagins, and death-associated proteins function in autophagy [116,117]. Autophagic and apoptotic cell death pathways crosstalk. The product of the tumor suppressor gene Beclin1 (the human homolog of the yeast autophagy gene APG6) interacts with the anti-apoptosis regulator Bcl-2 [118]. Autophagy can block apoptosis by sequestration of mitochondria. If the capacity for autophagy is reduced, stressed cells die by apoptosis, whereas inhibition or blockade of molecules that function in apoptosis can convert the cell death process into autophagy [119]. Thus, a continuum between autophagy and apoptosis could exist.
Cellular and Molecular Regulation of Apoptosis
Apoptosis is a structurally and biochemically organized form of cell death (Figure 2). Apoptotic molecular networks are conserved in yeast, hydra, nematode, fruit fly, zebra fish, mouse, and human [120]. The current understanding of the molecular mechanisms of apoptosis in cells is built on studies by Robert Horvitz and colleagues on PCD in a nematode Caenorhabditis elegans [121]. They pioneered the understanding of the genetic control of developmental cell death by showing that it is regulated predominantly by three genes (ced-3, ced-4, and ced-9) [121]. This seminal work led to the identification of several families of apoptosis-regulation genes (Table 2) in mammals, including the Bcl-2 family [37,38,122] and the caspase family of cysteine-containing, aspartate-specific proteases [123]. Other regulators of apoptotic cell death, most of which are mitochondrial proteins or influence mitochondria, are the p53 gene family, cell surface death receptors, cytochrome c, apoptosis inducing factor (AIF), second mitochondrial activator of caspases (Smac), the inhibitor of apoptosis protein (IAP) family, and HtrA2/Omi [100,124,125,126,127,128,129].
Specific organelles, including mitochondria and the ER, have been identified as critical for the apoptotic process. In seminal work by Li, Wang, and colleagues, it was discovered that the mitochondrion integrates death signals engaged by proteins in the Bcl-2 family and releases molecules residing in the mitochondrial intermembrane space, such as cytochrome c, that complexes with cytoplasmic proteins (e.g., apoptotic protease activating factor 1, Apaf1) to activate caspase proteases leading to internucleosomal cleavage of DNA (Figure 1 and Figure 2) [125,126]. The ER, which regulates intracellular Ca2+ levels, participates in a loop with mitochondria to modulate mPT and cytochrome c release through the actions of Bcl-2 protein family members (Figure 1) [130].
Table 2.
Some Molecular Regulators of Apoptosis Relevant to Neurodegeneration and Potential Drug Targeting for Neuroprotection.
| Bcl-2 Family | Caspase Family | IAP Family | Tumor Suppressor | |
| Anti-apoptotic proteins | Pro-apoptotic proteins | |||
| Bcl-2 | Bax | Apoptosis “initiators”: caspase-2, 8, 9, 10 | NAIP | p53 |
| Bcl-xL | Bak1 | Apollon | p63 | |
| Mcl-1 | Bcl-xS | Survivin | p73 | |
| Boo/Diva | Bad | Apoptosis “executioners”: caspase-2, 3, 6, 7 | IAP1 | |
| Bid | IAP2 | |||
| Bik | XIAP | |||
| Bim | Cytokine processors: caspase-1, 4, 5, 11, 12, 14 | |||
| Noxa | ||||
| Puma | ||||
Bcl-2 family of Cell Survival and Cell Death Proteins
Mitochondria can control cell death (Figure 1) using Bcl-2 family members to regulate apoptosis by modulating the release of cytochrome c from mitochondria into the cytosol. Two models can account for this process: the Bcl-2-associated X protein (Bax)/Bak1 channel model and the mitochondrial apoptosis-induced channel or MAC) (Figure 1). The bcl-2 proto-oncogene family is a large group of apoptosis regulatory genes encoding about 20 different proteins. These proteins are defined by at least one of four conserved B-cell lymphoma (Bcl) homology domains (BH1-BH4) in their amino acid sequence that function in protein-protein interactions [37,38,122]. Some of the proteins (e.g., Bcl-2, Bcl-xL, and Mcl-1) have all four BH1-BH4 domains and are anti-apoptotic (Table 2). Other proteins that are pro-apoptotic have BH1-BH3 sequences (e.g., Bax and Bak1) or only the BH3 domain (e.g., Bad, Bid, Bim, Bik, Noxa, and Puma) that contains the critical death domain (Table 2). Bcl-xL and Bax have α-helices resembling the pore-forming subunit of diphtheria toxin [131]; thus, Bcl-2 family members appear to function by conformation-induced insertion into the outer mitochondrial membrane to form channels or pores that can regulate release apoptogenic factors (Figure 1). Bcl-2 family members can form homodimers or heterodimers and higher-order multimers with other family members. Bax/Bak1 heterodimerization with either Bcl-2 or Bcl-xL neutralizes their pro-apoptotic activity. When Bax and Bak1 are present in excess, the anti-apoptotic activity of Bcl-2 and Bcl-xL is antagonized, and apoptosis is promoted.
The expression of many of these proteins is regulated developmentally, and they have differential tissue distributions and subcellular localizations. Most of these proteins are found in CNS. The subcellular distributions of Bax, Bak1, and Bad in healthy adult rodent CNS tissue [132] are consistent with what is known about these proteins in cultured mammalian non-neuronal cells [133,134]. Bax, Bad, and Bcl-2 reside primarily in the cytosol, whereas Bak1 resides primarily in mitochondria.
Release of cytochrome c from mitochondria (Figure 1) can occur through mechanisms that involve the formation of membrane channels comprised of Bax or Bak1 [135] and Bax and the VDAC [136]. In the Bax/Bak1 channel model (Figure 1, left), after specific cell death inducing stimuli Bax undergoes a conformation shift and translocates to the OMM where it inserts. Bak1 is a similar pro-apoptotic protein localized mostly to the OMM. Bax/Bak1 monomers physically interact to form oligomeric or heteromeric channels that are permeable to cytochrome c. The formation of these channels is blocked by Bcl-2 and Bcl-xL at multiple sites. BH3-only members (Bad, Bid, Noxa, Puma) are pro-apoptotic and can modulate the conformation of Bax/Bak1 to sensitize this channel, possibly by exposing its membrane insertion domain. The MAC could be a channel similar to the Bax/Bak1 channel, but it might also have additional components such as VDAC.
Released cytochrome c then triggers the assembly of the cytoplasmic apoptosome. The apoptosime is a protein complex of apoptotic protease activating factor 1 (Apaf1), cytochrome c, and procaspase-9; this is the engine that drives caspase-3 activation in mammalian cells (Figure 1) [125]. Bcl-2 and Bcl-xL block the release of cytochrome c [137,138] from mitochondria and thus the activation of caspase-3 (Figure 1) [125,126]. This Bcl-2 and Bcl-xL mediated retention of mitochondrial cytochrome c [126,139] is caused by inhibition of Bax channel-forming activity in the outer mitochondrial membrane [135] or by modulation mitochondrial membrane potential and volume homeostasis [139]. BH3-only proteins such as Bim, Bid, Puma, and Noxa appear to induce a conformational change in Bax or they serve as decoys for Bcl-xL that allow Bax to form pores in the outer mitochondrial membrane [140]. Cells without bax and bak genes are resistant to mitochondrial cytochrome c release during apoptosis [141].
Some anti-apoptotic proteins also have functions downstream of mitochondria. For, example Bcl-xL has anti-apoptotic activity by interacting with Apaf1 and caspase-9 and inhibiting Apaf1-mediated autocatalytic maturation of caspase-9 [142]. Boo can inhibit Bak- and Bik-induced apoptosis (but not Bax-induced cell death) possibly through heterodimerization and by interactions with Apaf1 and caspase-9 [143].
Protein phosphorylation regulates the functions of some Bcl-2 family members having downstream mitochondrial consequences. Bcl-2 loses its anti-apoptotic activity following serine phosphorylation, possibly because its antioxidant function is inactivated [144]. Bcl-2 can also associate with non-homologous proteins, including the protein kinase Raf-1 [145]. This association can target Raf-1 to mitochondrial membranes, allowing this kinase to phosphorylate Bad at serine residues [145]. The phosphatidylinositol 3-kinase (PI3-K) –Akt pathway also regulates the function of Bad [146,147] and caspase-9 [148] through phosphorylation. In the presence of sufficient trophic factors, Bad is phosphorylated. Phosphorylated Bad is sequestered in the cytosol by interacting with soluble protein 14-3-3 and, when bound to protein 14-3-3, Bad is unable to interact with Bcl-2 and Bcl-xL, thereby promoting cell survival [149]. Conversely, when Bad is dephosphorylated by calcineurin [150], it dissociates from protein 14-3-3 in the cytosol and translocates to the mitochondria where it exerts pro-apoptotic activity. Non-phosphorylated Bad heterodimerizes with membrane-associated Bcl-2 or Bcl-xL to liberate Bax from Bax-Bcl-2 and Bax-Bcl-XL dimers, thus promoting cell death [109]. In liver mitochondria, Bad and glucokinase exist in a complex that functions in mitochondrial-based glucokinase activity and mitochondrial respiration in response to glucose [152]. Glucose deprivation results Bad dephosphorylation and Bad-dependent cell death, thereby linking glucose metabolism to apoptosis [152].
Caspase Family of Cell Demolition Proteases
Caspases (cysteinyl aspartate-specific proteinases) are cysteine proteases that have a near absolute substrate requirement for aspartate in the P1 position of the peptide bond. Fourteen caspase genes have been identified in mammals [153]. Some caspases (e.g., caspase-12) in human and mouse function differently and have different contributions to cell death mechanisms. Caspases exist as constitutively expressed inactive pro-enzymes (30–50 kDa) in healthy cells. Caspase zymogens are found in different proportions at different subcellular locations. In HeLa cells, most caspase-3 pro-enzyme is found in the cytoplasm, while only 10% is found in mitochondria [154]. In rat heart and brain, 90% of caspase-9 pro-enzyme is mitochondrial [155]. The zymogens contain 3 domains: an amino-terminal pro-domain; a large subunit (~20 kDa); and a small subunit (~10 kDa). Caspases are activated through regulated proteolysis of pro-enzyme with “initiator” caspases activating “executioner” caspases (Table 1; Figure 1). Other caspase family members function in inflammation by processing cytokines (Table 1) [153].
The pro-domain of initiator caspases contains amino acid sequences that are caspase recruitment domains (CARD) or death effector domains (DED) that enable the caspases to interact with other molecules that regulate their activation. Activation of caspases involves proteolytic processing between domains, and then association of large and small subunits to form a heterodimer with both subunits contributing to the catalytic site. Two heterodimers associate to form a tetramer that has 2 catalytic sites that function independently. Some isoforms of caspases (e.g., caspase-9, isoform 2) are inactive proteolytically and function as dominant negative inhibitors of active forms.
Active caspases have many target proteins [112] that are cleaved during regulated and organized cell death. Caspases cleave nuclear proteins (e.g., DNases, poly(ADP) ribose polymerase, DNA-dependent protein kinase, heteronuclear ribonucleoproteins, transcription factors or lamins), cytoskeletal proteins (e.g., actin and fodrin), and cytosolic proteins (e.g., other caspases, protein kinases, Bid).
In human cell line models of apoptosis (Figure 1), activation of caspase-3 occurs when caspase-9 pro-enzyme (also known as Apaf3) is bound by Apaf1 that then oligomerizes in a process initiated by cytochrome c (identified as Apaf2) and either ATP or dATP [125]. Cytosolic ATP or dATP are required cofactors for cytochrome c-induced caspase activation [125]. Apaf1, a 130 kDa cytoplasmic protein, serves as a docking protein for procaspase-9 and cytochrome c [125]. Apaf1 becomes activated when ATP is bound and hydrolyzed, with the hydrolysis of ATP and the binding of cytochrome c promoting Apaf1 oligomerization [113]. This oligomeric complex recruits procaspase-9 (forming the apoptosome) and mediates the autocatalytic activation of caspase-9 that disassociates from the complex and becomes available to activate caspase-3 (Figure 1). Once activated, caspase-3 cleaves a protein with DNase activity (i.e., DFF-45), and this cleavage activates a process leading to the internucleosomal fragmentation of genomic DNA (Figure 2, top left) [103].
So far three caspase-related signaling pathways have been identified that can lead to apoptosis [103,125,126,157], but crosstalk among these pathways is possible. The intrinsic mitochondria-mediated pathway is controlled by Bcl-2 family proteins. It is regulated by cytochrome c release from mitochondria, promoting the activation of caspase-9 through Apaf1 and then caspase-3 activation. The extrinsic death receptor pathway involves the activation of cell-surface death receptors (see below), including Fas and tumor necrosis factor receptor, leading to the formation of the death-inducing signaling complex (DISC) and caspase-8 activation that in turn cleaves and activates downstream caspases such as caspase-3, -6, and -7. Caspase-8 can also cleave Bid leading to the translocation, oligomerization, and insertion of Bax or Bak1 into the mitochondrial membrane. Another pathway involves the activation of caspase-2 by DNA damage or ER stress as a pre-mitochondrial signal [158].
Inhibitor of Apoptosis Protein (IAP) Family
The activity of pro-apoptotic proteins is blocked to prevent untimely apoptosis in normal cells. Apoptosis can be antagonized by the IAP family in mammalian cells [159,160,161]. This family includes X chromosome-linked IAP (XIAP), IAP1, IAP2, neuronal apoptosis inhibitory protein (NAIP), Survivin, Livin, and Apollon. These proteins are characterized by 1 to 3 baculoviral IAP repeat domains consisting of a zinc finger domain of ~70–80 amino acids [160]. Apollon is a huge (530 kDa) protein that also has a ubiquitin-conjugating enzyme domain. The main identified anti-apoptotic function of IAPs is the suppression of caspase activity [161]. Procaspase-9 and procaspase-3 are major targets of several IAPs. IAPs reversibly interact directly with caspases to block substrate cleavage. Apollon also ubiquitinates and facilitates proteasomal degradation of active caspase-9 and second mitochondria-derived activator of caspases (Smac) [162]. However, IAPs do not prevent caspase-8-induced proteolytic activation of procaspase-3. IAPs can also block apoptosis by reciprocal interactions with the nuclear transcription factor NFκB [160].
Scant information is available on IAPs in the nervous system. Survivin is essential for nervous system development in mouse because conditional deletion of survivin gene in neuronal precursor cells causes reduced brain size and severe multifocal degeneration and death shortly after birth [163]. NAIP is expressed throughout the CNS in neurons [164]. XIAP is enriched highly in mouse spinal motor neurons [165]. The importance of the IAP gene family in pediatric neurodegeneration is underscored by the finding that NAIP is deleted partially in a significant proportion of children with spinal muscular atrophy [166].
Mitochondrial proteins exist that inhibit mammalian IAPs. A murine mitochondrial protein called Smac and its human ortholog DIABLO (for direct IAP-binding protein with low pI) inactivate the anti-apoptotic actions of IAPs and thus exert pro-apoptotic actions [167,168]. Smac/DIABLO are released into the cytosol to inactivate the anti-apoptotic actions of inhibitor of apoptosis proteins that inhibit caspases (Figure 1). These IAP inhibitors are 23 kDa mitochondrial proteins (derived from 29 kDa precursor proteins processed in the mitochondria) that are released into the cytosol from the intermembrane space to sequester IAPs. High temperature requirement protein A2 (HtrA2), also called Omi, is another mitochondrial serine protease that exerts pro-apoptotic activity by inhibiting IAPs [169]. HtrA2/Omi functions as a homotrimeric protein that cleaves IAPs irreversibly, thus facilitating caspase activity. The intrinsic mitochondrial-mediated cell death pathway is regulated by Smac and HtrA2/Omi [169]. Mutations in the htra2 gene, identified as PARK13 (Table 3), have been linked to the development of Parkinson’s disease [170], but this linkage is controversial [171].
Apoptosis Inducing Factor (AIF)
AIF is a mammalian cell mitochondrial protein identified as a flavoprotein oxidoreductase [172]. AIF has an N-terminal mitochondrial localization signal that is cleaved off to generate a mature protein of 57 kDa after import into the inter-mitochondrial membrane space. Under normal physiological conditions AIF might function as a ROS scavenger targeting H2O2 [127] or in redox cycling with nicotinamide adenine dinucleotide phosphate [173]. After some apoptotic stimuli, AIF is released from mitochondria (Figure 1) and translocates to the nucleus [172]. Over-expression of AIF in cultured cells induces cardinal features of apoptosis, including chromatin condensation, high molecular weight DNA fragmentation, and loss of mitochondrial transmembrane potential [172].
p53/p63/p73 Family of Tumor Suppressors
Cell death by apoptosis can be triggered by DNA damage. p53 and related DNA binding proteins identified as p73 and p63 are involved in this process [124]. p53, p73 and p63 function in apoptosis as well as growth arrest and repair. They can commit to death cells that have sustained DNA damage from ROS, irradiation, and other genotoxic stresses [124]. p53 and p73 have similar oligomerization and DNA sequence transactivation properties. p73 exists as a group of full-length isoforms (including p73α and p73β) and as truncated isoforms that lack the transactivation domain (∆N-p73). p53 is the most well studied of this family of proteins.
p53 is a short-lived protein with a half-life of ~5-20 min in most types of cells studied but can rapidly accumulate several-fold in response to DNA damage. This rapid regulation is mediated by posttranslational modification such as phosphorylation and acetylation as well as intracellular redox state [174]. The elevation in p53 protein levels occurs through stabilization and prevention of degradation. p53 is degraded rapidly in a ubiquitination-dependent proteasomal pathway [175]. Murine double minute 2 (Mdm2, the human homolog is Hdm2) has a crucial role in this degradation pathway [176]. Mdm2 functions in a feedback loop to limit the duration or magnitude of the p53 response to DNA damage. Expression of the mdm2 gene is controlled by p53 [176]. Mdm2 binds to the N-terminal transcriptional activation domain of p53 and regulates its DNA binding activity and stability by direct association. Mdm2 has ubiquitin ligase activity for p53 through the ubiquitin-conjugating enzyme E2. Stabilization of p53 is achieved through phosphorylation of serine15 resulting in inhibition of formation of Mdm2-p53 complexes. Activated p53 binds the promoters of several genes encoding proteins associated with growth control and cell cycle checkpoints (e.g., p21, growth-arrest and DNA damage-45, Mdm2) and apoptosis (e.g., Bax, Bcl-2, Bcl-xL, and Fas). The BH3-only proteins Puma and Noxa are critical mediators of p53-mediated apoptosis [137].
p53 can mediate cell death through extra-nuclear transcriptional-independent mechanisms. p53 can translocate rapidly to mitochondria in response to genotoxic, hypoxic, and oxidative stresses in non-neuronal cells [178] and in neurons [90]. This localization can mediate mitochondrial membrane permeabilization through direct physical interaction with Bax [179] and activation of Bak through disruption of the Bak-Mcl1 complex [180].
p53 can drive apoptosis in cultured sympathetic ganglion neurons in response to neurotrophin withdrawal [181] and in cultured mouse cortical neurons in response to DNA damage [90]. A small-molecule inhibitor of p53 binding to mitochondria protects against neuronal apoptosis in cultured mouse cortical neurons [90]. p53-mediated neuronal apoptosis in vitro can be blocked by the ∆N-p73 isoform by direct binding and inactivation of p53 [141]. In vivo experiments show that p53 gene ablation protects against neuronal apoptosis induced by axotomy and target deprivation in rodent brain and spinal cord [182,183].
Cell Surface Death Receptors
Cell death can also be initiated at the cell membrane by surface death receptors of the tumor necrosis factor (TNF) receptor family. Fas (CD95/Apo-1) and the 75-kDa neurotrophin receptor (p75NTR) are members of the large TNF receptor family [100]. Signals for apoptosis are initiated at the cell surface by aggregation (trimerization) of the death domain containing members of this receptor family by their specific ligand. Fas death receptor-mediated apoptosis is a well described pathway for death receptor signaling and is independent of new RNA or protein synthesis. Activation of Fas is induced by binding of the multivalent Fas ligand (FasL), a member of the TNF-cytokine family. FasL is expressed on activated T cells and natural killer cells. Clustering of Fas on the target cell by FasL recruits Fas-associated death domain (FADD), a cytoplasmic adapter molecule that functions in the activation of the caspase 8-Bid pathway, thus forming the DISC [185]. Signaling for apoptosis then proceeds via the extrinsic or intrinsic pathway. In the extrinsic pathway, active caspase-8 then directly cleaves caspase-3 [100]. Activation of the mitochondrial or intrinsic pathway proceeds via caspase 8 mediated cleavage of cytosolic Bid [185]. The truncated form of Bid then translocates to mitochondria, thereby functioning as a BH3-only transducer of Fas activation signal at the cell plasma membrane to mitochondria [185]. Bid translocation from the cytosol to mitochondrial membranes is associated with a conformational change in Bax (that is prevented by Bcl-2 and Bcl-xL) and is accompanied by release of cytochrome c from mitochondria [186].
Apoptosis can also be mediated by p75NTR [187]. Activation of p75NTR occurs by binding of nerve growth factor. When p75NTR is activated without tropomyosin receptor kinases, neurotrophin binding induces homodimer formation and generation of ceramide through sphingomyelin hydrolysis. Ceramide production is associated with the activation of Jun N-terminal kinase (JNK) that phosphorylates and activates c-Jun and other transcription factors. p75NTR mediates hippocampal neuron death in response to neurotrophin withdrawal, involving cytochrome c, Apaf1, and caspases-9, -6, and -3 (but not caspase-8), and thus is different from Fas-mediated cell death [187].
Evidence for the importance of these signaling pathways in experimental brain injury is growing. Activation of multiple components of the Fas death receptor signaling pathway have been found in rat and mouse models of motor neuron degeneration [188] and blocking Fas death receptor signaling by genetic means affords protection in these models [188]. Neuron degeneration caused by target deprivation in vivo appears to be driven in part by a death receptor-dependent pathway [18].
Excitotoxic Neuronal Cell Death
Neuronal death can be induced by excitotoxicity. This observation was made originally by Lucas and Newhouse in 1957 [189], formulated into a concept by John Olney after showing that glutamate can kill neurons in brain [190], and then examined mechanistically by Dennis Choi, Steven Rothman and others [191,192]. This concept has fundamental importance to neural mitochondria pathobiology and to a variety of acute neurological insults, such as cerebral ischemia and trauma, and possibly chronic neurodegenerative diseases [2,7,17,24,33,43,193,194,195]. Excitotoxicity is pathologic neurodegeneration mediated by excessive activation of glutamate-gated ion channel receptors and voltage-dependent ion channels. Increased cytosolic free Ca2+ causes activation of Ca2+-sensitive proteases, protein kinases/phosphatases, phospholipases, and NOS when glutamate receptors are stimulated. The excessive interaction of ligand with subtypes of glutamate receptors causes pathophysiological changes in intracellular ion concentrations, pH, protein phosphorylation, energy metabolism, and mitochondrial function and movement [194,195]. Intracellular Ca2+ elevations can halt anterograde movement of mitochondria through kinesin-1 and the atypical GTPase Miro [196]. The mPTP is also involved at least in cell culture models of excitotoxicity [195]. The precise mechanisms of excitotoxic cell death and its relationships to mitochondria are still being examined intensively, driven by the hope of identifying therapeutic targets for neurological/neurodegenerative disorders with putative excitotoxic components. Evidence that the uncompetitive, low-affinity, NMDA receptor open-channel blocker shows benefits in AD and other human neurodegenerative diseases [196] justifies continued work on the excitotoxicity concept, but other mechanisms need to be folded into excitotoxicity theory, including sodium-calcium exchangers, volume-regulated anion channels, and acid-sensing channels, to complete the concept. A more complete excitotoxicity theory might help to validate the cell death matrix concept (see below) and to explain why cell culture and animal experimental data are discordant with regard to whether excitotoxic neuronal death is apoptosis, necrosis, apoptosis-necrosis hybrids, autophagy, or perhaps even a peculiar form of cell death that is unique to excitotoxicity.
The contribution of apoptotic mechanisms to excitotoxic death of neurons has been examined in cultured neurons. Excitotoxicity can cause activation of endonucleases and specific internucleosomal DNA fragmentation in cultures of cortical neurons [197,198] and cerebellar granule cells [199,200]. Internucleosomal fragmentation of DNA was not observed in other experiments on cerebellar granule cell cultures [201]. Excitotoxic cell death in neuronal cultures is prevented [198] or unaffected [197,200,201] by inhibitors of RNA or protein synthesis and is sensitive [198,200] or insensitive [201] to the endonuclease inhibitor aurintricarboxylic acid. In primary cultures of mouse cortical cells, the non-NMDA glutamate receptor agonist kainic acid (KA) induces increased Bax protein, and bax gene ablation significantly protects cells against KA receptor toxicity [202]. However, NMDA receptor toxicity in mouse cerebellar granule cells [203] and mouse cortical cells [204] was not Bax-dependent. These results suggest that non-NMDA glutamate receptor excitotoxicity is more likely than NMDA receptor-mediated excitotoxicity to induce apoptosis or apoptosis-necrosis hybrid cell death [17,28,29]; however, species-specific responses might be operative. Glutamate (100 μM) stimulation of mouse cortical cells did not cause an increase in caspase activity [205], but NMDA treated rat cortical cells showed increased caspase activity [206]. In cerebellar granule neurons, glutamate (100 µM - 1 mM) did not activate caspase activity and adenoviral-mediated expression of IAPs did not influence excitotoxic cell death [207]. These conflicting results can also be related to the finding that activation of different subtypes of glutamate receptors appears to engage different modes of cell death [17,28,29].
The precise mechanisms of excitotoxic neuronal cell death in animals have not been identified. The morphology of excitotoxicity in many neurons in rodents and large animals injected intracerebrally with excitotoxins include somatodendritic swelling, mitochondrial damage, and chromatin condensation into irregular clumps (Figure 2) [17,28,29,190,208], features that are thought to be typical of cellular necrosis; however, in other neurons, excitotoxicity causes cytological features more like apoptosis [28,29,208]. Excitotoxic degeneration of hippocampal CA3 neurons in response to KA is increased in naip gene-deleted mice, supporting a contribution of capsase-dependent apoptosis [209]. Excitotoxic neurodegeneration in adult rat brain has been shown to be either sensitive [160] or insensitive [211] to protein synthesis inhibition.
In newborn rodents, injection of KA or the NMDA receptor agonist quniolinic acid into the forebrain causes copious apoptosis of cortical, hippocampal, and striatal neurons serving as models of apoptosis in neurons in vivo [212,213]. This apoptosis has been verified structurally with light microscopy and EM and by immunolocalization of cleaved caspase-3 [212,123]. Ubiquitous apoptosis is observed at 24 hours after the insult. DNA degradation by internucleosomal fragmentation further confirms the presence of apoptosis. Excitotoxic neuronal apoptosis is associated with rapid (within 2 hours after neurotoxin exposure) translocation of Bax and cleaved caspase-3 to mitochondria [212].
A study has revealed that the ratio of mitochondrial membrane-associated Bax to soluble Bax in normal developing striatum changes prominently with brain maturation [212]. Newborn rat striatum has a much greater proportion of Bax in the mitochondrial fraction relative to soluble Bax [212]. Mature rat striatum has a much larger proportion of Bax in the soluble fraction relative to Bax in the mitochondrial fraction [212]. With brain maturation there is a linear decrease in the ratio of mitochondrial Bax to soluble Bax [212]. This developmental subcellular redistribution of Bax might be a reason why immature rodent neurons exhibit a more robust classical apoptosis response compared to adult neurons after brain damage [212].
The Cell Death Continuum
Animal models of neurodegeneration have revealed that age or maturity of brain and the subtype of excitatory glutamate receptor that is activated appear to influence the cytological features and rate of neuronal cell death [17,24,28,29,212,213,214]. This structural and temporal diversity of neuronal cell death is seen with a variety of brain injuries including excitotoxicity, cerebral hypoxia-ischemia, target deprivation, and axonal trauma. Hence, injury-associated neuronal death is not the same in immature and mature CNS and can be pleiomorphic in neurons within the same brain (Figure 2).
To help explain these data the concept of the cell death continuum was formulated (Figure 2). In this concept cell death exists as a continuum of necrosis and apoptosis with numerous hybrid forms of degeneration manifesting between necrosis and apoptosis (Figure 2) [17,24,28,29]. A fundamental cornerstone of the cell death continuum concept is thought to be gradations in the responses of cells to stress, particularly gradations in mitochondrial dysfunction and mPTP activation [108,215]. Some specific mechanisms thought to drive the continuum are the developmental expression of different subtypes of glutamate receptors, mitochondrial bioenergetics and membrane protein composition (e.g., Bax and mPTP components), the propinquity of developing neurons to the cell cycle, neurotrophin requirements and extensiveness of axonal collateralization, DNA damage vulnerability, and DNA repair mechanism availability [216]. Although the molecular mechanisms that drive this cell death continuum in the intact CNS are uncertain currently, cell culture data hint that ATP levels [42], intracellular Ca2+ levels [36] and mPT [50,51] could be involved. Whole animal experiments suggest that the relative level of Bax in the OMM could regulate the cell death continuum in neurons [212].
The concept of the cell death continuum has been challenged and deemed as confusing [217,218,219]. Do morphology and underlying biochemical processes of cell death remain binary and discrete [217]? While this is the case at the extremes of the cell death continuum, absolute discreteness ignores the observable features of cell degeneration seen in the injured and diseased CNS. Cell death is more than binary, it is multi-valued. Other possibilities that might have bearing on the reality of the cell death continuum concept include: 1) excitotoxic neuronal death in vivo is necrotic, regardless of age, and 2) apoptosis of neurons in the adult nervous system is extremely infrequent [217]. Experiments done by us [17,28,29,106,107,108] and others [220,221,222,223] have shown that neuronal degeneration triggered by excitotoxicity and hypoxia-ischemia can be apoptosis, apoptosis-necrosis hybrids, necrosis, and autophagy; furthermore, entire populations of neurons in the adult rodent CNS can indeed undergo apoptosis after injury [182,183].
Rigid conceptualization of cellular pathology is not realistic and is misleading and can hinder the goal of identification of relevant molecular mechanisms of neurodegeneration in complex biological systems, such as the developing, ageing or injured CNS, and ultimately limit the realization of therapeutic opportunities. For example, motor neuron degeneration in amyotrophic lateral sclerosis (ALS) was not considered to be a variant of apoptosis until the concept of the cell death continuum was applied [224], and now anti-apoptosis therapies are in clinical trials for the treatment of ALS [225]. The concept of the cell death continuum (Figure 2) might also be applicable to cytopathology in general, when dealing with cells that are resistant to one form of cell death.
The Cell Death Matrix
Studies show that morphologic appearance of the dying cell is a valuable tool for providing hints about the biochemical and molecular events responsible for the cell death type [17,18]. When studying mechanisms of cell death in human disease and in animal and cell models of disease it can be helpful to embrace the idea that apoptosis, necrosis, autophagy, or non-apoptotic PCD are not strictly “black and white”. For the nervous system, overlay this complexity with cell death mechanisms that are influenced by brain maturity, post-mitotic state of neurons, capacities for protein/RNA synthesis and DNA repair, antioxidant/redox status, neurotrophin requirements, death receptor expression, location in brain and location relative to the primary sites of injury, as well as intensity of the insult and mPT (Figure 2).
To help better comprehend neurodegeneration and discover laws that determine causes and effects in neurodegenerative settings, the concept of the cell death continuum was extended to a hypothetical cell death matrix to embrace the ‘fuzziness’ of cell death in the injured CNS (Figure 2). A matrix might be a useful modeling tool for pathology in general and specifically for predicting the contributions of the different forms of cell death, and the possible identification of previously unrecognized forms of cell death in human neurological disorders and in their animal/cell models. The cell death matrix draws on the framework of biological space-time. It integrates space (location in brain, location of primary insult) and time into a continuum; thus, cell death manifests in a brain regional 3-dimensional context with time playing the role of a fourth dimension that is of a different context than the spatial dimension. By combining space and time into a single matrix we organize a large number of cell death phenotypes and potential mechanisms into a manageable frame of reference to reveal the potential early and delayed responses of the brain to stress/injury and therapeutic interventions.
We need to identify better the relationships between mechanisms of cell death and the structure of dying cells in human pathology, in developing and adult CNS, as well as in animal and cell models of neurotoxicity in undifferentiated immature and terminally differentiated cells. The concept of a cell death matrix could be important for understanding neuronal degeneration in a variety of pathophysiological settings, and thus may be important for mechanism-based neuroprotective treatments in neurological disorders in infants, children, and adults. If brain maturity and brain location dictate how and when neurons die relative to the insult [17,18], then the molecular mechanisms responsible for neuronal degeneration in different brain regions (and at different times after the injury) in infants and children might be different from the mechanisms of neuronal degeneration in adults; hence, therapeutic targets will differ, and, thus, therapies will need to be customized for different brain regions, post-insult times, and age groups.
A cell death matrix could also be useful for modeling outcomes and how drugs and other treatments for human disease will work. It will be extremely important to use clues from cell death structure following different types and degrees of brain injury to better understand which injuries are most likely to respond to anti-necrosis, anti-apoptosis, or combination therapies and whether these therapies actually ameliorate injury or simply delay or change the mode of cell death. We predict that apoptosis inhibitors alone will be inadequate to ameliorate neurodegeneration in most settings, because if the cell death continuum is real, then apoptosis inhibitor drugs could simply push cell degeneration from apoptosis to apoptosis-variant, autophagy, or necrotic cell death as seen in cultured fibroblastic cells treated with caspase inhibitors after chemical hypoxia [30]. Using the cell death matrix we predict that it will be difficult to pinpoint appropriate times for effective mechanism-based, spatially-directed drug therapy for neuroprotection.
Cell Death in Human Neurodegenerative Diseases
Alzheimer’s disease (AD)
AD is the most common cause of dementia occurring in middle and late life [226]. Population based surveys estimate that AD affects 7–10% of individuals >65 years of age and possibly 50–60% of people over 85 years of age [227,228]. AD now affects about 2% of the population, or about 4 million people in the USA and ~35 million people worldwide [229]. The prevalence of AD is increasing proportionally to increased life expectancy and estimates predict that the prevalence will reach ~107 million by 2050 [230].
Most cases of AD have unknown etiologies and are called sporadic and have a late onset; however, some cases, particularly those with early onset, are familial and are inherited as autosomal dominant disorders linked to mutations in the gene that encodes amyloid precursor protein (APP) [231,232,233,234] or genes that encode for presenilin proteins [235,236]. For late onset sporadic cases, a variety of risk factors have been identified in addition to old age [237]. The apolipoprotein E (ApoE) allele is a susceptibility locus with the ApoE4 type showing dose-dependent contributions [238]. Cardiovascular disease and head trauma are additional risk factors for AD [226].
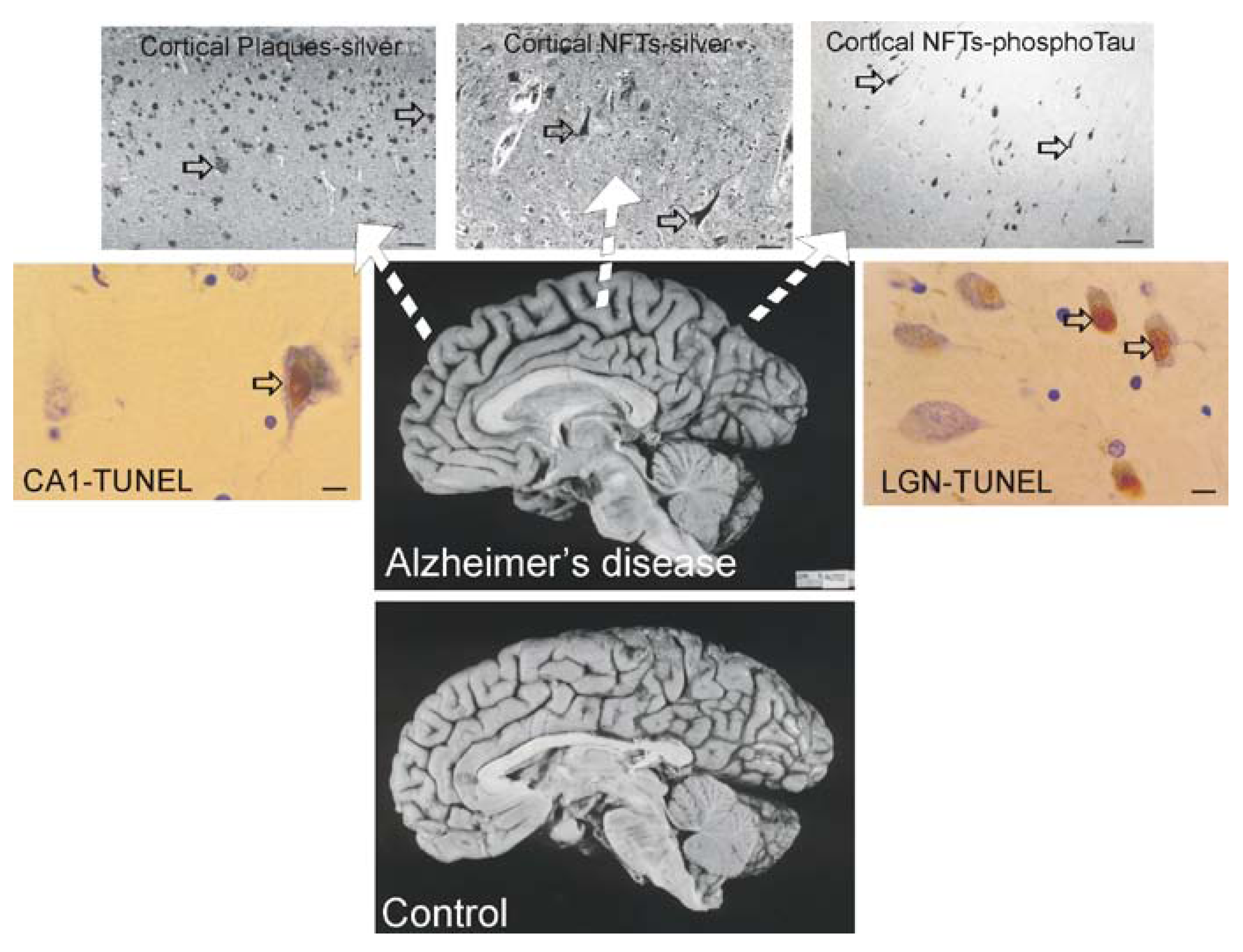
Figure 3.
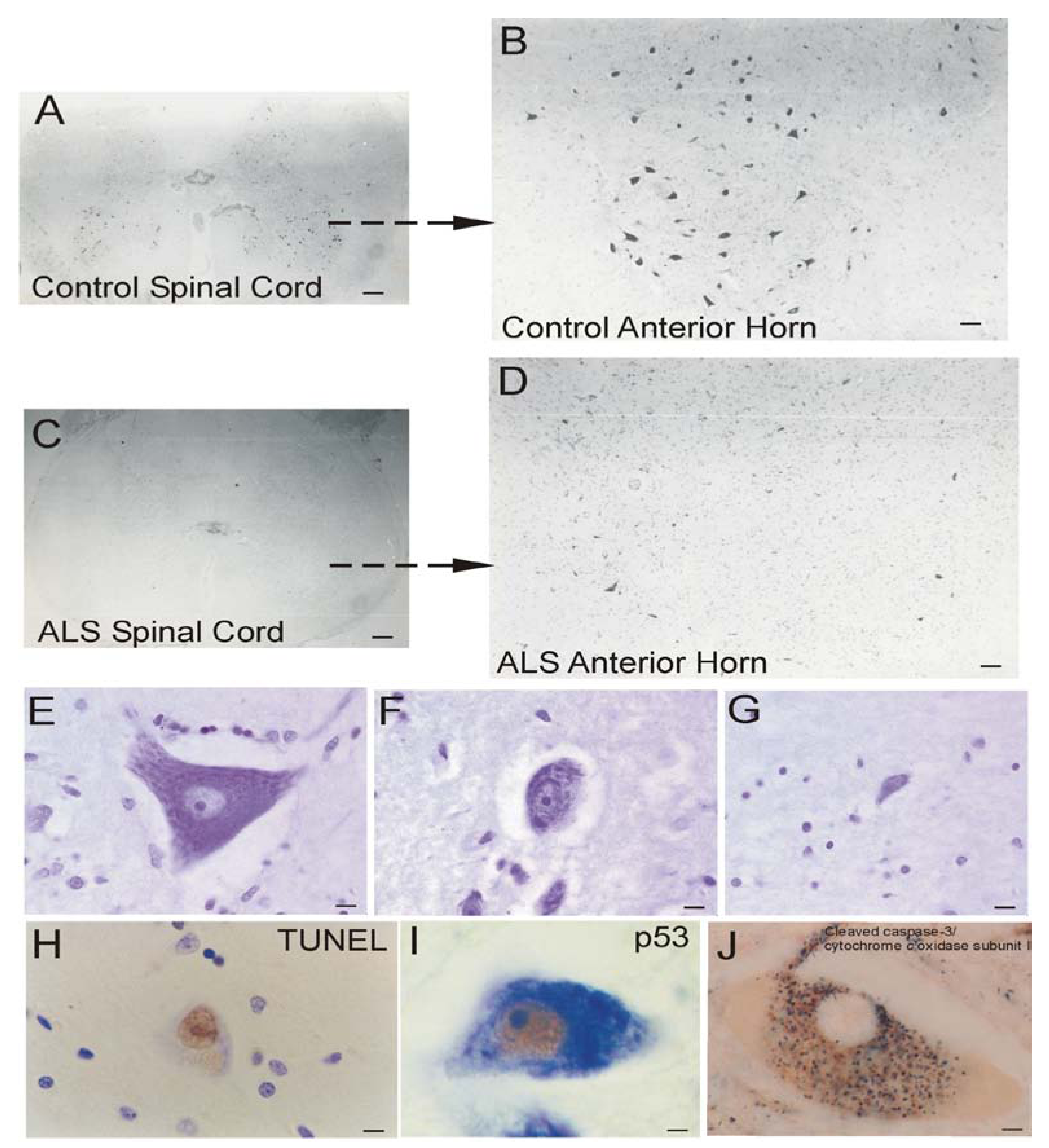
Brain atrophy and neurodegeneration in people with AD. Midsagittal views (center pictures) of the brains from an 85-year-old individual with AD and an 86-year-old normal control individual. The microscopic neuropathological hallmarks of AD are senile plaques (scale bar = 200 μm), neurofibrillary tangles (NFTs, scale bar = 50 μm.), and neuronal cell death determined by transferase-mediated biotin-dUTP nick-end labeling (TUNEL, brown nuclear staining, open arrows, scale bars = 10 μm) as seen in the hippocampus CA1 and in subcortical regions such as the thalamic lateral geniculate nucleus (LGN).
Figure 3.
Brain atrophy and neurodegeneration in people with AD. Midsagittal views (center pictures) of the brains from an 85-year-old individual with AD and an 86-year-old normal control individual. The microscopic neuropathological hallmarks of AD are senile plaques (scale bar = 200 μm), neurofibrillary tangles (NFTs, scale bar = 50 μm.), and neuronal cell death determined by transferase-mediated biotin-dUTP nick-end labeling (TUNEL, brown nuclear staining, open arrows, scale bars = 10 μm) as seen in the hippocampus CA1 and in subcortical regions such as the thalamic lateral geniculate nucleus (LGN).
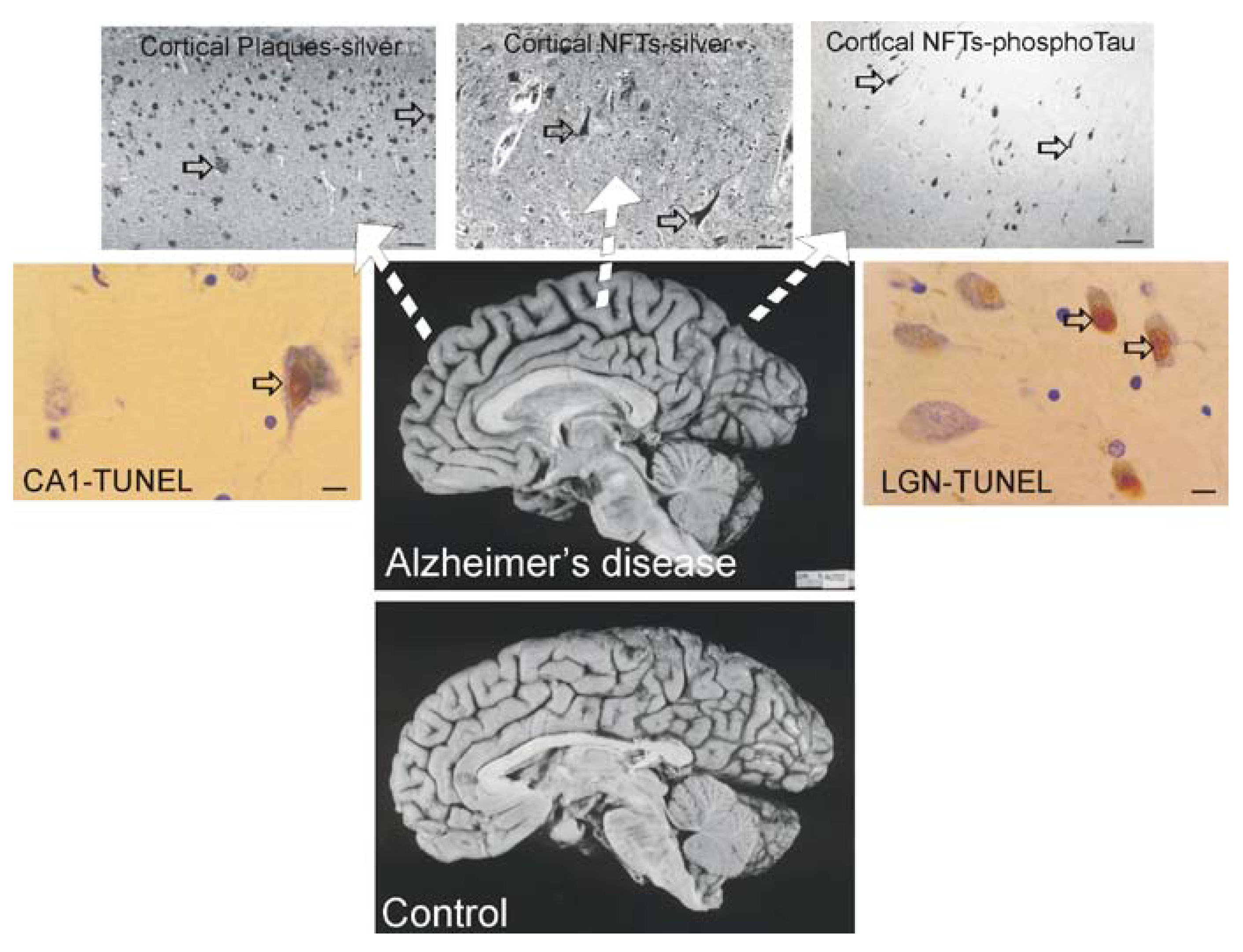
The dementia in AD is caused by severe atrophy of the cerebral cortex, as indicated by the widening of the sulci and narrowing of the gyri (Figure 3) while normal aged individuals have broad gyri and narrow sulci (Figure 3). Neurons in the neocortex, hippocampus, basal forebrain, and brainstem (e.g., dorsal raphe) are selectively vulnerable in AD [239,240,241,242,243]. The numerous lesions that are formed in the brains of AD patients are called senile plaques, containing abnormal extracellular deposits of Aβ amyloid protein (Figure 3, arrows), and NFTs which are abnormal intracellular aggregates of protein containing hyperphosphorylated tau (Figure 3, arrows).
It is useful to view the neurodegeneration in AD in the context of neural systems and stages [244]. Surprisingly, the classification of the neurodegeneration in AD is still not clear [245]. Dying neurons are found in cortical and subcortical regions in the AD brain (Figure 3). By TUNEL, an in situ DNA fragmentation/damage assay, subsets of neocortical, hippocampal, and thalamic neurons in the AD brain can be found with double-stranded DNA breaks, suggesting that these cells are in the process of dying (Figure 3). This neuronal death has been interpreted as apoptosis [246,247,248]; however, this DNA damage is not specifically indicative of apoptosis [17,24,249]. Other reports conclude that apoptosis does not have a major role in the neuronal degeneration of AD [250]. Experiments on changes in the levels of proteins in the Bcl-2 family in AD postmortem brain are difficult to interpret, with studies showing up-regulation of both anti-apoptotic and pro-apoptotic proteins or no changes [247,251,252]. AD brain degeneration might involve caspase-3 activation as determined by the imunohistochemical detection of cleaved caspase-3 [249,251,253] and caspase gene expression [254]. Cleaved caspase-3 [253], caspase-9 [255] and caspase-6 [256] have been found in NFT-bearing neurons in AD. However other studies have not shown evidence for the accumulation of cleaved caspase-3 in neurons in the AD brain [257], but changes seen in early and late AD may differ [253]. It is also noteworthy that immunodetection of cleaved caspase-3 is not always equivalent to caspase-3 activation as determined biochemically [258]. Furthermore, caspase-3 functions in processes other than cell death, including neuronal differentiation, migration, and plasticity [259,260].
Neuronal cell degeneration in AD occurs over a lengthy period. When considering the pathological classification of the primary neurodegeneration in AD it appears safe to conclude that based on morphology it is not classical apoptosis or necrosis. Autophagy could have a role in this neuronal cell death [249,261]. It might be more useful to consider neurofibrillary cell death separate from classical apoptosis and necrosis with some overlap in mechanisms according to the cell death matrix (Figure 2 and Figure 3). It could be important to know how neurons with neurofibrillary degeneration escape classical apoptosis and necrosis.
The mechanisms that cause the profound degeneration and loss of neurons in AD are not known, and existing information is incomplete. Abnormal processing or modification of APP and the cytoskeletal protein tau (a microtubule-associated protein) are involved in the pathogenesis (Figure 3) [262] resulting in amyloid (Aβ) deposits and neurofibrillary changes consisting of paired helical filaments, NFTs and dystrophic neurites (Figure 3) [262]. Cortical and hippocampal neuronal degeneration could be the consequence of a combination of several mechanisms including perturbations in protein metabolism, excitotoxicity, oxidative stress, mitochondrial perturbations, and inflammation. The possible specific mechanisms for neuronal degeneration in AD may involve dysfunction of NMDA receptors [264,265], dysregulation of Ca2+ and mitochondrial homeostasis [266,267], defects in synapses [268,269,270,271,272], abnormalities in the metabolism of APP and presenilin proteins, toxic actions of Aβ protein derived from APP [273,274], and cytoskeletal pathology [275,276].
There are possible disease links between intraneuronal Aβ and mitochondria suggesting an intracellular toxicity of Aβ [277,278,279]. Importantly, APP possesses a targeting sequence for mitochondria [277]. When over-expressed in cultured cells, APP interacts with mitochondrial import proteins, can arrest mitochondrial import, and can result in bioenergetic deficits [277]. In postmortem human brain samples, APP variants were found associated with mitochondria from the AD brain, but not mitochondria from control brain [278,280], and APP can interact with the translocase of the outer mitochondrial membrane (TOMM40) and TIMM23 [278]. The human AD autopsy brain shows evidence for mitochondrial impairments (for review see [267]). High mitochondrial APP levels mirror abnormalities in respiratory chain subunit levels and activity and enhanced ROS production [278]. Aβ can interact with the mitochondrial matrix protein Aβ-binding alcohol dehydrogenase in human AD brain and is believed to participate in mitochondrial dysfunction and oxidative stress [281]. A possible intraneuronal Aβ-mitochondria link was shown by EM in aged non-human primate neocortex [270].
In an APP transgenic (tg) mouse line (Tg2576) Aβ was also found to associate with mitochondria isolated from cerebral cortex [279]. It has also been reported that Aβ interacts with CyPD in mouse and human cerebral cortex mitochondria to potentiate synaptic stress [282]. Genetic deletion of CyPD (ppif) in human mutant APP tg mice (J-20 line) protects neurons from Aβ- and oxidative stress-induced cell death [282]. However, these abnormalities might not be related to the mPTP-driven cell death because these mice, and most other mouse models of AD, show scant or modest evidence for neurodegeneration resulting in neuronal cell death, despite tremendous brain burden of Aβ [283].
Most Tg Mouse Models of AD are not Useful to Study Neuronal Cell Death
Animal experiments should provide critical insight into to the mechanisms of neurodegeneration in AD. However, most human APP and/or presenilin tg mice show substantial Aβ deposits in hippocampus and cortex, but do not develop robust neuronal loss [284,285,286]. Analysis of a different tg mouse line (APP23) showed that Aβ deposition is accompanied by a modest loss of CA1 neurons but no cortical neuron loss, despite an Aβ burden similar to that seen in CA1 [287]. None of these studies have found the formation of NFTs. Cleaved caspase-3 was not found in mutant APPSwedish tg mice [288], but sparse cleaved caspase-3-positive cells were found in mutant APP/presenilin mice [289]. Triple-tg mice harboring mutant presenilin, APP, and tau transgenes have intraneuronal accumulations of Aβ and phosphorylated tau, but neuronal loss was not reported [290]. However, in tg mice over-expressing Aβ protein, extracellular deposition Aβ and neuronal cell death is observed [291]. The neuronal cell death mechanism in these mice may involve p53 [291], consistent with cell culture findings that Aβ neurotoxicity involves p53 [292].
Cell Culture Models of Cortical and Hippocampal Neuron Cell Death and Interactions between APP, Aβ, Tau, and Caspases
It has long been thought by many researchers that Aβ is the primary cause of AD [272]. Extracellular application of Aβ can induce apoptosis [293] or necrosis [294] in cultured neurons. Extracellularly applied Aβ25–53 or Aβ1–40 induces mitochondrial dysfunction in primary cortical neurons [295], enhanced production of free radicals, intracellular Ca2+ destabilization, and DNA damage [296,297]. Studies of the specific intracellular signaling pathways that are activated by Aβ to trigger cell death are only now appearing in the literature. In studies of rodent neurons exposed to extracellular Aβ, the induction of apoptosis involves the Fas death receptor-Jun N-terminal kinase (JNK) pathway [298], the p75NTR-JNK pathway [299], and caspase-12 [300]. Neuronal cultures exposed to Aβ can be protected from neurotoxicity by caspase-8 inhibition and expression of dominant-negative Fas-associated protein with death domain (FADD), both components of the Fas death receptor pathway [301]. Some in vitro experiments have implicated p53 in neuron death trigged by extracellular Aβ [302,303], but other in vitro studies with Aβ-treated cortical neurons reveal a p53-independent mechanism for cell death involving the transcription factor E2F1 [304]. It is noteworthy that results of cell culture experiments using extracellular application of Aβ are likely to be dependent on Aβ concentration and oligomeric state. When human neuron primary cultures are treated with Aβ at concentrations closer to physiological levels for up to 3 days, evidence for apoptosis is scarce; however, Bcl-2 levels are down-regulated and Bax levels are up-regulated [305]. Thus, extracellular Aβ at physiological concentrations might render neurons more sensitive to cell stress rather than kill them outright. Intracellular Aβ1-42 exposure (as little as 1 picomolar) is, in contrast, toxic to human cortical neurons, and this toxicity requires de novo protein synthesis, Bax, p53, and caspases, suggesting cell death by some form of apoptosis [292].
Experiments have been performed to link APP and presenilins to cell death. Over-expression and intracellular accumulation of APP activates caspase-3 [306]. APP is a target of caspase-3 [306] and caspase-6 [307], and APP cleavage by caspase-3 or caspase-6 may promote Aβ formation [307,308]. Thus, increased production of Aβ may be a consequence of neuronal apoptosis. Recent work has shown that shedded N-terminal fragments of APP are surface ligands that bind death receptor 6 (DR6) to trigger axon pruning through caspase-6 and neuron death through caspase-3 [309]. Neurotrophin deprivation engages the shedding of APP to activate DR6 [309]. Presenilin proteins can influence mitochondrial regulation of apoptosis, such as Bax activation and cytochrome c release, through interactions with Bcl-XL [310], and they are also substrates for caspase-3 [311]. However, over-expression of human wild-type or mutant presenilin-1 or presenilin-2 does not enhance apoptosis in neurons [312,313]. Other work indicates that the presenilin-1 mutation sensitizes neurons to DNA damage-induced apoptosis [314]. More cell culture work needs to be done on the basic mechanisms of human neuron degeneration and on APP and Aβ neurotoxicity mechanisms under basal conditions and in the presence of familial AD-related and tau gene mutations.
Parkinson’s Disease (PD)
PD is a chronically progressive, age-related, fatal neurological disease in humans described first by James Parkinson in 1817. Estimates indicate that 4 to 6 million people have been diagnosed with PD. It affects about 2% of the population at some time in life. The greatest prevalence occurs in the USA, with between 100 and 250 cases per 100,000 [315], placing PD as the 2nd most common neurodegenerative disease with an adult onset (after AD). Progressive resting tremor (4–7 Hz), rigidity, bradykinesia/akinesia, gait disturbance, and postural instability characterize PD clinically [316]. The disease progression is also associated with mood disturbances, dementia, sleep disturbances, and autonomic dysfunction [316]. There are currently no cures for PD. Medications and neurosurgery can relieve some of the symptoms.
A major neuropathological feature of PD is the degeneration and elimination of dopamine neurons in substantia nigra pars compacta (SNc) and in other brainstem regions which causes the movement disorder (Figure 4) [317]. The movement disorder in PD is thought to arise from reduced dopaminergic innervation of the striatum resulting from the loss of SNc neurons [317]. The effect of reduced dopaminergic input is over-activity of striatal neurons that project to and inhibit neurons in external globus pallidus (GPe), thus reducing the normal GPe inhibition of excitatory subthalamic neurons [2]. In addition, due to actions of dopamine on different dopamine receptor subtypes, there is also loss of normal dopaminergic excitation of striatal neurons that innervate internal GP (GPi) and SN reticularis, causing increased γ-aminobutyric acidergic inhibition of thalamic nuclei that are needed to drive cortical activation [317]. PD can thus be explained functionally by over-activity of the subthalamic nucleus and GPi [317]. PD should however be regarded as a multi-regional, multi-system neurodegenerative disorder in which the pathology appears in a regionally specific sequence, beginning in the dorsal motor nucleus of the vagus and olfactory bulbs and anterior nucleus followed by the locus coeruleus and then the SNc, at which time (when ~ 50% of SNc neurons are lost) a clinical diagnosis of PD becomes possible [318].
The degeneration of pigmented SNc neurons is characterized by chromatolysis (Figure 4A), nuclear condensation (Figure 4B), and severe soma attrition (Figure 4C). The neuronal chromatolysis (Figure 4A) is indicated by the eccentrically placed nucleus, pale cytoplasm, and peripheral margination of the Nissl substance. Glial/macrophage-like cells (Figure 4A, arrows) are laden with phagocytosed cellular debris. The nucleus of SNc neurons undergoes considerable condensation (Figure 4B, arrow) while the Nissl substance dissipates, but before appreciable somal shrinkage. The cell body of SNc neurons then becomes attritional (Figure 4C, arrow), resulting in residual neurons that are ~10–20% their normal size. Cells can be identified as atrophic neurons, rather than a debris-laden macrophage, because of the presence of a condensed nucleus with a single prominent nucleolus (Figure 4C). This degeneration pattern could be indicative of autophagy. The nuclear condensation stage of pigmented SNc neuron degeneration is characterized by the appearance of DNA double-strand breaks (Figure 4D), and in the chromatolytic stage SNc neurons accumulate cleaved caspase-3 immunoreactivity (Figure 4E) prior to their undergoing nuclear condensation and somal attrition. Another neuropathological feature of PD is the formation of eosinophilic proteinaceous intra-neuronal or intra-glial inclusions (Figure 4F, arrow), known as Lewy bodies (LBs), first described by Frederich Lewy in 1912. LBs are comprised of a dense core of filamentous material enshrouded by filaments 10–20 nm in diameter and are usually positive for ubiquitin and α-synuclein (α-Syn) [319]. It is not clear whether LBs are related causally to the disease process.
Figure 4.
Degeneration of SNc neurons in human PD. A-C. Hematoxylin-eosin (H&E) staining shows that SNc neuron degeneration is characterized by chromatolysis (A), nuclear condensation (B), and severe soma attrition (C). D. Pigmented SNc neurons accumulate DNA double-strand breaks (arrow, brown staining). E. SNc neurons accumulate cleaved caspase-3 immunoreactivity (arrows, brown staining). F. SNc neurons can form Lewy bodies (arrow). Scale bars: A, 20 μm (same for B-D, F); E, 45 μm.
Figure 4.
Degeneration of SNc neurons in human PD. A-C. Hematoxylin-eosin (H&E) staining shows that SNc neuron degeneration is characterized by chromatolysis (A), nuclear condensation (B), and severe soma attrition (C). D. Pigmented SNc neurons accumulate DNA double-strand breaks (arrow, brown staining). E. SNc neurons accumulate cleaved caspase-3 immunoreactivity (arrows, brown staining). F. SNc neurons can form Lewy bodies (arrow). Scale bars: A, 20 μm (same for B-D, F); E, 45 μm.
The molecular pathogenesis of PD is still not understood. At least 2 forms of PD exist: idiopathic (sporadic) and heritable (familial) [320]. The majority of PD cases are sporadic with no known genetic component. Epidemiological studies reveal several risk factors for developing idiopathic PD in addition to aging. Pesticides have now been linked convincingly to the development of PD [321]. Herbicides, well water (contaminated with pesticides), and industrial chemicals are possible neurotoxic agents related to the development of PD [320].
| Locus | Inheritance | Gene | Protein Name/ Function |
|---|---|---|---|
| PARK1/4q21 | autosomal dominant | α-syn | α-Syn/presynaptic maintenance? |
| PARK2/6q25.2-27 | autosomal recessive | parkin | Parkin/ubiquitin E3 ligase |
| PARK3/2p13 | autosomal dominant | ? | ? |
| PARK4/4p15 | autosomal dominant | α-syn | α-Syn/presynaptic maintenance? |
| PARK5/4p14 | autosomal dominant | UCHL1 | UCHL1/polyubiquitin hydrolase |
| PARK6/1p36 | autosomal recessive | PINK1 | PTEN-induced putative kinase-1/mitochondrial protein kinase |
| PARK7/1p36.33-36-12 | autosomal recessive | DJ-1 | DJ-1/mitochondrial antioxidant, chaperone |
| PARK8/12q12 | autosomal dominant | LRRK2 | Dardarin/multifunctional kinase/GTPase |
| PARK9/1p36 | autosomal recessive | ATP13A2 | Lysosomal type 5 P-ATPase |
| PARK10/1p32 | ? | ? | |
| PARK11/2q36-37 | autosomal dominant | GIGYF2? | Grb10-interacting GYP protein 2, modulates tyrosine kinase receptor signaling, including IGF-1 |
| PARK12/Xq21-q25 | X-linked | ? | ? |
| PARK13/2p12 | autosomal recessive susceptibility factor | Omi/HtrA2 | Omi/HtrA2, mitochondrial serine peptidase, inhibitor of IAPs |
| PARK14/22q13.1 | autosomal recessive | PLA2G6 | Phospholipase A2 group VI |
| PARK15/22q12-q13 | autosomal recessive | FBXO7 | F-box protein 7 |
Mutant Genes that Cause Some Forms of PD
About 5–10% of PD patients have familial patterns of inheritance [320]. Several genes have been identified in Mendelian forms of PD. Gene mutations with autosomal dominant or autosomal recessive inheritance patterns have been identified in familial forms of PD (Table 3). PD-linked mutations occur in the genes encoding α-Syn, Parkin, ubiquitin carboxy-terminal hydrolyase-L1 (UCH-L1), phosphatase and tensin homolog (PTEN)-induced putative kinase-1 (PINK1), DJ-1, and leucine-rich repeat kinase-2 (LRRK2). In rare autosomal dominant inherited forms of PD, missense mutations in the α-syn gene (PARK1) result in amino acid substitutions Ala-53→Thr, Ala-30→Pro, Glu-46→Lys; in addition, duplication and triplication mutations in the α-syn gene (PARK4) have been found [322,323,324]. A missense mutation in the UCH-L1 gene (PARK5), resulting in the amino acid substitution Ile-93→Met, can also cause very rare autosomal dominant PD [325]. Loss-of-function mutations due to large deletions and truncations and also missense or nonsense mutations in parkin (PARK2), PINK1 (PARK 6), and DJ-1 (PARK7) are the cause of autosomal-recessive inheritance of PD [326,327,328,329]. Several missense mutations in the LRRK2 gene (PARK8) have been found, resulting in amino acid substitutions Tyr-1654→Cys, Arg-1396→Gly, Tyr-1699→Cys, Arg-1441→Cys, Ile-1122→Val, Ile-2020→Thr, that cause more commonly occurring autosomal dominant PD and possibly ‘sporadic’ PD [330,331]. PARK9 has been ascribed to a deletion mutation (cytosine at nucleotide position 3057) or guanine-to-adenine transition at a splice site of exon 13 in the ATP13A2 gene that encodes a predominantly neuronal P-type lysosomal ATPase [332]. Potential lysosomal dysfunction related to ATP13A2 mutant proteins might tie into PD etiology through abnormalities in autophagy. Mutations of genes at other PD loci are more controversial (Table 3).
α-Syn
α-Syn is a relatively small (140 amino acids) very abundant protein (~1% of total protein) found in cells throughout the nervous system and is particularly enriched in neuronal axon terminals [333,334,335]. The growing evidence shows a role for α-Syn in neurotransmitter release. Mice without α-Syn have no overt phenotype [337], but neurons deficient in α-Syn have a reduction in the reserve pool of synaptic vesicles needed for responses to tetanic stimulation and show defective mobilization of dopamine and glutamate [335,338]. Without α-Syn neurons have impaired long-lasting enhancement of evoked and miniature neurotransmitter release [339]. α-Syn is highly mobile and rapidly dissociates from synaptic vesicle membranes after fusion in response to neuronal activity [340]. It appears that α-Syn acts as a molecular chaperone to assist cysteine-string protein-α (alias for DNAJC5) in the folding and refolding of SNARE synaptic proteins [341].
α-Syn is a soluble monomeric protein that can associate with mitochondrial membranes [342]. α-Syn can be induced to polymerize into insoluble fibrils due to a conformational change from an α-helical coil to a β-pleated sheet [343]. α-Syn is a major structural component of LBs (Figure 4F), forming the ~10-nm fibrils, but in most neurodegenerative diseases, LBs are associated with accumulation of wild-type, not mutant, α-Syn [319]. α-Syn mutations cause increased levels of protofibrils, possibly being the more toxic form of the protein [344]. α-Syn protofibrils might also be toxic by making membranes of cells more porous [345]. Over-expression of human wild-type or mutant α-Syn in cultured cells elevates the generation of intracellular ROS [346,347] and causes mitochondrial deficits [346]; moreover, expression of mutant α-Syn increases cytotoxicity to dopamine oxidation products [348]. Aggregation of wild-type and mutated α-Syn is associated with enhanced cell death in cultured cells [349]. Nitration of α-Syn, signifying the presence of potent reactive nitrogen species such as ONOO- or its free radical derivative nitrogen dioxide (NO2), is a major signature of human PD and other synuclinopathies and might be critical to the aggregation process [350,351].
UCH-L1 and Parkin
UCH-L1 is a very abundant protein (~1–2% total soluble protein in brain) that functions in the formation and recycling of ubiquitin monomers for the ubiquitin-proteasome pathway [352]. This pathway is important for intracellular protein turnover and degradation and functions generally in quality control of proteins in cells to eliminate misfolded, mutated, and damaged proteins [353]. Ubiquitin is an abundant small (~8.5 kDa) protein that is attached covalently to lysine aliphatic chains in proteins to mark them for degradation carried out by the 26S proteasome. UCH-L1 hydrolyses the C-terminus of fusion proteins containing polyubiquitin molecules and ribosomal protein, thus generating ubiquitin monomers. In vitro, PD-linked mutant UCH-L1 has reduced enzyme activity [354], and inhibition of UCH-L1 is associated with production of α-Syn aggregates [355], indicating that α-Syn is degraded by the proteasome.
The ubiquitination of proteins is catalyzed by the activities of three enzymes called ubiquitin-activing enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3) [353]. PARK2, encoding Parkin, a ubiquitin E3 ligase, causes juvenile-onset recessive PD (before 40 years of age) [328] with relatively confined neuronal loss in the SNc and locus coeruleus, but with an absence of LBs. Several substrates of Parkin have been identified, including α-Syn, synphilin-1 and other synaptic proteins [356]. Mutations in the parkin gene result in a loss-of-function of E3, thus possibly causing some substrates of Parkin to accumulate and aggregate within cells. One parkin mutation found in a Turkish patient (Gln-311→X), replacing a glutamine residue at position 311 with a stop codon), causes a C-terminal truncation of 155 amino acids of Parkin [357].
PINK1
The PARK6 locus contains the PINK1 gene [327,329]. PARK6 kindred have juvenile-onset PD and truncation mutations or missense mutations in the PINK1 gene resulting in truncation or single amino acid substitutions in the PINK1 protein (His-271→Gln; Gly-309→Ala; Leu-347→Pro; Glu-417→Gly) as well as mutations that are nonsense (Arg-246→X, where X is any other amino acid; Trp-437→X) or compound nonsense (Gln-309→X/Arg-492→X). Clues about the intracellular localization and normal functions of PINK1 are emerging. It is a 581 amino acid protein (~63 kDa) and contains a domain highly homologous to the serine/threonine protein kinases of the calcium/calmodulin family and a mitochondrial targeting motif [329]. Thus, PINK1 is a mitochondrial kinase. It is processed at the N-terminus in a manner consistent with mitochondrial import, but the mature protein is also present in the cytosol [358]. Both human wild-type and mutant PINK1 localize to mitochondria [359]. Interestingly, most of the reported mutations are in the putative kinase domain. PINK1 is expressed in many adult human tissues [360]. In adult rodents PINK1 is expressed throughout the brain [361].
PINK1 appears to function in mitochondrial trafficking by forming a multiprotein complex with the GTPase Miro and the adaptor protein Milton [362]. Speculation has PINK1 protecting human dopaminergic neuroblastoma cells (SH-SY5Y) against mitochondrial malfunction under conditions of cell stress [363]. In rat neuroblastoma cells, mutant PINK1 can induce abnormalities in mitochondrial Ca2+ influx and aggravate the cytopathology caused by mutant α-Syn in a mechanism that involves the mPTP [364]. It is unclear how PINK1 mutations cause the selective death of SNc neurons in PD.
DJ-1
The PARK7 locus contains the DJ-1 gene [326]. PARK7 kindred can have homozygous deletion of a large region within the DJ-1 gene causing complete loss of DJ-1 expression or homozygous missense mutations in the DJ-1 gene resulting in single amino acid substitutions in the DJ-1 molecule (Met-26→Ile; Glu-64→Asp; Leu-166→Pro) [326]. DJ-1 is a small (189-amino acid, ~20–25 kDa) protein with multiple apparent functions involving cellular transformation, male fertility, control of protein-RNA interaction, and oxidative stress response. The protein exists in vivo as a dimer [326], and information on its specific functions is growing. DJ-1 is expressed throughout the mouse nervous system [366], and it might act as a neuroprotective intracellular redox sensor that can localize to the cytoplasmic side of mitochondria [367]. The localization of DJ-1 to mitochondria is associated with protective actions against some mitochondrial poisons [367]. Some DJ-1 mutants have abnormalities in dimer formation ability and decreased stability [368,369]. It remains to be identified how DJ-1 mutations cause the selective death of SNc neurons in PD.
LRRK2
The PARK8 locus contains the LRRK2 gene [330,331]. Mutations in this gene are to date the most common in both familial and “sporadic” PD. The LRRK2 protein is a large multidomain protein (2527 amino acids, 286 kDa), also called dardarin (derived from the Basque word dardara, meaning tremor), that is expressed throughout the body. LRRK2 contains leucine-rich repeat domains, a Ras/small GTPase domain, a non-receptor tyrosine kinase-like domain, and a WD40 domain (~40 amino acid motifs often terminating in a Trp-Asp dipeptide), consistent with the architecture of multifunctional Ras/GTPases of the Ras of complex (ROC) family. The presence of leucine-rich and WD40 domains suggests that LRRK2 is capable of multiple protein-protein interactions. The GTPase activity indicates that LRRK2 functions as a molecular switch, possibly involved in cytoskeleton organization and vesicle trafficking. The kinase domain possibly belongs to the mitogen-activated protein kinase kinase kinase (MAPKKK or MEKK) family of kinases.
Currently it is not evident how LRRK2 gene mutations relate to the selective death of neurons that causes PD. Studies of rodent brain show little or no expression of LRRK2 in SNc neurons [370,371]; however, expression of LRRK2 is high in dopamine-innervated regions [370]. Recent work shows that LRRK2 can influence mitochondrial- and death receptor-mediated cell death in cultured cells [372,373]. These findings might be hints that the target of SNc neurons (i.e. the striatum) and SNc neuron target-deprivation are important to the understanding of LRRK2-related pathogenic mechanisms of PD.
Neuronal Cell Death in Human PD
Many groups of neurons are affected in PD [318], but the loss of dopaminergic neurons in the SNc is particularly disease manifesting. The mechanisms by which nigral neurons degenerate in human PD are not well understood and require further study for the development of effective cures. The pathology seen in autopsy tissue of the SNc in human PD shows atrophy, degeneration, and loss of large, multipolar melanin-containing neurons (Figure 4). Some studies report that apoptotic PCD contributes to the neurodegeneration in human PD based on the presence of cleaved caspase-3 (Figure 4E) [374], but other studies caution against such claims [375,376]. Nigral neurons in human PD do not degenerate with the morphology consistent with the process of classical apoptosis (Figure 4A-C); however, cell death exists as a morphological continuum [4,28,29] and non-apoptotic forms of PCD exist [27,46,47,48]. The pathological process in PD might involve autophagy [377,378]. Studies using in situ DNA-end labeling methods to detect dying cells with DNA fragmentation in the SNc of humans with PD are conflicting. Some studies report no labeling [375], but other experiments have found nuclear DNA fragmentation [374,379]. Using TUNEL, subsets of large, melanin-containing, atrophic neurons with DNA double-strand breaks can be found (Figure 4D). Other neurons without melanin are TUNEL-positive, including glial and macrophage-like cells.
Confusion about neuronal cell death mechanisms in the PD brain originates from how PCD is defined, the use of detection systems that do not distinguish between types of DNA-strand breaks associated with apoptosis or necrosis, arbitrary morphological interpretations of chromatin condensation, and uncertainty about the meaning of caspase immunoreactivities in cells [17,24,375]. Studies are warranted that utilize high-resolution techniques, such as laser capture microdissection, with unambiguous selectivity for specific neurons and proteomic approaches [380,381] to reveal quantitatively mechanisms of cell death, such as the activation of apoptotic, autophagic and programmed necrotic pathways, directly in SNc neurons in human autopsy tissue and optimally prepared animal model tissue.
PD α-Syn Tg Mice Develop Neuronal Mitochondrial Degeneration and Cell Death
Information gleaned from molecular genetic studies of human genes linked to familial PD drives experimental work on the generation of animal and cell models of PD. Parkin null mice appear to have a normal lifespan, do not develop any major neurological abnormalities, and have no loss of midbrain dopaminergic neurons and no formation of inclusions [382,383]. However, parkin-/- mice exhibit some evidence of dopaminergic presynaptic dysfunction in striatum and possible deficits in behavioral tests indicative of nigrostriatal dysfunction [382], although this finding has not been confirmed in another mouse line [383]. Parkin-/- mice have decreases in proteins involved in mitochondrial oxidative phosphorylation and oxidative stress in ventral midbrain and exhibit reduced mitochondrial respiration in striatum, but they have no mitochondrial ultrastructural abnormalities [384]. In contrast, tg mice expressing the Parkin Q311X truncation mutation develop a progressive hypokinetic disorder, degeneration of SNc neurons, and loss of striatal dopamine [385]. Thus, Parkin could be important for maintenance of mitochondrial function or mitochondrial turnover through autophagy and synaptic integrity distally within the SNc neuron target region.
Mice with null mutations in DJ-1 also have a normal lifespan and do not develop an overt phenotype or loss of dopaminergic neurons, but behavioral tests reveal age-dependent motor deficits and neurochemical assessment shows altered striatal dopamine content [386]. DJ-1 null mice also show altered D2 dopamine receptor-mediated function [387].
Several tg mouse lines have been made using different promoters to drive expression of human full-length wild-type or mutant α-Syn [349,388,389,390,391,392,393]. Of these mouse lines, tg mice expressing human A53T mutant α-Syn have a shortened lifespan and develop a severe movement disorder and synucleinopathy [349,390,393]. It is noteworthy that there have been no reports of robust dopamine SNc neuron degeneration in full-length α-Syn tg mice. However, tg mice expressing a truncation mutant of human α-Syn show a development-related loss of SNc neurons [394]. Cell death mechanisms or thresholds for cell death activation in human and mouse brain dopamine neurons might differ.
Despite the absence of prominent changes in the SNc, tg α-Syn mice do develop robust cell death and neuronal loss in other regions of brain and in spinal cord [395]. These tg mice express high levels of human wild-type or mutant (A53T and A30P) α-Syn under the control of the mouse prion protein promoter [390]. Mice expressing A53T α-Syn (lines G2-3 and H5), but not mice expressing wild-type (line I2-2) or A30P (line O2) α-Syn, develop adult-onset progressive motor deficits, including reduced spontaneous activity with bradykinesia, mild ataxia and dystonia, at ~10–15 months of age followed by rapidly progressive paralysis and death [390]. A53T mice develop intraneuronal inclusions, mitochondrial degeneration, and cell death in neocortex, brainstem, and spinal cord [395]. Brainstem neurons and spinal motor neurons display a prominent chromatolysis reaction and axonal spheroids, typical of that seen after axonal injury [396]. A53T mice form LB-like inclusions in neocortical and spinal motor neurons and have progressive, profound loss (~75%) of motor neurons that causes their paralysis [341,395].
Mitochondrial pathology in A53T mice involving mitochondrial DNA damage is seen frequently in the absence of nuclear DNA damage in large brainstem neurons and spinal motor neurons [395]. Subsets of mitochondria in brainstem and spinal cord cells in A53T mice appear dysmorphic, becoming shrunken, swollen, or vacuolated [395]. Human α-Syn is found bound to some mitochondria in degenerating neurons in A53T mice [395]. Some abnormal intracellular inclusions in these cells are degenerating mitochondria. A mitochondrial defect in A53T mice is further indicated by biochemical evidence revealing loss of complex IV activity [395].
The mechanisms for this mitochondrial DNA damage are possibly related to oxidative stress. The presence of oxidative stress in mutant α-Syn tg mice is shown by evidence that mitochondrial associated metabolic proteins are oxidized in A30P mice [397]. α-Syn can generate H2O2 [398] and •OH [399] in vitro upon incubation with Fe(II). There is evidence for ONOO--mediated oxidative/nitrative stress in A53T mouse motor neurons, as indicated by the presence of nitrated human synuclein [395]. Nitrated synuclein forms inclusions in motor neurons consistent with in vitro data showing that ONOO- promotes the formation of stable α-Syn oligomers [400,401]. The evidence for mitochondrial DNA damage is consistent with the presence of ONOO- or its derivatives near mitochondria, because ONOO- or products of ONOO- are directly genotoxic by causing single- and double-strand breaks in DNA [13]. Moreover, the loss of complex IV enzyme activity without a change in protein level [395] might be explained by inactivation of this mitochondrial enzyme by nitration. Overall, ONOO- mediated damage in mitochondria could be a key pathological mechanism leading to motor neuron degeneration in A53T mice.
The reasons for the vulnerability of mouse motor neurons to human A53T mutant α-Syn are not clear. These mice express high levels of mRNA and protein for human α-Syn in the forebrain, diencephalon, and midbrain [390], but these regions are much less vulnerable than spinal cord. A53T α-Syn causes axonopathy [349,393], thus motor neuron vulnerability could be related to their long myelinated axons and interactions with oligodendrocytes and Schwann cells for myelin support. Motor neuron vulnerability could also be related to their unusual expression of inducible NOS (iNOS) in mitochondria [381,402]. Moreover, distal axonopathy and muscle disease may have roles in the pathogenesis in A53T mice [395]. Prominent skeletal muscle denervation occurs in α-Syn tg mice [393,395]. This work is intriguing because the original goal was to develop a tg mouse model of PD, but the result is a mouse model of ALS. Thus, the mutant α-Syn A53T tg mouse is a new model to study mechanisms of motor neuron degeneration and could provide insight into the selective vulnerability of motor neurons in age-related disorders and the possible roles of α-Syn in synaptic maintenance and diseases of long-axon neurons [395].
ALS
ALS is a progressive and severely disabling neurological disease in humans characterized by initial muscle weakness, and then muscle atrophy, spasticity, and eventual paralysis and death typically 3 to 5 years after symptoms begin [403]. The cause of the spasticity, paralysis and death is progressive degeneration and elimination of upper motor neurons in cerebral cortex and lower motor neurons in brainstem and spinal cord (Figure 5) [403,404]. Degeneration and loss of spinal and neocortical interneurons has also been found in human ALS [405,406]. More than 5000 people in the USA are diagnosed with ALS each year (ALS Association, http://www.alsa.org), and, in parts of the United Kingdom, three people die every day from some form of motor neuron disease (http://www.mndassociation.org). Other than life support management, no effective treatments exist for ALS [225].
It is still not understood why specific neuronal populations are selectively vulnerable in ALS, such as certain somatic motor neurons and interneurons [404,405,406]. The molecular pathogenesis of ALS is understood poorly, contributing to the lack of appropriate target identification and effective mechanism-based therapies to treat even the symptoms of this disease. Two forms of ALS exist: idiopathic (sporadic) and heritable (familial). The majority of ALS cases are sporadic with few known genetic contributions, except for missense mutations in TAR-DNA binding protein [407]. Aging is a strong risk factor for ALS because the average age of onset is 55 (ALS Association, www.alsa.org). Familial forms of ALS have autosomal dominant or autosomal recessive inheritance patterns and make up ~10% or less of all ALS cases. ALS-linked mutations occur in the genes (Table 4) encoding SOD1 (ALS1), Alsin (ALS2), senataxin (ALS4), fused in sarcoma (FUS, ALS6), vesicle associated membrane protein (VAMP/synaptobrevin)-associated protein B (VAPB, ALS8), p150 dynactin (DCTN1), and TAR-DNA binding protein (TADBP or TDP43) [5,408]. Most recently, variations in the phosphoinositide phosphatase FIG4 gene cause ALS11 [409]. Several other genes are believed to be susceptibility factors for ALS (Table 4).
Figure 5.
Motor neurons in spinal cord degenerate in people with ALS. A, B. In normal individuals, the spinal cord contains many large, multipolar motor neurons (dark cells). C, D. In ALS, the anterior horn is depleted of large neurons (dark cells) and remaining neurons are atrophic. E-G. Nissl staining shows motor neurons that appear normal (E), attritional (F), and residual (G). H. ALS motor neurons accumulate DNA double-strand breaks in the nucleus (brown labeling). I. p53 (brown labeling) accumulates in the nucleus of motor neurons. J. ALS motor neurons are immunopositive for cleaved caspase-3 (black-dark green labeling), and discrete mitochondria (brown-orange labeling, detected with antibody to cytochrome c oxidase subunit I) accumulate around the nucleus (pale circle). Scale bars: A, 500 μm (same for C); B, 76 μm (same for D); E, 6 μm (same for F,G); H, 12 μm (same for I,J).
Figure 5.
Motor neurons in spinal cord degenerate in people with ALS. A, B. In normal individuals, the spinal cord contains many large, multipolar motor neurons (dark cells). C, D. In ALS, the anterior horn is depleted of large neurons (dark cells) and remaining neurons are atrophic. E-G. Nissl staining shows motor neurons that appear normal (E), attritional (F), and residual (G). H. ALS motor neurons accumulate DNA double-strand breaks in the nucleus (brown labeling). I. p53 (brown labeling) accumulates in the nucleus of motor neurons. J. ALS motor neurons are immunopositive for cleaved caspase-3 (black-dark green labeling), and discrete mitochondria (brown-orange labeling, detected with antibody to cytochrome c oxidase subunit I) accumulate around the nucleus (pale circle). Scale bars: A, 500 μm (same for C); B, 76 μm (same for D); E, 6 μm (same for F,G); H, 12 μm (same for I,J).
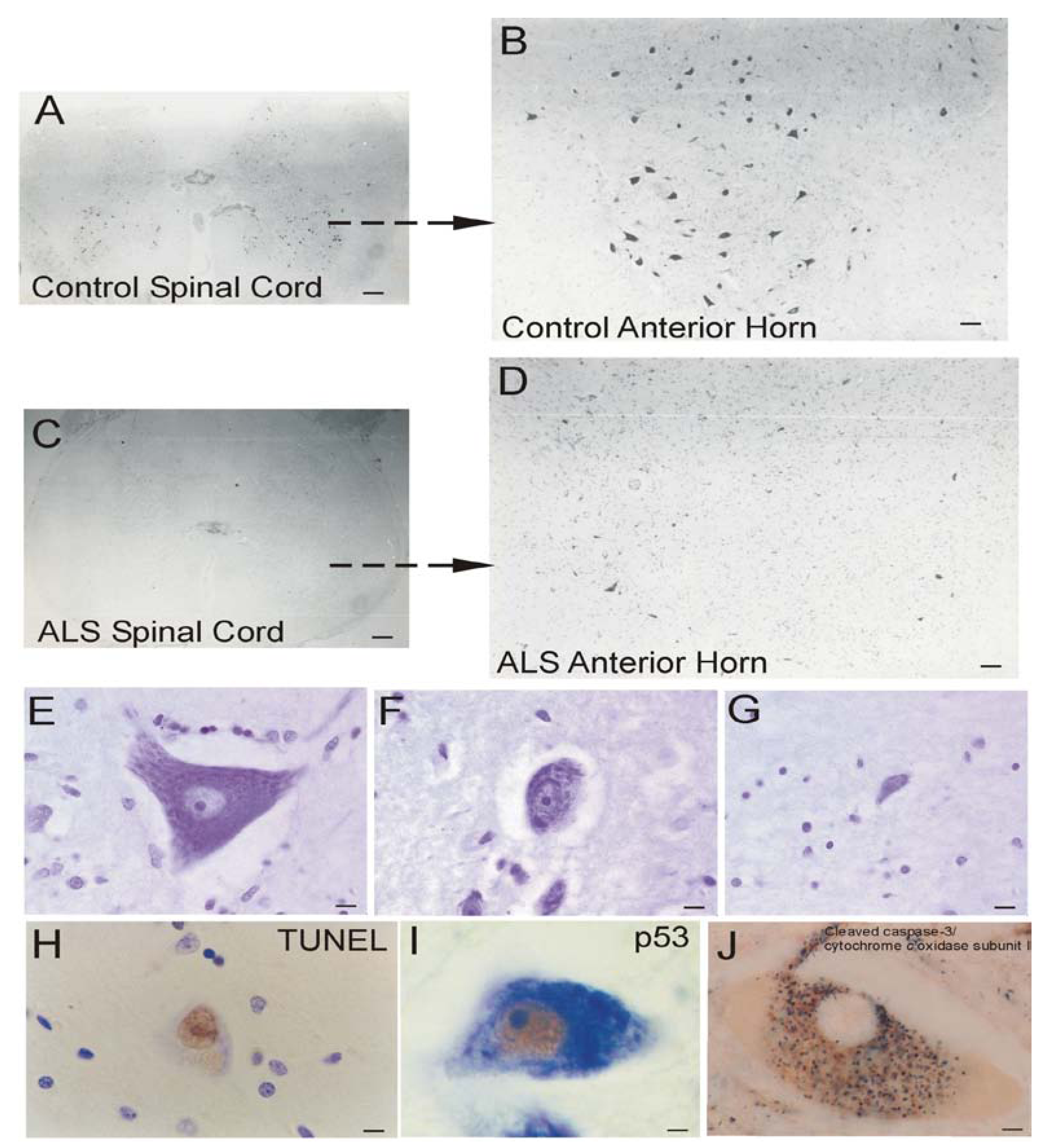
| Locus | Inheritance | Gene | Protein Name/ Function |
|---|---|---|---|
| ALS1/21q22 | autosomal dominant (adult onset) | SOD1 | Cu/Zn superoxide dismutase/ dismutation of superoxide |
| ALS2/2q33.2 | autosomal recessive (juvenile onset primary lateral sclerosis) | Alsin | Alsin/guanine exchange factor for RAB5A and Rac1 |
| ALS4/9q34 | autosomal dominant (adult onset) | SETX | Senataxin/helicase, RNA processing |
| ALS6/16q12 | autosomal recessive (adult onset) | FUS | Fused in sarcoma, component of heterogeneous nuclear ribonuclear protein complex; RNA/DNA binding protein |
| ALS8/20q13.33 | autosomal dominant | VAPB | VAMP-associated protein B/part of SNARE complex |
| 2q13 | autosomal dominant (adult onset, atypical ALS) | DCTN1 | Dynactin p150glued/axonal transport, link between dynein and microtubule network |
| ALS10/1p36.22 | autosomal dominant | TARDBP | TAR DNA binding protein, DNA and RNA binding protein, regulates RNA splicing |
| ALS11/6q21 | autosomal recessive | FIG4 | FIG4 homolog, SAC1 lipid phosphatase domain containing protein; regulates phosphotidylinositol turnover |
| 14q11.1-q11.2 | susceptibility factor | ANG | Angiogenin; angiogenesis; stimulates production of rRNA |
| 22q12.2 | susceptibility factor | NEFH | Neurofilament, heavy polypeptide; neurofilament subunit |
| 12q12-q13 | susceptibility factor | PRPH | Peripherin; intermediate filament formation |
| 5q13 | susceptibility factor | SMN | Survival motor neuron; RNA processing |
| 7q36.6 | susceptibility factor? | DPP6 | Dipeptidyl-peptidase 6; S9B serine protease, binds voltage-gated potassium channels |
Mitochondrial Dysfunction in Human ALS
Mitochondrial abnormalities have been found in human ALS. Structural abnormalities in mitochondria are seen by electron microscopy in skeletal muscle, liver, spinal motor neurons and motor cortex of ALS patients [410,411]. A mutation in cytochrome c oxidase subunit I was found in a patient with a motor neurons disease phenotype [412]. Another patient with motor neuron disease had a mutation in a mitochondrial tRNA gene [413]. One type of mitochondrial DNA (mtDNA) mutation, called the common mtDNA deletion (mtDNA4977), is found non-uniformly within different human brain areas; the highest levels are detected in the striatum and SN [414,415]. However, no significant accumulation of the 5 kb common deletion in mtDNA has been found by single-cell analysis of motor neurons from sporadic ALS cases [416]. Some ALS patients with defects in skeletal muscle mitochondrial oxidative phosphorylation have a novel SOD1 gene mutation [417]. More work on human ALS and animal/cell models needs to be done to determine conclusively if mitochondrial abnormalities participate in disease-causing mechanisms of human ALS.
Intracellular Ca2+ abnormalities and excitotoxicity are suspected links to mitochondrial dysfunction and oxidative stress in ALS. Mitochondria function in the regulation of intracellular Ca2+ levels [7,418]. Skeletal muscle biopsies of patients with sporadic ALS show ultrastructural changes indicative of elevated Ca2+ in motor neuron terminals, with some mitochondria showing an augmented Ca2+ signal [419]. Utilizing specific transport systems, mitochondria can pump Ca2+ from the cytosol into the matrix by the Ca2+ uniporter and eject Ca2+ from the matrix via the Na+/Ca2+ exchanger [7] and more catastrophically through the mPTP [50]. Under conditions of elevated cytoplasmic Ca2+, whenever the local free Ca2+ concentration rises above a set-point of ~0.5 μM, mitochondria avidly accumulate Ca2+ to a fixed capacity [7]. The electrical gradient across the IMM, the ΔΨm, established by electron transport chain activity (Figure 2), provides the driving force for the accumulation of Ca2+ into the mitochondrial matrix [418]. Cytosolic Ca2+ concentrations above set-point levels are believed to be achieved during tetanic stimulation and by activation of glutamate receptors on the plasma membrane [7]. In pathological settings of excitotoxicity, resulting from excessive overstimulation of glutamate receptors [190,191], Ca2+ overload in neurons is significant and causes cell death [420]. When mitochondria become overloaded with Ca2+, they undergo mPT resulting in osmotic swelling and rupture of the OMM (Figure 1). Mitochondria within synapses appear to be more susceptible than non-synaptic mitochondria to Ca2+ overload [421].
Excitotoxicity has been implicated in the pathogenesis of ALS for a long time [422] and is another possible mechanism of motor neuron damage in ALS [420]. While many drugs targeting excitotoxicity as a mechanism have failed in ALS clinical trials, the anti-excitotoxic drug riluzole is currently approved by the Food and Drug Administration for ALS treatment. Many sporadic ALS patients have reduced levels of synaptosomal high-affinity glutamate uptake [422] and astroglial glutamate transporter EAAT2 (excitatory amino acid transporter 2 or GLT1) in motor cortex and spinal cord [423]. Reductions in levels of activity of EAAT2 in spinal cord could increase the extracellular concentrations of glutamate at synapses on motor neurons. Motor neurons might be particularly sensitive to glutamate excitotoxicity because they have a low proportion of GluR2-edited or under-edited AMPA subtype glutamate receptor on their surfaces, predisposing these cells to risk of excess Ca2+ entry and mitochondrial perturbations (Figure 1) [424,425]. Cell culture studies show that excess glutamate receptor activation in neurons can cause increased intracellular Ca2+, mitochondrial ROS production, bioenergetic failure, and mitochondrial trafficking abnormalities [426]. Ca2+-induced generation of ROS in brain mitochondria is mediated by mPT (Figure 1) [427]. Motor neurons are particularly affected by inhibition of mitochondrial metabolism which can cause elevated cytosolic Ca2+ levels and increased excitability [428].
Markers of oxidative stress and ROS damage are elevated in ALS tissues [429]. In human sporadic and familial ALS, protein carbonyls are elevated in motor cortex [430]. Tyrosine nitration (Figure 1) is elevated in human ALS nervous tissues [431,432,433]. Studies of respiratory chain enzyme activities are discrepant. Studies have shown increases in complex I, II, and III activities in vulnerable and non-vulnerable brain regions in patients with familial ALS-mutant SOD1 [434], but other studies show decreased complex IV activity in spinal cord ventral horn [435] and skeletal muscle [436] of sporadic ALS cases. In sporadic ALS skeletal muscle, reductions in activity of respiratory chain complexes with subunits encoded by the mitochondrial genome are associated with decreased neuronal NOS levels [437]. Alterations in skeletal muscle mitochondria are progressive [438] and could be intrinsic to skeletal muscle and not due merely to neurogenic atrophy, as assumed commonly.
Human ALS and Mitochondrial-Orchestrated PCD Involving p53
PCD appears to contribute to the selective degeneration of motor neurons in human sporadic and familial ALS, albeit seemingly as a non-classical form differing from apoptosis (Figure 2 and Figure 5) [224]. Motor neurons appear to pass through sequential stages of chromatolysis (suggestive of initial axonal injury [396]), somatodendritic attrition without extensive cytoplasmic vacuolation, and then nuclear DNA fragmentation, nuclear condensation, and cell death (Figure 5) [224]. Motor neurons in people that have died from sporadic ALS and familial ALS show the same patterns of degeneration [224]. This cell death in human motor neurons is defined by genomic DNA fragmentation (determined by DNA agarose gel electrophoresis and in situ DNA nick-end labeling) and cell loss and is associated with accumulation of mitochondria, cytochrome c, and cleaved caspase-3 (Figure 5) [380,439]. p53 levels increase in vulnerable CNS regions in people with ALS, and p53 accumulates specifically in the nucleus of lower and upper motor neurons (Figure 5) [440]. This p53 is active functionally because it is phosphorylated at serine392 and has increased DNA binding activity [24,440]. However, the morphology of this cell death is distinct from classical apoptosis, despite the nuclear condensation [17,24,224]. Nevertheless, Bax and Bak1 protein levels are increased in mitochondria-enriched fractions of selectively vulnerable motor regions (spinal cord anterior horn and motor cortex gray matter), but not in regions unaffected by the disease (somatosensory cortex gray matter) [224]. In marked contrast, Bcl-2 protein is depleted severely in mitochondria-enriched fractions of affected regions and is sequestered in the cytosol (Figure 1) [224]. Although these western blot results lacked direct specificity for motor neuron events [224], subsequent immunohistochemistry for cleaved capsase-3 has shown numerous positive motor neurons in human ALS spinal cord (Figure 5) [439], but another study reported negative findings [441]. More recently, laser capture microdissection of motor neurons combined with mass spectroscopy-protein profiling have confirmed the presence of intact active caspase-3 in human ALS motor neurons [380].
These studies [24,224,380,439,441] support the concept of an aberrant re-emergence of an atypical PCD mechanism, involving p53 activation and redistributions of mitochondrial cell death proteins, participating in the pathogenesis of motor neuron degeneration in human ALS. The morphological and biochemical changes seen in human ALS are modeled robustly and faithfully at structural and molecular levels in axotomy models of motor neuron degeneration in adult mouse [442] but not in the commonly used human mutant SOD1 tg mouse models [381,439]. A role for mitochondria in human ALS pathogenesis is further supported by recent work suggesting that lithium treatment may be beneficial in ALS through mitochondrial autophagy [443].
Mitochondrial Pathobiology in Cell and Mouse Models of ALS
A common genetic mutation in human SOD1 that is linked to familial ALS (Table 4) is the substitution of glycine by alanine at position 93 (G93A) [444]. SOD1 is a metalloenzyme of 153 amino acids (~16 kDa) that binds one copper ion and one zinc ion per subunit (Figure 1) [9]. This enzyme, functioning as a ~32 kDa non-covalently linked homodimer, is responsible for the detoxification and maintenance of intracellular O2•- concentration in the low femtomolar range by catalyzing its dismutation [9,445]. SOD1 is ubiquitous (intracellular SOD concentrations are typically ~10–40 micromolar) in most tissues and possibly greater in neurons [446].
Data from cell culture studies reveal mitochondrial dysfunction, possibly motor neuron selective, in the presence of human mutant SOD1 (mSOD1) [447]. Expression of several mSOD1 variants increases mitochondrial O2•- levels (Figure 1) and causes toxicity in cultured primary embryonic motor neurons [448], neuroblastoma cells [449], and NSC-34 cells (a hybrid cell line with some motor neuron-like characteristics, produced by fusion of motor neuron-enriched embryonic mouse spinal cord cells with mouse neuroblastoma cells) [450]. These responses can be attenuated by over-expression of MnSOD [449]. ALS-mSOD1 variants, compared to human wild-type SOD1, associate more with mitochondria in NSC-34 cells and appear to form cross-linked oligomers that shift the mitochondrial glutathione/oxidized glutathione ratio toward oxidation [447].
Gurney and colleagues were the first to develop tg mice that express human mSOD1 [451,452]. Now, these mice are used widely as an animal model of ALS [439,444]. Human mSOD1 is expressed ubiquitously in these mice by its endogenous promoter in a tissue/cell non-selective pattern against a background of normal mouse SOD1 [451]. Effects of this human mutant gene in mice are profound. Hemizygous tg mice expressing high copy number of the G93A variant of human SOD1 become completely paralyzed and die at ~16–18 weeks of age [451]. G93A-mSOD1 mice with reduced transgene copy number have a much slower disease progression and die at ~8–9 months of age [19,451].
Spinal motor neurons and interneurons in mice expressing G93Ahigh-mSOD1 undergo prominent degeneration; about 80% of lumbar motor neurons are eliminated by end-stage disease [381,453]. Subsets of spinal interneurons degenerate before motor neurons in G93Ahigh-mSOD1 [381] and some are the glycinergic Renshaw cells [453]. Unlike the degeneration of motor neurons in human ALS (Figure 5) [224], motor neurons in these mice do not degenerate with a morphology resembling any form of apoptosis [215,381,439,454]. The motor neurons degeneration seen in G93Ahigh-mSOD1 mice more closely resembles a prolonged necrotic-like cell death process [381] involving early-occurring mitochondrial damage, cellular swelling, and dissolution [215,381,439,453,454]. Biochemically, the death of motor neurons in G93Ahigh-mSOD1 is characterized by somal and mitochondrial swelling and formation of DNA single-strand breaks prior to DNA double-strand breaks occurring in nuclear DNA and mtDNA [381]. The motor neuron death in G93Ahigh-mSOD1 mice is independent of activation of caspases-1 and -3, and also appears to be independent of capsase-8 and AIF activation within motor neurons [381]. Indeed, caspase-dependent and p53-mediated apoptosis mechanisms might be blocked actively in G93Ahigh-mSOD1 mouse motor neurons, possibly by upregulation of IAPs and blockade in the nuclear import of p53 [381]. More work is needed on the cell death in G93Alow-mSOD1 mice, because these mice could be more relevant physiologically and clinically to the human disease compared to G93Ahigh-mSOD1 mouse.
Mitochondrial pathology has been implicated in the mechanisms of mouse ALS [5], but until recently, most evidence is circumstantial. In different mSOD1 mouse models of ALS, mitochondria in spinal cord neurons exhibit structural pathology [81,381,439,453,454,455,456] and some of the mitochondrial degeneration occurs very early in the course of the disease [381,454]. Mitochondrial ultrastructural microvacuolar damage in motor neurons emerges by 4 weeks of age in G93Ahigh mice [381]. It has been argued that mitochondrial damage in G93Ahigh-mSOD1 mice is related to supra-normal levels of SOD1 and might not be related causally to the disease process because tg mice expressing high levels of human wild-type SOD1 show some mitochondrial pathology [457], but mitochondrial abnormalities have been found histologically also in G93Alow-mSOD1 mice [458]. Thus, mitochondria could be primary sites of human SOD1 toxicity in tg mice irrespective of transgene copy number and expression level of human SOD1, but direct, unequivocal causal relationships have been lacking.
Human mSOD1 proteins appear to acquire a toxic property or function, rather than having diminished O2•- scavenging activity [459,460,461], and wild-type SOD1 can gain toxic properties through loss of Zn [448] and oxidative modification [462,463]. A gain in aberrant oxidative chemistry could contribute to the mechanisms of mitochondriopathy in G93Ahigh mice (Figure 1) [12,464]. G93A-mSOD1 has enhanced free radical-generating capacity compared to wild-type enzyme [461] and can catalyze protein oxidation by hydroxyl-like intermediates and carbonate radical (Figure 1) [465]. G93Ahigh mice have increased protein carbonyl formation in total spinal cord tissue extracts at pre-symptomatic disease [466]. Protein carbonyl formation in mitochondrial membrane-enriched fractions of spinal cord is a robust signature of incipient disease [81]. A mass spectroscopy study of G93Ahigh mice identified proteins in total spinal cord tissue extracts with greater than baseline carbonyl modification, including SOD1, translationally controlled tumor protein, and UCH-L1 [467]. Nitrated and aggregated cytochrome c oxidase subunit-I and α-Syn accumulate in G93Ahigh mouse spinal cord [381]. Nitrated MnSOD accumulates also in G93Ahigh mouse spinal cord [381]. Toxic properties of mSOD1 could also be mediated through protein binding or aggregation. Wild-type SOD1 and human mSOD1 associate with mitochondria [468,469]. Human SOD1 mutants associate with spinal cord mitochondria in mSOD1 mice and can bind Bcl-2 [470,471], thus potentially being decoys or dominant negative regulators of cell survival molecules (Figure 1, Table 1), but it is not known if this process is occurring specifically in motor neurons. Binding of mSOD1 (and perhaps its low-mobility species) to mitochondria has been reported to be spinal cord-selective and age-dependent [472], but this work also lacks cellular resolution. A recent biochemical in vitro study has shown that endogenous SOD1 in the mitochondrial intermembrane space controls cytochrome c-catalyzed peroxidation and that G93A-mSOD1 mediates greater ROS production in the intermembrane space compared to wild-type SOD1 [473]. Human SOD1 mutants can also shift mitochondrial redox potential when expressed in cultured cells [447]. Nevertheless, the direct links between the physicochemical changes in wild-type and mutant SOD1 and the mitochondrial functional and structural changes associated with ALS and motor neuron degeneration remain uncertain.
EM studies of motor neurons in G93Ahigh mice have shown that the OMM remains relatively intact to permit formation of mega-mitochondria [81,381]. Moreover, early in the disease of these mice, mitochondria in dendrites in spinal cord ventral horn undergo extensive cristae and matrix remodeling, while few mitochondria in motor neuron cell bodies show major structural changes [81]. Thus, the disease might start distally in mitochondria of the motor neuron processes [81,381]. Another interpretation of ultrastructural findings is that the mSOD1 causes mitochondrial degeneration by inducing OMM extension and leakage and intermembrane space expansion [474]. Mechanisms for this damage could be related to mSOD1 gaining access to the mitochondrial intermembrane space [473,474]. This mitochondrial conformation seen by EM might favor the formation of the mPTP (Figure 1); indeed, evidence for increased contact sites between the OMM and IMM in dendritic mitochondria in G93Ahigh mice has been found [81]. Another feature of motor neurons in young G93Ahigh mice, before symptoms emerge, is apparent fission of ultrastructurally normal mitochondria in cell bodies and fragmentation of abnormal mitochondria [81]. It is not clear if the cristae and matrix remodeling and the apparent fragmentation and fission mitochondria are related or independent events and if these abnormalities interfere with mitochondrial trafficking; nevertheless, morphological observations enforce the idea that mitochondria could be important to the pathobiology of mSOD1 toxicity to motor neurons in G93Ahigh mice.
The possibility of changes in mitochondrial trafficking in motor neurons of mSOD1 mice is mostly unexplored. Some data support the novel idea that mitochondria might act as messengers from distal regions of motor neurons in mSOD1 mice [5]. G93Ahigh-mSOD1 mouse motor neurons accumulate mitochondria from the axon terminals and generate higher levels of O2•-, •NO, and ONOO- than motor neurons in tg mice expressing human wild-type SOD1 [381]. This mitochondrial accumulation occurs at a time when motor neuron cell body volume is increasing, suggestive of ongoing abnormalities with ATP production or plasma membrane Na+,K+ ATPase [381]. G93A-mSOD1 perturbs anterograde axonal transport of mitochondria in cultured primary embryonic motor neurons [475] making it possible that retrogradely transported mitochondria with toxic properties from the neuromuscular junction fail to be returned to distal processes [5,381]. Mitochondria with enhanced toxic potential from distal axons and terminals could therefore have a “Trojan horse” role in triggering degeneration of motor neurons in ALS via retrograde transport from diseased skeletal muscle.
Motor neurons in G93Ahigh-mSOD1 mice also accumulate higher levels of intracellular Ca2+ than motor neurons in human wild-type SOD1 tg mice [381]. The intracellular Ca2+ signal in motor neurons is very compartmental and mitochondrial-like in its appearance [188,381]. Abnormal elevations intracellular Ca2+ in G93Ahigh-mSOD1 mouse motor neurons have been seen also by different Ca2+ detection methods [476,477]. Recent work on a mouse neuromuscular junction preparation has shown that mitochondrial Ca2+ accumulation is accompanied by greater IMM depolarization, specifically within motor neuron terminals of human mutant SOD1 tg mice [478].
NO signaling mechanisms in mitochondria (Figure 1) of ALS mice appear to be involved in the pathogenesis. Motor neurons seem to be unique regarding NO production because they express constitutively low levels of inducible NO synthase (iNOS) [188,381,402]. G93Ahigh-mSOD1 mouse motor neurons accumulate nicotinamide adenine dinucleotide phosphate diaphorase and iNOS-like immunoreactivity [381,402]. iNOS is also up-regulated aberrantly in human sporadic ALS motor neurons [479]. iNOS gene knockout [381] and iNOS inhibition with 1400W [402] extend significantly the lifespan of G93Ahigh-mSOD1 mice. Thus, mitochondrial oxidative stress, Ca2+ dysregulation, iNOS activation, protein nitration, and protein aggregation (not necessarily SOD1 though) are all likely intrinsic, cell automonous mechanisms in the process of motor neuron degeneration caused by mSOD1 in mice [381]. The mechanistic basis for the differences between human ALS and mSOD1 mice, regarding cell death phenotype [19,439], is not yet clear, but could be related to the extreme non-physiological expression of toxic mSOD1 or to fundamental differences in cell death mechanisms in human and mouse neurons [439] or tissue inflammation that drive motor neurons in mSOD1 tg mice to necrotic-like death according to the cell death matrix (Figure 2). Another contributing factor for this difference between human and mouse motor neurons is that mitochondria are functionally diverse and have species-specific activities and molecular compositions, including the makeup of the mPTP [480]. These possibilities allow for skepticism regarding the suitability of existing tg mSOD1 mouse lines to model human ALS.
The mPTP Contributes to the Disease Mechanisms of ALS in Mice
Despite the implication of toxic effects of mSOD1 on mitochondria in mouse ALS, cause-effect relationships between abnormal functioning of mitochondria and initiation and progression of disease have been uncertain. These relationships need to be known because this knowledge could lead to new mechanism-based treatments for ALS. One specific target of investigation for mitochondria in disease causality is the mPTP (Figure 1).
The mPTP was first implicated in ALS pathogenesis using pharmacological approaches. Cyclopsorine A treatment of G93Ahigh mice, delivered intracerebroventricularly or systemically to mice on a multiple drug resistance type 1a/b background, improved outcome modestly [481,482,483]. These studies were confounded by the immunosuppressant actions of cyclopsorine A through calcineurin inhibition. Pharmacological studies using CyPD inhibitors devoid of effects on calcineurin need to be done on ALS mice. Another study showed that treatment with cholest-4-en-3-one oxime (TRO19622), a drug that binds VDAC and the 18 kDa translocator protein (TSPO, or peripheral benzodiazepine receptor), improved motor performance, delayed disease onset, and extended survival of G93Ahigh mice [484]. However, another study using a different TSPO ligand (Ro-4864) did not show positive effects with G93Ahigh mice [485].
CyPD and ANT are targets of nitration in ALS mice (Figure 1) [81]. CyPD nitration is elevated in early- to mid-symptomatic stages, but declines to baseline at end-stage disease [81]. ANT nitration is pertinent because it occurs in pre-symptomatic and symptomatic stages but not at end-stage disease or in tg mice expressing human wild-type SOD1 [81]. The ANT is important in the context of age-related neurodegenerative disease because it undergoes carbonyl modification during normal aging in housefly flight muscle [486] and rat brain [487]. In vitro cell-free and cell experiments have shown that NO and ONOO- can act directly on the ANT to induce mitochondrial permeabilization in a cyclosporine A-sensitive manner [488]. Oxidative stress enhances the binding of CyPD to ANT [489]. Some SOD1 mutants are unstable and lose copper [464,465], and interestingly, copper interactions with ANT and thiol modification of ANT can cause mPTP opening [490,491]. Together these data and future work could reveal that oxidative and nitrative damage to proteins, some of which are core components of the mPTP, in G93Ahigh mice is targeted rather than stochastic and could impinge on the functioning of the mPTP.
The role of CyPD in the process of motor neuron disease in ALS mice has been examined through gene-ablation [81]. G93Ahigh-mSOD1 mice without CyPD show markedly delayed disease onset and lived significantly longer than tg mice with CyPD. The effect of CyPD deletion was much more prominent in females than in males [81]. Remarkably, female mice showed positive effects with only haplo-deletion of CyPD. Ppif gene ablation in tg mice with much lower levels of human mSOD1 expression and a slower disease progression (G93Alow-mSOD1 mice) also show significantly delayed disease onset and lived significantly longer than tg mice with CyPD [81]. Thus, some form of mPTP pathobiology is occurring regardless of whether transgene expression of G93A is high or low.
Nevertheless, most G93A-mSOD1 mice without CyPD develop eventually motor neuron disease and die. Other work on CypD null mice has shown that high concentrations of Ca2+ (2 mM) can still lead to mPTP activation without CyPD and that cell deaths caused by Bid, Bax, DNA damage and TNF still occur without CyPD [492]. The effects of CyPD deficiency on motor neuron cell mechanisms thus need detailed examination, but the cell death phenotype might switch or convert to another form with the attenuation of mitochondrial swelling. A switch in the cell death morphology and molecular mechanisms in motor neurons of mSOD1 mice without CyPD is an outcome consistent with the cell death matrix concept (Figure 2) [194].
Summary and Outlook
Mitochondria have diverse functions and properties and could be critically important for the development of human adult-onset neurodegenerative disorders, including AD, PD, and ALS [7]. Structural and biochemical data from studies of human autopsy CNS as well as cell and animal models of these neurodegenerative disorders suggest that mitochondrial dysfunction is a trigger or propagator of neurodegeneration. Novel mechanisms for mitochondriopathy and neurodegeneration could involve the mPTP (Figure 1 and Figure 2). There is precedence for this logic in mouse models of AD [282], multiple sclerosis [493], stroke [494], and ALS [81]. Using ALS as one example, the mPTP actively participates in the mechanisms of motor neuron death in ALS mice in a gender-preferential pattern [81]. Thus, mPTP activation is a possible triggering event for motor neuron degeneration, and motor neuron selective vulnerability in ALS could be related to amount, composition, and trafficking of mitochondria in these cells. Further study of mitochondria in neurons, glial cells, and skeletal myocytes can define new mechanisms of cell death and disease and can lead to the identification of molecular mechanism-based therapies for treating AD, PD, and ALS.
Abbreviations
| Aβ | amyloid beta protein |
| AD | Alzheimer’s disease |
| AIF | apoptosis-inducing factor |
| ALS | amyotrophic lateral sclerosis |
| ANT | adenine nucleotide translocator |
| Apaf | apoptotic protease activating factor |
| APP | amyloid precursor protein |
| CNS | central nervous system |
| Cu/ZnSOD | copper/zinc superoxide dismutase (also SOD1) |
| CyPD | cyclophilin D |
| DISC | death-inducing signaling complex |
| EM | electron microscopy |
| ER | endoplasmic reticulum |
| GPe | globus pallidus external |
| GPi | globus pallidus internal |
| HtrA2 | high temperature requirement protein A2 |
| IAP | inhibitor of apoptosis protein |
| IMM | inner mitochondrial membrane |
| KA | kainic acid |
| LB | Lewy body |
| LGN | lateral geniculate nucleus |
| LRRK2 | leucine-rich repeat kinase 2 |
| mnSOD | manganese SOD (also SOD2) |
| mPT | mitochondrial permeability transition |
| mPTP | mitochondrial permeability transition pore |
| mSOD1 | mutant SOD1 |
| mtDNA | mitochondrial DNA |
| NAIP | neuronal apoptosis inhibitory protein |
| NFT | neurofibrillary tangle |
| NMDA | N-methy-D-aspartate |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| O2•- | superoxide radical |
| OMM | outer mitochondrial membrane |
| ONOO- | peroxynitrite |
| PCD | programmed cell death |
| PD | Parkinson’s disease |
| PINK1 | phosphatase and tensin homolog-induced putative kinase-1 |
| ROS | reactive oxygen species |
| SNc | substantia nigra compacta |
| Syn | α-synuclein |
| Tg | transgenic |
| TIMM | translocase of inner mitochondrial membrane |
| TOMM | translocase of outer mitochondrial membrane |
| TSPO | translocator protein 18 kDa (peripheral benzodiazepine receptor) |
| TNF | tumor necrosis factor |
| TUNEL | terminal transferase-mediated biotin-dUTP nick-end labeling |
| UCH-L1 | ubiquitin carboxy-terminal hydrolyase-L1 |
| VDAC | voltage-dependent anion channel |
Acknowledgments
The author thanks all of the individuals in his lab for their hard work, particularly Yan Pan and Ann Price for data generated on human AD, PD and ALS autopsy brain and spinal cord, the A53T α-Syn tg mouse, and the G93A-mSOD1 tg mouse. This work was supported by grants from the U.S. Public Health Service, NIH-NINDS (NS065895, NS034100, NS052098) and NIH-NIA (AG016282).
References
- Virchow, R. Cellular Pathology as Based Upon Physiological and Pathological Histology; John Churchill: London (Translation), UK, 1860. [Google Scholar]
- Martin, L.J. Neurodegenerative disorders of the human brain and spinal cord. In Encyclopedia of the Human Brain; Ramachandran, V.S., Ed.; Elsevier Science Academic Press: San Diego, USA, 2002; Volume 3, pp. 441–463. [Google Scholar]
- Rich, T.; Allen, R.L.; Wyllie, A.H. Defying death after DNA damage. Nature 2000, 407, 777–783. [Google Scholar]
- Zheng, T.S. Death by design: the big debut of small molecules. Nat. Cell Biol. 2001, 3, E1–E3. [Google Scholar]
- Martin, L.J. Mitochondriopathy in Parkinson’s disease and amyotrophic lateral sclerosis. J. Neuropathol. Exp. Neurol. 2006, 65, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Isave, N.K.; Plotnikov, E.Y.; Zorova, L.D.; Stelmashook, E.V.; Vasileva, A.K.; Arkhagelskaya, A.A.; Khrjapenkova, T.G. The mitochondrion as Janus Bifrons. Biochemistry (Moscow) 2007, 72, 1115–1126. [Google Scholar] [CrossRef]
- Nicholls, D.G. Mitochondrial function and dysfunction in the cell: its relevance to aging and aging-related disease. Intl. J. Biochem. Cell Biol. 2002, 34, 1372–1381. [Google Scholar]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn of evolutionary medicine. Annu. Rev. Genet. 2005, 39, 359–407. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar]
- Halliwell, B. Role of free radicals in the neurodegenerative diseases. Drugs Aging 2001, 18, 685–716. [Google Scholar]
- Mungrue, I.N.; Bredt, D.S.; Stewart, D.J.; Husain, M. From molecules to mammals: what’s NOS got to do with it? Acta Physiol. Scand. 2003, 179, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S.; Carson, M.; Smith, C.D.; Koppenol, W.H. ALS, SOD and peroxynitrite. Nature 1993, 364, 548. [Google Scholar]
- Martin, L.J.; Liu, Z. DNA damage profiling in motor neurons: a single-cell analysis by comet assay. Neurochem. Res. 2002, 27, 1089–1100. [Google Scholar]
- Giulini, C. Characterization and function of mitochondrial nitric-oxide synthase. Free Radic. Biol. Med. 2003, 34, 397–408. [Google Scholar]
- Brown, G.C.; Borutaite, V. Nitric oxide, cytochrome c, and mitochondria. Biochem. Soc. Sym. 1999, 66, 17–25. [Google Scholar]
- Delettre, C.; Lenaers, G.; Pelloquin, L.; Belenguer, P.; Hamel, C.P. OPA1 (Kjer type) dominant optic atrophy: a novel mitochondrial disease. Mol. Genet. Metab. 2002, 75, 97–107. [Google Scholar]
- Martin, L.J.; Al-Abdulla, N.A.; Brambrink, A.M.; Kirsch, J.R.; Sieber, F.E.; Portera-Cailliau, C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res. Bull. 1998, 46, 281–309. [Google Scholar] [CrossRef] [PubMed]
- Northington, F.J.; Graham, E.M.; Martin, L.J. Apoptosis in perinatal hypoxic-ischemic brain injury: how important is it and should it be inhibited? Brain Res. Rev. 2005, 50, 244–257. [Google Scholar] [CrossRef]
- Martin, L.J. The mitochondrial permeability transition pore: a molecular target for amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2010, 1802, 186–197. [Google Scholar] [PubMed]
- Waldmeier, P.C.; Zimmermann, K.; Qian, T.; Tintelnot-Blomley, M.; Lemasters, J.J. Cyclophilin D as a drug target. Curr. Med. Chem. 2003, 10, 1485–1506. [Google Scholar]
- Crompton, M. Mitochondria and aging: a role for the permeability transition? Aging Cell 2004, 3, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P.; Krauskopf, A; Basso, E.; Petronilli, V.; Blalchy-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [PubMed]
- Martin, L.J. Neuronal cell death in nervous system development, disease, and injury. Int. J. Mol. Med. 2001, 7, 455–478. [Google Scholar] [PubMed]
- Lockshin, R.A.; Zakeri, Z. Caspase-independent cell deaths. Curr. Opin. Cell Biol. 2002, 14, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S. Developmental Biology; Sinauer Associates: Sunderland, MA, USA, 2006. [Google Scholar]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef] [PubMed]
- Portera-Cailliau, C.; Price, D.L.; Martin, L.J. Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J. Comp. Neurol. 1997, 378, 70–87. [Google Scholar]
- Portera-Cailliau, C.; Price, D.L.; Martin, L.J. Non-NMDA and NMDA receptor-mediated excitotoxic neuronal deaths in adult brain are morphologically distinct: further evidence for an apoptosis-necrosis continuum. J. Comp. Neurol. 1997, 378, 88–104. [Google Scholar]
- Formigli, L.; Papucci, L.; Tani, N.; Schiavone, N.; Tempestini, A.; Orlandini, G.E.; Capaccioli, S.; Zecchi Orlandini, S. Aponecrosis: morphological and biochemical exploration of a syncretic process of cell death sharing apoptosis and necrosis. J Cell. Physiol. 2000, 182, 41–49. [Google Scholar]
- Lennon, S.V.; Martin, S.J.; Cotter, T.G. Dose-dependent induction of apoptosis in human tumour cell lines by widely diverging stimuli. Cell Prolif. 1991, 24, 203–214. [Google Scholar]
- Fernandes, R.S.; Cotter, T.G. Apoptosis or necrosis: intracellular levels of glutathione influence mode of cell death. Biochem. Pharmacol. 1994, 48, 675–681. [Google Scholar]
- Bonfoco, E.; Krainc, D.; Ankarcrona, M.; Nicotera, P.; Lipton, S.A. Apoptosis and necrosis: two distinct events induced, respectively, by mild and intense insults with N-methyl-D-aspartate or nitric oxide/superoxide in cortical cell culture. Proc. Natl. Acad. Sci. USA 1995, 92, 7162–7166. [Google Scholar]
- Raffray, M.; Cohen, G.M. Apoptosis and necrosis in toxicology: a continuum or distinct modes of cell death? Pharmacol. Ther. 1997, 75, 153–177. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Lipinski, M.; Degterev, A. Diversity in the mechanisms of neuronal cell death. Neuron 2003, 40, 401–413. [Google Scholar]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: the calcium-apoptosis link. Nat. Rev. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The Bcl-2 protein family: opposing activities that mediate cell death. Nat. Rev. 2008, 9, 47–59. [Google Scholar]
- Merry, D.E.; Korsmeyer, S.J. Bcl-2 gene family in the nervous system. Ann. Rev. Neurosci. 1997, 20, 245–267. [Google Scholar]
- Trump, B.F.; Berezesky, I.K. The role of altered [Ca2+]i regulation in apoptosis, oncosis, and necrosis. Biochim. Biophys. Acta 1996, 1313, 173–178. [Google Scholar] [CrossRef]
- Majno, G.; Joris, I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am. J. Pathol. 1995, 146, 3–15. [Google Scholar]
- Trump, B.J.; Goldblatt, P.J.; Stowell, R.E. Studies on necrosis of mouse liver in vitro. Ultrastructural alterations in the mitochondria of hepatic parenchymal cells. Lab. Invest. 1964, 14, 343–371. [Google Scholar]
- Leist, M.; Single, B.; Castoldi, A.F.; Kuhnles, S.; Nicotera, P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J. Exp. Med. 1997, 185, 1481–1486. [Google Scholar]
- Martin, L.J.; Brambrink, A.M.; Price, A.C.; Kaiser, A.; Agnew, D.M.; Ichord, R.N.; Traystman, R.J. Neuronal death in newborn striatum after hypoxia-ischemia is necrosis and evolves with oxidative stress. Neurobiol. Dis. 2000, 7, 169–191. [Google Scholar]
- Golden, W.C.; Brambrink, A.M.; Traystman, R.J.; Martin, L.J. Failure to sustain recovery of Na,K ATPase function is a possible mechanism for striatal neurodegeneration in hypoxic-ischemic newborn piglets. Mol. Brain Res. 2001, 88, 94–102. [Google Scholar] [CrossRef]
- Castro, J.; Ruminot, I.; Porras, O.H.; Flores, C.M.; Hermosilla, T.; Verdugo, E.; Venegas, F.; Hartel, S.; Michea, L.; Barros, L.F. ATP steal between cation pumps: a mechanism linking Na+ influx to the onset or necrotic Ca2+ overload. Cell Death Diff. 2006, 13, 1675–1685. [Google Scholar] [CrossRef]
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Gabai, V.L. Necrosis: a specific form of programmed cell death. Exp. Cell Res. 2003, 283, 1–16. [Google Scholar]
- Kim, Y-S.; Morgan, M.J.; Choksi, S.; Lu, Z-G. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell 2007, 26, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, J.; Christofferson, D.E.; Ng, A.; Yao, J.; Degterev, A.; Xavier, R.J.; Yuan, J. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008, 135, 1311–1323. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.C.; Snyder, S.H. Poly(ADP-ribose) polymerase-1 in the nervous system. Neurobiol. Dis. 2000, 7, 225–239. [Google Scholar]
- Zoratti, M.; Szabo, I. The mitochondrial permeability transition. Biochem. Biophys. Acta 1995, 1241, 139–176. [Google Scholar]
- Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 1999, 341, 233–249. [Google Scholar]
- Van Gurp, M.; Festjens, N.; van Loo, G.; Saelens, X.; Vandenabeele, P. Mitochondrial intermembrane proteins in cell death. Biochem. Biophys. Res. Comm. 2003, 304, 487–497. [Google Scholar]
- Leung, A.W.C.; Halestrap, A.P. Recent progress in elucidating the molecular mechanism of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 2008, 1777, 946–952. [Google Scholar] [CrossRef] [PubMed]
- Shoshan-Barmatz, V.; Israelson, A.; Brdiczka, D.; Sheu, S.S. The voltage-dependent anion channel (VDAC): function in intracellular signaling, cell life and cell death. Curr. Pharm. Des. 2006, 12, 2249–2270. [Google Scholar]
- Rostovtseva, T.K.; Tan, W.; Colombini, M. On the role of VDAC in apoptosis: fact and fiction. J. Bioenerget. Biomembr. 2005, 37, 129–142. [Google Scholar]
- Granville, D.J.; Gottlieb, R.A. The mitochondrial voltage-dependent anion channel (VDAC) as a therapeutic target for initiating cell death. Curr. Med. Chem. 2003, 10, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Huizing, M.; Ruitenbeek, W.; van den Heuvel, L.P.; Dolce, V.; Iacobazzi, V.; Smeitink, J.A.M.; Palmieri, F.; Trijbels, J.M.F. Human mitochondrial transmembrane metabolite carriers: tissue distribution and its implication for mitochondrial disorders. J. Bioenerg. Biomembr. 1998, 30, 277–284. [Google Scholar]
- Wu, S.; Sampson, M.J.; Decker, W.K.; Craigen, W.J. Each mammalian mitochondrial outer membrane porin protein is dispensable: effects on cellular respiration. Biochem. Biophys. Acta 1999, 1452, 68–78. [Google Scholar] [CrossRef]
- Anflous, K.; Armstrong, D.D.; Craigen, W.J. Altered sensitivity for ADP and maintenance of creatine-stimulated respiration in oxidative striated muscles from VDAC1-deficient mice. J. Biol. Chem. 2001, 276, 1954–1960. [Google Scholar] [PubMed]
- Sampson, M.J.; Decker, W.K.; Beaudet, A.L.; Ruitenbeek, W.; Armstrong, D.; Hicks, M.J.; Craigen, W.J. Immotile sperm and infertility in mice lacking mitochondrial voltage-dependent anion channel type 3. J. Biol. Chem. 2001, 276, 39206–39212. [Google Scholar]
- Cheng, E.H.; Sheiko, T.V.; Fisher, J.K.; Craigen, W.J.; Korsemeyer, S.J. VDAC2 inhibits Bak activation and mitochondrial apoptosis. Science 2003, 301, 513–517. [Google Scholar]
- Chandra, D.; Choy, G.; Daniel, P.T.; Tang, D.G. Bax-dependent regulation of Bak by voltage-dependent anion channel 2. J. Biol. Chem. 2005, 280, 19051–19061. [Google Scholar]
- Baines, C.P.; Kaiser, R.A.; Sheiko, T.; Craigen, W.J.; Molkentin, J.D. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat. Cell Biol. 2007, 9, 550–555. [Google Scholar]
- Karachitos, A.; Galganska, H.; Wojtkowska, M.; Budzinska, M.; Stobienia, O.; Bartosz, G.; Kimita, H. Cu,Zn-superoxide dismutase is necessary for proper function of VDAC in Saccharomyces cerevisiae cells. FEBS Lett. 2009, 583, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Halestrap, A.P.; Brenner, C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr. Med. Chem. 2003, 10, 1507–1525. [Google Scholar] [CrossRef] [PubMed]
- Graham, B.H.; Waymire, K.G.; Cottrell, B.; Trounce, I.A.; MacGregor, G.R.; Wallace, D.C. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat. Genet. 1997, 16, 226–234. [Google Scholar]
- Stepien, G.; Torroni, A.; Chung, A.B.; Hodge, J.A.; Wallace, D.C. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J. Bio. Chem. 1992, 267, 14592–14597. [Google Scholar]
- Vyssokikh, M.Y.; Katz, A.; Rueck, A.; Wuensch, C.; Dorner, A.; Zorov, D.B.; Brdiczka, D. Adenine nucleotide translocator isoforms 1 and 2 are differently distributed in the mitochondrial inner membrane and have distinct affinities to cyclophilin D. Biochem. J. 2001, 358, 349–358. [Google Scholar]
- Kikoszka, J.E.; Waymire, K.G.; Levy, S.E.; Sligh, J.E.; Cai, J.; Jones, D.P.; MacGregor, G.R.; Wallace, D.C. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004, 427, 461–465. [Google Scholar] [PubMed]
- Machida, K.; Hayashi, Y.; Osada, H. A novel adenine nucleotide translocase inhibitor, MT-21, induces cytochrome c release through a mitochondrial permeability transition-independent mechanisms. J. Biol. Chem. 2002, 277, 31243–31248. [Google Scholar] [PubMed]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, H.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn II, G.W.; Robbins, J.; Molkentin, J.D. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar]
- Woodfield, K.; Rück, A.; Brdiczka, D.; Halestrap, A.P. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem. J. 1998, 336, 287–290. [Google Scholar]
- Johnson, N.; Khan, A.; Virji, S.; Ward, J.M.; Crompton, M. Import and processing of heart mitochondrial cyclophilin D. Eur. J. Biochem. 1999, 263, 353–359. [Google Scholar] [CrossRef] [PubMed]
- McEnery, M.W.; Dawson, T.M.; Verma, A.; Gurley, D.; Colombini, M.; Snyder, S.H. Mitochondrial voltage-dependent anion channel. J. Biol. Chem. 1993, 268, 23289–23296. [Google Scholar]
- Shoshan-Barmatz, V.; Zalk, R.; Gincel, D.; Vardi, N. Subcellular localization of VDAC in mitochondria and ER in the cerebellum. Biochem. Biophys. Acta 2004, 1657, 105–114. [Google Scholar] [CrossRef]
- Akanda, N.; Tofight, R.; Brask, J.; Tamm, C.; Elinder, F.; Ceccatelli, S. Voltage-dependent anion channels (VDAC) in the plasma membrane play a critical role in apoptosis in differentiated hippocampal neurons but not in neural stem cells. Cell Cycle 2008, 7, 3225–3234. [Google Scholar]
- Yu, W.H.; Wolfgang, W.; Forte, M. Subcellular localization of human voltage-dependent anion channel isoforms. J. Biol. Chem. 1995, 270, 13998–14006. [Google Scholar]
- Buck, C.R.; Jurynec, M.J.; Upta, D.E.; Law, A.K.T.; Bilger, J.; Wallace, D.C.; McKeon, R.J. Increased adenine nucleotide translocator 1 in reactive astrocytes facilitates glutamate transport. Exp. Neurol. 2003, 181, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Hazelton, J.L.; Petrasheuskaya, M.; Fiskum, G.; Kristian, T. Cyclophilin D is expressed predominantly in mitochondria of γ-aminobutyric acidergic interneurons. J. Neurosci. Res. 2009, 87, 1250–1259. [Google Scholar]
- Naga, K.K.; Sullivan, P.G.; Geddes, J.W. High cyclophilin D content of synaptic mitochondria results in increased vulnerability to permeability transition. J. Neurosci. 2007, 27, 7469–7475. [Google Scholar]
- Martin, L.J.; Gertz, B.; Pan, Y.; Price, A.C.; Molkentin, J.D.; Chang, Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp. Neurol. 2009, 218, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Freedman, R.B. Peptidyl prolyl cis-trans-isomerase activity associated with the lumen of the endoplasmic reticulum. Biochem. J. 1994, 300, 865–870. [Google Scholar] [PubMed]
- Sullivan, P.G.; Rabchevsky, A.G.; Keller, J.N.; Lovell, M.; Sodhi, A.; Hart, R.P.; Scheff, S.W. Intrinsic differences in brain and spinal cord mitochondria: implications for therapeutic interventions. J. Comp. Neurol. 2004, 474, 524–534. [Google Scholar] [CrossRef] [PubMed]
- Morota, S.; Hansson, M.J.; Ishii, N.; Kudo, Y.; Elmer, E.; Uchino, H. Spinal cord mitochondria display lower calcium retention capacity compared with brain mitochondria without inherent differences in sensitivity to cyclophilin D inhibition. J. Neurochem. 2007, 103, 2066–2076. [Google Scholar]
- Collins, T.J.; Bootman, M.D. Mitochondria are morphologically heterogeneous within cells. J. Exp. Biol. 2003, 206, 1993–2000. [Google Scholar]
- Jensen, R.E. Control of mitochondrial shape. Curr. Opin. Cell Biol. 2005, 17, 384–388. [Google Scholar]
- Hamberger, A.; Blomstrand, C.; Lehninger, A.L. Comparative studies of mitochondria isolated from neuron-enriched and glia-enriched fractions of rabbit and beef brain. J. Cell Biol. 1970, 45, 221–234. [Google Scholar]
- Tata, J.R. Requirement for RNA and protein synthesis for induced regression of tadpole tail in organ culture. Dev. Biol. 1966, 13, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Chipuk, J.E.; Green, D.R. p53’s believe it or not: lessons on transcription-independent death. J. Clin. Immunol. 2003, 23, 355–361. [Google Scholar]
- Martin, L.J.; Liu, Z.; Pipino, J.; Chestnut, B.; Landek, M.A. Molecular regulation of DNA damage-induced apoptosis in neurons of cerebral cortex. Cereb. Cortex 2009, 19, 1273–1293. [Google Scholar]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar]
- Schwartz, L.M.; Smith, S.W.; Jones, M.E.; Osborne, B.A. Do all programmed cell deaths occur via apoptosis? Proc. Natl. Acad. Sci. USA 1993, 90, 980–984. [Google Scholar]
- Amin, F.; Bowen, I.D.; Szegedi, Z.; Mihalik, R.; Szende, B. Apoptotic and non-apoptotic modes of programmed cell death in MCF-7 human breast carcinoma cells. Cell Biol. Intl. 2000, 24, 253–260. [Google Scholar]
- Jacobson, M. Developmental Neurobiology; Plenum Press: New York, NY, USA, 1991. [Google Scholar]
- Glücksmann, A. Cell deaths in normal vertebrate ontogeny. Biol. Rev. 1951, 26, 59–86. [Google Scholar]
- Lockshin, R.A.; Williams, C.M. Programmed cell death: II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J. Insect Physiol. 1964, 10, 643–649. [Google Scholar] [CrossRef]
- Saunders, J.W. Death in embryonic systems. Science 1966, 154, 604–612. [Google Scholar]
- Bursch, W.; Paffe, S.; Putz, B.; Barthel, G.; Schulte-Hermann, R. Determination of the length of the histological stages of apoptosis in normal liver and in altered hepatic foci of rats. Carcinogenesis 1990, 11, 847–853. [Google Scholar]
- Wyllie, A.H.; Kerr, J.F.R.; Currie, A.R. Cell death: the significance of apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar]
- Nagata, S. Fas ligand-induced apoptosis. Annu. Rev. Genet. 1999, 33, 29–55. [Google Scholar]
- Clarke, P.G.H. Developmental cell death: morphological diversity and multiple mechanisms. Anat. Embryol. 1990, 181, 195–213. [Google Scholar]
- Wyllie, A.H. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature 1980, 284, 555–556. [Google Scholar]
- Liu, X.; Zou, H.; Slaughter, C.; Wang, X. DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 1997, 89, 175–184. [Google Scholar]
- Sakahira, H.; Enari, M.; Ohsawa, Y.; Uchiyama, Y.; Nagata, S. Apoptotic nuclear morphological change without DNA fragmentation. Curr. Biol. 1999, 9, 543–546. [Google Scholar]
- Pilar, G.; Landmesser, L. Ultrastructural differences during embryonic cell death in normal and peripherally deprived ciliary ganglia. J. Cell Biol. 1976, 68, 339–356. [Google Scholar]
- Nakajima, W.; Ishida, A.; Lange, M.S.; Gabrielson, K.L.; Wilson, M.A.; Martin, L.J.; Blue, M.E.; Johnston, M.V. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J. Neurosci. 2000, 20, 7994–8004. [Google Scholar]
- Northington, F.J.; Ferriero, D.M.; Graham, E.M.; Traystman, R.J.; Martin, L.J. Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol. Dis. 2001, 8, 207–219. [Google Scholar]
- Northington, F.J.; Zelaya, M.E.; O’Riordan, D.P.; Blomgren, K.; Flock, D.L.; Hagberg, H.; Ferriero, D.M.; Martin, L.J. Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience 2007, 149, 822–833. [Google Scholar]
- Schweichel, J.U.; Merker, H.J. The morphology of various types of cell death in prenatal tissues. Teratology 1973, 7, 253–266. [Google Scholar]
- Xue, L. Z.; Fletcher, G. C.; Tolkovsky, A. M. Autophagy is activated by apoptotic signaling in sympathetic neurons: an alternative mechanism of death execution. Mol. Cell. Neurosci. 1999, 14, 180–198. [Google Scholar]
- Yue, Z.; Horton, A.; Bravin, M.; DeJager, P.L.; Selimi, F.; Heintz, N. A novel protein complex linking the δ2 glutamate receptor and autophagy: implications for neurodegeneration in Lurcher mice. Neuron 2002, 35, 921–933. [Google Scholar] [PubMed]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; Misushima, N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006, 44, 885–889. [Google Scholar]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; Tanaka, K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006, 44, 880–884. [Google Scholar]
- Nakendra, D.; Tanaka, A.; Suen, D-F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Todde, V.; Veenhuis, M.; van der Klei, I.J. Autophagy: principles and significance in health and disease. Biochim. Biophys. Acta 2009, 1792, 3–13. [Google Scholar] [PubMed]
- Bursch, W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Diff. 2001, 8, 569–581. [Google Scholar]
- Inbal, B.; Bialik, S.; Sabanay, I.; Shani, G.; Kimchi, A. DAP kinase and DRP-1 mediate membrane blebbing and the formation of autophagic vesicles during programmed cell death. J. Cell Biol. 2002, 157, 455–468. [Google Scholar]
- Liange, X.H.; Kleeman, L.K.; Jiang, H.H.; Gordon, G.; Goldman, J.E.; Berry, G.; Herman, B.; Levine, B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J. Virol. 1998, 72, 8586–8596. [Google Scholar]
- Ogier-Denis, E.; Codogno, P. Autophagy: a barrier or an adaptive response to cancer. Biochim. Biophys. Acta 2003, 1603, 113–128. [Google Scholar] [PubMed]
- Ameisen, J.C. On the origin, evolution, and nature of programmed cell death: a timeline of four billion years. Cell Death Diff. 2002, 9, 367–393. [Google Scholar] [CrossRef]
- Metzstein, M.M.; Stanfield, G.M.; Horvitz, H.R. Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet. 1998, 14, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Adams, J.M. The bcl-2 family: regulators of the cellular life-or-death switch. Nat. Rev. 2002, 2, 647–656. [Google Scholar]
- Wolf, B.B.; Green, D.R. Suicidal tendencies: apoptotic cell death by caspase family proteinases. J. Biol. Chem. 1999, 274, 20049–20052. [Google Scholar]
- Levrero, M.; De Laurenzi, V.; Costanzo, A.; Sabatini, S.; Gong, J.; Wang, J.Y.J.; Melino, G. The p53/p63/p73 family of transcription factors: overlapping and distinct functions. J. Cell Sci. 2000, 113, 1661–1670. [Google Scholar]
- Li, P.; Nijhawan, D.; Budihardjo, I.; Srinivasula, S.M.; Ahmad, M.; Alnemri, E.S.; Wang, X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 1997, 91, 479–489. [Google Scholar]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar]
- Klein, J.A.; Longo-Guess, C.M.; Rossmann, M.P.; Seburn, R.E.; Hurd, R.E.; Frankel, W.N.; Bronson, R.T.; Ackerman, S.L. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature 2002, 419, 367–374. [Google Scholar]
- Hegde, R.; Srinivasula, S.M.; Zhang, Z.; Wassell, R.; Mukattash, R.; Cilentei, L.; DuBois, G.; Lazebnik, Y.; Zervos, A.S.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 2002, 277, 4432–4538. [Google Scholar]
- Liston, P.; Roy, N.; Tamai, K.; Lefebvre, C.; Baird, S.; Cherton-Horvat, G.; Farahani, R.; McLean, M.; Ikeda, J-E.; MacKenzie, A.; Korneluk, R.G. Suppression of apoptosis in mammalian cells by NAIP and a related family of IAP genes. Nature 1996, 379, 349–353. [Google Scholar] [PubMed]
- Scorrano, L.; Oakes, S.A.; Opferman, T.J.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. Bax and Bak regulation of endoplasmic reticulum Ca2+: a control point for apoptosis. Science 2003, 300, 135–139. [Google Scholar]
- Muchmore, S.W.; Sattler, M.; Liang, H.; Meadows, R.P.; Harlan, J.E.; Yoon, H.S.; Nettesheim, D.; Chang, B.S.; Thompson, C.B.; Wong, S.L.; Fesik, S.W. X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 1999, 381, 335–341. [Google Scholar]
- Martin, L.J.; Price, A.C.; McClendon, K.B.; Al-Abdulla, N.A.; Subramaniam, J.R.; Wong, P.C.; Liu, Z. Early events of target deprivation/axotomy-induced neuronal apoptosis in vivo: oxidative stress, DNA damage, p53 phosphorylation and subcellular redistribution of death proteins. J. Neurochem. 2003, 85, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Wolter, K.G.; Hsu, Y-T.; Smith, C.L.; Nechushtan, A.; Xi, X-G.; Youle, R.L. Movement of Bax from the cytosol to mitochondria during apoptosis. J. Cell Biol. 1997, 139, 1281–1292. [Google Scholar] [PubMed]
- Nechushtan, A.; Smith, C.L.; Lamensdorf, I.; Yoon, S-H.; Youle, R.J. Bax and Bak coalesce into novel mitochondria-associated clusters during apoptosis. J. Cell Biol. 2001, 153, 1265–1276. [Google Scholar] [PubMed]
- Antonsson, B.; Conti, F.; Ciavatta, A.; Montessuit, S.; Lewis, S.; Martinou, I.; Bernasconi, L.; Bernard, A.; Mermod, J-J.; Mazzei, G.; Maundrell, K.; Gambale, F.; Sadoul, R.; Martinou, J-C. Inhibition of bax channel-forming activity by bcl-2. Science 1997, 277, 370–372. [Google Scholar] [PubMed]
- Shimizu, S.; Ide, T.; Yanagida, T.; Tsujimoto, Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 2000, 275, 12321–12325. [Google Scholar]
- Kluck, R.M.; Bossy-Wetzel, E.; Green, D.R.; Newmeyer, D.D. The release of cytochrome c from mitochondria: a primary site for bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar]
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T-I.; Jones, D.P.; Wang, X. Prevention of apoptosis by bcl-2: release of cytochrome c from mitochondria blocked. Science 1997, 275, 1129–1132. [Google Scholar]
- Vander Heiden, M.G.; Chandel, N.S.; Williamson, E.K.; Schumacker, P.T.; Thompson, C.B. Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell 1997, 91, 627–637. [Google Scholar]
- Letai, A.; Bassik, M.C.; Walensky, L.D.; Sorcinelli, M.D.; Weiler, S.; Korsmeyer, S.J. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2002, 2, 183–192. [Google Scholar]
- Wei, M.C.; Zong, W-X.; Cheng, E.H.-Y.; Lindsten, T.; Panoutsakopoulou, V.; Ross, A.J.; Roth, K.A.; MacGregor, G.R.; Thompson, C.B.; Korsmeyer, S.J. Proapoptotic Bax and Bak: a requisite gateway to mitochondrial dysfunction and death. Science 2001, 292, 727–730. [Google Scholar] [PubMed]
- Hu, Y.; Benedict, M.A.; Wu, D.; Inohara, N.; Núñez, G. Bcl-xL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc. Natl. Acad. Sci. USA 1998, 95, 4386–4391. [Google Scholar]
- Song, Q.; Kuang, Y.; Dixit, V.M.; Vincenz, C. Boo, a negative regulator of cell death, interacts with Apaf-1. EMBO J. 1999, 18, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Haldar, S.; Jena, N.; Croce, C.M. Inactivation of Bcl-2 by phosphorylation. Proc. Natl. Acad. Sci. USA 1995, 92, 4507–4511. [Google Scholar]
- Wang, H-G.; Rapp, U.R.; Reed, J.C. Bcl-2 targets the protein kinase raf-1 to mitochondria. Cell 1996, 87, 629–638. [Google Scholar] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of Bad couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar]
- del Peso, L.; Gonzalez-Garcia, M.; Page, C.; Herrera, R.; Nunez, G. Interleukin-3-induced phosphorylation of bad through the protein kinase Akt. Science 1997, 278, 687–689. [Google Scholar]
- Cardone, M.H.; Roy, N.; Stennicke, H.R.; Salvensen, G.S.; Franke, T.F.; Stanbridge, E.; Frisch, S.; Reed, J.C. Regulation of cell death protease caspase-9 by phosphorylation. Science 1998, 282, 1318–1321. [Google Scholar]
- Zha, J.; Harada, H.; Yang, E.; Jockel, J.; Korsmeyer, S.J. Serine phosphorylation of death agonist Bad in response to survival factor results in binding to 14-3-3 not Bcl-xL. Cell 1996, 87, 619–628. [Google Scholar]
- Wang, H-G.; Pathan, N.; Ethell, I.M.; Krajewski, S.; Yamaguchi, Y.; Shibasaki, F.; McKeon, F.; Bobo, T.; Franke, T.F.; Reed, J.C. Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD. Science 1999, 284, 339–343. [Google Scholar] [PubMed]
- Yang, E.; Zha, J.; Jockel, J.; Boise, L.H.; Thompson, C.B.; Korsmeyer, S.J. Bad, a heterodimeric partner for Bcl-xL and Bcl-2, displaces Bax and promotes cell death. Cell 1995, 80, 285–291. [Google Scholar] [PubMed]
- Danial, N.N.; Gramm, C.F.; Scorrano, L.; Zhang, C-Y.; Krauss, S.; Ranger, A.M.; Datta, S.R.; Greenberg, M.E.; Licklider, L.J.; Lowell, B.B.; Gygi, S.P.; Korsmeyer, S.J. Bad and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 2003, 424, 952–956. [Google Scholar] [PubMed]
- Chowdhury, I.; Tharakan, B.; Bhat, G.H. Caspases- an update. Comp. Biochem. Physiol. 2008, 151, 10–27. [Google Scholar]
- Mancini, M.; Nicholson, D.W.; Roy, S.; Thornberry, N.A.; Peterson, E.P.; Casciola-Rosen, L.A.; Rosen, A. The caspase-3 precursor has a cytosolic and mitochondrial distribution: implications for apoptotic signaling. J. Cell Biol. 1998, 140, 1485–1495. [Google Scholar]
- Krajewski, S.; Krajewska, M.; Ellerby, L.M.; Welsh, K.; Xie, Z.; Deveraus, Q.L.; Salvesen, G.S.; Bredesen, D.E.; Rosenthal, R.E.; Fiskum, G.; Reed, J.C. Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia. Proc. Natl. Acad. Sci. USA 1999, 96, 5752–5757. [Google Scholar]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An Apaf-1-cytochrome c multimeric complex is functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar]
- Li, H.; Zhu, H.; Xu, C-J.; Yuan, J. Cleavage of Bid by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [PubMed]
- Robertson, J. D.; Enoksson, M.; Suomela, M.; Zhivotovsky, B.; Orrenius, S. Caspase-2 acts upstream of mitochondria to promote cytochrome c release during etoposide-induced apoptosis. J. Biol. Chem. 2002, 277, 29803–29809. [Google Scholar] [PubMed]
- LaCasse, E.C.; Baird, S.; Korneluk, R.G.; MacKenzie, A.E. The inhibitors of apoptosis (IAPs) and their emerging role in cancer. Oncogene 1998, 17, 3247–3259. [Google Scholar]
- Holcik, M. The IAP proteins. Trends Gen. 2002, 18, 537–538. [Google Scholar]
- Deveraux, Q.L.; Roy, N.; Stennicke, H.R.; Van Arsdale, T.; Zhou, Q.; Srinivasula, S.M.; Alnemri, E.S.; Salvesen, G.S.; Reed, J.C. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998, 17, 2215–2223. [Google Scholar] [PubMed]
- Hao, Y.; Sekine, K.; Kawabata, A; Nakamura, H.; Ishioka, T.; Ohata, H.; Katayama, R.; Hashimoto, C.; Zhang, X.; Noda, T.; Tsuruo, T.; Naito, M. Apollon ubiquitinates SMAC and caspase-9, and has an essential cytoprotection function. Nat. Cell Biol. 2004, 6, 849–860. [Google Scholar] [PubMed]
- Jiang, Y.; de Bruin, A.; Caldas, H.; Fangusaro, J.; Hayes, J.; Conway, E.M.; Robinson, M.L.; Altura, A. Essential role for survivin in early brain development. J. Neurosci. 2005, 25, 6962–6970. [Google Scholar] [PubMed]
- Xu, D.G.; Korneluk, R.G.; Tamai, K.; Wigle, N.; Hakim, A.; MacKenzie, A.; Robertson, G.S. Distribution of neuronal apoptosis inhibitory protein-like immunoreactivity in the rat central nervous system. J. Comp. Neurol. 1997, 382, 247–259. [Google Scholar]
- McPhail, L.T.; Vanderluit, J.L.; McBride, C.B.; Oschipok, L.W.; crocker, S.J.; Xu, D.; Thompson, C.S.; Liston, P.; Holcik, M.; Robertson, G.S.; Tetzlaff, W. Endogenous expression of inhibitor of apoptosis proteins in facial motoneurons of neonatal and adult rats following axotomy. Neuroscience 2003, 117, 567–575. [Google Scholar]
- Roy, N.; Mahadevan, M.S.; McLean, M.; Shutler, G.; Yaraghi, Z.; Farahani, R.; Baird, S.; Besner-Johnston, A.; Lefebvre, C.; Kang, X.; Salih, M.; Aubry, H.; Tamai, K.; Guan, X.; Ioannou, P.; Crawford, T.O.; de Jong, P.J.; Surh, L.; Ikeda, J.-E.; Korneluk, R.G.; Mackenzie, A. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 1995, 80, 167–178. [Google Scholar]
- Du, C.; Fang, M.; Li, Y.; Li, L.; Wang, X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000, 102, 33–42. [Google Scholar]
- Verhagen, A.M.; Ekert, P.G.; Pakusch, M.; Silke, J.; Connolly, L.M.; Reid, G.E.; Moritz, R.L.; Simpson, R.J.; Vaux, D.L. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell 2000, 102, 43–53. [Google Scholar]
- Suzuki, Y.; Imai, Y.; Nakayama, H.; Takahashi, K; Takio, K; Takahashi, R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol. Cell 2001, 8, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Bogaerts, V.; Nuytemans, K.; Reumers, J.; Pals, R.; Engelborghs, S.; Pickut, B.; Corsmit, E.; Peeters, K.; Schymkowizt, J.; De Deyn, P.P.; Cras, P.; Rousseau, F.; Theuns, J.; Van Broeckhoven, C. Genetic variability in the mitochondrial serine protease HTRA2 contributes to risk for Parkinson’s disease. Hum. Mut. 2008, 29, 832–840. [Google Scholar]
- Simon-Sanchez, J.; Singleton, A.B. Sequencing analysis of OMI/HTRA2 shows previously reported pathogenic mutations in neurologically normal controls. Hum. Mol. Gen. 2008, 17, 1988–1993. [Google Scholar]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Constantini, P.; Loeffler, M.; Larochette, N.; Goodlett, D.R.; Aebersold, R.; Siderovski, D.P.; Penninger, J.M.; Kroemer, G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar]
- Mate, M.J.; Ortiz-Lombardia, M.; Boitel, B.; Haouz, A.; Tello, D.; Susin, S.A.; Penninger, J.; Kroemer, G.; Alzari, P.M. The crystal structure of the mouse apoptosis-inducing factor AIF. Nature Struct. Biol. 2002, 9, 442–446. [Google Scholar]
- Giaccia, A.J.; Kastan, M.B. The complexity of p53 modulation: emerging patterns from divergent signals. Genes Develop. 1998, 12, 2973–2983. [Google Scholar]
- Maki, C.G.; Huibregtse, J.M.; Howley, P.M. In vivo ubiquitination and proteasome-mediated degradation of p53. Cancer Res. 1996, 56, 2649–2654. [Google Scholar] [PubMed]
- Shieh, S-Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [PubMed]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Müllauer, F.; Böck, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins Puma and Noxa. Science 2003, 302, 1036–1038. [Google Scholar]
- Erster, S.; Moll, U.M. Stress-induced p53 runs a transcription-independent death program. Biochem Biophys. Res. Comm. 2005, 331, 843–850. [Google Scholar]
- Chipuk, J.E.; Kuwana, T.; Bouchier-hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar]
- Leu, J. I-J.; Dumont, P.; Hafey, M.; Murphy, M.E.; George, D.L. Mitochondrial p53 activates Bak and causes disruption of a bak-Mcl1 complex. Nat. Cell Biol. 2004, 6, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Aloyz, R.S.; Bamji, S.X.; Pozniak, C.D.; Toma, J.G.; Atwal, J.; Kaplan, D.R.; Miller, F.D. p53 is essential for developmental neuron death regulated by the TrkA and p75 neurotrophin receptors. J. Cell. Biol. 1998, 143, 1691–1703. [Google Scholar]
- Martin, L.J.; Kaiser, A.; Yu, J.W.; Natale, J.E.; Al-Abdulla, N.A. Injury-induced apoptosis of neurons in adult brain is mediated by p53-dependent and p53-independent pathways and requires Bax. J. Comp. Neurol. 2001, 433, 299–311. [Google Scholar] [PubMed]
- Morrison, R.S.; Kinoshita, Y.; Johnson, M.D.; Guo, W.; Garden, G.A. p53-dependent cell death signaling in neurons. Neurochem. Res. 2003, 28, 15–27. [Google Scholar]
- Pozniak, C.D.; Radinovic, S.; Yang, A.; McKeon, F.; Kaplan, D.R.; Miller, F.D. An anti-apoptotic role for the p53 family member, p73, during developmental neuron death. Science 2000, 289, 304–306. [Google Scholar] [PubMed]
- Li, L.; Liu, F.; Salmonsen, R.A.; Turner, T.K.; Litofsky, N.S.; Di Cristofano, A.; Pandolfi, P.P.; Jones, S.N.; Recht, L.D.; Ross, A.H. PTEN in neural precursor cells: regulation of migration, apoptosis, and proliferation. Mol. Cell. Neurosci. 2002, 20, 21–29. [Google Scholar] [PubMed]
- Desagher, S.; Osen-Sand, A.; Nichols, A.; Eskes, R.; Montessuit, S.; Lauper, S.; Maundrell, K.; Antonsson, B.; Martinou, J-C. Bid-induced conformational change of bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell. Biol. 1999, 144, 891–901. [Google Scholar]
- Troy, C.M.; Friedman, J. E.; Friedman, W. J. Mechanisms of p75-mediated death of hippocampal neurons. Role of caspases. J. Biol. Chem. 2002, 277, 34295–34302. [Google Scholar] [PubMed]
- Martin, L.J.; Chen, K.; Liu, Z. Adult motor neuron apoptosis is mediated by nitric oxide and Fas death receptor linked by DNA damage and p53 activation. J. Neurosci. 2005, 25, 6449–6459. [Google Scholar] [CrossRef] [PubMed]
- Lucas, D.R.; Newhouse, J.P. The toxic effect of sodium L-glutamate on the inner layers of the retina. Arch. Ophthal. 1957, 58, 193–201. [Google Scholar]
- Olney, J.W. Glutamate-induced neuronal necrosis in the infant mouse hypothalamus. An electron microscopic study. J. Neuropathol. Exp. Neurol. 1971, 30, 75–90. [Google Scholar]
- Choi, D.W. Excitotoxic cell death. J. Neurobiol. 1992, 23, 1261–1276. [Google Scholar]
- Martin, L.J. The apoptosis-necrosis cell death continuum in CNS development, injury and disease: contributions and mechanisms. In Neuroprotection; Lo, E.H., Marwah, J., Eds.; Prominent Press: Scottsdale, AR, USA, 2002; pp. 379–412. [Google Scholar]
- Martin, L.J.; Sieber, F.E.; Traystman, R.J. Apoptosis and necrosis occur in separate neuronal populations in hippocampus and cerebellum after ischemia and are associated with alterations in metabotropic glutamate receptor signaling pathways. J. Cereb. Blood Flow. Metab. 2000, 20, 153–167. [Google Scholar]
- Martin, L.J. Excitotoxicity. In Encyclopedia of Neuroscience; Adelman, G., Smith, B.H., Eds.; Elsevier Science: Amsterdam, The Netherland, 2004; CD-ROM. [Google Scholar]
- Reynolds, I.J. Mitochondrial membrane potential and the permeability transition in excitotoxicity. Ann. N.Y. Acad. Sci. 1999, 893, 33–41. [Google Scholar]
- Sonkusare, S.K.; Kaul, C.L.; Ramarao, P. Dementia of Alzheimer’s disease and other neurodegenerative disorders- memantine, a new hope. Pharma. Res. 2005, 51, 1–17. [Google Scholar]
- Gwag, B.J.; Koh, J.Y.; DeMaro, J.A.; Ying, H.S.; Jacquin, M.; Choi, D.W. Slowly triggered excitotoxicity occurs by necrosis in cortical cultures. Neuroscience 1997, 77, 393–401. [Google Scholar]
- Kure, S.; Tominaga, T.; Yoshimoto, T.; Tada, K.; Narisawa, K. Glutamate triggers internucleosomal DNA cleavage in neuronal cells. Biochem. Biophys. Res. Commun. 1991, 179, 39–45. [Google Scholar]
- Ankarcrona, M.; Dypbukt, J.M.; Bonfoco, E.; Zhivotovsky, B.; Orrenius, S.; Lipton, S.A.; Nicotera, P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron 1995, 15, 961–973. [Google Scholar]
- Simonian, N.A.; Getz, R.L.; Leveque, J.C.; Konradi, C.; Coyle, J. T. Kainate induces apoptosis in neurons. Neuroscience 1996, 74, 675–683. [Google Scholar]
- Dessi, F.; Charriaut-Marlangue, C.; Khrestchatisky, M.; Ben-Ari, Y. Glutamate-induced neuronal death is not a programmed cell death in cerebellar culture. J. Neurochem. 1993, 60, 1953–1955. [Google Scholar]
- Xiang, H.; Kinoshita, Y.; Knudson, C.M.; Korsmeyer, S.J.; Schwartzkroin, P.A.; Morrison, R.S. Bax involvement in p53-mediated neuronal cell death. J. Neurosci. 1998, 18, 1363–1373. [Google Scholar]
- Miller, T.M.; Moulder, K.L.; Knudson, C.M.; Creedon, D.J.; Deshmukh, M.; Korsmeyer, S.J.; Johnson, E.M., Jr. Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J. Cell. Biol. 1997, 139, 205–217. [Google Scholar]
- Dargusch, R.; Piasecki, D.; Tan, S.; Liu, Y.; Schubert, D. The role of Bax in glutamate-induced nerve cell death. J. Neurochem. 2001, 76, 295–301. [Google Scholar]
- Johnson, M.D.; Kinoshita, Y.; Xiang, H.; Ghatan, S.; Morrison, R.S. Contribution of p53-dependent caspase activation to neuronal cell death declines with neuronal maturation. J. Neurosci. 1999, 19, 2996–3006. [Google Scholar]
- Tenneti, L.; Lipton, S.A. Involvement of activated caspase-3-like proteases in N-methyl-D-aspartate-induced apoptosis in cerebrocortical neurons. J. Neurochem. 2001, 74, 134–142. [Google Scholar] [CrossRef]
- Simons, M.; Beinroth, S.; Gleichmann, M.; Liston, P.; Korneluk, R.G.; MacKenzie, A.E.; Bahr, M.; Klockgether, T.; Robertson, G.S.; Weller, M.; Schulz, J.B. Adenovirus-mediated gene transfer of inhibitors of apoptosis proteins delays apoptosis in cerebellar granule neurons. J. Neurochem. 1999, 72, 292–301. [Google Scholar]
- van Lookeren Campagne, M.; Lucassen, P.J.; Vermeulen, J.P.; Balázs, R. NMDA and kainate induced internucleosomal DNA cleavage associated with both apoptotic and necrotic cell death in the neonatal rat brain. Eur. J. Neurosci. 1995, 7, 1627–1640. [Google Scholar]
- Holcik, M.; Thompson, C.S.; Yaraghi, Z.; Lefebvre, C.A.; MacKenzie, A.E.; Korneluk, R.G. The hippocampal neurons of neuronal apoptosis inhibitory protein 1 (NAIP1)-deleted mice display increased vulnerability to kainic acid-induced injury. Proc. Natl. Acad. Sci. USA 1999, 97, 2286–2290. [Google Scholar]
- Schreiber, S.S.; Tocco, G.; Najm, I.; Thompson, R.F.; Baudry, M. Cycloheximide prevents kainate-induced neuronal death and c-fos expression in adult rat brain. J. Mol. Neurosci. 1993, 4, 149–159. [Google Scholar]
- Leppin, C.; Finiels-Marlier, F.; Crawley, J.N.; Montpied, P.; Paul, S.M. Failure of a protein synthesis inhibitor to modify glutamate receptor-mediated neurotoxicity in vivo. Brain Res. 1992, 581, 168–170. [Google Scholar]
- Lok, J.; Martin, L.J. Rapid subcellular redistribution of Bax precedes caspase-3 and endonuclease activation during excitotoxic neuronal apoptosis in rat brain. J. Neurotrauma 2002, 19, 815–828. [Google Scholar]
- Mueller, D.; Shamblott, M.J.; Fox, H.F.; Gearhart, J.D.; Martin, L.J. Transplanted human embryonic germ cell-derived neural stem cells replace neurons and oligodendrocytes in the forebrain of neonatal mice with excitotoxic brain damage. J. Neurosci. Res. 2005, 82, 592–608. [Google Scholar]
- Al-Abdulla, N.A.; Portera-Cailliau, C.; Martin, L.J. Occipital cortex ablation in adult rat causes retrograde neuronal death in the lateral geniculate nucleus that resembles apoptosis. Neuroscience 1998, 86, 191–209. [Google Scholar]
- Yang, Y.; Xie, Y.; Chai, H.; Fan, M.; Liu, S.; Liu, H.; Bruce, I.; Wu, W. Microarray analysis of gene expression patterns in adult spinal motoneurons after different types of axonal injuries. Brain Res. 2006, 1075, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Al-Abdulla, N.A.; Martin, L.J. Apoptosis of retrogradely degenerating neurons occurs in association with the accumulation of perikaryal mitochondria and oxidative damage to the nucleus. Am. J. Pathol. 1998, 153, 447–456. [Google Scholar]
- Fujikawa, D.G. Confusion between neuronal apoptosis and activation of programmed cell death mechanisms in acute necrotic insults. Trends Neurosci. 2000, 23, 410–411. [Google Scholar]
- Ishimaru, M.J.; Ikonomidou, C.; Tenkova, T.I.; Der, T.C.; Dikranian, K; Sesma, M.A.; Olney, J.W. Distinguishing excitotoxic from apoptotic neurodegeneration in the developing rat brain. J. Comp. Neurol. 1999, 408, 461–476. [Google Scholar] [CrossRef] [PubMed]
- Sloviter, R.S. Apoptosis: a guide for the perplexed. Trends Pharmacol. Sci. 2002, 23, 19–24. [Google Scholar]
- Baille, V.; Clarke, P.G.; Brochier, G.; Dorander, F.; Verna, J.M.; Four, E.; Lallement, G.; Carpentier, P. Soman-induced convulsions: the neuropathology revisited. Toxicology 2005, 215, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, X.; Xu, F.; Bahr, B.A.; Shibata, M.; Uchiyama, Y.; Hagberg, H.; Blomgren, K. The influence of age on apoptotic and other mechanisms of cell death after cerebral hypoxia-ischemia. Cell Death Differ. 2005, 12, 162–176. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Hall, J.J.; Noble, L.J.; Ferriero, D.M. Delayed cell death in neonatal mouse hippocampus from hypoxia-ischemia is neither apoptotic nor necrotic. Neurosci. Lett. 2001, 304, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Ying, D.J.; Cui, L.; Lungsdorf, J.; Yu, S.P. Necrosis, apoptosis and hybrid death in the cortex and thalamus after barrel cortex ischemia in rats. Brain Res. 2004, 1022, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J. Neuronal death in amyotrophic lateral sclerosis is apoptosis: possible contribution of a programmed cell death mechanism. J. Neuropathol. Exp. Neurol. 1999, 58, 459–471. [Google Scholar]
- Zoccolella, S.; Santamato, A.; Lamberti, P. Current and emerging treatments for amyotrophic lateral sclerosis. Neuropsychiatr. Dis. Treat. 2009, 5, 577–595. [Google Scholar]
- Katzman, R. Education and the prevalence of dementia and Alzheimer’s disease. Neurology 1993, 43, 13–20. [Google Scholar]
- Evans, D.A.; Funkenstein, H.H.; Albert, M.S.; Scherr, P.A.; Cook, N.R.; Chown, M.J.; Hebert, L.E.; Hennekens, C.H.; Taylor, J.O. Prevalence of Alzheimer’s disease in a community population of older persons. Higher than previously reported. JAMA 1989, 262, 2551–2556. [Google Scholar] [PubMed]
- McKhann, G.; Drachman, D.; Folstein, M.; Katzman, R.; Price, D.; Stadlan, E.M. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA work group under the auspices of the Department of Health and Human Services task force on Alzheimer’s disease. Neurology 1984, 34, 939–944. [Google Scholar]
- Olshansky, S.J.; Carnes, B.A.; Cassel, C.K. The aging of the human species. Sci. Am. 1993, 268, 46–52. [Google Scholar]
- Minati, L.; Edginton, T.; Bruzzone, M.G.; Giaccone, G. Current concepts in Alzheimer's disease: A multidisciplinary review. Am. J. Alz. Dis. Other Demen. 2009, 24, 95–121. [Google Scholar]
- Chartier-Harlin, M.-C.; Crawford, F.; Houlden, H.; Warren, A.; Hughes, D.; Fidani, L.; Goate, A.; Rossor, M.; Roques, P.; Hardy, J.; Mullan, M. Early-onset Alzheimer's disease caused by mutations at codon 717 of the β-amyloid precursor protein gene. Nature 1991, 353, 844–846. [Google Scholar]
- Tilley, L.; Morgan, K.; Kalsheker, N. Genetic risk factors for Alzheimer’s disease. J. Clin. Pathol. Mol. Pathol. 1998, 51, 293–304. [Google Scholar]
- Goate, A.; Chartier-Harlin, M.-.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; Mant, R.; Newton, P.; Rooke, K.; Roques, P.; Talbot, C.; Pericak-Vance, M.; Roses, A.; Williamson, R.; Rossor, M.; Owen, M.; Hardy, J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature 1991, 349, 704–706. [Google Scholar] [PubMed]
- Naruse, S.; Igarashi, S.; Kobayashi, H.; Aoki, K.; Inuzuka, T.; Kaneko, K.; Shimizu, T.; Iihara, K.; Kojima, T.; Miyatake, T.; Tsuji, S. Mis-sense mutation Val->Ile in exon 17 of amyloid precursor protein gene in Japanese familial Alzheimer's disease. Lancet 1991, 337, 978–979. [Google Scholar]
- Campion, D.; Flaman, J.M.; Brice, A.; Hannequin, D.; Dubois, B.; Martin, C.; Moreau, V.; Charbonnier, F.; Didierjean, O.; Tardieu, S.; Penet, C.; Puel, M.; Pasquier, F.; Ledoze, F.; Bellis, G.; Calenda, A.; Heilig, R.; Martinez, M.; Mallet, J.; Bellis, M.; Clergetdarpoux, F.; Agid, Y.; Frebourg, T. Mutations of the presenilin 1 gene in families with early-onset Alzheimer's disease. Hum. Mol. Genet. 1995, 4, 2373–2377. [Google Scholar]
- Sherrington, R.; Rogaev, E.I.; Liang, Y.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Chi, H.; Lin, C.; Li, G.; Holman, K.; Tsuda, T.; Mar, L.; Foncin, J.-F.; Bruni, A.C.; Montesi, M.P.; Sorbi, S.; Rainero, I.; Pinessi, L.; Nee, L.; Chumakov, I.; Pollen, D.; Brookes, A.; Sanseau, P.; Polinsky, R.J.; Wasco, W.; Da Silva, H.A.R.; Haines, J.L.; Pericak-Vance, M.A.; Tanzi, R.E.; Roses, A.D.; Fraser, P.E.; Rommens, J.M.; St George-Hyslop, P.H. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature 1995, 375, 754–760. [Google Scholar]
- Kalaria, R.N. Dementia comes of age in the developing world. Lancet 2003, 361, 888–889. [Google Scholar]
- Roses, A.D. Apolipoprotein E alleles as risk factors in Alzheimer's disease. Annu. Rev. Med. 1996, 47, 387–400. [Google Scholar]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; DeLong, M.R. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar]
- Gomez-Isla, T.; Price, J.L.; McKeel, D.W., Jr.; Morris, J.C.; Growdon, J.H.; Hyman, B.T. Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J. Neurosci. 1996, 16, 4491–4500. [Google Scholar]
- Mouton, P.R.; Martin, L.J.; Calhoun, M.E.; Dal Forno, G.; Price, D.L. Cognitive decline strongly correlates with cortical atrophy in Alzheimer’s disease. Neurobiol. Aging 1998, 19, 371–377. [Google Scholar] [CrossRef] [PubMed]
- West, M.J.; Kawas, C.H.; Martin, L.J.; Troncoso, J.C. The CA1 region of the human hippocampus is a hot spot in Alzheimer’s disease. Ann. N.Y. Acad. Sci. 2000, 908, 255–259. [Google Scholar] [PubMed]
- Pelvig, D.P.; Pakkenberg, H.; Regeur, L.; Oster, S.; Pakkenberg, B. Neocortical glial cell numbers in Alzheimer's disease. A stereological study. Dement. Geriatr. Cogn. Disord. 2003, 16, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological staging of Alzheimer’s disease-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar]
- Kermer, P.; Liman, J; Weishaupt, J.H.; Bahr, M. Neuronal apoptosis in neurodegenerative diseases: from basic research to clinical application. Neurodeg. Dis. 2004, 1, 9–19. [Google Scholar]
- Smale, G.; Nichols, N.R.; Brady, D.R.; Finch, C.E.; Horten, W.E., Jr. Evidence for apoptotic cell death in Alzheimer's disease. Exp. Neurol. 1995, 133, 225–230. [Google Scholar]
- Anderson, A.J.; Su, J.H.; Cotman, C.W. DNA damage and apoptosis in Alzheimer's disease: colocalization with c-jun immunoreactivity, relationship to brain area, and effect of postmortem delay. J. Neurosci. 1996, 16, 1710–1719. [Google Scholar]
- Adamec, E.; Vonsattel, J.P.; Nixon, R.A. DNA strand breaks in Alzheimer's disease. Brain Res. 1999, 849, 67–77. [Google Scholar]
- Stadelmann, C.; Deckwerth, T.L.; Srinivasan, A.; Bancher, C.; Brock, W.; Lassmann, H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer's disease. Evidence for apoptotic cell death. Am. J. Pathol. 1999, 155, 1459–1466. [Google Scholar] [CrossRef] [PubMed]
- Lucassen, P.J.; Chung, W.C.J.; Kamphorst, W.; Swaab, D.F. DNA damage distribution in the human brain as shown by in situ end labeling; area-specific differences in aging and Alzheimer's disease in the absence of apoptotic morphology. J. Neuropathol. Exp. Neurol. 1997, 56, 887–900. [Google Scholar]
- Su, J.H.; Zhao, M.; Anderson, A.J.; Srinivasan, A.; Cotman, C.W. Activated caspase-3 expression in Alzheimer's and aged control brain: correlation with Alzheimer pathology. Brain Res. 2001, 898, 350–357. [Google Scholar]
- Kitamura, Y.; Shimohama, S.; Kamoshima, W.; Ota, T.; Matsuoka, Y.; Nomura, Y.; Smith, M.A.; Perry, G.; Whitehouse, P.J.; Taniguchi, T. Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP32, in Alzheimer's disease. Brain Res. 1998, 780, 260–269. [Google Scholar] [CrossRef] [PubMed]
- Gastard, M.C.; Troncoso, J.C.; Koliatsos, V.E. Caspase activation in the limbic cortex of subjects with early Alzheimer's disease. Ann. Neurol. 2003, 54, 393–398. [Google Scholar]
- Pompl, P.N.; Yemul, S.; Xiang, Z.; Ho, L.; Haroutunian, V.; Purohit, D.; Mohs, R.; Pasinetti, G.M. Caspase gene expression in the brain as a function of the clinical progression of Alzheimer's disease. Arch. Neurol. 2003, 60, 369–376. [Google Scholar]
- Rohn, T.T.; Rissman, R.A.; Davis, M.C.; Kim, Y.E.; Cotman, C.W.; Head, E. Caspase-9 activation and caspase cleavage of tau in the Alzheimer's disease brain. Neurobiol. Dis. 2002, 11, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Albrecht, S.; Bourdeau, M.; Petzke, T.; Bergeron, C.; LeBlanc, A.C. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am. J. Pathol. 2004, 165, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Selznick, L.A.; Holtzman, D.M.; Han, B.H.; Gokden, M.; Srinivasan, A.N.; Johnson, E.M., Jr.; Roth, K.A. In situ immunodetection of neuronal caspase-3 activation in Alzheimer's disease. J. Neuropath. Exp. Neurol. 1999, 58, 1020–1026. [Google Scholar] [CrossRef]
- Lesuisse, C.; Martin, L.J. Immature and mature neurons engage different apoptotic mechanisms involving caspase-3 and the mitogen-activated protein kinase pathway. J. Cereb. Blood Flow Metabol. 2002, 22, 935–950. [Google Scholar]
- Shimohama, S.; Tanino, H.; Fujimoto, S. Differential subcellular localization of caspase family protein in the adult rat brain. Neurosci. Lett. 2001, 315, 125–128. [Google Scholar]
- Yan, X.X.; Najbauer, J.; Woo, C.C.; Dashtipour, K.; Ribak, C.E.; Leon, M. Expression of active caspase-3 in mitotic and postmitotic cells or rat forebrain. J. Comp. Neurol. 2001, 433, 4–22. [Google Scholar]
- Troncoso, J.C.; Cataldo, A.M.; Nixon, R.A.; Barnett, J.L.; Lee, M.K.; Checler, F.; Fowler, D.R.; Smialek, J.E.; Crain, B.; Martin, L.J.; Kawas, C.H. Neuropathology of preclinical and clinical late-onset Alzheimer’s disease. Ann. Neurol. 1998, 43, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar]
- Kosik, K.S.; Joachim, C.L.; Selkoe, D.J. Microtubule-associated protein tau is a major antigenic component of paired helical filaments in Alzheimer's disease. Proc. Natl. Acad. Sci. USA 1986, 83, 4044–4048. [Google Scholar]
- Sze, C.-I.; Bi, H.; Kleinschmidt-DeMasters, B.K.; Filley, C.M.; Martin, L.J. N-Methyl-D-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer’s disease. J. Neurol. Sci. 2001, 182, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Kemp, J.A.; McKernan, R.M. NMDA receptor pathways as drug targets. Nat. Neurosci. 2002, 5, 1039–1042, (suppl). [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Cheng, B.; Davis, D.; Bryant, K.; Lieberburg, I.; Rydel, R.E. β-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J. Neurosci. 1993, 12, 376–389. [Google Scholar]
- Reddy, P.H.; Beal, M.F. Amyloid beta, mitochondrial dysfunction and synaptic damage: implications for cognitive decline in aging and Alzheimer’s disease. Trends Mol. Med. 2008, 14, 45–53. [Google Scholar]
- DeKosky, S.T.; Scheff, S.W. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann. Neurol. 1990, 27, 457–464. [Google Scholar]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katsman, R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar]
- Martin, L.J.; Pardo, C.A.; Cork, L.C.; Price, D.L. Synaptic pathology and glial responses to neuronal injury precede the formation of senile plaques and amyloid deposits in the aging cerebral cortex. Am. J. Pathol. 1994, 145, 1358–1381. [Google Scholar]
- Sze, C.-I.; Troncoso, J.C.; Kawas, C.; Mouton, P.; Price, D.L.; Martin, L.J. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 1997, 56, 933–994. [Google Scholar]
- Selkoe, D.J. Alzheimer's disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar]
- Yankner, B.A.; Dawes, L.R.; Fisher, S.; Villa-Komaroff, L.; Oster-Granite, M.L.; Neve, R.L. Neurotoxicity of a fragment of the amyloid precursor associated with Alzheimer's disease. Science 1989, 245, 417–420. [Google Scholar]
- Younkin, S.G. Evidence that Abeta 42 is the real culprit in Alzheimer's disease. Ann. Neurol. 1995, 37, 287–288. [Google Scholar]
- Fath, T.; Eidenmuller, J.; Brandt, R. Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer's disease. J. Neurosci. 2002, 22, 9733–9741. [Google Scholar]
- Rapoport, M.; Dawson, H.N.; Binder, L.I.; Vitek, M.P.; Ferreira, A. Tau is essential to β-amyloid-induced neurotoxicity. Proc. Natl. Acad. Sci. USA 2002, 99, 6364–6369. [Google Scholar]
- Anandatheerthavarada, H.K.; Biswas, G.; Robin, M.A.; Avadhani, N.G. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J. Cell Biol. 2003, 161, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef] [PubMed]
- Manczak, M.; Anekonda, T.S.; Henson, E.; Park, B.S.; Quinn, J.; Reddy, P.H. Mitochondria are a direct site of Aβ accumulation in Alzheimer’s disease neurons: implications for free radical generation and oxidative damage in disease progression. Hum. Mol. Gen. 2006, 15, 1437–1449. [Google Scholar] [CrossRef]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Aβ: a potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [PubMed]
- Lustbader, J.W.; Cirilli, M.; Lin, C.; Xu, H.W.; Takuma, K.; Wang, N.; Caspersen, C.; Chen, X.; Pollak, S.; Chaney, M.; Trinchese, F.; Liu, S.; Gunn-Moore, F.; Lue, L.-F.; Walker, D.G.; Kuppsamy, P.; Zewier, Z.L.; Arancio, O.; Stern, D.; Yan, S.S.; Wu, H. ABAD directly links Aβ to mitochondrial toxicity in Alzheimer’s disease. Science 2004, 304, 448–452. [Google Scholar] [PubMed]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D.; Gunn-Moore, F.J.; Vonsattel, J.P.; Aranico, O.; Chen, J.X.; Yan, S.D. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar]
- Duyckaerts, C.; Potier, M.C.; Delatour, B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008, 115, 5–38. [Google Scholar] [PubMed]
- Irizarry, M.C.; McNamara, M.; Fedorchak, K.; Hsiao, K.; Hyman, B.T. APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J. Neuropathol. Exp. Neurol. 1997, 56, 965–973. [Google Scholar]
- Irizarry, M.C.; Soriano, F.; McNamara, M.; page, K.J.; Schenk, D.; Games, D.; Hyman, B.T. Aβ deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyoid precursor protein V717F (PDAPP) transgenic mouse. J. Neurosci. 1997, 17, 7053–7059. [Google Scholar] [PubMed]
- Takeuchi, A.; Irizarry, M.C.; Duff, K.; Saido, T.C.; Hsiao-Ashe, K.; Hasegawa, M.; Mann, D.M.A.; Hyman, B.T.; Iwatsubo, T. Age-related amyloid β deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid β precursor protein Swedish mutant is not associated with global neuronal loss. Am. J. Pathol. 2000, 157, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Calhoun, M.E.; Wiederhold, K.H.; Abramowski, D.; Phinney, A.L.; Probst, A.; Sturchler-Pierrat, C.; Staufenbiel, M.; Sommer, B.; Jucker, M. Neuron loss in APP transgenic mice. Nature 1998, 395, 755–756. [Google Scholar]
- Selznick, L.A.; Holtzman, D.M.; Han, B.H.; Gokden, M.; Srinivasan, A.N.; Johnson, E.M., Jr.; Roth, K.A. In situ immunodetection of neuronal caspase-3 activation in Alzheimer's disease. J. Neuropath. Exp. Neurol. 1999, 58, 1020–1026. [Google Scholar]
- Yang, D.-S.; Kumar, A.; Stavrides, P.; Peterson, J.; Peterhoff, C.M.; Pawlik, M.; Levy, E.; Cataldo, A.M.; Nixon, R.A. Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer’s disease. Am. J. Pathol. 2008, 173, 665–681. [Google Scholar] [CrossRef] [PubMed]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar]
- LaFerla, F.M.; Hall, C.K.; Ngo, L.; Jay, G. Extracellular deposition of β-amyloid upon p53-dependent neuronal cell death in transgenic mice. J. Clin. Invest. 1996, 98, 1626–1632. [Google Scholar]
- Zhang, Y.; McLaughlin, R.; Goodyer, C.; LeBlanc, A. Selective cytotoxicity of intracellular amyloid β peptide1-42 through p53 and Bax in cultured primary human neurons. J. Cell Biol. 2002, 156, 519–529. [Google Scholar]
- Loo, D.T.; Copani, A.; Pike, C.J.; Whittemore, E.R.; Walencewicz, A.J.; Cotman, C.W. Apoptosis is induced by β-amyloid in cultured central nervous system neurons. Proc. Natl. Acad. Sci. USA 1993, 90, 7951–7955. [Google Scholar]
- Behl, C.; Davis, J.B.; Klier, F.G.; Schubert, D. Amyloid beta peptide induces necrosis rather than apoptosis. Brain Res. 1994, 645, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Casley, C.S.; Land, J.M.; Sharpe, M.A.; Clark, J.B.; Duchen, M.R.; Canevari, L. β-amyloid fragment 25-35 causes mitochondrial dysfunction in primary cortical neurons. Neurobiol. Dis. 2002, 10, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Duker, N.J.; Sperling, J.; Soprano, K.J.; Druin, D.P.; Davis, A.; Ashworth, R. β-Amyloid protein induces the formation of purine dimers in cellular DNA. J. Cell Biochem. 2001, 81, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Kuperstein, F; Yavin, E. ERK activation and nuclear translocation in amyloid-beta peptide- and iron-stressed neuronal cell cultures. Eur. J. Neurosci. 2002, 16, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Morishima, Y.; Gotoh, Y.; Zieg, J.; Barrett, T.; Takano, H.; Flavell, R.; Davis, R.J.; Shirasaki, Y.; Greenberg, M.E. β-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J. Neurosci. 2001, 21, 7551–7560. [Google Scholar] [PubMed]
- Yaar, M.; Zhai, S.; Fine, R.E.; Eisenhauer, P.B.; Arble, B.L.; Stewart, K.B.; Gilchrest, B.A. Amyloid β binds trimers as well as monomers of the 75-kDa neurotrophin receptor and activates receptor signaling. J. Biol. Chem. 2002, 277, 7720–7725. [Google Scholar]
- Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar]
- Ethell, D.W.; Kinloch, R.; Green, D.R. Metalloproteinase shedding of Fas ligand regulates β-amyloid neurotoxicity. Curr. Biol. 2002, 12, 1595–1600. [Google Scholar]
- Culmsee, C.; Zhu, X.; Yu, Q.S.; Chan, S.L.; Camandola, S.; Guo, Z.; Greig, N.H.; Mattson, M.P. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J. Neurochem. 2001, 77, 220–228. [Google Scholar]
- Ma, L.; Ohyagi, Y.; Miyoshi, K.; Sakae, N.; Motomaura, K.; Taniwaki, T.; Furuya, H.; Takeda, K.; Tabira, T.; Kira, J. Increase in p53 protein levels by presenilin 1 gene mutations and its inhibition by secretase inhibitors. J. Alz. Dis. 2009, 16, 565–575. [Google Scholar]
- Giovanni, A.; Keramaris, E.; Morris, E.J.; Hou, S.T.; O'Hare, M.; Dyson, N.; Robertson, G.S.; Slack, R.S.; Park, D.S. E2F1 mediates death of β-amyloid-treated cortical neurons in a manner independent of p53 and dependent on Bax and caspase 3. J. Biol. Chem. 2000, 275, 11553–11560. [Google Scholar]
- Paradis, E.; Douillard, H.; Koutroumanis, M.; Goodyer, C.; LeBlanc, A. Amyloid β peptide of Alzheimer's disease downregulates Bcl-2 and upregulates Bax expression in human neurons. J. Neurosci. 1997, 16, 7533–7539. [Google Scholar]
- Weidemann, A.; Paliga, K.; Durrwang, U.; Reinhard, F.B.M.; Schuckert, O.; Evin, G.; Masters, C.L. Proteolytic processing of the Alzheimer’s disease amyloid precursor protein within its cytoplasmic domain by caspase-like proteases. J. Biol. Chem. 1999, 274, 5823–5829. [Google Scholar] [PubMed]
- LeBlanc, A.; Liu, H.; Goodyer, C.; Bergeron, C.; Hammond, J. Caspase-6 role in apoptosis of human neurons, amyloidogenesis, and Alzheimer’s disease. J. Biol. Chem. 1999, 274, 23426–23436. [Google Scholar] [PubMed]
- Gervais, F.G.; Xu, D.; Robertson, G.S.; Vaillancourt, J.P.; Zhu, Y.; Huang, J.; LeBlanc, A.; Smith, D.; Rigby, M.; Shearman, M.S.; Clarke, E.E.; Zheng, H.; van der Ploeg, L.H.; Ruffolo, S.C.; Thornberry, N.A.; Xanthoudakis, S.; Zamboni, R.J.; Roy, S.; Nicholson, D.W. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 1999, 97, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.M.; Tessier-Lavign, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar]
- Passer, B.J.; Pellegrini, L.; Vito, P.; Ganjei, J.K.; D'Adamio, L. Interaction of Alzheimer’s presenilin-1 and presenilin-2 with Bcl-X(L). A potential role in modulating the threshold of cell death. J. Biol. Chem. 1999, 274, 24007–24013. [Google Scholar] [PubMed]
- Kim, T.-W.; Pettingell, W.H.; Jung, Y.-K.; Kovacs, D.M.; Tazi, R.E. Alternative cleavage of Alzheimer-associated presenilins during apoptosis by a caspase-3 family protein. Science 1997, 277, 373–376. [Google Scholar] [PubMed]
- Bursztajn, S.; DeSouza, R.; McPhie, D.L.; Berman, S.A.; Shioi, J.; Robakis, N.K.; Neve, R.L. Overexpression in neurons of human presenilin-1 or a presenilin-1 familial Alzheimer disease mutant does not enhance apoptosis. J. Neurosci. 1998, 18, 9790–9799. [Google Scholar]
- Gamliel, A.; Teicher, C.; Hartmann, T.; Beyreuther, K.; Stein, R. Overexpression of wild-type presenilin 2 or its familial Alzheimer's disease-associated mutant does not induce or increase susceptibility to apoptosis in different cells. Neuroscience 2003, 117, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.L.; Culmsee, C.; Haughey, N.; Klapper, W.; Mattson, M.P. Presenilin-1 mutations sensitize neurons to DNA damage-induced death by a mechanism involving perturbed calcium homeostasis and activation of calpains and caspase-12. Neurobiol. Dis. 2002, 11, 2–19. [Google Scholar]
- Van Den Eeden, S.K.; Tanner, C.M.; Bernstein, A.L.; Fross, R.D.; Leimpeter, A.; Bloch, D.A.; Nelson, L.M. Indicence of Parkinson's disease: variations by age, gender and race ethnicity. Am. J. Epidemol. 2003, 157, 1015–1022. [Google Scholar]
- Jankovic, J. Parkinson’s disease: clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar]
- Lowe, J.; Lennox, G.; Leigh, P.N. Disorders of movement and system degeneration. In Greenfields Neuropathology; Graham, D.I., Lantos, P.L., Eds.; London: Arnold, 1997; pp. 281–366. [Google Scholar]
- Braak, H.; Del Tredici, K.; Rüb, U.; d Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H.V. Etiology of Parkinson's disease. Neurology 2006, 66 (Suppl. 4), S10–S23. [Google Scholar] [PubMed]
- Ascherio, A.; Chen, H.; Weisskopf, M.G.; O’Reilly, E.; McCullough, M.L.; Calle, E.E.; Schwarzchild, M.A.; Thun, M.J. Pesticide exposure and risk for Parkinson’s disease. Ann. Neurol. 2006, 60, 197–203. [Google Scholar]
- Krüger, R.; Kuhn, W.; Müller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Reiss, O. Ala30Pro mutation in the gene encoding α-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; Stenroos, E.S.; Chandrasekharappa, S.; Athanassiadou, A.; Papapetropoulos, T.; Johnson, W.G.; Lazzarini, A.M.; Duvoisin, R.C.; Di Iorio, G.; Golbe, L.I.; Nussbaum, R.L. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; Lincoln, S.; Crawley, A.; Hanson, M.; Maraganore, D.; Adler, C.; Cookson, M.R.; Muenter, M.; Baptista, M.; Miller, D.; Blancato, J.; Hardy, J.; Gwinn-Hardy, K. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [PubMed]
- Leroy, E.; Boyer, R.; Auburger, G.; Leube, B.; Ulm, G.; Mezey, E.; Harta, G.; Browstein, M.J.; Jonnalagada, S.; Chernova, T.; Dehejia, A.; Lavedan, C.; Gasser, T.; Steinbach, P.J.; Wilkinson, K.D.; Polymeropoulos, M.H. The ubiquitin pathway in Parkinson's disease. Nature 1998, 395, 451–452. [Google Scholar]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; van Dongen, J.W.; Vanacore, N.; van Swieten, J.C.; Brice, A.; Meco, G.; van Duijn, C.M.; Oostra, B.A.; Heutink, P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar]
- Hatano, Y.; Li, Y.; Sato, K.; Asakawa, S.; Yamamura, Y.; Tomiyama, H.; Yoshino, H.; Asahina, M.; Kobayashi, S.; Hassin-Baer, S.; Lu, C.S.; Ng, A.R.; Rosales, P.L.; Shimizu, N.; Toda, T.; Mizuno, Y.; Hattori, N. Novel PINK1 mutations in early-onset parkinsonism. Ann. Neurol. 2004, 56, 424–427. [Google Scholar]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, A.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; Albanese, A.; Nussbaum, R.; Gonzalez-Maldonado, R.; Deller, T.; Salvi, S.; Cortelli, P.; Gilks, W.P.; Latchman, D.S.; Harvey, R.J.; Dallapiccola, B.; Auburger, G.; Wood, N.W. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar]
- Paisán-Ruίz, C.; Jain, S.; Whitney Evans, E.; Gilks, W.P.; Simón, J.; van der Brug, M.; López de Munain, A.; Aparicio, S.; Martίnez-Gil, A.; Khan, N.; Johnson, J.; Ruiz Martinez, J.; Nicholl, D.; Marti Carrera, I.; Saénz Peňa, A.; de Silva, R.; Lees, A.; Martί-Massó, J.F.; Pérez-Tur, J.; Wood, N.W.; Singleton, A.B. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron 2004, 44, 595–600. [Google Scholar] [PubMed]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; Stoessi, J.; Pfeiffer, R.F.; Patenge, N.; Carballo Carbajal, I.; Vieregge, P.; Asmus, F.; Müller-Myhsok, B.; Dickson, D.W.; Meitinger, T.; Storm, T.M.; Wszolek, Z.K.; Gasser, T. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 2004, 44, 601–607. [Google Scholar]
- Ramirez, A.; Heimbach, A.; grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A,F.; Wriekat, A.-L.; Roeper, J.; Al-Din, A.; Hillmer, A.M.; Karsak, M.; Liss, B.; Woods, C.G.; Behrens, M.I. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Gen. 2006, 38, 1184–1191. [Google Scholar]
- Lesuisse, C.; Martin, L.J. Long-term culture of mouse cortical neurons as a model for neuronal development, aging, and death. J. Neurobiol. 2002, 51, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: a neuron-specific protein localized to the nucleus and presynaptic nerve terminals. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar]
- Murphy, D.D.; Rueter, S.M.; Trojanowski, J.Q.; Lee, V.M.Y. Synucleins are developmentally expressed, and α-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J. Neurosci. 2000, 20, 3214–3220. [Google Scholar]
- Nakamura, K.; Nemani, V.M.; Wallender, E.K.; Kaehlcke, K.; Ott, M.; Edwards, R.H. Optical reporters for the conformation of α-synuclein reveal a specific interaction with mitochondria. J. Neurosci. 2008, 28, 12305–12317. [Google Scholar] [PubMed]
- Chandra, S.; Fornai, F.; Kwon, H.B.; Yazdani, U.; Atasoy, D.; Liu, X.; Hammer, R.E.; Battaglia, G.; German, D.C.; Castillo, P.E.; Sudhof, T.C. Double knockout mice for α- and β-synucleins: effect on synaptic functions. Proc. Natl. Acad. Sci. USA 2004, 101, 14966–14971. [Google Scholar]
- Gurevicine, I.; Gurevicius, K.; Tanila, H. Role of α-synuclein in synaptic glutamate release. Neurobiol. Dis. 2007, 28, 83–89. [Google Scholar]
- Liu, S.; Fa, M.; Ninan, I.; Trinchese, F.; Dauer, W.; Aranico, O. α-Synuclein involvement in hippocampla synaptic plasticity: role of NO, cGMP, cGK and CAMKII. Eur. J. Neurosci. 2007, 25, 3583–3596. [Google Scholar] [CrossRef] [PubMed]
- Fortin, D.L.; Nemani, V.M.; Voglmaier, S.M.; Anthony, M.D.; Ryan, T.A.; Edwards, R.H. Neural activity control the synaptic accumulation of α-synuclein. J. Neurosci. 2005, 25, 10913–10921. [Google Scholar] [PubMed]
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudholf, T.C. α-Synuclein cooperates with CSPα in preventing neurodegeneration. Cell 2005, 123, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, G.; Schluter, O.M.; Sudhof, T.C. A molecular pathway of neurodegeneration linking α-synuclein to ApoE and Aβ peptides. Nat. Neurosci. 2008, 11, 301–308. [Google Scholar] [PubMed]
- Serpell, L.C.; Berriman, J.; Jakes, M.; Goedert, M.; Crowther, R.A. Fiber diffraction of synthetic alpha synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. USA 2000, 97, 4897–4902. [Google Scholar]
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrilization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576. [Google Scholar]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibril, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410. [Google Scholar]
- Junn, E.; Mouradian, M.M. Human α-synuclein over-expression increases intracellular reactive oxygen species levels and susceptibility to dopamine. Neurosci. Lett. 2002, 320, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Orth, M.; Wilkinson, J.M.; Taanman, J.W.; Warner, T.T.; Cooper, J.M.; Schapira, A.H. Expression of mutant alpha-synuclein causes increased susceptibility to dopamine toxicity. Hum. Mol. Genet. 2000, 9, 2683–2689. [Google Scholar]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, W.M-Y. Neuronal α-synucleinopathy with severe movement disorder in mice expressing A53T human α-synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.J.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, JQ.; Lee, V.M-Y. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989. [Google Scholar] [PubMed]
- Ischiropoulos, H. Oxidative modification of alpha-synuclein. Ann. N.Y. Acad. Sci. 2003, 991, 93–100. [Google Scholar]
- Wilkinson, K.D.; Lee, K.M.; Deshpande, S.; Duerken-Hughes, P.; Boss, J.M.; Pohl, J. The neuron-specific protein PGP 9.5 is a ubiquitin carboxyl terminal hydrolase. Science 1989, 246, 670–673. [Google Scholar] [PubMed]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar]
- Lansbury, P.T., Jr.; Brice, A. Genetics of Parkinson's disease and biochemical studies of implicated gene products. Curr. Opin. Cell Biol. 2002, 14, 653–660. [Google Scholar]
- McNaught, K.S.; Mytilineou, C.; Jnobaptiste, R.; Yabut, J.; Shahidharan, P.; Jennert, P.; Olanow, C.W. Impairment of the ubiquitin-proteasome system causes dopaminergic cell death and inclusion body formation in ventral mesencephalic cultures. J. Neurochem. 2002, 81, 301–306. [Google Scholar]
- Imai, Y.; Takahashi, R. How do Parkin mutations result in neurodegeneration? Curr. Opin. Neurobiol. 2004, 14, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Hattori, N.; Matsumine, H.; Asakawa, S.; Kitada, T.; Yoshino, H.; Elibol, B; brookes, A.J.; Yamamura, Y.; Kobayashi, T.; Wang, M.; Yoritaka, A.; Minoshima, S.; Shimizu, N.; Mizuno, Y. Point mutations (Thr240Arg and Gln311Stop) in the Parkin gene. Biochem. Biophys. Res. Commun. 1998, 249, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Beilina, A.; Van Der Brug, M.; Ahmad, R.; Kesavapany, S.; Miller, D.W.; Petsko, G.A.; Cookson, M.R. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc. Natl. Acad. Sci. USA 2005, 102, 5703–5708. [Google Scholar]
- Silvestri, L.; Caputo, V.; Bellacchio, E.; Atorino, L.; Dallapiccola, B.; Valente, E.M.; Casari, G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum. Mol. Genet. 2005, 14, 3477–3492. [Google Scholar]
- Unoki, M.; Nakamura, Y. Growth-suppressive effects of BPOZ and RGR2, two genes involved in the PTEN signaling pathway. Oncogene 2001, 20, 4457–4465. [Google Scholar] [CrossRef] [PubMed]
- Taymans, J-M.; Van den Haute, C.; Baekelandt, V. Distribution of PINK1 and LRRK2 in rat and mouse brain. J. Neurochem. 2006, 98, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Weihofen, A.; Thomas, K.J.; Ostazewski, B.L.; Cooksen, M.R.; Selkoe, D.J. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 2009, 48, 2045–2052. [Google Scholar] [PubMed]
- Deng, H.; Jankovic, J.; Guo, Y.; Xie, W.; Le, W. Small interfering RNA targeting the PINK1 induces apoptosis in dopaminergic cells SH-SY5Y. Biochem. Biophys. Res. Commun. 2005, 337, 1133–1138. [Google Scholar]
- Marongiu, R.; Spencer, B.; Crews, L.; Adame, A.; Patrick, C.; Trejo, M.; dallapiccola, B.; Valente, E.M.; Masliah, E. Mutant Pink1 induces mitochondrial dysfunction in a neuronal cell model of Parkinson’s disease by disturbing calcium flux. J. Neurochem. 2009, 108, 1561–1574. [Google Scholar]
- Wilson, M.A.; Collins, J.L.; Hod, Y.; Ringe, D.; Petsko, G.A. The 1.1-A resolution crystal structure of DJ-1, the protein mutated in autosomal recessive early onset Parkinson's disease. Proc. Natl. Acad. Sci. USA 2003, 100, 9256–9261. [Google Scholar]
- Shang, H.; Lang, D.; Jean-Marc, B.; Kaelin-Lang, A. Localization of DJ-1 mRNA in the mouse brain. Neurosci. Lett. 2004, 367, 273–277. [Google Scholar]
- Canet-Aviles, R.M.; Wilson, M.A.; Miller, D.W.; Ahmad, R.; McLendon, C.; Bandyopadhyay, S.; Baptista, M.J.; Ringe, D.; Petsko, G.A.; Cookson, M.R. The Parkinson's disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc. Natl. Acad. Sci. USA 2004, 101, 9103–9108. [Google Scholar]
- Miller, D.W.; Ahmad, R.; Hague, S.; Baptista, M.J.; Canet-Aviles, R.; McLendon, C.; Carter, D.M.; Zhu, P.P.; Stadler, J.; Chandran, J.; Klinefelter, G.R.; Blackstone, C.; Cookson, M.R. L166P mutant DJ-1, causative for recessive Parkinson's disease, is degraded through the ubiquitin-proteasome system. J. Biol. Chem. 2003, 278, 36588–36595. [Google Scholar] [PubMed]
- Takahashi-Niki, K.; Niki, T.; Taira, T.; Iguchi-Ariga, S.M.; Ariga, H. Reduced anti-oxidative stress activities of DJ-1 mutants found in Parkinson's disease patients. Biochem. Biophys. Res. Commun. 2004, 320, 389–397. [Google Scholar]
- Galter, D.; Westerlund, M.; Carmine, A.; Lindqvist, E.; Sydow, O.; Olson, L. LRRK2 expression linked to dopamine-innervated areas. Ann. Neurol. 2006, 59, 714–719. [Google Scholar]
- Melrose, H.; Lincoln, S.; Tyndall, G.; Dickson, D.; Farrer, M. Anatomical localization of leucine-rich repeat kinase 2 mouse brain. Neuroscience 2006, 139, 791–794. [Google Scholar]
- Iaccarino, C.; Crosio, C.; Vitale, C.; Sanna, G.; Carri, M.T.; Barone, P. Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum. Mol. Gen. 2007, 16, 1319–1326. [Google Scholar]
- Ho, C. C-Y.; Rideout, H.J.; Ribe, E.; Troy, C.M.; Dauer, W.T. The Parkinson’s disease protein leucine-rich repeat kinase 2 transduces death signals via Fas-associated protein with death domain and caspase-8 in a cellular model of neurodegeneration. J. Neurosci. 2009, 29, 1011–1016. [Google Scholar] [PubMed]
- Tatton, N.A.; MaClean-Fraser, A.; Tatton, W.G.; Perl, D.P.; Olnanow, C.W. A fluorescent double-labeling method to detect and confirm apoptotic nuclei in Parkinson’s disease. Ann. Neurol. 1998, 44, S142–S148. [Google Scholar]
- Jellinger, K.A. Is there apoptosis in Lewy body disease? Acta Neuropathol. 1999, 97, 413–415. [Google Scholar] [CrossRef] [PubMed]
- Wüllner, U.; Kornhuber, J.; Weller, M.; Schulz, J.B.; Loschmann, P.A.; Riederer, P.; Klockgether, T. Cell death and apoptosis regulating proteins in Parkinson’s disease- a cautionary note. Acta Neuropathol. 1999, 97, 408–412. [Google Scholar]
- Anglade, P.; Vyas, S.; Javoy-Agid, F.; Herrero, M.T.; Michel, P.P.; Marguez, J.; Mouatt-Prigent, A.; Ruberg, M.; Hirsch, E.C.; Agid, Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol. 1997, 12, 25–31. [Google Scholar]
- Martin, L.J.; Liu, Z.; Troncoso, J.C.; Price, D.L. Neuronal cell death in human neurodegenerative diseases and their animal/cell models. In Apoptosis in Health and Disease; Holcik, M., LaCasse, E., Korneluk, R., MacKenzie, A., Eds.; Cambridge University Press: Cambridge, UK, 2005; pp. 242–315. [Google Scholar]
- Tompkins, M.M.; Basgall, E.J.; Zamrini, E.; Hill, W.D. Apoptotic-like changes in Lewy-body-associated disorders and normal aging in substantia nigra neurons. Am. J. Pathol. 1997, 150, 119–131. [Google Scholar]
- Ginsberg, S.D.; Hemby, S.E.; Mufson, E.J.; Martin, L.J. Cell and tissue microdissection in combination with genomic and proteomic profiling. In Neuroanatomical Tract-Tracing 3, Molecules, Neurons, and Systems; Zaborszky, L., Wouterlood, F.G., Lanciego, J.L., Eds.; Springer: New York, NY, USA, 2006; pp. 109–141. [Google Scholar]
- Martin, L.J; Liu, Z.; Chen, K.; Price, A.C.; Pan, Y.; Swaby, J.A.; Golden, W.C. Motor neuron degeneration in amyotrophic lateral sclerosis mutant superoxide dismutase-1 transgenic mice: mechanisms of mitochondriopathy and cell death. J. Comp. Neurol. 2007, 500, 20–46. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Fleming, S.M.; Palacino, J.J.; Capedam, C.; Lam, H.A.; Bhatnagar, A.; Meloni, E.G.; Wu, N.; Ackerson, L.C.; Klapstein, G.J.; Gajendiram, M.; Roth, B.L.; Chesselet, M.F.; Maidment, N.T.; Levine, M.S.; Shen, J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J. Biol. Chem. 2003, 278, 43628–43635. [Google Scholar] [PubMed]
- Perez, F.A.; Palmiter, R.D. Parkin-deficient mice are not a robust model of parkinsonism. Proc. Natl. Acad. Sci. USA 2005, 102, 2174–2179. [Google Scholar]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar]
- Lu, X-H.; Fleming, S.M.; Meurers, B.; Ackerson, L.C.; Mortazavi, F.; Lo, V.; Hernandez, D,; Sulzer, D.; Jackson, G.R.; Maidment, N.T.; Chesselet, M-F.; Yang, X.W. Bacterial artificial chromosome transgenic mice expressing a truncated mutant Parkin exhibit age-dependent hypokinetic motor deficits, dopaminergic neuron degeneration, and accumulation of proteinase K-resistant α-synuclein. J. Neurosci. 2009, 29, 1962–1976. [Google Scholar] [PubMed]
- Chen, L.; Cagniard, B.; Mathews, T.; Jones, S.; Koh, H.C.; Ding, Y.; Carvey, P.M.; Ling, Z.; Kang, U.J.; Zhuang, X. Age-dependent motor deficits and dopaminergic dysfunction in DJ-1 null mice. J. Biol. Chem. 2005, 280, 21418–21426. [Google Scholar]
- Goldberg, M.S.; Pisani, A.; Haburcak, M.; Vortherms, T.A.; Kitada, Y.; Costa, C.; Tong, Y.; Martella, G.; Tscherter, A.; Martins, A.; Bernardi, G.; Roth, B.L.; Pothos, E.N.; Calabresi, P.; Shen, J. Nigrostriatal dopaminergic deficits and hypokinesia caused by inactivation of the familial parkinsonism-linked gene DJ-1. Neuron 2005, 45, 489–496. [Google Scholar]
- Gispert, S.; Del Turco, D.; Garrett, L.; Chen, A.; Bernard, D.J.; Hamm-Clement, J.; Korf, H.W.; Deller, T.; Braak, H.; Auburger, G.; Nussbaum, R.L. Transgenic mice expressing mutant A53T human alpha-synuclein show neuronal dysfunction in the absence or aggregate formation. Mol. Cell. Neurosci. 2003, 24, 419–429. [Google Scholar]
- Kahle, P.J.; Neumann, M.; Ozmen, L.; Muller, V.; Jacobsen, H.; Schindzielorz, A.; Okochi, M.; Leimer, U.; van der Putten, H.; Probst, A.; Kremmer, E.; Kretzschmar, H.A.; Haassm, C. Subcellular localization of wild-type and Parkinson’s disease-associated mutant alpha-synuclein in human and transgenic mouse brain. J. Neurosci. 2000, 20, 6365–6373. [Google Scholar] [PubMed]
- Lee, M.K.; Stirling, W.; Xu, Y.; Xu, X.; Qui, D.; Mandir, A.S.; Dawson, T.M.; Copeland, N.G.; Jenkins, N.A.; Price, D.L. Human α-synuclein-harboring familial Parkinson’s disease-linked Ala-53 → Thr mutation causes neurodegenerative disease with α-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8968–8973. [Google Scholar]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Mallory, M.; Hashimoto, M.; Takeda, A.; Sagara, Y.; Sisk, A.; Mucke, L. Dopaminergic loss and inclusion body formation in α-synuclein mice: implications for neurodegenerative disorders. Science 2000, 287, 1265–1269. [Google Scholar]
- Richfield, E.K.; Thiruchelvam, M.J.; Cory-Slechta, D.A.; Wuetzer, C.; Gainetdinov, R.R.; Caron, M.G.; Di Monte, D.A.; Federoff, H.J. Behavioral and neurochemical effects of wild-type and mutated human alpha-synuclein in transgenic mice. Exp. Neurol. 2002, 175, 35–48. [Google Scholar]
- van der Putten, H.; Wiederhold, K-H.; Probst, A.; Barbieri, S.; Mistl, C.; Danner, S.; Kauffmann, S.; Hofele, K.; Spooren, W.P.; Ruegg, M.A.; Lin, S.; Caroni, P.; Sommer, B.; Tolnay, M.; Bilbe, G. Neuropathology in mice expressing human α-synuclein. J. Neurosci. 2000, 20, 6021–6029. [Google Scholar] [PubMed]
- Wakamatsu, M.; Ishii, A.; Iwata, S.; Sakagami, J.; Ukai, Y.; Ono, M.; Kanbe, D.; Muramatsu, S-i.; Kabayashi, K.; Iwatsubo, T.; Yoshimoto, M. Selective loss of nigral dopamine neurons induced by overexpression of truncated human α-synuclein. Neurobiol. Aging 2008, 29, 547–585. [Google Scholar]
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50. [Google Scholar]
- Lieberman, A.R. The axon reaction: a review of the principal features of perikaryal responses to axon injury. Int. Rev. Neurobiol. 1971, 14, 49–124. [Google Scholar]
- Poon, H.F.; Frasier, M.; Shreve, N.; Calabrese, V.; Wolozin, B.; Butterfield, D.A. Mitochondrial associated metabolic proteins are selectively oxidized in A30P α-synuclein transgenic mice- a model of familial Parkinson’s disease. Neurobiol. Dis. 2005, 18, 492–498. [Google Scholar]
- Turnbull, S.; Tabner, B.J.; El-Agnaf, O.M.; Moore, S.; Davies, Y.; Allsop, D. Alpha-synuclein implicated in Parkinson’s disease catalyses the formation of hydrogen peroxide in vitro. Free Radic. Biol. Med. 2001, 30, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Tabrizi, S.J.; Orth, M.; Wilkinson, J.M.; Taanman, J.W.; Warner, T.T.; Cooper, J.M.; Schapira, A.H. Expression of mutant alpha-synuclein causes increased susceptibility to dopamine toxicity. Hum. Mol. Genet. 2000, 9, 2683–2689. [Google Scholar]
- Paxinou, E.; Chen, Q.; Weisse, M.; Giasson, B.I.; Norris, E.H.; Rueter, S.M.; Trojanowski, J.Q.; Lee, VM-Y.; Ischiropoulos, H. Induction of alpha-synuclein aggregation by intracellular nitrative insult. J. Neurosci. 2001, 21, 8053–8061. [Google Scholar] [PubMed]
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, VM-Y.; Ischiropoulos, H. Dityrosine cross-linking promotes formation of stable α-synuclein polymers. J. Biol. Chem. 2000, 365, 18344–18349. [Google Scholar]
- Chen, K.; Northington, F.J.; Martin, L.J. Inducible nitric oxide synthase is present in motor neuron mitochondria and Schwann cells and contributes to disease mechanisms in ALS mice. Brain Struct. Funct. 2010, in press.. [Google Scholar]
- Rowland, L.P.; Shneider, N.A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar]
- Sathasivam, S.; Ince, P.G.; Shaw, P.J. Apoptosis in amyotrophic lateral sclerosis: a review of the evidence. Neuropathol. Appl. Neurobiol. 2001, 27, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Stephens, B.; Guiloff, R.J.; Navarrete, R.; Newman, P.; Nikhar, N.; Lewis, P. Widespread loss of neuronal populations in spinal ventral horn in sporadic motor neuron disease. A morphometric study. J. Neurol. Sci. 2006, 244, 41–58. [Google Scholar] [CrossRef] [PubMed]
- Maekawa, S.; Al-Sarraj, S.; Kibble, M.; Landau, S.; Parnavelas, J.; Cotter, D.; Everall, I.; Leigh, P.N. Cortical selective vulnerability in motor neurons disease: a morphometric study. Brain 2004, 127, 1237–1251. [Google Scholar]
- Kabashi, E.; Valdmains, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; Pradat, P-F.; Camu, W.; Meininger, V.; Dupre, N.; Rouleau, G.A. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [PubMed]
- Schymick, J.C.; Talbot, K.; Traynor, G.J. Genetics of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2007, 16, R233–R242. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.Y.; Lander, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; Figlewicz, D.; Brown, R.H.; Meisler, M.H. Deleterious variants of FIG4, a phosphoinositade phosphatase, in patients with ALS. Am. J. Human Gen. 2009, 84, 85–88. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Ultrastructural changes of synapses of Betz cell in patients with amyotrophic lateral sclerosis. Neurosci. Lett. 1999, 268, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Ince, P.G.; Shaw, P.J. Mitochondrial involvement in amyotrophic lateral sclerosis. Neurochem. Intl. 2002, 40, 543–551. [Google Scholar] [CrossRef]
- Comi, G.P.; Bordoni, A.; Salani, S.; Franeschina, L.; Sciacco, M.; Prelle, A.; Fortunato, F.; Zeviani, M.; Napoli, L.; Bresolin, N.; Moggio, M.; Ausenda, C.D.; Taanman, J.W.; Scarlato, G. Cytochrome c oxidase subunit I microdeletion in a paitent with motor neuron disease. Ann. Neurol. 1998, 43, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Borthwick, G.M.; Taylo, R.W.; Walls, T.J.; Tonska, K.; Taylor, G.A.; Shaw, P.J.; Ince, P.G.; Turnbull, D.M. Motor neuron disease in a patient with a mitochondrial tRNAIle mutation. Ann. Neurol. 2006, 59, 570–574. [Google Scholar]
- Soong, N.W.; Hinton, D.R.; Cortopassi, G.; Arnheim, N. Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat. Genet. 1992, 2, 318–323. [Google Scholar]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar]
- Mawrin, C.; Kirches, E.; Krause, G.; Wiedemann, F.R.; Vorwerk, C.K.; Bogerts, B.; Schildhaus, H.U.; Dietzmann, K.; Schneider-Stock, R. Single-cell analysis of mtDNA levels in sporadic amyotrophic lateral sclerosis. NeuroReport 2004, 15, 939–943. [Google Scholar]
- Corti, S.; Donadonu, C.; Ronchi, D.; Bordoni, A.; Fortunato, F.; Santoro, D.; Del Bo, R.; Lucchini, V.; Crugnola, V.; Papadimitriou, D.; Salani, S.; Moggio, M.; Bresolin, N.; Comi, G.P. Amyotrophic lateral sclerosis linked to a novel SOD1 mutation with muscle mitochondrial dysfunction. J. Neurol. Sci. 2009, 276, 170–174. [Google Scholar]
- Babcock, D.; Hille, B. Mitochondrial oversight of cellular Ca2+ signaling. Curr. Opin. Neurobiol. 1998, 8, 398–404. [Google Scholar]
- Siklos, L.; Engelhardt, J.; Harat, Y.; Smith, R.G.; Joo, F.; Appel, S.H. Ultrastructural evidence for altered calcium in motor nerve terminals in amyotrophic lateral sclerosis. Ann. Neurol. 1996, 39, 203–216. [Google Scholar]
- Choi, D.W. Cellular defences destroyed. Nature 2005, 433, 696–698. [Google Scholar]
- Brown, M.R.; Sullivan, P.G.; Geddes, J.W. Synaptic mitochondria are more susceptible to Ca2+ overload than nonsynaptic mitochondria. J. Biol. Chem. 2006, 281, 11658–11668. [Google Scholar]
- Rothstein, J.D.; Martin, L.J.; Kuncl, R.W. Decreased glutamate transport by brain and spinal cord in amyotrophic lateral sclerosis. N. Engl. J. Med. 1992, 326, 1464–1468. [Google Scholar]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar]
- Heath, P.R.; Tomkins, J.; Ince, P.G.; Shaw, P.J. Quantitative assessment of AMPA receptor mRNA in human spinal motor neurons isolated by laser capture microdissection. NeuroReport 2002, 13, 1753–1757. [Google Scholar]
- Kwak, S.; Kawahara, Y. Deficient RNA editing of GluR2 and neuronal death in amyotrophic lateral sclerosis. J. Mol. Med. 2005, 83, 110–120. [Google Scholar]
- Chang, D.T.W.; Reynolds, I.J. Mitochondrial trafficking and morphology in healthy and injured neurons. Prog. Brain Res. 2006, 80, 241–268. [Google Scholar]
- Hansson, M.J.; Mansson, R.; Morota, S.; Uchino, H.; Kallur, T.; Sumi, T.; Ishii, N.; Shimazu, M.; Keep, M.F.; Jegorov, A.; Elmer, E. Calcium-induced generation of reactive oxygen species in brain mitochondria is mediated by permeability transition. Free Radic. Biol. Med. 2008, 45, 284–294. [Google Scholar]
- Bergmann, F.; Keller, B.U. Impact of mitochondrial inhibition on excitability and cytosolic Ca2+ levels in brainstem motoneurones. J. Physiol. 2004, 555, 45–59. [Google Scholar]
- Beal, M.F. Oxidatively modified protein in aging and disease. Free Radic. Biol. Med. 2002, 32, 797–803. [Google Scholar]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar]
- Abe, K.; Pan, L-H.; Watanabe, M.; Kato, T.; Itoyama, Y. Induction of nitrotyrosine-like immunoreactivity in the lower motor neuron of amyotrophic lateral sclerosis. Neurosci. Lett. 1995, 199, 152–154. [Google Scholar] [PubMed]
- Beal, M.F.; Ferrante, R.J.; Browne, S.E.; Matthews, R.T.; Kowall, N.W.; Brown, R.H., Jr. Increased 3-nitrotyrosine in both sporadic and familial amyotrophic lateral sclerosis. Ann. Neurol. 1997, 42, 644–654. [Google Scholar]
- Sasaki, S.; Warita, H.; Abe, K.; Iwata, M. Inducible nitric oxide synthase (iNOS) and nitrotyrosine immunoreactivity in the spinal cords of transgenic mice with mutant SOD1 gene. J. Neuropathol. Exp. Neurol. 2001, 60, 839–846. [Google Scholar]
- Browne, S.E.; Bowling, A.C.; Baik, M.J.; Gurney, M.; Brown, R.H., Jr; Beal, M.F. Metabolic dysfunction in familial, but not sporadic, amyotrophic lateral sclerosis. J. Neurochem. 1998, 71, 281–287. [Google Scholar] [PubMed]
- Borthwick, G.M.; Johnson, M.A.; Ince, P.G.; Shaw, P.J.; Turnbul, D.M. Mitochondrial enzyme activity in amyotrophic lateral sclerosis: implications for the role of mitochondria in neuronal cell death. Ann. Neurol. 1999, 46, 787–790. [Google Scholar]
- Vielhaber, S.; Kunz, D.; Winkler, K.; Wiedemann, F.R.; Kirches, E.; Feistner, H.; Heinze, H.J.; Elger, C.E.; Schubert, W.; Kunz, W.S. Mitochondrial DNA abnormalities in skeletal muscle of patients with sporadic amyotrophic lateral sclerosis. Brain 2000, 123, 1339–1348. [Google Scholar]
- Soraru, G.; Vergani, L.; Fedrizzi, L.; D’Ascenzo, C.; Polo, A.; Bernazzi, B.; Angelini, C. Activities of mitochondrial complexes correlate with nNOS amount in muscle from ALS patients. Neuropath. Appl. Neurobiol. 2007, 33, 204–211. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Zoll, J.; Ponsot, E.; N’Guessan, B.; Tranchant, C.; Loeffler, J-P.; Lampert, E. Muscular mitochondrial function in amyotrophic lateral sclerosis is progressively altered as the disease develops: a temporal study in man. Exp. Neurol. 2006, 198, 25–30. [Google Scholar] [PubMed]
- Martin, L.J.; Liu, Z. Opportunities for neuroprotection in ALS using cell death mechanism rationales. Drug Discov. Today 2004, 1, 135–143. [Google Scholar] [CrossRef]
- Martin, L.J. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol. Dis. 2000, 7, 613–622. [Google Scholar]
- Yamazaki, M.; Esumi, E.; Nakani, I. Is motoneuronal death in amyotrophic lateral sclerosis apoptosis? Neuropathology 2005, 25, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.J.; Liu, Z. Injury-induced spinal motor neuron apoptosis is preceded by DNA single-strand breaks and is p53- and Bax-dependent. J. Neurobiol. 2002, 50, 181–197. [Google Scholar]
- Fornai, F.; Longone, P.; Ferrucci, M.; Lenzi, P.; Isidoro, C.; Ruggieri, S.; Paparelli, A. Autophagy and amyotrophic lateral sclerosis. Autophagy 2008, 4, 527–530. [Google Scholar]
- Turner, B.J.; Talbot, K. Transgenics, toxicity and therapeutics in rodent models of mutant SOD1-mediated familial ALS. Prog. Neurobiol. 2008, 85, 94–134. [Google Scholar]
- McCord, J.M.; Fridovich, I. Superoxide dismutase, an enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar]
- Rakhit, R.; Crow, J.P.; Lepock, J.R.; Kondejewski, L.H.; Cashman, N.R.; Chakrabartty, A. Monomeric Cu, Zn-superoxide dismutase is a common misfolding intermediate in the oxidation models of sporadic and familial amyotrophic sclerosis. J. Biol. Chem. 2004, 279, 15499–15504. [Google Scholar]
- Ferri, A.; Cozzolino, M.; Crosio, C.; Nencini, M.; Casciati, A.; Gralla, E.B.; Rotilio, G.; Valentine, J.S.; Carri, M.T. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc. Natl. Acad. Sci. USA 2006, 103, 13860–13865. [Google Scholar] [Green Version]
- Estévez, A.G.; Crow, J.P.; Sampson, J.B.; Reiter, C.; Zhuang, Y.; Richardson, G.J.; Tarpey, L. ; Barbeito, M.M.; Beckman, J.S. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science 1999, 286, 2498–2500. [Google Scholar] [PubMed]
- Flanagan, S.W.; Anderson, R.D.; Ross, M.A.; Oberley, L.W. Overexpression of manganese superoxide dismutase attenuates neuronal death in human cells expressing mutant (G37R) Cu/Zn-superoxide dismutase. J. Neurochem. 2002, 81, 170–177. [Google Scholar]
- Bilsland, L.G.; Nirmalananthan, N.; Yip, J.; Greensmith, L.; Duhcen, M.R. Expression of mutant SOD1G93A in astrocytes induces functional deficits in motoneuron mitochondria. J. Neurochem. 2008, 107, 1271–1283. [Google Scholar]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; Chen, W.; Zhai, P.; Sufit, R.L.; Siddique, T. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar]
- Dal Canto, M.C.; Gurney, M.E. Development of central nervous system pathology in a murine transgenic model of human amyotrophic lateral sclerosis. Am. J. Pathol. 1994, 145, 1271–1279. [Google Scholar]
- Chang, Q.; Martin, L.J. Glycinergic innervation of motoneurons is deficient in amyotrophic lateral sclerosis mice: a confocal quantitative analysis. Am. J. Path. 2009, 174, 574–585. [Google Scholar]
- Bendotti, C.; Calvaresi, N.; Chiveri, L.; Prelle, A.; Moggio, M.; Braga, M.; Silani, V.; De Biasi, S. Early vacuolization and mitochondrial damage in motor neurons of FALS mice are not associated with apoptosis or with changes in cytochrome oxidase histochemical reactivity. J. Neurol. Sci. 2001, 191, 25–33. [Google Scholar]
- Wong, P.C.; Pardo, C.A.; Borchelt, D.R.; Lee, M.K.; Copeland, N.G.; Jenkins, N.A.; Sisodia, S.S.; Cleveland, D.W.; Price, D.L. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 1995, 14, 1105–1116. [Google Scholar]
- Kong, J.; Xu, Z. Massive mitochondrial degeneration in motor neurons triggers the onset of amyotrophic lateral sclerosis in mice expressing a mutant SOD1. J. Neurosci. 1998, 18, 3241–3250. [Google Scholar] [PubMed]
- Jaarsma, D.; Rognoni, F.; van Duijn, W.; Verspaget, H.W.; Haasdijk, E.D.; Holstege, J.C. CuZn superoxide dismutase (SOD1) accumulates in vacuolated mitochondria in transgenic mice expressing amyotrophic lateral sclerosis-linked SOD1 mutations. Acta Neuropathol. 2001, 102, 293–305. [Google Scholar]
- Sasaki, S.; Warita, H.; Murakami, T.; Abe, K.; Iwata, M. Ultrastructural study of mitochondria in the spinal cord of transgenic mice with a G93A mutant SOD1 gene. Acta Neuropathol. 2004, 107, 461–474. [Google Scholar]
- Deng, H-X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W-Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; Warner, C.; Deng, G.; Soriano, E.; Smyth, C.; Parge, H.E.; Ahmed, A.; Roses, A.D.; Hallewell, R.A.; Pericak-Vance, M.A.; Siddique, T. Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [PubMed]
- Borchelt, D.R.; Lee, M.K.; Slunt, H.H.; Guarnieri, M.; Xu, Z-S.; Wong, P.C.; Brown, R.H., Jr.; Price, D.L.; Sisodia, S.S.; Cleveland, D.W. Superoxide dismutase 1 with mutations linked to familial amyotrophic lateral sclerosis possesses significant activity. Proc. Natl. Acad. Sci. USA 1994, 91, 8292–8296. [Google Scholar]
- Yim, M.B.; Kang, J-H.; Yim, H-S.; Kwak, H-S.; Chock, P.B.; Stadtman, E.R. A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: an enhancement of free radical formation due to a decrease in Km for hydrogen peroxide. Proc. Natl. Acad. Sci. USA 1996, 93, 5709–5714. [Google Scholar]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Rouleau, G.A. Oxidized/misfolded superoxide dismutase-1: the cause of all amyotrophic lateral sclerosis? Ann. Neurol. 2007, 62, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Ezzi, S.A.; Urushitani, M.; Julien, J-P. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J. Neurochem. 2007, 102, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I.; Fridovich, I. Mutant Cu,Zn superoxide dismutases and familial amyotrophic lateral sclerosis: evaluation of oxidative hypotheses. Free Radic. Biol. Med. 2003, 34, 1383–1389. [Google Scholar]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar]
- Andrus, P.K.; Fleck, T.J.; Gurney, M.E.; Hall, E.D. Protein oxidative damage in a transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 1998, 71, 2041–2048. [Google Scholar]
- Poon, H.F.; Hensley, K.; Thongboonkerd, V.; Merchant, M.L.; Lynn, B.C.; Pierce, W.M.; Klein, J.B.; Calabrese, V.; Butterfield, D.A. Redox proteomics analysis of oxidatively modified proteins in G93A-SOD1 transgenic mice- a model of familial amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2005, 39, 435–462. [Google Scholar]
- Higgins, C.M.J.; Jung, C.; Ding, H.; Xu, Z. Mutant Cu, Zn Superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS. J. Neurosci. 2002, 22, RC215:1–RC215:6. [Google Scholar]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide (SOD) in rat liver. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar]
- Liu, J.; Lillo, C.; Jonsson, A.; Vande Velde, C.; Ward, C.W.; Miller, T.M.; Subramaniam, J.R.; Rothstein, J.D.; Marklund, S.; Anderson, P.M.; Brannstrom, T.M.; Gredal, O.; Wong, P.C.; Williams, D.S.; Cleveland, D.W. Toxicity of familial ALS-linked SOD1 mutants from selective recruitment to spinal mitochondria. Neuron 2004, 43, 5–15. [Google Scholar]
- Pasinelli, P.; Belford, M.E.; Lennon, N.; Bacskai, B.J.; Hyman, B.T.; Trotti, D.; Brown, R.H., Jr. Amyotrophic lateral sclerosis-associated SOD1 mutant protein bind and aggregate with Bcl-2 in spinal cord mitochondria. Neuron 2004, 43, 19–30. [Google Scholar]
- Vande Velde, C.; Miller, T.M.; Cashman, N.R.; Cleveland, D.W. Selective association of misfolded ALS-linked mutant SOD1 with the cytoplasmic face of mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 4022–4027. [Google Scholar]
- Goldsteins, G.; Keksa-Goldsteine, V.; Ahtiniemi, T.; Jaronen, M.; Arens, E.; Akerman, K.; Chan, R.H.; Koistinaho, J. Deleterious role of superoxide dismutase in the mitochondrial intermembrane space. J. Biol. Chem. 2008, 283, 8446–8452. [Google Scholar]
- Higgins, C.M.; Jung, C.; Xu, Z. ALS-associated mutant SOD1G93A causes mitochondrial vacuolation by expansion of the intermembrane space and by involvement of SOD1 aggregation and peroxisomes. BMC Neurosci. 2003, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K-F.; Browlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; Shaw, C.E.; Leigh, P.N.; Miller, C.C. J.; Grierson, A.J. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondrial content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [PubMed]
- Siklos, L.; Engelhardt, J.I.; Alexianu, M.E.; Gurney, M.E.; Siddique, T.; Appel, S.H. Intracellular calcium parallels motoneuron degeneration in SOD-1 mutant mice. J. Neuropath. Exp. Neurol. 1998, 57, 571–587. [Google Scholar]
- Jaiswal, M.K.; Keller, B.U. Cu/Zn superoxide dismutase typical for familial amyotrophic lateral sclerosis increases the vulnerability of mitochondria and perturbs Ca2+ homeostasis in SOD1G93A mice. Mol. Pharmacol. 2009, 75, 478–489. [Google Scholar]
- Nguyen, K.T.; Garcia-Chacon, L.E.; Barrett, J.N.; Barrett, E.F.; David, G. The ψm depolarization that accompanies mitochondrial Ca2+ uptake is greater in mutant SOD1 than in wild-type mouse motor terminals. Proc. Natl. Acad. Sci. USA 2009, 106, 2007–2011. [Google Scholar]
- Sasaki, S.; Shibata, N.; Komori, T.; Iwata, M. iNOS and nitrotyrosine immunoreactivity in amyotrophic lateral sclerosis. Neurosci. Lett. 2000, 291, 44–48. [Google Scholar]
- Kunz, W.S. Different metabolic properties of mitochondrial oxidative phosphorylation in different cell types- important implications for mitochondrial cytopathies. Exp. Physiol. 2003, 88.1, 149–154. [Google Scholar] [CrossRef]
- Keep, M.; Elmér, E.; Fong, K.S.K.; Csiszar, K. Intrathecal cyclosporin prolongs survival of late-stage ALS mice. Brain Res. 2001, 894, 327–331. [Google Scholar]
- Karlsson, J.; Fong, K.S.; Hansson, M.J.; Elmer, E.; Csiszar, K.; Keep, M.F. Life span extension and reduced neuronal death after weekly intraventricular cyclosporine injections in the G93A transgenic mouse model of amyotrophic lateral sclerosis. J. Neurosurg. 2004, 101, 128–137. [Google Scholar]
- Kirkinezos, I.G.; Hernandez, D.; Bradley, W.G.; Moraes, C.T. An ALS mouse model with a permeable blood-brain barrier benefits from systemic cyclosporine A treatment. J. Neurochem. 2004, 88, 821–826. [Google Scholar]
- Bordet, T.; Buisson, B.; Michaud, M.; Drouot, C.; Galea, P.; Delaage, P.; Akentieva, N.P.; Evers, A.S.; Covey, D.F.; Ostuni, M.A.; lacapere, JJ-J.; Massaad, C.; Schmacher, M.; Steidl, E-M.; Maux, D.; Delaage, M.; Henderson, C.E.; Pruss, R.M. Identification and characterization of Cholest-4-en-3-one, oxime (TRO19622), a novel drug candidate for amyotrophic lateral sclerosis. J. Pharmacol. Exp. Ther. 2007, 322, 709–720. [Google Scholar] [PubMed]
- Mills, C.; Makwana, M.; Wallace, A.; Benn, S.; Schmidt, H.; Tegeder, I.; Costigan, M.; Brown, R.H., Jr; Raivich, G.; Woolf, C. Ro5-4864 promotes neonatal motor neuron survival and nerve regeneration in adult rats. Eur. J. Neurosci. 2008, 27, 937–946. [Google Scholar]
- Yan, L-J.; Sohal, R.S. Mitochondrial adenine nucleotide translocase is modified oxidatively during aging. Proc. Natl. Acad. Sci. USA 1998, 95, 12896–12901. [Google Scholar]
- Prokai, L.; Yan, L-J; Vera-Serrano, J.L.; Stevens, S.M., Jr; Forster, M.J. Mass spectrometry-based survey of age-associated protein carbonylation in rat brain mitochondria. J. Mass Spectrom. 2007, 42, 1583–1589. [Google Scholar] [CrossRef] [PubMed]
- Vieira, H.L.A.; Belzacq, A-S.; Haouzu, D.; Bernassola, F.; Cohen, I.; Jacotot, E.; Ferri, K.F.; Hamel, C.E.; Bartle, L.M.; Melino, G.; Brenner, C.; Goldmacher, V.; Kroemer, G. The adenine nucleotide translocator: a target of nitric oxide, peroxynitrite, and 4-hydroxynonenal. Oncogene 2001, 20, 4305–4316. [Google Scholar] [PubMed]
- McStay, G.P.; Clarke, S.J.; Halestrap, A.P. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem. J. 2002, 367, 541–548. [Google Scholar]
- Costantini, P.; Belzacq, A-S.; Vieira, H.L.A.; Larochette, N.; de Pablo, M.A.; Zamzami, N.; Susin, S.A.; Brenner, C.; Kroemer, G. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene 2000, 19, 307–314. [Google Scholar] [PubMed]
- García, N.; Martínez-Abundis, E.; Pavón, N.; Correa, F.; Chávez, E. Copper induces permeability transition through its interaction with the adenine nucleotide translocase. Cell Biol. Int. 2007, 31, 893–899. [Google Scholar]
- Grimm, S.; Brdiczka, D. The permeability transition pore in cell death. Apoptosis 2007, 12, 841–855. [Google Scholar]
- Forte, M.; Gold, B.G.; Marracci, G.; Chaudhary, P.; Basso, E.; Johnsen, D.; Yu, Z.; Fowlkes, J.; Rahder, M.; Stern, K.; Bernardi, P.; Bourdette, D. Cyclophilin D inactivation protects axons in experimental autoimmune encephalomyelitis, an animal model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2007, 104, 7558–7563. [Google Scholar]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hertz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Martin, L.J. Mitochondrial and Cell Death Mechanisms in Neurodegenerative Diseases. Pharmaceuticals 2010, 3, 839-915. https://doi.org/10.3390/ph3040839
AMA Style
Martin LJ. Mitochondrial and Cell Death Mechanisms in Neurodegenerative Diseases. Pharmaceuticals. 2010; 3(4):839-915. https://doi.org/10.3390/ph3040839
Chicago/Turabian StyleMartin, Lee J. 2010. "Mitochondrial and Cell Death Mechanisms in Neurodegenerative Diseases" Pharmaceuticals 3, no. 4: 839-915. https://doi.org/10.3390/ph3040839